

Summary
Hepatitis C virus (HCV) is one of the major etiologic agents that causes hepatocellular carcinoma (HCC) by generating an inflammatory, fibrogenic, and carcinogenic tissue microenvironment in the liver. HCV-induced HCC is a rational target for cancer preventive intervention because of the clear-cut high-risk condition, cirrhosis, associated with high cancer incidence (1% to 7% per year). Studies have elucidated direct and indirect carcinogenic effects of HCV, which have in turn led to identification of candidate HCC chemoprevention targets. Selective molecular targeted agents may enable personalized strategies for HCC chemoprevention. In addition, multiple experimental and epidemiological studies suggest the potential value of generic drugs or dietary supplements targeting inflammation, oxidant stress, or metabolic derangements as possible HCC chemopreventive agents. While the successful use of highly effective direct-acting antiviral agents will make important inroads into reducing long term HCC risk, there will remain an important role for HCC chemoprevention even after viral cure, given the persistence of HCC risk in persons with advanced HCV fibrosis as shown in recent studies. The successful development of cancer preventive therapies will be more challenging compared to cancer therapeutics because of the requirement for larger and longer clinical trials and the need for a safer toxicity profile given its use as a preventive agent. Molecular biomarkers to selectively identify high-risk population could help mitigate these challenges. Genome-wide, unbiased molecular characterization, high-throughput drug/gene screening, experimental model-based functional analysis, and systems-level in silico modeling are expected to complement each other to facilitate discovery of new HCC chemoprevention targets and therapies.
Keywords: Hepatitis C virus, hepatocellular carcinoma, prevention, carcinogenesis
Introduction
Liver cancer, predominantly hepatocellular carcinoma (HCC), is the second most deadly cancer worldwide (GLOBOCAN 2012, http://globocan.iarc.fr). HCC is the most rapidly increasing cause of cancer-related mortality in the U.S. In contrast to developing countries in the Asia-Pacific regions and sub-Saharan Africa, where hepatitis B virus (HBV) is the major risk factor for HCC, chronic infection with hepatitis C virus (HCV) has been responsible for the increasing HCC incidence in developed countries [1]. It is estimated that approximately 3% of the world population is chronically infected with HCV (WHO, www.who.int). More than one million individuals, representing the “baby boomer” population, are estimated to develop HCV-related cirrhosis, hepatic decompensation, or HCC by 2020, and estimated costs for management of the patients reaches $8.6 billion (non-pharmacological cost only) by 2015 in the U.S. [2]. In Canada, total health care costs associated with HCV are expected to increase by 60% until they peak in 2032 [3]. Given the extremely frequent tumor recurrence even after aggressive treatment (70% after 5 years of surgical resection) and limited treatment options available for advanced-stage liver disease, including liver transplantation, a costly proposition, prevention of HCC development in patients with advanced liver fibrosis may be the most effective strategy to substantially impact patient survival [4]. Prevention of exposure to the risk factors (primary prevention) with vaccination has shown to be an effective measure in reducing HBV-related HCC, although no analogous vaccine is available for HCV [5]. Efforts have been made to prevent HCC in individuals who have already acquired the risk factors (secondary prevention) with no substantial success as of yet. Prevention of HCC recurrence after curative therapies (tertiary prevention) has also been explored because the patients are still at risk for new HCC [4].
In patients with chronic HCV infection, the risk of HCC gradually increases as liver fibrosis progresses. Once cirrhosis is established, the annual incidence of HCC is extremely high (1%-7% per year), although HCC rarely develops in less fibrotic livers [6, 7]. The emergence of highly effective direct-acting antivirals (DAAs) for HCV is expected to reduce HCV-related HCC [8]. However, HCV eradication does not eliminate the risk of HCC, especially when the patients already have advanced liver fibrosis [9]. Although molecular mechanisms of HCV-induced HCC development have not been fully elucidated, these epidemiological observations suggest that the major role of HCV in carcinogenesis is to create a cirrhotic tissue microenvironment that serves as a carcinogenic milieu. In addition, direct carcinogenic effects of HCV proteins have been suggested in a variety of experimental models as additional drivers of HCV-induced HCC development [10]. These findings may lead to discovery of targets for secondary/tertiary HCC prevention strategies. Targets in the mechanisms of fibrosis/cirrhosis-driven carcinogenesis may also be relevant to other etiologies, including HBV, alcohol, and non-alcoholic fatty liver diseases (NAFLD).
In this article, we review the current knowledge regarding molecular mechanisms of HCV-induced hepatocarcinogenesis that potentially provide clues about preventive therapies, and discuss strategies to translate the knowledge into clinical practice to ultimately prevent poor prognosis of HCV-related HCC.
Molecular targets in HCV-induced hepatocarcinogenesis
As HCV is an RNA virus with little potential for integration of its genetic material into the host genome, it is generally assumed that HCV contributes to HCC development in an indirect way, through induction of chronic inflammation, and directly, by means of viral factors. HCV-induced HCC development is a multi-step process that involves establishment of chronic HCV infection, persistent chronic hepatic inflammation, progressive liver fibrogenesis, initiation of neoplastic clones accompanied by irreversible somatic genetic/epigenetic alterations, and progression of the malignant clones in a carcinogenic tissue microenvironment. This process could take 20–40 years (Figure 1), and each step in the process could be a target for prevention of HCC.
Figure 1. Natural history and biological processes in HCV-induced HCC development.
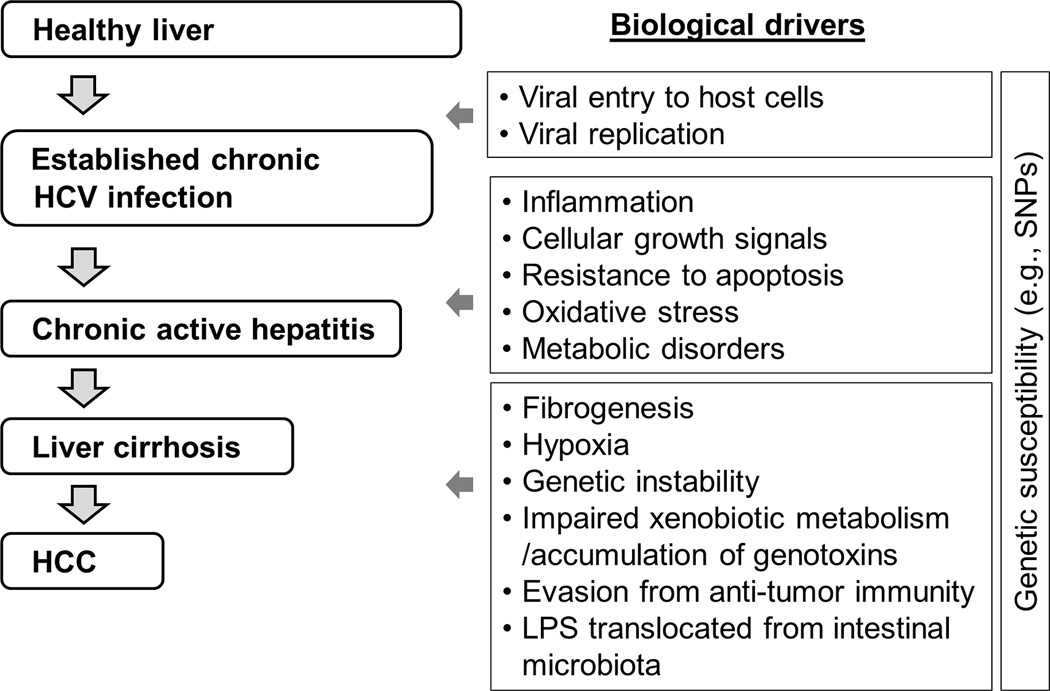
HCV: hepatitis C virus, HCC: hepatocellular carcinoma, LPS: lipopolysaccharide, SNP: single nucleotide polymorphism.
