

Abstract
The inhibitor of differentiation (ID) proteins are helix-loop-helix transcriptional repressors with established roles in stem cell self-renewal, lineage commitment, and niche interactions. While deregulated expression of ID proteins in cancer was identified more than a decade ago, emerging evidence has revealed a central role for ID proteins in neoplastic progression of multiple tumor types that often mirrors their function in physiological stem and progenitor cells. ID proteins are required for the maintenance of cancer stem cells, self-renewal, and proliferation in a range of malignancies. Furthermore, ID proteins promote metastatic dissemination through their role in remodeling the tumor microenvironment and by promoting tumor-associated endothelial progenitor cell proliferation and mobilization. Here, we discuss the latest findings in this area and the clinical opportunities that they provide.
The inhibitor of differentiation (ID) family of proteins are critical regulators in a wide range of developmental and cellular processes. They regulate stem cell homeostasis and fate commitment in various cell types, including neuronal,1 hematopoietic,2,3 and embryonic4 cells, where they function by both inhibiting cell-autonomous differentiation programs4 and by coordinating the cell's interaction with the extracellular milieu and its niche.5,6 In this review, we will focus on recent discoveries demonstrating that these functions of IDs are retained by many cancers to promote proliferation and self-renewal and to facilitate signaling from the tumor microenvironment.
The four members of the vertebrate ID family (ID1, ID2, ID3, and ID4) belong to the basic helix-loop-helix (bHLH) family of transcription factors. All four members share the highly conserved bHLH region and have similar molecular weights of between 13–20 kDa.7,8 Outside the HLH domain, there are extensive sequence differences among the four members of the ID proteins. Different members of the ID proteins are expressed in distinct expression patterns in a tissue-specific and stage-dependent manner, hence controlling different cellular and physiological processes.9,10,11 The bHLH transcription factors are key regulators of lineage- and tissue-specific gene expression and act as obligate dimers binding DNA through composite basic domains to regulate the transcription of target genes containing E-boxes (CANNTG) in their promoters. ID proteins dimerize with bHLH proteins, but because ID proteins lack a basic DNA-binding domain, ID-bHLH heterodimers fail to bind DNA, thereby inhibiting the transcriptional activity of the bHLH proteins. As such, ID proteins are dominant negative regulators of bHLH function.12 ID proteins interact with the ubiquitously expressed E protein transcription factors (E12, E47, E2-2, and HEB) which can act as homodimers (in B cells) or as heterodimers with tissue-restricted bHLH proteins such as MyoD (muscle) and NeuroD (nerve). A number of reports demonstrate noncanonical functions for ID proteins, including binding to non-HLH transcription factors such as Rb-family pocket proteins,13 Ets factors,14 or RNA15 although the broader significance of these findings to ID protein biology is yet to be explored. The biochemical mechanisms of ID protein activity remain largely unelucidated and comprise an area of intensive investigation.
Deregulation of IDs in Human Cancer
ID family members exhibit unique spatio-temporal patterns of tissue expression during development16 and malignancy,17 although evidence suggests biochemical redundancy in vitro.8 The mechanisms governing ID protein expression are complex—an extensive body of literature shows that ID gene transcription is exquisitely sensitive to signals from the extracellular environment, including transforming growth factor-β (TGF-β),18,19 steroid hormones,20 receptor tyrosine kinases,21,22 and oncoproteins23 (Figure 1). The stability of IDs is also tightly controlled by the APC/Cdh1 E3 ubiquitin ligase complex,24 resulting in short half-lives for ID proteins in most tissues. In certain physiological and malignant stem cell populations, ID proteins are stabilized by the ubiquitin-specific peptidase 1 deubiquitinase which counters ubiquitin-mediated ID destruction.25 Ubiquitin-specific peptidase 1 is overexpressed in a subset of primary osteosarcomas, where it stabilizes ID1, ID2, and ID3, leading to repression of p21 and the osteogenic differentiation program.25
Figure 1.

Regulation of inhibitor of differentiation (ID) expression and their function in cancer biology. (a) ID proteins are sensitive to a diverse array of extracellular signals, including steroid hormones, growth factors, and members of the TGF-β superfamily. ID proteins are also downstream of well-established oncogenic pathways such as RAS-Egr1, MYC, and Src-PI3k as well as tumor suppressors RB p53 and KLF17. (b) ID proteins regulate cellular pathways that are essential to the development and progression of cancer. IDs regulate self-renewal and cell-cycle through a number of known stem and proliferation factors such as Notch, Sox2/4, LIF, cyclin genes and the CDK inhibitors p21waf1 and p16INK4A. In addition, IDs remodel the tumor microenvironment by inducing the expression of pro-angiogenic cytokines such as IL6 and CXCL1 which increase endothelial cell proliferation and migration and that might influence the biological properties of other cell types in the tumor microenvironment. ID proteins have also been shown to promote invasion by degrading the extracellular matrix through induction of several members of the maxtrix metalloproteinase (MMP) protein family such as MMP-2, MMP-9, and MMP-14. ID genes control a stem cell-intrinsic transcriptional program that preserves stem cell adhesion to the niche in neural stem cells and in glioma. ID proteins activate the Ras-related protein RAP1 by suppressing the GTPase activating protein RAP1GAP, thereby promoting adhesion of cells to a supportive endothelial niche.
Analysis of clinical specimens has shown that high expression of ID proteins, particularly ID1, correlates with aggressive clinical behavior and poor patient outcome in many cancers (Table 1) Furthermore, data from our group shows ID1 expression is upregulated between primary breast cancers and their matched brain metastases (unpublished data) suggesting a functional role for ID1 in the metastatic process. However, analysis of ID1 expression in human cancers is complicated by several factors. ID1 can be expressed by rare neoplastic cells in subsets of breast,26 glioma,27 and bladder17 cancers, which can be difficult to identify using tissue microarrays. Furthermore, tumor-associated endothelial cells often show high ID1 expression, confounding expression analysis in whole tumor extracts.
Table 1. ID protein expression in primary human malignancies.