A major obstacle for the understanding of the mechanisms linking HCV infection, inflammation and carcinogenesis is the lack of efficient and convenient model systems to study disease biology. While tremendous progress has been made in recent years regarding the establishment of novel cell culture models to study HCV-host interactions, there are limited in vitro models to study virus-induced liver disease. Moreover, the very narrow host range of HCV, infecting only humans and chimpanzees, so far precludes the study of HCV infection in conventional small animal models. Different mouse models, including HCV transgenic mice, immunodeficient human liver chimeric mice and immunocompetent humanized mice have been developed to study defined aspects of HCV pathogenesis. While these mouse models provided first insights into HCV-induced fibrosis and carcinogenesis, a mouse model that closely mimics human liver disease including HCC is still lacking [11, 12].
Oncogenic effects of HCV proteins
HCV is a single-strand RNA virus in the Flaviviridae family that encodes structural (core, E1, E2) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) [13]. The viral particle is formed by a nucleocapsid comprising the core protein and viral genome and an envelope consisting of envelope glycoproteins E1 and E2. Following viral infection the cellular expression of the nucleocapsid core protein localizes in the cytosol, lipid droplets, endoplasmic reticulum/Golgi apparatus, mitochondria and nuclei, and has been suggested to affect a variety of cellular functions. The envelope glycoproteins (E1 and E2) are involved in interaction with host cells and viral entry, and potential targets for vaccine development [14, 15]. NS3 has serine protease and helicase activities, and cleaves downstream NS proteins together with NS4A. NS4B is a component of a membrane-associated cytoplasmic HCV replication complex. NS5A is an indispensable factor in the HCV replication complex and virion assembly. NS5B, an RNA-dependent RNA polymerase, synthesizes viral RNA. Due to inability to stably integrate into the host genome, in contrast to HBV, HCV requires continuous replication for its viability. There are several clinical data suggesting the role of HCV viral factors in disease progression such as more frequent steatosis in genotype 3 and more frequent HCC development in genotype 1b, although some of the evidences are conflicting [16–19]. Nevertheless, several experimental models have suggested direct oncogenic effects of HCV proteins (Figure 2).
Figure 2. Interactions of HCV with cellular components in cirrhotic tissue microenvironment that promote hepatocarcinogenesis.

Potential HCC chemoprevention targets are extracted from the broader pathogenic involvement of HCV in the development of hepatitis, fibrosis, and cirrhosis. HCV proteins directly or indirectly promote cellular proliferation and survival, induce inflammation, metabolic pathway deregulation leading to steatosis, oxidative stress, endoplasmic reticulum (ER) stress, DNA damage and genetic instability, expansion of tumor-initiating cell (TIC)/cancer stem cell (CSC), fibrogenesis by activating hepatic stellate cells, and attenuate immune cell response leading to immune evasion. Genes/proteins and pathways for which pharmacological interventions have been clinically evaluated (not limited to liver diseases) are highlighted in bold. HCV: hepatitis C virus, HCC: hepatocellular carcinoma, ECM: extracellular matrix.
Cellular proliferation and survival pathways
Artificial over-expression of HCV proteins, e.g., core, NS3, and NS5A promotes cellular proliferation, transformation, anchorage-independent growth, and/or tumor formation in mice, suggesting their direct contribution in activating oncogenic molecular pathways [20–23]. The core protein inhibits tumor suppressor genes TP53, TP73, and RB1 as well as negative regulator of cell cycle such as CDKN1A (also known as p21/CIP) through physical interaction, modulation of regulatory networks, or post-translational modifications [24–27]. NS3 and NS5A also inhibit TP53 [28, 29], and NS5B inhibited RB1 [30]. HCV core, E2, NS5A, and NS5B activate cellular proliferative RAF/MAPK/ERK kinase pathways and E2F1 pathway, which are associated with more aggressive biological phenotype of HCC tumors [26, 30–33]. HCV proteins such as core are known to induce generation of reactive oxygen species (ROS) and transactivate MAPK and AP1 pathways [34]. Insulin-like growth factor signaling is activated via IGF1R in early stage HCV-related HCC [35]. NS5A was found to be involved in activation of PI3K/AKT and beta-catenin/WNT pathways, and evasion from apoptosis by caspase-3 inhibition [36]. Transforming growth factor-beta (TGF-beta) is elevated in the serum of chronic hepatitis C patients [37]. HCV core directly interacts with Smad3 and inhibits the tumor suppressor activity of TGF-beta pathway [38]. YAP1 and IGF2BP3 expressed in TLR4/NANOG-dependent tumor-initiating stem-like cells (TICs) also inhibit the tumor suppressing role of TGF-beta pathway in HCV-related HCC [39]. NS5A inhibits TGF-beta signaling by preventing nuclear translocation of Smad proteins, resulting in down-regulation a tumor suppressor CDKN1A [40]. NS5A down-regulates abnormal spindle-like, microcephaly-associated (ASPM), a regulator of mitotic spindle, and induces mitotic dysregulation and chromosomal instability [41]. NS5A inhibits tumor necrosis factor-alpha (TNF-alpha) mediated apoptosis [42]._ENREF_118 HCV induces cancer stem cell-like gene signatures in cell culture and murine tumor xenografts through DCLK1 [43].
Retinoid X receptor-alpha (RXR-alpha), activated by RAF/MAPK signaling, is a nuclear receptor for retinoids, vitamin A analogs, involved in cell growth, differentiation, and apoptosis [44]. Acyclic retinoid counteracts this process and induces apoptosis. Silymarin, a herbal flavonoid, induces cell cycle arrest and apoptosis in HCC cells, suppresses N-nitrosodiethylamine (NDEA)-induced hepatocarcinogenesis in rats, and shows anti-HCV activity [45, 46]. An observational study showed that silymarin use was associated with reduced fibrosis progression, but an association with HCC incidence was not obvious during the follow up of 5.5 years [47].
Genetic instability
Structural alterations of host genomic DNA, including somatic oncogenic mutations and deletions of tumor suppressor genes, are major drivers of carcinogenesis. HCV core inhibits mitotic spindle checkpoint function by reducing RB1, and increases chromosomal polyploidy [27]. Chronic oxidative stress induced by the core also leads to mitochondrial and chromosomal DNA damage, leading to HCC development [34]. NS3/4A interacts with ATM, a cell cycle checkpoint kinase, and impairs DNA damage repair [48]. Perturbations of the endoplasmic reticulum (ER) lead to an evolutionarily conserved cell stress response called the unfolded protein response (UPR) to compensate for damage or eventually trigger cell death when ER dysfunction is severe or prolonged. HCV has been shown to induce ER stress [49]. It has been hypothesized that persistent ER stress induction could predispose a cell to mutagenesis secondary to intracellular and extracellular accumulation of DNA-damaging factors.