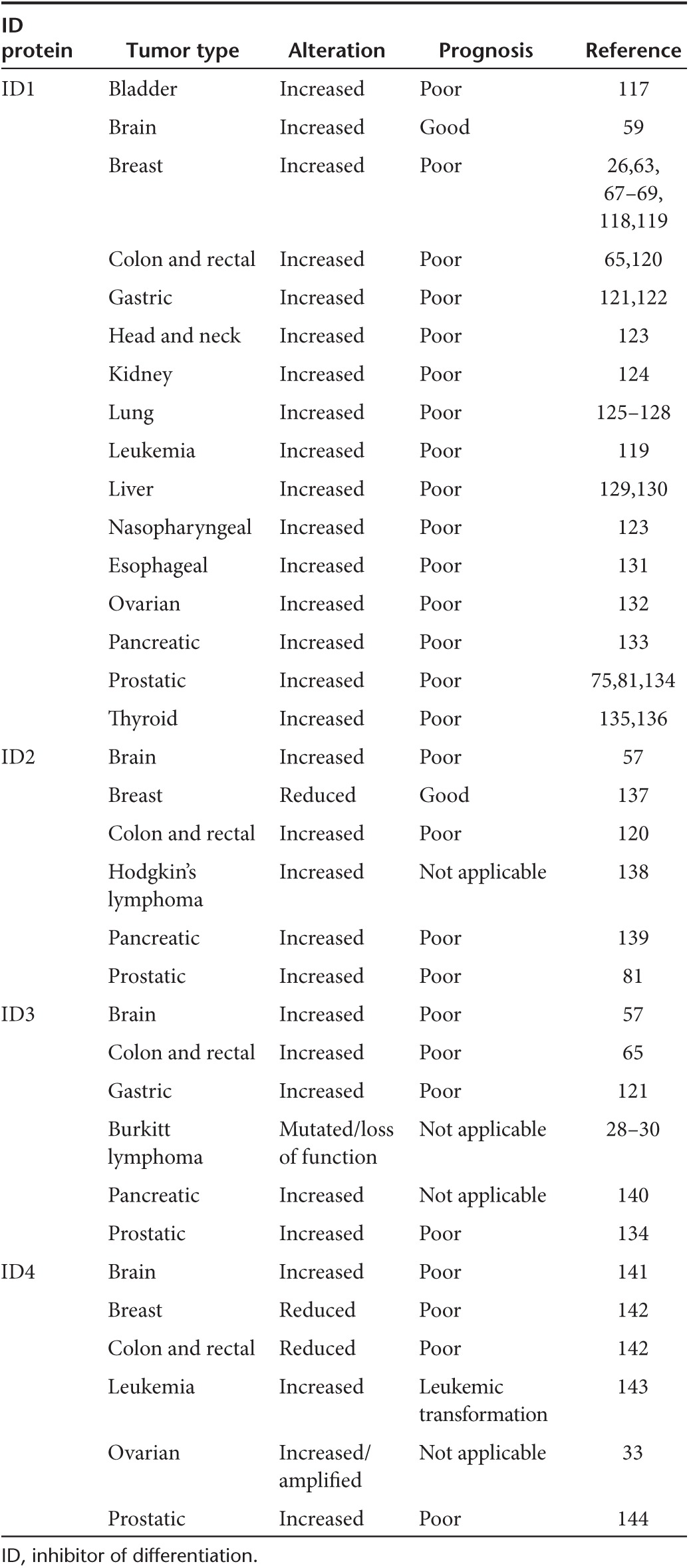
In contrast to ID1, other ID proteins possess diverse patterns of expression and function in cancer. Recent data from several groups demonstrates that ID3 is a tumor suppressor in Burkitt's lymphoma, and is inactivated through somatic mutation in up to 68% of cases.28,29,30 ID3 inactivation promotes tumor cell survival through ligand-independent signaling by the B-cell receptor to the PI3K pathway. Interestingly, ID3 is not mutated in other B-cell lymphomas, perhaps reflecting ID3's role in specific phases of B-cell maturation.29 Similarly, ID4 is epigenetically silenced through promoter hypermethylation in subsets of cancers, including human leukemia,31,32 suggesting a tumor suppressive function. Conversely, ID4 acts as a proto-oncogene in serous ovarian cancer where it is genomically amplified, overexpressed, and required for SOC cell line proliferation.33 ID4 has been identified as a negative regulator of BRCA1 which is the most commonly mutated gene in familial breast and ovarian cancer.34 In an unbiased ribozyme-based screen ID4 was discovered to inversely regulate expression of BRCA1 in an ovarian and breast cancer cell line.35 In a separate study by Welcsh et al., overexpression of BRCA1 increases the expression level of ID4, suggesting a negative feed-back loop in BRCA1 signalling.36 Therefore, depending on the cell type and development stage, ID proteins can have different functional outcomes as oncogenes or tumor suppressors. These data demonstrate the importance of context in understanding ID protein function in cancer. IDs also serve as downstream targets of several known oncogenic pathways. Regulation of ID proteins has been shown to be mediated by several well-established oncoproteins such as MYC, RAS, RB, and SRC (Figure 1).13,23,37 Tumor-suppressor genes that cause repression of ID transcription include p53, FOXO3, and growth inhibitory signals by TGF-β–SMAD.38,39,40 ID proteins have been shown to regulate central hallmarks of cancer such as proliferation, cellular senescence, and survival, as reviewed previously by Perk et al.41 Aberrant levels of ID proteins have been associated with the upregulation of a number of proliferation and pro-survival factors such as cyclins D1 and E,42 PI3K–AKT,43,44,45,46 and nuclear factor-κB (NF-κB),47 and the inhibition of cyclin-dependent kinase inhibitors (CKI) p16INK4A, p21waf1, p27Kip1,48,49,50 and pRb51 (Figure 1).
Recent studies have provided critical insights into novel cellular and molecular events controlled by ID proteins, revealing complex roles within neoplastic cells and their local microenvironment. In particular, ID expression is critical in controlling cancer stem cell (CSC) phenotypes and is associated with the activation of angiogenesis, induction of cancer cell invasion and metastasis, and remodeling of the microenvironment.
IDs Regulate the CSC Phenotype
There is now compelling evidence that specific subpopulations of tumor cells, known as CSC, drive the tumorigenic and metastatic potential of many tumors.52 In many cases, signaling pathways active in tissue stem and progenitor cells are important in the maintenance of CSC pools. Recent data demonstrates an important role for ID proteins in tissue stem cells, which appears to be conserved in their malignant counterparts. Tissue stem cells reside in anatomical niches and lineage commitment of these cells is often coordinated with departure of stem cells from their niche. Hematopoietic3 and adult neural stem cells1 are identified by high expression of Id1 and recent work has identified a critical role for ID proteins in niche interactions and self renewal of hematopoietic stem cell and neural stem cell. Increasing evidence supports a role for bone remodeling cells in providing a niche for HSC. Mice deficient in Id1 exhibit reduced long-term hemaotopoietic repopulating activity and enhanced myeloid differentiation,3 associated with altered bone formation53 and altered bone stromal cytokine expression.5 Niola et al. have recently identified that Id1, Id2, and Id3 maintain adhesion of adult neural stem cells to an endothelial niche.6 ID proteins promote adhesion by activating RAP1, a GTPase involved in integrin-dependent adhesion, by suppressing the expression of its negative regulator RAPGAP1. Compound deletion of Id genes leads to exit of neural stem cells from the niche.6
A significant body of literature supports a key role for ID proteins in high-grade glioma (HGG), which is the most frequent adult brain tumor and which remains practicably incurable, with median survival of ~15 months for patients with the highest grade tumors, known as glioblastoma multiforme. HGGs fall into four molecular subtypes: proneural, neural, classical, and mesenchymal, each with unique clinical features and genomic defects. Numerous studies demonstrate that HGG are maintained by a minor subset of glioma-initiating cells (GICs) that share many traits with neural stem cells, including self-renewal, multilineage differentiation, and a dependence on a perivascular niche.54 GICs are commonly identified by the capacity to form spheres in suspension culture and/or through expression of cell surface stem cell markers CD133 or CD44.
Gliomas express high levels of ID proteins55 and various studies have provided evidence that ID proteins regulate the genesis and maintenance of GICs. GICs are enriched for co-expression of ID1 and CD44, which preferentially localize to the perivascular environment.27 Furthermore, coexpression of ID1 and CD44 predicts poor prognosis in glioblastoma multiforme, albeit weakly.27 The cytokine TGF-β is a major player in the regulation of Id proteins in HGG, where it upregulates expression of Id1 and promotes the self renewal capacity of GICs27 (Figure 2a). This result is in contrast to “normal” epithelial cells where TGF-β is known to repress ID1 expression through engagement of the Smad protein corepressor ATF3.19 In glioblastoma multiforme, ATF3 is commonly epigenetically silenced,56 switching the TGF-β regulation of Id1 from repression to induction. Further in vitro studies support a role for ID1 and ID3 in GIC biology as pharmacological inhibition of TGF-β or genetic ablation of ID1 and ID3 reduces human HGG sphere formation and invasiveness in vitro,27,57 which is in turn associated with reduced expression of the neural stem cell marker Sox2.58 However, Barrett et al. provide evidence that in vitro sphere-forming assays must be interpreted with caution. By crossing a knock-in mouse model in which the Id1 promoter drives GFP expression to two distinct transgenic mouse models of HGG, the authors demonstrate that while Id1hi cells are enriched for in vitro sphere-forming capacity compared to Id1low cells, they are not enriched for the capacity to transplant disease into naive recipients.59 Surprisingly, the authors found that the Idlow glioma cells have greater tumorigenic potential than the Idhigh glioma cells (which have greater self-renewal capacity). Though both populations are capable of transmitting disease, the more proliferative Idlow glioma cells are more tumorigenic. One of the caveats of this study is the lack long-term transplantation assay and could imply that Id1 does not mark the CSC in this model. Accordingly, deletion of Id1 and Id3 in vivo led to a very modest increase in survival in these models. In keeping with the animal data, low ID1 expression associates with poorer outcome in the proneural subtype of HGG,57,59 although these data are based on analysis of mRNA expression in total tumor extracts, which has caveats as previously discussed.