Immune response, inflammation pathways
Interferon pathway activation is a well-known innate immune response to HCV infection, and recent studies have elucidated its role in anti-tumor immunity [50]. The nuclear factor kappa-B (NF-kappaB) pathway was implicated in HCC development especially during progression of initiated tumor clones [51], although there is somewhat conflicting evidence regarding its role in hepatocarcinogenesis [52]. HCV core protein inhibits NF-kappaB-mediated immune responses [53]. The c-Jun N-terminal kinase (JNK) pathway, activated in non-parenchymal liver cells by proinflammatory signals such as reactive oxygen species (ROS), generates an inflammatory hepatic microenvironment that supports HCC development [54]. NS5A activates the JNK pathway through interaction with TRAF2 [55]. A JNK inhibitor, SP600125, suppressed HCC development in diethylnitrosamine-treated rats [56]. Selective inhibition of cyclooxygenase-2 (COX2) prevents HCC in an experimental animal model [57]. Liver-specific expression of lymphotoxin (LT)-alpha and beta in mice caused hepatic inflammation and HCC, which was suppressed by inhibition of LT beta receptor [58].
Viral proteins also appear to subvert innate immune pathways. NS3 suppresses innate immunity by cleavage of a mitochondrial antiviral signaling protein (MAVS) responsible for induction of type-I interferon [59]. The _ENREF_61inhibition of natural killer cells by E2 may contribute to immune evasion and establishment of chronic infection [60]. Interleukin-6 (IL6) is a multifunctional cytokine involved in estrogen-regulated liver carcinogenesis [61]. Extracellular HCV core protein was suggested to impair antigen-presenting cells via IL-6 pathway [62].
Metabolic pathways
Clinically, HCV-related HCC is often accompanied by steatosis within the tumors and non-tumor liver, suggesting modulation of metabolic pathways [63]. HCV core protein co-localizes with apolipoprotein A2 on the surface of triglyceride in vitro and in vivo, suggesting its association with lipid metabolism [64]. Transgenic mice that express core protein develop progressive steatosis in the liver and then HCC [23]_ENREF_70. Insulin resistance is another feature of the HCV core transgenic mice, which results in lipid accumulation in the liver [65]. HCV core protein suppresses microsomal triglyceride transfer protein (MTTP) activity and interferes with hepatic assembly and secretion of triglyceride-rich very low density lipoproteins (VLDL), further contributing to steatosis [66]. HCV core protein interacts with RXR-alpha and peroxisome proliferator-activated receptor-alpha (PPAR-alpha), and modulates cell differentiation, proliferation and fatty acid transport and catabolism in mice [67]. PPAR-alpha generally ameliorates steatosis, but in the presence of HCV core-induced mitochondrial dysfunction, it exacerbates steatosis, induces oxidative stress, and increases cell growth signals [68].
Cellular senescence
Hepatocyte proliferation is generally decreased at the stage of cirrhosis after many rounds of regeneration accompanied by telomere shortening that triggers cellular senescence though ATM, TP53, and CDKN1A as a safeguard to prevent carcinogenesis [69]. Activating somatic mutations in telomerase reverse-transcriptase (TERT) promoter is a frequent early neoplastic event in HCC with mixed etiologies including HCV [70]. HCV core protein overcomes stress-induced hepatocyte senescence by down-regulating CDKN2A expression via DNA methylation [71]. Senescence of hepatic stellate cells has also been shown to limit liver fibrosis [72]. HCV does not infect stellate cells but could have an indirect role in this process.
Fibrogenic pathways
Irrespective of the etiology, established cirrhosis serves as a milieu/microenvironment that fosters initiation and promotion of neoplastic clones by facilitating genetic aberrations and cellular transformation, which is often referred to as “field cancerization” or “field effect” [73]. Liver fibrosis is an excessive wound healing response to chronic liver injury that results in increased production and deposition of extracellular matrix (ECM). Dynamic balancing between fibrogenesis and fibrolysis determines liver fibrosis as a result of a complex interplay between various cell types in the liver, including hepatic stellate cell, Kupffer cell, hepatocytes, cholangiocytes, sinusoidal endothelial cells, and infiltrating immune cells. Severity of liver fibrosis is tightly correlated with increasing risk of HCC in patients with chronic HCV infection, suggesting that cirrhosis-driven carcinogenesis is the major mechanism in the development of HCV-related HCC [6, 74]. Although sustained virological response (SVR) from HCV improves histological fibrosis, a subset of patients is still at risk of fibrosis progression and HCC development [75], indicating the necessity of anti-fibrotic therapies to prevent HCC [76].
Activation of the hepatic stellate cell, or myofibroblast, is the major driver of liver fibrogenesis [77]. HCV broadly infects hepatocytes, monocytes, lymphocytes and other secretory cells, and contributes to stellate cell activation. HCV core and nonstructural proteins stimulate profibrogenic mediators such as TGF-beta [78]. HCV infection induces TGFB1 through ROS production, p38 MAPK, JNK, ERK, and NF-kappaB pathways [79], although concerns regarding toxicities have been raised about targeting the TGF-beta pathway exclusively [80]. Platelet-derived growth factor (PDGF) is the most potent mitogenic signal, inducing expression of beta PDGF receptor expression in stellate cells together with other cell surface receptors of growth signaling such as integrins [81]. Transgenic mice expressing PDGF-C develop liver fibrosis and HCC [82], and an acyclic retinoid, peretinoin, represses fibrosis and HCC development in the model [83].
In cell culture models, HCV non-structural proteins and, to a lesser extent, core protein stimulate production of proinflammatory chemokines such as IL-8, MCP-1 and RANTES and induce expression of ICAM-1, a cell adhesion molecule known to activate T cells [78]. JNK pathway activation by the proinflammatory cytokine IL1-beta can shift TGF-beta signaling from tumor suppression to oncogenesis through accelerated fibrogenesis [84, 85]. IL32 expression in hepatocytes is associated with hepatic inflammation and fibrosis in HCV infection [86]. Hepatocyte death serves as a stimuli activating stellate cells [87], and is a potential therapeutic target in chronic hepatitis C [88]. Apoptotic bodies with HCV infection could amplify fibrogenic signals [89]. Bacterial lipopolysaccharide (LPS) permeabilized from intestinal microbiota elicits fibrogenic response and carcinogenesis through Toll-like receptor 4 (TLR4) expressed on stellate cells by inducing TGF-beta, which can be prevented by gut sterilization [90]. Multiple variants in TLR4 modulate risk of fibrosis in HCV-infected Caucasian patients [91]. Matrix metalloproteinase-2 (MMP2), a major ECM-degrading enzyme, is induced by interaction of E2 with CD81, a member of receptor complex for HCV cellular internalization, which may exacerbate inflammatory infiltration and parenchymal damage [92].
Adipokines including leptin, adiponectin, and resistin are implicated in liver fibrogenesis in hepatitis C and NAFLD [93]. Suppression of heat shock protein (Hsp) 47, a collagen-specific chaperon, by siRNA in stellate cells reduced fibrosis in rodent models of fibrosis, and is now under early clinical evaluation [94]. Renin-angiotensin system (RAS) is suggested to be involved in hepatocarcinogenesis [95]. Inhibition of angiotensin-II (AT-II) by angiotensin-converting enzyme inhibitor (ACE-I) down regulates angiogenic factors such as VEGF, and ACE-I administration combined with branched-chain amino acids (BCAA) has been shown to attenuate insulin resistance-related hepatocarcinogenesis in a diabetic rat model [96].