Figure 2.
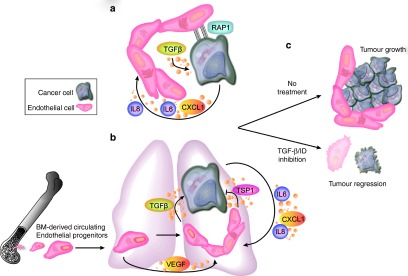
A proposed conserved cellular mechanism for inhibitor of differentiation (ID) proteins in tumorigenesis and metastasis. (a) IDs control the glioma cancer stem cell niche in a cell autonomous as well as paracrine manner. TGF-β, expressed by endothelial cells, upregulates the expression of ID proteins in glioma stem cells, thereby maintaining glioma stem cell self renewal via RAP1-dependent adhesion to the niche. ID expression may also re-inforce the vascular niche by the induction of pro-angiogenic factors like IL6 and CXCL1. (b) ID proteins may play a similar role in coordinating an endothelial niche for disseminated breast cancer cells in the lung. Endothelial cells can enforce quiescence of disseminated breast cancer cells in the lung through the expression of thrombospondin-1 (TSP1). In contrast, activated or sprouting endothelial cells downregulate TSP1- and instead express TGF-β which promotes the escape of disseminated tumor cells from dormancy, in an ID-protein dependent manner. ID proteins in turn can activate the expression of proangiogenic factors CXCL1 and IL8 to drive endothelial activation and sprouting. Endothelial activation can be further promoted by the recruitment from the bone marrow to nascent metastatic sites of Id+ EPCs, which require ID1 for mobilization and proliferation. EPCs support angiogenesis by the expression of VEGF and through direct luminal incorporation. ID signaling in activated endothelial cells further represses TSP-1. (c) Targeting ID expression and their associated pathways in tumor cells and endothelial cells from bone marrow and tumor blood vessels will disrupt both cell-autonomous and cell-nonautonomous programs, which may eventually produce additive or even synergistic antitumor effects. VEGF, vascular endothelial growth factor.
Several studies suggest that ID2, ID3, and perhaps ID4 may also be critical in controlling GIC malignancy. Id360 and Id461 are able to reprogram Ink4a/Arf-deficient mouse astrocytes into GIC-like cells through a variety of proposed mechanisms. These include cell autonomous derepression of Sox2, cyclin E, and Notch signaling by Id4,62 and by induction of the pro-angiogenic factors IL6, IL8, and CXCL1 by Id360 (Figures 1 and 2a), which may promote the development of a vascular niche for GICs, which will be discussed later. To further explore the functional redundancy between Id proteins, Niola et al. first created a new transplantable mouse model in which p53 loss cooperates with expression of oncogenic h-Ras to generate murine tumors with transcriptional signatures resembling human mesenchymal HGG.57 By further engineering inducible Id1, Id2, and Id3 silencing, they showed that coordinated loss of three Id proteins leads to marked reductions in proliferation and stem cell marker expression, inhibition of in vitro sphere-forming capacity and a dramatic improvement in animal survival. This apparent discrepancy with the limited phenotype observed following deletion of Id1 and Id3 by Barrett et al. may be explained by differences in the requirement for Id proteins between different models of HGG or by compensation between family members, requiring deletion of 3 or more ID members to reveal a phenotype.
Mirroring the effects of Id deletion in neural stem cells,6 deletion of Ids in HGG is associated with increased Rap1Gap expression, decreased Rap1 activity and loss of adhesion to endothelial cells (Figure 2a). In support of the generality of this result, RapGap1 expression is dramatically downregulated in human HGG and a five-gene expression signature including ID2, ID3, and Rap1Gap strongly predicts outcome in HGG.57 This suggests that the mechanism of ID function in neural stem cells may be maintained by CSCs.
There is increasing evidence for ID proteins, in particular ID1 and ID3, in the promotion of the CSC phenotype of epithelial cancers. We have shown that Id1 cooperates with oncogenic h-Ras in transformation of mammary epithelia to generate highly metastatic breast cancers.63 Id1 was required for tumor maintenance, as inactivation of the Id1 transgene in established tumors led to growth arrest followed by regression associated with widespread cellular senescence, which is essentially the irreversible loss of self-renewal and proliferative capacity.63 Similarly, Stankic et al. recently demonstrated that Id1 expression generates breast cancer cells with tumor-initiating properties independent of an epithelial-to-mesenchymal transition (EMT) program.64 ID1 and ID3 are required for the re-initiation of proliferative capacity in experimental breast cancer lung metastases.26 John Dick et al. recently reported that ID1 and ID3 are also required for the self renewal and tumor-initiating capacity of colon CSCs through cell-cycle restriction driven by the cell-cycle inhibitor p21.65 In these studies, silencing of Id1 and Id3 sensitized colon cancer-initiating cells to the chemotherapeutic agent oxaliplatin. Taken together, there is a recurring role for ID proteins across diverse tumor types in the maintenance of the CSC phenotype. Although by and large the mechanistic details remain to be determined, these studies identify self-renewal pathways controlled by ID1 and ID3 as potential targets for the development of therapies to eradicate cancer-initiating cells.
IDs Regulate Tumor Metastasis
The metastatic cascade is a complex multistep process involving intravasation, dissemination of the cancer cells into circulation, extravasation followed by initiation and outgrowth in distant metastatic organs. In many solid cancers, cancer cells disseminate early in the life of a tumor, before clinical presentation, and surgical resection,66 followed by a long period of dormancy prior to metastatic relapse. ID genes have demonstrated roles in many facets of metastatic dissemination, including tissue remodeling and invasion, initiation of proliferative capacity at the distant site, as well as functions in host endothelial cells that contribute to angiogenesis.
Cancer Cell-Autonomous Roles for ID Proteins in Metastasis
To investigate the transcriptional changes associated with metastatic dissemination, Minn et al.67 used murine xenografts of MDA-MB-231 human breast cancer cells to select cell subpopulations highly metastatic to lung. Transcriptional profiling analysis revealed 95 genes differentially expressed by lung-tropic sublines, among which ID1 was the sole transcription regulator. Functional studies by Minn and others now clearly show that ID1 and ID3 are required for breast tumor initiating functions, both in primary tumor formation and during metastatic colonization of distant organs in breast, gastric, and pancreatic cancer models.26,67,68,69,70,71
Emerging evidence suggests that activation of the EMT program can promote invasion and metastasis by promoting the acquisition of a CSC-like state.72,73 As such, EMT may confer on epithelial cells a set of characteristics that enable them to disseminate from primary tumors and seed metastases.74 ID1 is highly expressed in metaplastic breast cancers,17 a rare subtype with a poorly differentiated mesenchymal-like phenotype. There is evidence that ID1 may regulate EMT, both directly through interaction with Cav-1 in prostate cancer cells,75 induction of cadherin-switching in immortalized esophageal epithelial cells,76 suppression of E-cadherin and ZO-1 in human kidney cells,77 and indirectly through loss of KLF1769 in breast cancer cells. ID1 and ID3 also regulate MMPs, a major protein family associated with EMT that regulates remodeling of the ECM, degradation of the basement membrane and stromal cell layers, allowing the infiltration of cancer cells during invasion.78,79 Desprez et al.80 originally reported that constitutive expression of ID1 results in upregulation of a novel MMP protein, MT1-MMP, and invasion through basement membrane (Figure 1b). High expression of ID1 also induces increased secretion of MMP-2 in prostate cancer81 and MMP-9 in leukemia.82
However, several recent studies have demonstrated that epithelial–mesenchymal plasticity is crucial at different stages of metastasis, in particular, the reversal of EMT is necessary for efficient metastatic colonization.83 Stankic et al. have recently shown that ID1, under the control of TGF-β signaling, mediates epithelial–mesenchymal plasticity and that ID1 expression is associated with an epithelial phenotype in breast cancer lung metastases.64 ID1 induces a mesenchymal-to-epithelial transition at the metastatic site by antagonizing the activity of Twist, a bHLH transcription factor, but not at the primary site, where this state is controlled by the zinc finger protein Snail1. This observation has also shed light on a possible role of Id proteins in regulating the dynamic interactions among epithelial and mesenchymal gene programs.