However, it is important to note that most of these findings have been derived from experimental cell culture or animal models over-expressing individual proteins. Since a robust infectious small animal model recapitulating the virus-induced carcinogenesis is not yet available [11], the functional relevance of these observations for hepatocarcinogenesis in humans is still unclear and needs to be confirmed. The development of immunocompetent animal models fully recapitulating the viral life cycle and virus-induced liver disease in combination with studies in liver tissue from HCV-infected patients will ultimately be required to validate these findings and concepts. Also, ancillary assessment of HCC development as an additional endpoint in clinical trials of the anti-fibrotic agents may provide insight into their potential role as HCC chemoprevention therapies [97].
Host factors affecting susceptibility to HCV-related HCC
Growth signaling pathways
Kinase signaling pathways represent druggable/targetable molecular pathways that have been extensively studied. In HCV-related HCC, genome-wide profiling of genomic DNA variants as well as RNA transcripts has identified several candidate genes and pathways. Epidermal growth factor (EGF) is a mitogen involved in cellular growth, proliferation, differentiation, and carcinogenesis. In rodent models of cirrhosis-driven HCC, the EGF pathway was activated in hepatic stellate cells, and pharmacological inhibition with a small molecule EGF receptor (EGFR) inhibitor, erlotinib, regressed fibrosis and inhibited HCC development [98]. Interestingly, there was no inhibition of EGF pathway in the tumors, suggesting that the HCC preventive effect was through regression of the cirrhotic tissue microenvironment that supports initiation of neoplastic clones. In contrast, another small molecule EGFR inhibitor, gefitinib, suppressed growth of initiated HCC clones in rats [99]. EGFR was recently identified as a co-factor for HCV cellular entry, and erlotinib inhibited HCV infection, suggesting its role as anti-HCV drug [100, 101]. The role of EGF pathway in HCV-related liver diseases might be complicated though because HCV infection induces the expression of other EGFR ligands such as amphiregulin (AREG) and heparin-binding EGF-like growth factor (HBEGF), and while AREG enhances liver fibrosis, HBEGF suppresses liver fibrosis [102–105]. A multi-kinase inhibitor, sorafenib, improved portal hypertension in cirrhosis patients, supposedly due to its anti-angiogenic activity [106]. Sorafenib showed anti-HCC effect by blocking paracrine hepatocyte growth factor (HGF) from stromal cells in response to vascular endothelial growth factor-A (VEGFA) secreted from HCC cells [107]. Selective inhibitors of these growth signaling pathways have been clinically evaluated mostly as cancer therapeutics. There may be opportunities to repurpose this class of drugs for HCC chemoprevention if the toxicity concern is satisfactorily addressed.
Immune pathways
An IL28B variant (rs12979860), initially identified as an interferon response predictor [108], may be associated with increased risk of HCV-related HCC [109]. Interferon effector genes (IEGs) such as BCHE were identified through high-throughput RNAi screening [110]. A genome-wide association study (GWAS) comparing HCV-related HCC patients with chronic hepatitis C patients in Japan identified a SNP in MHC class I polypeptide-related sequence A (MICA) (rs2596542), which is involved in response of dendritic cells to type-I interferon in chronic hepatitis C [111, 112]. Another SNP in MICA promoter (rs2596538) was associated with increased serum soluble MICA protein [113]. Because the controls are patients without cirrhosis, it is possible that the variants indirectly contribute to carcinogenesis through increased inflammation and/or fibrogenesis [114]. A subsequent study in Caucasian hepatitis C patients in Switzerland did not replicate the association with HCC for this locus, but for a nearby locus in HCP5 (rs2244546), suggesting that the MICA/HCP5 region contains a potential susceptibility locus [115]. Additional GWAS-identified locus in another Japanese patient series is in DEPDC5 (rs1012068) [116], which was not replicated in the Caucasian patients [115].
Metabolic pathways
A SNP in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene (rs738409) associated with alcoholic and non-alcoholic steatohepatitis may have weak association with HCV-related HCC [117]. In patients with chronic hepatitis C with advanced fibrosis, positive association between liver iron deposition and higher incidence of HCC and poor prognosis was observed [118]. Hepatic iron overload was associated with elevated levels of 8-hydroxy-2’-deoxyguanosine (8-OHdG), which signifies hepatic oxidative DNA damage in patients with chronic hepatitis C [119]. With an excess iron diet, transgenic mice expressing HCV polyprotein showed development of hepatic steatosis, ultrastructural alterations of mitochondria, and HCC accompanied with elevated levels of hepatic 8-OHdG [120]. HFE gene mutations, in particular H63D, were associated with increased SVR [121].
microRNA
microRNAs (miRNAs) are small non-coding RNA that negatively regulate gene expression by binding to complementary sites within the 3’UTR of multiple target protein-coding mRNAs. miRNA expression profiling of HCV-related HCC tissues revealed deregulated miRNAs including MIR122, MIR100, MIR10A, MIR198, MIR145, and MIR517A as well as distinct expression pattern compared to HBV-related HCC [122–124]. MIR122 is a liver-specific miRNA that promotes replication of HCV [125]. In contrast, MIR122 is under-expressed in HCC and associated with more aggressive biological phenotype including over-expression of alpha-fetoprotein [126]. Therapeutic delivery of MIR122 inhibit MYC-driven mouse HCC [127]. Infection of HCV genotypes 1a, 1b, and 2a in primary human hepatocytes revealed that MIR141 targets a tumor suppressor gene DLC1 [128].
Prevention of HCV-induced HCC
It has been noted that early detection and prevention are the most effective and rational approach to substantially impact prognosis of cancer patients rather than starting the treatment at advanced/terminal stage [129]. However, development of cancer prevention therapies is more challenging compared to cancer therapeutics due to the requirement for larger and longer clinical trials because of the lower incidence of clinical events. In addition, a safer toxicity profile is required as preventive medicine administered to asymptomatic, cancer-free patients potentially for long durations. HCV-related HCC is one of the most rational targets for cancer preventive intervention because of the well-established risk factor, HCV infection and cirrhosis, which in fact enabled conduction of cancer chemoprevention trials with significantly smaller sample size compared to other cancer types [130–133]. Although the trials failed to demonstrate satisfactory effect and toxicity profile as a standard of care, the HCC preventive effect in patients with established or more advanced cirrhosis provides the proof of concept of HCC chemoprevention as a valid option for further exploration.
Molecular biomarkers of HCC risk in HCV-related cirrhosis
Molecular biomarkers of HCC risk and/or poor prognosis will enable further enrichment of the high-risk population and boost statistical power in HCC chemoprevention trials [134]. HCC risk biomarkers will also significantly contribute to improvement of early HCC detection. The current practice guidelines recommend regular tumor surveillance with biannual ultrasound to increase the opportunity to identify lesions at a stage where potentially curative radical therapies can be applied [135]. However, the sizable cirrhosis population poses a challenge in implementing the surveillance program: only 12% of new HCV-related HCC patients are diagnosed through the surveillance in the U.S. [136] Growing numbers of early-stage, asymptomatic cirrhotics identified by non-invasive fibrosis detection methods such as elastography will also add to the HCC screening burden [137]. Clinical variable-based prediction models for HCC development have been explored, although their performance is limited and none of them has been established in practice [138, 139].
Numerous germline SNPs have been reported as HCC risk variants, although very few of them are replicated in independent patient series/cohorts [140]. The EGF 61*G allele (rs4444903) was associated with HCC risk in a prospective cohort of patients with HCV-related advanced fibrosis or cirrhosis with a hazard ratio (HR) of 2.10 for G/G genotype in comparison to A/A (Table 1) [141, 142]. Despite diverse allele frequency across patient populations, association between the EGF genotype and HCC risk remains significant and independent of patient race [143]. A SNP in an antioxidant enzymes, myeloperoxidase, (MPO −463*G, rs2333227) was associated with HCC risk in a prospective study (HR=2.80) [144]. A panel of 7 SNPs (Cirrhosis Risk Score) was shown to be associated with the risk of fibrosis progression in male Caucasian patients with chronic hepatitis C, although association with longer term outcomes including HCC is yet to be determined [145]. A 186-gene-expression signature was associated with HCC risk in prospectively followed patients with early-stage HCV-related cirrhosis (HR=2.65) [146]. Annual HCC incidence in patients with poor-prognosis signature (5.8%) was nearly 4 times higher than the incidence in patients with good-prognosis signature (1.5%). The signature reflects activation of NF-kappa-B, IL6, EGF, and interferon pathways, suppression of DNA damage repair genes such as GSNOR, and hepatic stellate cell activation. In rodent models of cirrhosis-driven HCC, the signature was induced from the inception of liver fibrosis, reversed in response to an EGFR inhibitor, erlotinib, and accompanied its HCC chemopreventive effect, suggesting its role as a pharmacogeomic companion biomarker [98]. With the recent emergence of highly selective molecular targeted agents, tissue-based assessment of predictive biomarker for response is now recommended in practice guidelines [135]. Circulating cells or biomolecules such as miRNAs may be alternative sources to obtain similar molecular information less invasively [147]. In addition, molecular imaging of collagen could potentially be used to monitor fibrosis regression, which may correlate with decreased HCC risk [148].
Table 1.
Molecular biomarkers of HCV-related HCC risk.
| Molecular biomarker | Type | No. of patients |
HR | Race/ethnicity | Reference |
|---|---|---|---|---|---|
| EGF 61*G (rs4444903) | SNP | 816 | 2.10 | White, Hispanic, Black, Asian |
[141] |
| MPO −463*G (rs2333227) | SNP | 205 | 2.80 | White | [143] |
| CAT −262*C (rs1001179) | SNP | 205 | 1.74 | White | [143] |
| 186-gene poor prognosis signature |
Gene expression | 216 | 2.65 | White, Hispanic, Black, Asian |
[145] |
HCV: hepatitis C virus, HCC: hepatocellular carcinoma, HR: hazard ratio
Molecular biomarkers demonstrating HR>1.50 in independent prospective or prospective-retrospective cohort (n>100) are shown.
rs numbers indicate accession numbers in NCBI dbSNP database (www.ncbi.nlm.nih.gov/snp).
Strategies to prevent HCV-related HCC
Primary prevention, i.e., HCV vaccination, is as of yet unavailable due to the high variability in the viral genomic structure and envelope proteins, the large number of quasispecies, and the lack of neutralizing antibody. Secondary prevention aims at preventing HCC development in established HCV-related advanced fibrosis or cirrhosis. To date, several relatively large phase 3 trials have been conducted, which demonstrated limited efficacy and utility of the tested therapies [130–132]. Tertiary prevention targets recurrence of de novo second primary HCC after curative treatment of initial primary HCC, but available evidence is still scarce [149–151]. Theoretically, secondary and tertiary prevention could be achieved by anti-HCV therapies and/or non-etiology-specific therapies targeting inflammation, fibrogenesis, and/or carcinogenesis, which have been extensively studied in the past decades. However, there are still several undetermined study design issues, including appropriate sample size, study duration, and elusive primary and surrogate study endpoints according to the preventive strategies. These points need to be clarified to streamline and facilitate design and conduction of HCC chemoprevention trials in HCV cirrhosis. Targeted disease stage/severity, e.g., compensated or decompensated cirrhosis, should be specified in inclusion criteria especially in secondary prevention trials because of the distinct difference in expected outcome. Enrichment of high-risk patients with the use of HCC risk biomarkers and/or prognostic indices is critical to boost HCC incidence and keep required sample size and study duration within practically feasible range. Testing candidate chemoprevention therapies in the setting of tertiary, instead of secondary, prevention could be a way to further boost HCC incidence because post-surgical recurrence is approximately three times more frequent compared to the first HCC in cirrhosis, although diagnosis of de novo HCC recurrence should be unambiguously determined based on explicit criteria [152]. Also, a consensus needs to be developed on acceptable toxicities in the context of preventive intervention in patients with advanced fibrosis or cirrhosis.
Anti-HCV therapies
Recent clinical trials have reported SVR rates greater than 90% with the use of DAA-based interferon-free regimens even in patients with cirrhosis [153, 154]. Interferon-based therapies have shown that SVR is consistently associated with gradual regression of fibrosis and lower risk of HCC in retrospective studies [9, 155]. However, the clinical utility of achieving SVR with the use of anti-HCV therapies in the context of HCC prevention needs to be clarified especially in patients with comorbid conditions, e.g., decompensated cirrhosis and older age, in future studies. It also needs to be determined whether DAAs have any role in tertiary prevention. Nevertheless, the cost of DAAs could be prohibitive in their use as preventive drugs. Also, because the patients are still at risk of HCC even after SVR, additional measures of secondary/tertiary prevention are needed. In liver transplantation for HCV-related HCC, HCV reinfection in grafted liver could lead to progressive fibrosis and de novo HCC, which may be prevented by inhibition of HCV entry [100].
Non-etiology-specific HCC chemoprevention
Anti-inflammatory, immune therapies
Suppression of hepatic inflammation could delay disease progression and reduce HCC risk; biochemical response, i.e., normalization of liver enzymes such as alanine aminotransferase (ALT), achieved by either glycyrrhizin or ursodeoxycholic acid (UDCA), have been suggested to reduce HCC risk [4]. Interferon has been extensively evaluated as a chemopreventive agent in HCV-related HCC. In two relatively large randomized trials of maintenance low-dose interferon, HCC risk was modestly reduced in patients with more advanced fibrosis/cirrhosis (HALT-C trial), and composite of first liver-related clinical events was reduced in patients with portal hypertension (EPIC3 trial) in post hoc subgroup analyses [131, 156]. However, the modest effects and poor tolerability (nearly 40% drop out and excess mortality in HALT-C trial) of Peg-interferon preclude its wide application as standard of care. The HCC suppressive effect in these studies was not evident during the first two to three years of treatment, which may reflect latent period for newly initiated cancer clones to be clinically detected. Interferon has been also assessed as tertiary prevention in retrospective and prospective studies, which consistently showed a trend of reducing post-treatment recurrence or death [4]. Immunosuppression after liver transplantation with sirolimus, an mTOR inhibitor, reduced HCC recurrence and improved survival [157]. Result of an ongoing multicenter trial of sirolimus (SiLVER study) is anticipated (Table 2). Aspirin may elicit cancer preventive effect through inhibition of COX2, although there are conflicting data about HCC chemopreventive effect with COX2 inhibition [158].
Table 2.