ID Control of the Metastatic Microenvironment
In addition to their cell-autonomous roles in metastatic dissemination, ID proteins may play a role in stromal control of metastatic progression. Recent studies suggest the existence of a perivascular niche in which lung-metastatic breast cancer cell dormancy is enforced by the expression of thrombospondin-1 (TSP-1), an antiangiogenic factor, by endothelial cells.84 Ghajar et al. demonstrated that metastatic cells can be activated to proliferate by vessel remodeling and sprouting via a TGF-β-dependent mechanism84 (Figure 2b), which bears striking parallels to the endothelial niche in which glioma stem cells reside (Figure 2a). Although the involvement of ID proteins in the secretion of TSP-1 or the response of disseminated breast cancer cells to TGF-β is yet to be explored in this setting, ID1 is known to suppress TSP-1 expression85,86,87 so it is tempting to speculate that ID signaling in activated endothelial cells may be responsible for the suppression of TSP-1 in the escape from metastatic dormancy.
ID proteins also play a role in the expression of proangiogenic factors by neoplastic cells. ID4 and ID3 promote tumor angiogenesis via secretion of IL8 and CXCL1 by glioma60,88 and breast cancer cells,15 which may support tumor growth via the promotion of vessel remodeling and reinforcement of the cancer niche, whether it be glioma growing in the brain or breast cancer cells growing in the lung (Figure 2a,b). Given the proven role for ID proteins in niche interactions in primary cancers, exemplified by glioma, the role of ID proteins in establishing niche interactions in metastatic sites is an important area of future study.
ID Expression and Function in Angiogenesis
Angiogenesis is a rate limiting step in tumor progression required to provide nutrients necessary for tumor growth and access to the circulation for metastatic cells. ID proteins are commonly expressed by endothelial cells and are upregulated in tumor-associated vessels. The mechanism controlling Id expression in tumor vasculature are not fully elucidated, however bone morphogenic proteins (BMPs), members of the TGF-β superfamily, induce ID1 expression in endothelial cells through receptor-mediated activation of Smad1, Smad4, and Smad5 binding to bone morphogenic protein response elements in the ID1 promoter.89,90 Bone morphogenic protein-dependent upregulation of ID1 expression stimulates endothelial cell proliferation and sprouting in vitro and angiogenesis in vivo.91,92 This activity of ID1 may be dependent on binding to the HLH protein E2-2; ID1 binds to E2-2 in vitro and can reverse the blockade of experimental angiogenesis in vivo resulting from E2-2 overexpression.93 In addition, vascular endothelial growth factor secreted by tumors activates the mitogen-activated protein kinase pathway and may impinge on the ID1 and ID3 promoters of endothelial cell at the EGR-1 site, as has been demonstrated in other cell types.94,95 Id2 loss leads to downregulation of vascular endothelial growth factor in pituitary tumors suggesting a possible feedback loop between Ids and vascular endothelial growth factor-mediated by Hif-1α.96,97
Mice lacking Id1, Id2, and Id3 show extensive embryonic hemorrhaging,98 suggesting a strict requirement for ID proteins in endothelial biology. In xenograft models of tumor growth, animals with reduced Id1 and Id3 dosages showed a significant loss of tumor vascular integrity and fail to develop metastatic lesions.99 However, spontaneous models of solid tumors, including breast (MMTV-Neu), prostate (TRAMP), and multiple cancers (PTEN deficiency) demonstrate a more varied and complex dependency on ID proteins for angiogenesis.100,101,102 In these models, the expression of ID proteins in the tumor vessels varies with tumor type and grade: e.g., ID is expressed in the neovessels of breast, intrauterine, and poorly differentiated prostate cancers, but not in pheochromocytomas or well-differentiated prostate tumors.100,101 Interestingly, while ID deficiency led to the development of hypoxia and/or hemorrhage in tumors with high endothelial ID expression, it did not significantly impact on tumor progression. However, in the MMTV-Her2/neu model, partial loss of Id function in combination with chemical inhibition of stress-activated pathways lead to dramatic regression of aggressive tumors.102
These models revealed a functional contribution for bone marrow (BM)-derived endothelial progenitor cells (EPCs) in tumor angiogenesis, which increased with grade.101 vascular endothelial growth factor secreted by tumors leads to a dramatic upregulation of Id1 and Id3 proteins in the BM, presumably in BM-derived EPCs which results in their mobilization into the circulation and subsequently into tumors.103 EPCs provide stability to nascent vessels by direct luminal incorporation into sprouting vessels but also regulate the angiogenic switch at a critical early stage of tumor growth via paracrine secretion of pro-angiogenic growth factors104 (Figure 2b). While the contributions of EPCs to neovessel formation in primary and metastatic tumors have been reported to be variable,105 remarkably, specific ablation of EPCs in vivo results in severe angiogenesis inhibition and impaired tumor growth.106
EPCs from tumor-bearing mice104,107 and isolated from cancer patients108 express high levels of ID1. The Id1 KO mice were critical in demonstrating that BM-derived progenitors are the source of tumor endothelium, as Id1 KO mice failed to mobilize these progenitors, while BM transplantation of Id1 KO mice with wild type BM rescued the observed vascular defects.103 Following these observations, acute and conditional shRNA-mediated silencing of Id1 in the adult BM resulted in EPC mobilization defects associated with severe angiogenesis inhibition, impaired primary tumor growth, and progression of micrometastases to macrometastases, suggesting a critical role for these cells in angiogenesis-mediated tumor growth.104 Importantly, ID1 deficiency reduced levels of mobilized EPCs and not other hematopoietic cells, as observed in Id KO animals which maintain normal hematopoiesis.109 The selectivity of Id1 gene expression for EPCs has been exploited to specifically express transgenes in EPCs in vivo.107 Use of the Id1 promoter to drive expression of suicide genes, or shRNAs targeting EPC-intrinsic factors including VEGFR2, reduced circulating EPCs and showed significant defects in angiogenesis-mediated tumor growth.
How IDs control the generation of EPCs is also beginning to be explored. ID1 is required for the maintenance of long term repopulating hematopoietic stem cells (lin− Sca+ kit+ CD34−)2,3 in the BM through suppression of p21. Loss of ID1 upregulates p21, which in turn drives the stem cells towards a more committed myeloid state, as assessed by gene expression profiling, an event that is associated with the depletion of cells capable of endothelial cell fate commitment.110 These results suggest that ID1 is required in early hematopoietic stem cells to restrain the commitment to the myeloid lineage and preserve a pool of cells that give rise to endothelial progenitors in response to vasculogenic growth signals. To understand signaling mechanisms responsible for Id1-mediated EPC functions, a recent study has demonstrated the role of Id1/PI3K/Akt/NF-κB/survivin signaling pathway in EPC proliferation.111
ID Signaling Pathways as Therapeutic Targets in Cancer
The transcriptional regulators ID1 and ID3 are attractive targets for cancer therapy as they are required for angiogenesis, tumor invasiveness, and metastasis. Furthermore, ID proteins are undetectable in most normal tissues, but are highly expressed in many cancer cells and cancer-associated blood vessels suggesting that targeting ID proteins may provide a large therapeutic window in which to treat cancers while minimizing toxicity (Figure 2c). ID proteins have cell-autonomous roles in proliferation and CSC homeostasis, and genetic studies in glioblastoma,27,57 breast,26,63 and colon cancer65 demonstrate that Id activity is required for the maintenance of certain cancers. A novel systemic siRNA delivery system was used to demonstrate the feasibility of systemic Id protein targeting. Knockdown of ID4 in established ovarian cancer xenografts led to long-term tumor remission in 80% of mice.33 While these data suggest that inactivation of Id proteins may serve as a novel therapeutic strategy, transcription factors are notoriously difficult to target with small molecule inhibitors. Several groups have used a variety of alternative methods to disrupt Id factor protein complexes in cancer cells. For example, treatment with cell permeable peptides aimed at disrupting the interaction of ID1/ID3 with the bHLH protein E47 led to marked activation of E47 transcriptional activity and the induction of cell cycle arrest and apoptosis in breast112 and ovarian113 cancer cells. The broader effectiveness of targeting ID protein binding depends on the nature of the biochemical or transcriptional complexes in which ID proteins act, which in many instances has not been elucidated and remains a major knowledge gap in this field.