On-going HCC chemoprevention trails relevant to HCV-related HCC.
| Trial number | Agent | Type of agent | Phase | Type of prevention |
Participants | Comple tion |
|---|---|---|---|---|---|---|
| NCT00513461 | S- adenosylmethionine (SAMe) |
Dietary supplement |
2 | Secondary | Advanced chronic hepatitis C |
Dec 2013 |
| NCT01956864 | High-dose vitamin D |
Dietary supplement |
1 | Secondary | Cirrhosis without HCC |
Sept 2014 |
| MAY2013-02-02 | Erlotinib | Kinase (EGFR) inhibitor |
1 | Secondary /Tertiary |
HCC after resection |
2015- 2016 |
| NCT00355862 | Sirolimus (SiLVER trial) |
Immune modulator |
3 | Tertiary | HCC after transplantation |
May 2014 |
| NCT01924624* | Thalidomide | Immune modulator, anti- angiogenesis |
n/a | Tertiary | HCC after resection |
Dec 2019 |
| NCT01717066* | Ginsenoside Rg3 |
Chemo- sensitizer, anti- angiogenesis |
n/a | Tertiary | HCC after resection |
May 2015 |
| NCT01770431* | Huaier Granule | Traditional herbal medicine |
4 | Tertiary | HCC after resection |
Dec 2014 |
| NCT01964001* | Vitamin B6, Coenzyme Q10 |
Dietary supplement |
2/3 | Tertiary | HCC after resection |
Dec 2015 |
HCC: hepatocellular carcinoma, HCV: hepatitis C virus, EGFR: epidermal growth factor.
Likely enroll mainly hepatitis B virus-infected patients.
From www.ClinicalTrials.gov and cancerpreventionnetwork.org accessed May 2014. Verified trials after 2012 are shown.
NCT: National Clinical Trials ID, MAY: Cancer Prevention Network DCP protocol ID.
Treatment of metabolic disorders, dietary supplements
Statins, HMG-CoA reductase inhibitors, have been suggested to have anti-proliferative effect through inhibition of RAS/MAPK and cell cycle pathways and pro-apoptotic effect. Observational studies suggest HCC preventive effect by statins, which is not yet verified in a clinical trial [159]. Diabetes is associated with prognosis in HCV-related cirrhosis, and an anti-diabetic drug, metformin, inhibits mTOR pathway by activating AMPK, may reduce HCC risk and improve survival [160]. Coffee and a green tea polyphenol, epigallocatechin gallate (EGCG) show modest HCC preventive effect supposedly by activating anti-oxidant and detoxification pathways in experimental and epidemiological studies [4, 161]. EGCG is also reported to inhibit HCV entry [162]. S-adenosylmethionine (SAMe), a major methyl donor inhibiting hepatocyte growth factor (HGF), is being tested in HCV-related HCC for AFP reduction in a phase 2 trial (Table 2). Other phytochemicals such as curcumin, resveratrol, silymarin, and genistein showed HCC preventive effect in animal models, but clinical evidence in HCV-infected patients is limited [4]. HCC preventive effect of this type of drugs is generally expected to be modest. Therefore, enrichment of high-risk patients as well as utilization of epidemiological data/resources will be critical in determining their clinical utility.
Molecular targeted agents
Given the rapidly expanding inventory of selective molecular targeted agents newly synthesized or identified through high-throughput screening, molecular targeted cancer chemoprevention is now an increasingly feasible option. An acyclic retinoid, peretinoin, was tested in a large-scale phase 3 trial enrolling HCV-related cirrhosis, which showed modest HCC preventive effect [132]. Interestingly, the HCC reduction was observed after 2 years of enrollment as seen in the previous interferon trials. The multi-kinase inhibitor, sorafenib, was tested in the setting of tertiary prevention, although no net HCC preventive effect was observed (Table 2). It is assumed that the “all-comer” approach without biomarker-based enrichment is the major basis for failure [163]. Nevertheless, a post hoc exploration of predictive biomarkers is currently underway. An EGFR inhibitor, erlotinib, is being tested in a phase 1 trial, in which the 186-gene signature is assessed as a companion biomarker (Table 2). A clinical trial of another EGFR inhibitor, gefitinib, is also registered.
New chemopreventive targets in HCV-related HCC
Genome-wide profiling of various biomolecules and high-throughput drug screening have facilitated unbiased, large-scale survey of new molecular targets and therapeutics [164]. in vivo high-throughput RNAi screening will be another powerful tool to identify functional targets [165]. Recent advancement in in vitro and in vivo modeling of HCV infection has allowed more physiological and functional assessment of HCV-host interactions and the viral life cycle to identify and verify candidate target genes and pathways [13]. Transcriptome signatures have been successfully utilized to identify new drugs or indications, i.e., drug repurposing, in variety of diseases [166]. Regulatory transcriptome network analysis could be a complementary approach in identifying key driver genes in hepatocaricinogenesis [167]. Genome-scale mathematical metabolic modeling of hepatocyte led to identification of serine deficiency as potential target in non-alcoholic steatohepatitis-related HCC [168]. This may suggest potential utility for the construction of a model of the HCV-infected hepatocyte to explore HCC chemoprevention targets.
Conclusions
HCV-related HCC will remain a major health problem in the coming decades. Although prevention of HCV-induced HCC is not yet established, direct and indirect oncogenic roles of HCV and candidate targets genes and molecular pathways have been suggested in experimental and clinical studies. Integration of genome-wide characterization, high-throughput and unbiased target/drug screening against libraries of RNAi/selective targeted agents, and more physiological HCV infection and liver disease models is expected to facilitate development of molecularly targeted HCC chemoprevention, which may be widely applicable to cirrhosis-driven HCC caused by other etiologies as well as inflammation-driven cancer in other organs such as gastric, cervical, and colon cancers. Clinical assessment of antiviral, anti-inflammatory, and anti-fibrosis drugs in the context of HCC chemoprevention will be a challenge. Molecular biomarkers that could be used to select target patients and/or predict response will be the key in designing clinically feasible trials of HCC chemoprevention therapies.
Key points.
HCV-induced HCC is a model of chronic inflammation-driven cancer, where complex interactions between multiple cell types form carcinogenic tissue microenvironment that foster and promote progression of neoplastic clones.
Recent clinical data suggest that HCV eradication does not eliminate the risk of HCC development especially when the patients have more advanced fibrosis, indicating necessity to develop HCC prevention therapies to improve patient prognosis.
Direct and indirect oncogenic effects of HCV have been identified as potential targets to prevent disease progression to HCC development by using various, mostly cell culture-based, experimental systems.
Better in vitro, in vivo, and ex vivo experimental models of HCV infection are needed to study molecular mechanisms of HCV-induced hepatocarcinogenesis under more physiological conditions.
Molecular biomarkers of HCC risk will help clinical translation of molecular targeted chemoprevention therapies for HCV-induced HCC.
Acknowledgments
Financial support:
This research was supported by National Institute of Health (DK099558 to Y.H., CA140861 to B.C.F., DK078772 to R.T.C.), European Commission Framework Programme 7 (Heptromic, proposal number 259744 to Y.H.), French Cancer Foundation (ARC IHU201301187 to T.F.B.), French Research Agency (LABEX ANR-10-LAB-28 to T.F.B.), European Commission (ERC-2008-AdG-233130-HEPCENT, INTERREG-IV-Rhin Supérieur-FEDER-Hepato-Regio-Net 2012 to T.F.B.)
Footnotes
Conflict of interest
The authors declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
References
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson IM, Davis GL, El-Serag H, Negro F, Trepo C. Prevalence and challenges of liver diseases in patients with chronic hepatitis C virus infection. Clin Gastroenterol Hepatol. 2010;8:924–933. doi: 10.1016/j.cgh.2010.06.032. quiz e117. [DOI] [PubMed] [Google Scholar]
- 3.Myers RP, Krajden M, Bilodeau M, Kaita K, Marotta P, Peltekian K, et al. Burden of disease and cost of chronic hepatitis C infection in Canada. Canadian journal of gastroenterology & hepatology. 2014;28:243–250. doi: 10.1155/2014/317623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoshida Y, Fuchs BC, Tanabe KK. Prevention of hepatocellular carcinoma: potential targets, experimental models, and clinical challenges. Curr Cancer Drug Targets. 2012;12:1129–1159. [PMC free article] [PubMed] [Google Scholar]
- 5.Chang MH, Shau WY, Chen CJ, Wu TC, Kong MS, Liang DC, et al. Hepatitis B vaccination and hepatocellular carcinoma rates in boys and girls. Jama. 2000;284:3040–3042. doi: 10.1001/jama.284.23.3040. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida H, Shiratori Y, Moriyama M, Arakawa Y, Ide T, Sata M, et al. Interferon therapy reduces the risk for hepatocellular carcinoma: national surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group Inhibition of Hepatocarcinogenesis by Interferon Therapy. Ann Intern Med. 1999;131:174–181. doi: 10.7326/0003-4819-131-3-199908030-00003. [DOI] [PubMed] [Google Scholar]
- 7.Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung RT, Baumert TF. Curing chronic hepatitis C--the arc of a medical triumph. N Engl J Med. 2014;370:1576–1578. doi: 10.1056/NEJMp1400986. [DOI] [PubMed] [Google Scholar]
- 9.van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. Jama. 2012;308:2584–2593. doi: 10.1001/jama.2012.144878. [DOI] [PubMed] [Google Scholar]
- 10.Koike K. Molecular basis of hepatitis C virus-associated hepatocarcinogenesis: lessons from animal model studies. Clin Gastroenterol Hepatol. 2005;3:S132–S135. doi: 10.1016/s1542-3565(05)00700-7. [DOI] [PubMed] [Google Scholar]
- 11.Mailly L, Robinet E, Meuleman P, Baumert TF, Zeisel MB. Hepatitis C virus infection and related liver disease: the quest for the best animal model. Frontiers in microbiology. 2013;4:213. doi: 10.3389/fmicb.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. Animal models for hepatitis C. Current topics in microbiology and immunology. 2013;369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 13.Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nature reviews Microbiology. 2013;11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 14.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature. 2014;509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeisel MB, Felmlee DJ, Baumert TF. Hepatitis C virus entry. Current topics in microbiology and immunology. 2013;369:87–112. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- 16.Ishiguro S, Inoue M, Tanaka Y, Mizokami M, Iwasaki M, Tsugane S. Impact of viral load of hepatitis C on the incidence of hepatocellular carcinoma: A population-based cohort study (JPHC Study) Cancer Lett. 2011;300:173–179. doi: 10.1016/j.canlet.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Lee MH, Yang HI, Lu SN, Jen CL, Yeh SH, Liu CJ, et al. Hepatitis C virus seromarkers and subsequent risk of hepatocellular carcinoma: long-term predictors from a community-based cohort study. J Clin Oncol. 2010;28:4587–4593. doi: 10.1200/JCO.2010.29.1500. [DOI] [PubMed] [Google Scholar]
- 18.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Male PJ, Mentha G, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol. 2000;33:106–115. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 19.Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol. 2009;50:1142–1154. doi: 10.1016/j.jhep.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 20.Fukutomi T, Zhou Y, Kawai S, Eguchi H, Wands JR, Li J. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology. 2005;41:1096–1105. doi: 10.1002/hep.20668. [DOI] [PubMed] [Google Scholar]
- 21.Zemel R, Gerechet S, Greif H, Bachmatove L, Birk Y, Golan-Goldhirsh A, et al. Cell transformation induced by hepatitis C virus NS3 serine protease. J Viral Hepat. 2001;8:96–102. doi: 10.1046/j.1365-2893.2001.00283.x. [DOI] [PubMed] [Google Scholar]
- 22.Arima N, Kao CY, Licht T, Padmanabhan R, Sasaguri Y. Modulation of cell growth by the hepatitis C virus nonstructural protein NS5A. J Biol Chem. 2001;276:12675–12684. doi: 10.1074/jbc.M008329200. [DOI] [PubMed] [Google Scholar]
- 23.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 24.Kao CF, Chen SY, Chen JY, Wu Lee YH. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene. 2004;23:2472–2483. doi: 10.1038/sj.onc.1207368. [DOI] [PubMed] [Google Scholar]
- 25.Alisi A, Giambartolomei S, Cupelli F, Merlo P, Fontemaggi G, Spaziani A, et al. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene. 2003;22:2573–2580. doi: 10.1038/sj.onc.1206333. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi J, Aoki H, Kajino K, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology. 2000;32:958–961. doi: 10.1053/jhep.2000.19343. [DOI] [PubMed] [Google Scholar]
- 27.Machida K, Liu JC, McNamara G, Levine A, Duan L, Lai MM. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. Journal of virology. 2009;83:12590–12600. doi: 10.1128/JVI.02643-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng L, Nagano-Fujii M, Tanaka M, Nomura-Takigawa Y, Ikeda M, Kato N, et al. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. The Journal of general virology. 2006;87:1703–1713. doi: 10.1099/vir.0.81735-0. [DOI] [PubMed] [Google Scholar]
- 29.Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. Journal of virology. 2001;75:1401–1407. doi: 10.1128/JVI.75.3.1401-1407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munakata T, Nakamura M, Liang Y, Li K, Lemon SM. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2005;102:18159–18164. doi: 10.1073/pnas.0505605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS, et al. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Experimental cell research. 2005;305:23–32. doi: 10.1016/j.yexcr.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 32.Aoki H, Hayashi J, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. Journal of virology. 2000;74:1736–1741. doi: 10.1128/jvi.74.4.1736-1741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan SL, Nakao H, He Y, Vijaysri S, Neddermann P, Jacobs BL, et al. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc Natl Acad Sci U S A. 1999;96:5533–5538. doi: 10.1073/pnas.96.10.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koike K, Hepatitis C. virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol. 2007;22(Suppl 1):S108–111. doi: 10.1111/j.1440-1746.2006.04669.x. [DOI] [PubMed] [Google Scholar]
- 35.Tovar V, Alsinet C, Villanueva A, Hoshida Y, Chiang DY, Sole M, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52:550–559. doi: 10.1016/j.jhep.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Street A, Macdonald A, McCormick C, Harris M. Hepatitis C virus NS5A–mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. Journal of virology. 2005;79:5006–5016. doi: 10.1128/JVI.79.8.5006-5016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson DR, Gonzalez-Peralta RP, Qian K, Xu Y, Marousis CG, Davis GL, et al. Transforming growth factor-beta 1 in chronic hepatitis C. J Viral Hepat. 1997;4:29–35. doi: 10.1046/j.1365-2893.1997.00124.x. [DOI] [PubMed] [Google Scholar]
- 38.Hopp SL, Sinnott JM, Owren MJ, Petersen MR. Differential sensitivity of Japanese macaques (Macaca fuscata) and humans (Homo sapiens) to peak position along a synthetic coo call continuum. J Comp Psychol. 1992;106:128–136. doi: 10.1037/0735-7036.106.2.128. [DOI] [PubMed] [Google Scholar]
- 39.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-beta in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Choi SH, Hwang SB. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J Biol Chem. 2006;281:7468–7478. doi: 10.1074/jbc.M512438200. [DOI] [PubMed] [Google Scholar]
- 41.Wu SC, Chang SC, Wu HY, Liao PJ, Chang MF. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J Biol Chem. 2008;283:29396–29404. doi: 10.1074/jbc.M802821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh AK, Majumder M, Steele R, Meyer K, Ray R, Ray RB. Hepatitis C virus NS5A protein protects against TNF-alpha mediated apoptotic cell death. Virus Res. 2000;67:173–178. doi: 10.1016/s0168-1702(00)00141-6. [DOI] [PubMed] [Google Scholar]