Others have targeted ID1 expression rather than function. Anido et al. used clinically approved small molecule inhibitiors of TGF-β receptor (TGFβRI) to downregulate Id1 and Id3 in a mouse model of glioblastoma multiforme, leading to reduced tumor initiation and tumor growth in an ID-dependent manner.27 Treatment of mice bearing orthotopic HGG tumors with a small molecule known to downregulate Id1, cannabidiol, led to marked inhibition of tumor growth in vivo.58 Similarly, Mistry et al. show that small molecule inhibitors of the ubiquitin specific protease ubiquitin-specific peptidase 1 promote degradation of ID1 and inhibition of acute myeloid leukemia cell line growth and survival in vitro and in vivo.114 The relative importance of ID protein downregulation to the therapeutic efficacy of cannabidiol and ubiquitin-specific peptidase 1 inhibitors remains to be determined.
Since ID proteins play a role in supporting vasculature and BM-derived EPCs, several groups have targeted the critical roles for ID proteins in cancer-associated endothelial cells which form the stem cell niche and provide nutrients to growing metastatic tumors.115 To model the efficacy of therapeutic targeting of Id1+ EPCs, Mellick et al. targeted the toxic thymidine kinase gene to EPCs using the Id1 promoter, leading to potent inhibition of angiogenic-mediated tumor growth.107 Systemic delivery of Id1 antisense oligonucleotide fused to an endothelial delivery peptide downregulated Id1 in tumor endothelial cells in vivo and led to remarkable inhibition of primary tumor growth and metastasis.116 Targeting of ID proteins in appropriate tumor types, such as high grade serous ovarian cancer, basal breast cancer and glioma may deplete the CSC pool, its niche and supporting vasculature, opening new therapeutic opportunities in these aggressive cancers (Figure 2c).
Conclusions and Future Directions
The studies highlighted in this review demonstrate the complex context-dependency of ID protein expression and function in cancer, where ID proteins can play opposing roles at times in neoplastic progression. ID proteins have fundamental roles in sensing and integrating extracellular cues to control the phenotype of cancers, via cell autonomous and extrinsic pathways, such as formation of the metastatic niche, increased proliferation and self renewal, metastasis, and angiogenesis. The corruption of the normal functions of ID proteins to maintain a more undifferentiated state in a cancer cell (CSCs) make ID proteins an attractive therapeutic target. A detailed understanding of the mechanism of action of ID proteins in different cancer subtypes will be essential to exploit their therapeutic potential.
While ID genes are deregulated through genetic or epigenetic events in a subset of cancers, emerging evidence suggests that ID expression may be required by a broader range of cancers where they are not necessarily genomically altered or overexpressed, a process known as lineage-dependency. In such cancers, ID proteins and associated molecular networks may be promising new therapeutic targets, permitting simultaneous targeting of neoplastic cells, and their supportive microenvironment.
ID proteins are also valuable tools in deconvoluting tumor heterogeneity, and their association with CSCs may lead us to therapies directed at the CSC subpopulation of cancers. Many questions remain to translate better biological insights of ID function into therapeutics. In particular, further studies into the role of IDs in the escape from metastatic dormancy and their relationship to the cell of origin of cancer promise to yield exciting biological breakthroughs and valuable insights in the clinical management of cancer progression.
Acknowledgments
The authors thank Kim Moran-Jones and Mohammed Bensellam for critical reading of this manuscript. We would like to acknowledge funding from Cancer Council New South Wales, International Postgraduate Research Scholarship and Beth Yarrow Award (W.S.T.), National Health and Medical Research Council of Australia (NHMRC) and Colin Biggers & Paisley, Sydney (A.S.), and NIH grant R01-CA107429 (V.M.). A.S. is a Career Development Fellow of the NHMRC. Kate Patterson (Medipics and Prose) prepared the figures. The authors declare no conflict of interest.
References
- Nam HS, Benezra R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem Cell. 2009;5:515–526. doi: 10.1016/j.stem.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, Nimer SD. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci USA. 2007;104:1260–1265. doi: 10.1073/pnas.0607894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SS, Zhao Y, Nie L, Cochrane SW, Huang Z, Sun XH. Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood. 2007;110:2351–2360. doi: 10.1182/blood-2007-01-069914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Lanman EE, Pavlovic S, Amlani B, Chin Y, Benezra R. Id1 maintains embryonic stem cell self-renewal by up-regulation of Nanog and repression of Brachyury expression. Stem Cells Dev. 2012;21:384–393. doi: 10.1089/scd.2011.0428. [DOI] [PubMed] [Google Scholar]
- Suh HC, Ji M, Gooya J, Lee M, Klarmann KD, Keller JR. Cell-nonautonomous function of Id1 in the hematopoietic progenitor cell niche. Blood. 2009;114:1186–1195. doi: 10.1182/blood-2008-09-179788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niola F, Zhao X, Singh D, Castano A, Sullivan R, Lauria M, et al. Id proteins synchronize stemness and anchorage to the niche of neural stem cells. Nat Cell Biol. 2012;14:477–487. doi: 10.1038/ncb2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RW, Wu JM. Molecular mechanisms regulating expression and function of transcription regulator inhibitor of differentiation 3. Acta Pharmacol Sin. 2005;26:1409–1420. doi: 10.1111/j.1745-7254.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- Biggs J, Murphy EV, Israel MA. A human Id-like helix-loop-helix protein expressed during early development. Proc Natl Acad Sci USA. 1992;89:1512–1516. doi: 10.1073/pnas.89.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann V, van Crüchten I, Sablitzky F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994;22:749–755. doi: 10.1093/nar/22.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Iavarone A, Israel MA. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol Cell Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, et al. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Fontemaggi G, Dell'Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol. 2009;16:1086–1093. doi: 10.1038/nsmb.1669. [DOI] [PubMed] [Google Scholar]
- Jen Y, Manova K, Benezra R. Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn. 1997;208:92–106. doi: 10.1002/(SICI)1097-0177(199701)208:1<92::AID-AJA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Perk J, Gil-Bazo I, Chin Y, de Candia P, Chen JJ, Zhao Y, et al. Reassessment of id1 protein expression in human mammary, prostate, and bladder cancers using a monospecific rabbit monoclonal anti-id1 antibody. Cancer Res. 2006;66:10870–10877. doi: 10.1158/0008-5472.CAN-06-2643. [DOI] [PubMed] [Google Scholar]
- Kowanetz M, Valcourt U, Bergström R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241–4254. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massagué J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Lin CQ, Singh J, Murata K, Itahana Y, Parrinello S, Liang SH, et al. A role for Id-1 in the aggressive phenotype and steroid hormone response of human breast cancer cells. Cancer Res. 2000;60:1332–1340. [PubMed] [Google Scholar]
- Tam WF, Gu TL, Chen J, Lee BH, Bullinger L, Fröhling S, et al. Id1 is a common downstream target of oncogenic tyrosine kinases in leukemic cells. Blood. 2008;112:1981–1992. doi: 10.1182/blood-2007-07-103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat Immunol. 2001;2:165–171. doi: 10.1038/84273. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Stegmüller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, et al. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442:471–474. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, et al. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011;146:918–930. doi: 10.1016/j.cell.2011.07.040. [DOI] [PubMed] [Google Scholar]
- Gupta GP.Perk J.Acharyya S.de Candia P.Mittal V.Todorova-Manova K.et al. (2007ID genes mediate tumor reinitiation during breast cancer lung metastasis Proc Natl Acad Sci USA 10419506–19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA.et al.(2010TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma Cancer Cell 18655–668. [DOI] [PubMed] [Google Scholar]
- Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–1325. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B.et al. ICGC MMML-Seq Project 2012Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing Nat Genet 441316–1320. [DOI] [PubMed] [Google Scholar]
- Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–120. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet. 2005;37:265–274. doi: 10.1038/ng1521. [DOI] [PubMed] [Google Scholar]
- Chen SS, Claus R, Lucas DM, Yu L, Qian J, Ruppert AS, et al. Silencing of the inhibitor of DNA binding protein 4 (ID4) contributes to the pathogenesis of mouse and human CLL. Blood. 2011;117:862–871. doi: 10.1182/blood-2010-05-284638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, et al. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Sci Transl Med. 2012;4:147ra112. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness' in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- Beger C, Pierce LN, Kruger M, Marcusson EG, Robbins JM, Welcsh P, et al. Identification of Id4 as a regulator of BRCA1 expression by using a ribozyme-library-based inverse genomics approach. Proc Natl Acad Sci USA. 2001;98:130–135. doi: 10.1073/pnas.98.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcsh PL.Lee MK.Gonzalez-Hernandez RM.Black DJ.Mahadevappa M.Swisher EM.et al.(2002BRCA1 transcriptionally regulates genes involved in breast tumorigenesis Proc Natl Acad Sci USA 997560–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi O, Tepper CG, Purnell PR, Izumiya Y, Evans CP, Green TP, et al. Regulation of Id1 expression by SRC: implications for targeting of the bone morphogenetic protein pathway in cancer. Cancer Res. 2008;68:2250–2258. doi: 10.1158/0008-5472.CAN-07-6403. [DOI] [PubMed] [Google Scholar]
- Birkenkamp KU.Essafi A.van der Vos KE.da Costa M.Hui RC.Holstege F.et al. (2007FOXO3a induces differentiation of Bcr-Abl-transformed cells through transcriptional down-regulation of Id1 J Biol Chem 2822211–2220. [DOI] [PubMed] [Google Scholar]
- Paolella BR, Havrda MC, Mantani A, Wray CM, Zhang Z, Israel MA. p53 directly represses Id2 to inhibit the proliferation of neural progenitor cells. Stem Cells. 2011;29:1090–1101. doi: 10.1002/stem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- Swarbrick A, Akerfeldt MC, Lee CS, Sergio CM, Caldon CE, Hunter LJ, et al. Regulation of cyclin expression and cell cycle progression in breast epithelial cells by the helix-loop-helix protein Id1. Oncogene. 2005;24:381–389. doi: 10.1038/sj.onc.1208188. [DOI] [PubMed] [Google Scholar]
- Ling MT, Wang X, Ouyang XS, Xu K, Tsao SW, Wong YC. Id-1 expression promotes cell survival through activation of NF-kappaB signalling pathway in prostate cancer cells. Oncogene. 2003;22:4498–4508. doi: 10.1038/sj.onc.1206693. [DOI] [PubMed] [Google Scholar]
- Li B, Tsao SW, Li YY, Wang X, Ling MT, Wong YC.et al. (2009Id-1 promotes tumorigenicity and metastasis of human esophageal cancer cells through activation of PI3K/AKT signaling pathway Int J Cancer 1252576–2585. [DOI] [PubMed] [Google Scholar]
- Lin J, Guan Z, Wang C, Feng L, Zheng Y, Caicedo E.et al. (2010Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways Clin Cancer Res 1677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kang MB, Jang SH, Qian T, Kim HJ, Kim CH, et al. Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene. 2009;28:824–831. doi: 10.1038/onc.2008.451. [DOI] [PubMed] [Google Scholar]
- Kim H, Chung H, Kim HJ, Lee JY, Oh MY, Kim Y.et al. (2008Id-1 regulates Bcl-2 and Bax expression through p53 and NF-kappaB in MCF-7 breast cancer cells Breast Cancer Res Treat 112287–296. [DOI] [PubMed] [Google Scholar]
- Tang J, Gordon GM, Nickoloff BJ, Foreman KE. The helix-loop-helix protein id-1 delays onset of replicative senescence in human endothelial cells. Lab Invest. 2002;82:1073–1079. doi: 10.1097/01.lab.0000022223.65962.3a. [DOI] [PubMed] [Google Scholar]
- Alani RM, Young AZ, Shifflett CB. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc Natl Acad Sci USA. 2001;98:7812–7816. doi: 10.1073/pnas.141235398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff BJ, Chaturvedi V, Bacon P, Qin JZ, Denning MF, Diaz MO. Id-1 delays senescence but does not immortalize keratinocytes. J Biol Chem. 2000;275:27501–27504. doi: 10.1074/jbc.C000311200. [DOI] [PubMed] [Google Scholar]
- Alani RM, Hasskarl J, Grace M, Hernandez MC, Israel MA, Münger K. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc Natl Acad Sci USA. 1999;96:9637–9641. doi: 10.1073/pnas.96.17.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Stem cells and cancer—the promise and puzzles. Mol Oncol. 2010;4:369–372. doi: 10.1016/j.molonc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AS, Jensen KK, Skokos D, Doty S, Lederman HK, Kaplan RN, et al. Id1 represses osteoclast-dependent transcription and affects bone formation and hematopoiesis. PLoS One. 2009;4:e7955. doi: 10.1371/journal.pone.0007955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- Vandeputte DA, Troost D, Leenstra S, Ijlst-Keizers H, Ramkema M, Bosch DA, et al. Expression and distribution of id helix-loop-helix proteins in human astrocytic tumors. Glia. 2002;38:329–338. doi: 10.1002/glia.10076. [DOI] [PubMed] [Google Scholar]
- Gargiulo G, Cesaroni M, Serresi M, de Vries N, Hulsman D, Bruggeman SW, et al. In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell. 2013;23:660–676. doi: 10.1016/j.ccr.2013.03.030. [DOI] [PubMed] [Google Scholar]
- Niola F, Zhao X, Singh D, Sullivan R, Castano A, Verrico A.et al. (2013Mesenchymal high-grade glioma is maintained by the ID-RAP1 axis J Clin Invest 123405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L, Murase R, Limbad C, Singer E, Allison J, Adrados I, et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013;73:1559–1569. doi: 10.1158/0008-5472.CAN-12-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, et al. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell. 2012;21:11–24. doi: 10.1016/j.ccr.2011.11.025. [DOI] [PubMed] [Google Scholar]
- Jin X, Yin J, Kim SH, Sohn YW, Beck S, Lim YC, et al. EGFR-AKT-Smad signaling promotes formation of glioma stem-like cells and tumor angiogenesis by ID3-driven cytokine induction. Cancer Res. 2011;71:7125–7134. doi: 10.1158/0008-5472.CAN-11-1330. [DOI] [PubMed] [Google Scholar]
- Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, Park HJ.et al. (2008Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling Genes Dev 222028–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon HM, Sohn YW, Oh SY, Oh SY, Kim SH, Beck S, et al. ID4 imparts chemoresistance and cancer stemness to glioma cells by derepressing miR-9*-mediated suppression of SOX2. Cancer Res. 2011;71:3410–3421. doi: 10.1158/0008-5472.CAN-10-3340. [DOI] [PubMed] [Google Scholar]
- Swarbrick A, Roy E, Allen T, Bishop JM. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc Natl Acad Sci USA. 2008;105:5402–5407. doi: 10.1073/pnas.0801505105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013;5:1228–1242. doi: 10.1016/j.celrep.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CA, Kreso A, Ryan P, Hermans KG, Gibson L, Wang Y, et al. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell. 2012;21:777–792. doi: 10.1016/j.ccr.2012.04.036. [DOI] [PubMed] [Google Scholar]
- Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong S, Itahana Y, Sumida T, Singh J, Coppe JP, Liu Y.et al. (2003Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis Proc Natl Acad Sci USA 10013543–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, et al. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nat Cell Biol. 2009;11:1297–1304. doi: 10.1038/ncb1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya T, Okaji Y, Tsuno NH, Sakurai D, Tsuchiya N, Kawai K, et al. Targeting Id1 and Id3 inhibits peritoneal metastasis of gastric cancer. Cancer Sci. 2005;96:784–790. doi: 10.1111/j.1349-7006.2005.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuno Y, Tsuno NH, Okaji Y, Tsuchiya T, Sakurai D, Nishikawa T.et al. (2010Id1/Id3 knockdown inhibits metastatic potential of pancreatic cancer J Surg Res 16176–82. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ling MT, Wang Q, Lau CK, Leung SC, Lee TK.et al. (2007Identification of a novel inhibitor of differentiation-1 (ID-1) binding partner, caveolin-1, and its role in epithelial-mesenchymal transition and resistance to apoptosis in prostate cancer cells J Biol Chem 28233284–33294. [DOI] [PubMed] [Google Scholar]
- Cheung PY, Yip YL, Tsao SW, Ching YP, Cheung AL. Id-1 induces cell invasiveness in immortalized epithelial cells by regulating cadherin switching and Rho GTPases. J Cell Biochem. 2011;112:157–168. doi: 10.1002/jcb.22911. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang J, Luo JH, Dedhar S, Liu Y. Tubular epithelial cell dedifferentiation is driven by the helix-loop-helix transcriptional inhibitor Id1. J Am Soc Nephrol. 2007;18:449–460. doi: 10.1681/ASN.2006030236. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprez PY, Lin CQ, Thomasset N, Sympson CJ, Bissell MJ, Campisi J. A novel pathway for mammary epithelial cell invasion induced by the helix-loop-helix protein Id-1. Mol Cell Biol. 1998;18:4577–4588. doi: 10.1128/mcb.18.8.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Itahana Y, Moore DH, Bennington JL, Desprez PY. Id-1 and Id-2 proteins as molecular markers for human prostate cancer progression. Clin Cancer Res. 2004;10:2044–2051. doi: 10.1158/1078-0432.ccr-03-0933. [DOI] [PubMed] [Google Scholar]
- Nieborowska-Skorska M, Hoser G, Rink L, Malecki M, Kossev P, Wasik MA, et al. Id1 transcription inhibitor-matrix metalloproteinase 9 axis enhances invasiveness of the breakpoint cluster region/abelson tyrosine kinase-transformed leukemia cells. Cancer Res. 2006;66:4108–4116. doi: 10.1158/0008-5472.CAN-05-1584. [DOI] [PubMed] [Google Scholar]
- Brabletz T. To differentiate or not–routes towards metastasis. Nat Rev Cancer. 2012;12:425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H.et al. (2013The perivascular niche regulates breast tumour dormancy Nat Cell Biol 15807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpert OV, Pili R, Sikder HA, Nelius T, Zaichuk T, Morris C, et al. Id1 regulates angiogenesis through transcriptional repression of thrombospondin-1. Cancer Cell. 2002;2:473–483. doi: 10.1016/s1535-6108(02)00209-x. [DOI] [PubMed] [Google Scholar]
- Straume O, Akslen LA. Strong expression of ID1 protein is associated with decreased survival, increased expression of ephrin-A1/EPHA2, and reduced thrombospondin-1 in malignant melanoma. Br J Cancer. 2005;93:933–938. doi: 10.1038/sj.bjc.6602792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalas W, Yu JL, Milsom C, Rosenfeld J, Benezra R, Bornstein P, et al. Oncogenes and Angiogenesis: down-regulation of thrombospondin-1 in normal fibroblasts exposed to factors from cancer cells harboring mutant ras. Cancer Res. 2005;65:8878–8886. doi: 10.1158/0008-5472.CAN-05-1479. [DOI] [PubMed] [Google Scholar]
- Martini M, Cenci T, D'Alessandris GQ, Cesarini V, Cocomazzi A, Ricci-Vitiani L, et al. Epigenetic silencing of Id4 identifies a glioblastoma subgroup with a better prognosis as a consequence of an inhibition of angiogenesis. Cancer. 2013;119:1004–1012. doi: 10.1002/cncr.27821. [DOI] [PubMed] [Google Scholar]
- Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- López-Rovira T, Chalaux E, Massagué J, Rosa JL, Ventura F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem. 2002;277:3176–3185. doi: 10.1074/jbc.M106826200. [DOI] [PubMed] [Google Scholar]
- Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, et al. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci. 2007;120 Pt 6:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Itoh F, Nishiyama K, Takezawa T, Kurihara H, Itoh S, et al. Inhibition of endothelial cell activation by bHLH protein E2-2 and its impairment of angiogenesis. Blood. 2010;115:4138–4147. doi: 10.1182/blood-2009-05-223057. [DOI] [PubMed] [Google Scholar]
- Mechtcheriakova D, Schabbauer G, Lucerna M, Clauss M, De Martin R, Binder BR.et al. (2001Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor up-regulation via EGR-1 in endothelial cells FASEB J 15230–242. [DOI] [PubMed] [Google Scholar]
- Mechtcheriakova D, Wlachos A, Holzmüller H, Binder BR, Hofer E. Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. Blood. 1999;93:3811–3823. [PubMed] [Google Scholar]
- Kim HJ, Chung H, Yoo YG, Kim H, Lee JY, Lee MO, et al. Inhibitor of DNA binding 1 activates vascular endothelial growth factor through enhancing the stability and activity of hypoxia-inducible factor-1alpha. Mol Cancer Res. 2007;5:321–329. doi: 10.1158/1541-7786.MCR-06-0218. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Rothschild G, Yokota Y, Russell RG, Iavarone A. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol. 2005;25:3563–3574. doi: 10.1128/MCB.25.9.3563-3574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, et al. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science. 2004;306:247–252. doi: 10.1126/science.1102612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- Ruzinova MB, Schoer RA, Gerald W, Egan JE, Pandolfi PP, Rafii S, et al. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell. 2003;4:277–289. doi: 10.1016/s1535-6108(03)00240-x. [DOI] [PubMed] [Google Scholar]
- Li H, Gerald WL, Benezra R. Utilization of bone marrow-derived endothelial cell precursors in spontaneous prostate tumors varies with tumor grade. Cancer Res. 2004;64:6137–6143. doi: 10.1158/0008-5472.CAN-04-1287. [DOI] [PubMed] [Google Scholar]
- de Candia P, Solit DB, Giri D, Brogi E, Siegel PM, Olshen AB.et al. (2003Angiogenesis impairment in Id-deficient mice cooperates with an Hsp90 inhibitor to completely suppress HER2/neu-dependent breast tumors Proc Natl Acad Sci USA 10012337–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- Kerbel RS, Benezra R, Lyden DC, Hattori K, Heissig B, Nolan DJ, et al. Endothelial progenitor cells are cellular hubs essential for neoangiogenesis of certain aggressive adenocarcinomas and metastatic transition but not adenomas. Proc Natl Acad Sci USA. 2008;105:E54; author reply E55. doi: 10.1073/pnas.0804876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan DJ, Ciarrocchi A, Mellick AS, Jaggi JS, Bambino K, Gupta S, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–1558. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellick AS, Plummer PN, Nolan DJ, Gao D, Bambino K, Hahn M, et al. Using the transcription factor inhibitor of DNA binding 1 to selectively target endothelial progenitor cells offers novel strategies to inhibit tumor angiogenesis and growth. Cancer Res. 2010;70:7273–7282. doi: 10.1158/0008-5472.CAN-10-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Zheng L, Wang Q, Bao J, Cai Z, Liu A. The PI3K/Akt pathway upregulates Id1 and integrin α4 to enhance recruitment of human ovarian cancer endothelial progenitor cells. BMC Cancer. 2010;10:459. doi: 10.1186/1471-2407-10-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol Cell Biol. 1997;17:7317–7327. doi: 10.1128/mcb.17.12.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarrocchi A, Jankovic V, Shaked Y, Nolan DJ, Mittal V, Kerbel RS, et al. Id1 restrains p21 expression to control endothelial progenitor cell formation. PLoS One. 2007;2:e1338. doi: 10.1371/journal.pone.0001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, et al. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2012;363:135–145. doi: 10.1007/s11010-011-1166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mern DS, Hoppe-Seyler K, Hoppe-Seyler F, Hasskarl J, Burwinkel B. Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res Treat. 2010;124:623–633. doi: 10.1007/s10549-010-0810-6. [DOI] [PubMed] [Google Scholar]
- Mern DS, Hasskarl J, Burwinkel B. Inhibition of Id proteins by a peptide aptamer induces cell-cycle arrest and apoptosis in ovarian cancer cells. Br J Cancer. 2010;103:1237–1244. doi: 10.1038/sj.bjc.6605897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–273. doi: 10.1016/j.ccr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke E, Perk J, Vider J, de Candia P, Chin Y, Solit DB, et al. Peptide-conjugated antisense oligonucleotides for targeted inhibition of a transcriptional regulator in vivo. Nat Biotechnol. 2008;26:91–100. doi: 10.1038/nbt1366. [DOI] [PubMed] [Google Scholar]
- Ding Y, Wang G, Ling MT, Wong YC, Li X, Na Y.et al. (2006Significance of Id-1 up-regulation and its association with EGFR in bladder cancer cell invasion Int J Oncol 28847–854. [PubMed] [Google Scholar]
- Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007;67:11721–11731. doi: 10.1158/0008-5472.CAN-07-2318. [DOI] [PubMed] [Google Scholar]
- Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer. 2011;11:417. doi: 10.1186/1471-2407-11-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JW, Deed RW, Inoue T, Balzi M, Becciolini A, Faraoni P, et al. Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res. 2001;61:8803–8810. [PubMed] [Google Scholar]
- Yang HY, Liu HL, Liu GY, Zhu H, Meng QW, Qu LD.et al. (2011Expression and prognostic values of Id-1 and Id-3 in gastric adenocarcinoma J Surg Res 167258–266. [DOI] [PubMed] [Google Scholar]
- Iwatsuki M, Fukagawa T, Mimori K, Nakanishi H, Ito S, Ishii H.et al. (2009Bone marrow and peripheral blood expression of ID1 in human gastric carcinoma patients is a bona fide indicator of lymph node and peritoneal metastasis Br J Cancer 1001937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Guo MM, Han P, Lin JZ, Liang FY, Tan GM, et al. Id-1 and the p65 subunit of NF-κB promote migration of nasopharyngeal carcinoma cells and are correlated with poor prognosis. Carcinogenesis. 2012;33:810–817. doi: 10.1093/carcin/bgs027. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Z, Xin D, Chua CW, Wong YC, Leung SC, et al. Prognostic significance of Id-1 and its association with EGFR in renal cell cancer. Histopathology. 2007;50:484–490. doi: 10.1111/j.1365-2559.2007.02637.x. [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Kowalski J, Larson AR, Brock M, Alani RM. Id1 promotes tumor cell migration in nonsmall cell lung cancers. J Oncol. 2010;2010:856105. doi: 10.1155/2010/856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Rizwani W, Li X, Rawal B, Nair S, Schell MJ.et al. (2011ID1 facilitates the growth and metastasis of non-small cell lung cancer in response to nicotinic acetylcholine receptor and epidermal growth factor receptor signaling Mol Cell Biol 313052–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañon E, Bosch-Barrera J, López I, Collado V, Moreno M, López-Picazo JM, et al. Id1 and Id3 co-expression correlates with clinical outcome in stage III-N2 non-small cell lung cancer patients treated with definitive chemoradiotherapy. J Transl Med. 2013;11:13. doi: 10.1186/1479-5876-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponz-Sarvisé M, Nguewa PA, Pajares MJ, Agorreta J, Lozano MD, Redrado M.et al. (2011Inhibitor of differentiation-1 as a novel prognostic factor in NSCLC patients with adenocarcinoma histology and its potential contribution to therapy resistance Clin Cancer Res 174155–4166. [DOI] [PubMed] [Google Scholar]
- Ding R, Han S, Lu Y, Guo C, Xie H, Zhang N, et al. Overexpressed Id-1 is associated with patient prognosis and HBx expression in hepatitis B virus-related hepatocellular carcinoma. Cancer Biol Ther. 2010;10:299–307. doi: 10.4161/cbt.10.3.12454. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Yamagiwa S, Takamura M, Honda Y, Ishimoto Y, Ichida T, et al. Overexpressed Id-1 is associated with a high risk of hepatocellular carcinoma development in patients with cirrhosis without transcriptional repression of p16. Cancer. 2005;104:1037–1044. doi: 10.1002/cncr.21259. [DOI] [PubMed] [Google Scholar]
- Luo KJ, Wen J, Xie X, Fu JH, Luo RZ, Wu QL.et al. (2012Prognostic relevance of Id-1 expression in patients with resectable esophageal squamous cell carcinoma Ann Thorac Surg 931682–1688. [DOI] [PubMed] [Google Scholar]
- Schindl M, Schoppmann SF, Ströbel T, Heinzl H, Leisser C, Horvat R.et al. (2003Level of Id-1 protein expression correlates with poor differentiation, enhanced malignant potential, and more aggressive clinical behavior of epithelial ovarian tumors Clin Cancer Res 9779–785. [PubMed] [Google Scholar]
- Maruyama H, Kleeff J, Wildi S, Friess H, Büchler MW, Israel MA, et al. Id-1 and Id-2 are overexpressed in pancreatic cancer and in dysplastic lesions in chronic pancreatitis. Am J Pathol. 1999;155:815–822. doi: 10.1016/S0002-9440(10)65180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Patel D, Chaudhary J. Id1 and Id3 expression is associated with increasing grade of prostate cancer: Id3 preferentially regulates CDKN1B. Cancer Med. 2012;1:187–197. doi: 10.1002/cam4.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarrocchi A, Piana S, Valcavi R, Gardini G, Casali B. Inhibitor of DNA binding-1 induces mesenchymal features and promotes invasiveness in thyroid tumour cells. Eur J Cancer. 2011;47:934–945. doi: 10.1016/j.ejca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Kebebew E, Peng M, Treseler PA, Clark OH, Duh QY, Ginzinger D.et al. (2004Id1 gene expression is up-regulated in hyperplastic and neoplastic thyroid tissue and regulates growth and differentiation in thyroid cancer cells J Clin Endocrinol Metab 896105–6111. [DOI] [PubMed] [Google Scholar]
- Stighall M, Manetopoulos C, Axelson H, Landberg G. High ID2 protein expression correlates with a favourable prognosis in patients with primary breast cancer and reduces cellular invasiveness of breast cancer cells. Int J Cancer. 2005;115:403–411. doi: 10.1002/ijc.20875. [DOI] [PubMed] [Google Scholar]
- Renné C, Martin-Subero JI, Eickernjäger M, Hansmann ML, Küppers R, Siebert R.et al. (2006Aberrant expression of ID2, a suppressor of B-cell-specific gene expression, in Hodgkin's lymphoma Am J Pathol 169655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J, Ishiwata T, Friess H, Büchler MW, Israel MA, Korc M. The helix-loop-helix protein Id2 is overexpressed in human pancreatic cancer. Cancer Res. 1998;58:3769–3772. [PubMed] [Google Scholar]
- Lee SH, Hao E, Kiselyuk A, Shapiro J, Shields DJ, Lowy A, et al. The Id3/E47 axis mediates cell-cycle control in human pancreatic ducts and adenocarcinoma. Mol Cancer Res. 2011;9:782–790. doi: 10.1158/1541-7786.MCR-10-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Rushing EJ, Hartmann DP, Azumi N. Increased inhibitor of differentiation 4 (id4) expression in glioblastoma: a tissue microarray study. J Cancer. 2010;1:1–5. doi: 10.7150/jca.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umetani N, Mori T, Koyanagi K, Shinozaki M, Kim J, Giuliano AE, et al. Aberrant hypermethylation of ID4 gene promoter region increases risk of lymph node metastasis in T1 breast cancer. Oncogene. 2005;24:4721–4727. doi: 10.1038/sj.onc.1208538. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang XQ, Xu XP, Lin GW. ID4 methylation predicts high risk of leukemic transformation in patients with myelodysplastic syndrome. Leuk Res. 2010;34:598–604. doi: 10.1016/j.leukres.2009.09.031. [DOI] [PubMed] [Google Scholar]
- Yuen HF, Chua CW, Chan YP, Wong YC, Wang X, Chan KW. Id proteins expression in prostate cancer: high-level expression of Id-4 in primary prostate cancer is associated with development of metastases. Mod Pathol. 2006;19:931–941. doi: 10.1038/modpathol.3800602. [DOI] [PubMed] [Google Scholar]