- 43.Ali N, Allam H, May R, Sureban SM, Bronze MS, Bader T, et al. Hepatitis C virus-induced cancer stem cell-like signatures in cell culture and murine tumor xenografts. Journal of virology. 2011;85:12292–12303. doi: 10.1128/JVI.05920-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu M, Takai K, Moriwaki H. Strategy and mechanism for the prevention of hepatocellular carcinoma: phosphorylated retinoid X receptor alpha is a critical target for hepatocellular carcinoma chemoprevention. Cancer Sci. 2009;100:369–374. doi: 10.1111/j.1349-7006.2008.01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DY. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappaB signaling, and HCV infection by standardized Silymarin. Gastroenterology. 2007;132:1925–1936. doi: 10.1053/j.gastro.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 46.Reddy KR, Belle SH, Fried MW, Afdhal N, Navarro VJ, Hawke RL, et al. Rationale, challenges, and participants in a Phase II trial of a botanical product for chronic hepatitis C. Clin Trials. 2012;9:102–112. doi: 10.1177/1740774511427064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freedman ND, Curto TM, Morishima C, Seeff LB, Goodman ZD, Wright EC, et al. Silymarin use and liver disease progression in the Hepatitis C Antiviral Long-Term Treatment against Cirrhosis trial. Aliment Pharmacol Ther. 2011;33:127–137. doi: 10.1111/j.1365-2036.2010.04503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai CK, Jeng KS, Machida K, Cheng YS, Lai MM. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology. 2008;370:295–309. doi: 10.1016/j.virol.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 49.Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. Journal of virology. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends in immunology. 2013;34:67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 52.Luedde T, Schwabe RF. NF-kappaB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joo M, Hahn YS, Kwon M, Sadikot RT, Blackwell TS, Christman JW. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. Journal of virology. 2005;79:7648–7657. doi: 10.1128/JVI.79.12.7648-7657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–3953. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park KJ, Choi SH, Choi DH, Park JM, Yie SW, Lee SY, et al. 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2003;278:30711–30718. doi: 10.1074/jbc.M209623200. [DOI] [PubMed] [Google Scholar]
- 56.Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–1953. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- 57.Nagahara T, Okano J, Fujise Y, Abe R, Murawaki Y. Preventive effect of JTE-522, a selective cyclooxygenase-2 inhibitor, on DEN-induced hepatocarcinogenesis in rats. Biomed Pharmacother. 2010;64:319–326. doi: 10.1016/j.biopha.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 58.Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43–49. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 62.Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. 2011;286:10847–10855. doi: 10.1074/jbc.M110.217653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang SJ, Lee SD. Hepatic steatosis and hepatitis C: Still unhappy bedfellows? J Gastroenterol Hepatol. 2011;26(Suppl 1):96–101. doi: 10.1111/j.1440-1746.2010.06542.x. [DOI] [PubMed] [Google Scholar]
- 64.Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci U S A. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koike K, Moriya K. Metabolic aspects of hepatitis C viral infection: steatohepatitis resembling but distinct from NASH. J Gastroenterol. 2005;40:329–336. doi: 10.1007/s00535-005-1586-z. [DOI] [PubMed] [Google Scholar]
- 66.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest. 2008;118:683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koike K. Steatosis, liver injury, and hepatocarcinogenesis in hepatitis C viral infection. J Gastroenterol. 2009;44(Suppl 19):82–88. doi: 10.1007/s00535-008-2276-4. [DOI] [PubMed] [Google Scholar]
- 69.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 70.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim JS, Park SH, Jang KL. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012;321:154–161. doi: 10.1016/j.canlet.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 72.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aihara T, Noguchi S, Sasaki Y, Nakano H, Imaoka S. Clonal analysis of regenerative nodules in hepatitis C virus-induced liver cirrhosis. Gastroenterology. 1994;107:1805–1811. doi: 10.1016/0016-5085(94)90824-9. [DOI] [PubMed] [Google Scholar]
- 74.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–S50. doi: 10.1053/j.gastro.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 75.Lee YA, Friedman SL. Reversal, maintenance or progression: What happens to the liver after a virologic cure of hepatitis C? Antiviral research. 2014;107C:23–30. doi: 10.1016/j.antiviral.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–527. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 78.Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529–540. doi: 10.1053/j.gastro.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 79.Lin W, Tsai WL, Shao RX, Wu G, Peng LF, Barlow LL, et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–2518. doi: 10.1053/j.gastro.2010.03.008. 2518 e2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294–1306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]
- 81.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okada H, Honda M, Campbell JS, Sakai Y, Yamashita T, Takebuchi Y, et al. Acyclic retinoid targets platelet-derived growth factor signaling in the prevention of hepatic fibrosis and hepatocellular carcinoma development. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-0028. [DOI] [PubMed] [Google Scholar]
- 84.Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 85.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, Barrie HD, et al. IL-32: A new proinflammatory cytokine involved in HCV-related liver inflammation and fibrosis. Hepatology. 2011 doi: 10.1002/hep.24285. [DOI] [PubMed] [Google Scholar]
- 87.Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Semin Liver Dis. 2010;30:402–410. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, Afdhal NH, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46:324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 89.Gieseler RK, Marquitan G, Schlattjan M, Sowa JP, Bechmann LP, Timm J, et al. Hepatocyte apoptotic bodies encasing nonstructural HCV proteins amplify hepatic stellate cell activation: implications for chronic hepatitis C. J Viral Hepat. 2011;18:760–767. doi: 10.1111/j.1365-2893.2010.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Chang M, Abar O, Garcia V, Rowland C, Catanese J, et al. Multiple variants in toll-like receptor 4 gene modulate risk of liver fibrosis in Caucasians with chronic hepatitis C infection. J Hepatol. 2009;51:750–757. doi: 10.1016/j.jhep.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mazzocca A, Sciammetta SC, Carloni V, Cosmi L, Annunziato F, Harada T, et al. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem. 2005;280:11329–11339. doi: 10.1074/jbc.M410161200. [DOI] [PubMed] [Google Scholar]
- 93.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 94.Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26:431–442. doi: 10.1038/nbt1396. [DOI] [PubMed] [Google Scholar]
- 95.Oakley F, Teoh V, Ching ASG, Bataller R, Colmenero J, Jonsson JR, et al. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology. 2009;136:2334–2344. doi: 10.1053/j.gastro.2009.02.081. e2331. [DOI] [PubMed] [Google Scholar]
- 96.Yoshiji H, Noguchi R, Kaji K, Ikenaka Y, Shirai Y, Namisaki T, et al. Attenuation of insulin-resistance-based hepatocarcinogenesis and angiogenesis by combined treatment with branched-chain amino acids and angiotensin-converting enzyme inhibitor in obese diabetic rats. J Gastroenterol. 2010;45:443–450. doi: 10.1007/s00535-009-0158-z. [DOI] [PubMed] [Google Scholar]
- 97.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59:1577–1590. doi: 10.1002/hep.26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schiffer E, Housset C, Cacheux W, Wendum D, Desbois-Mouthon C, Rey C, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41:307–314. doi: 10.1002/hep.20538. [DOI] [PubMed] [Google Scholar]
- 100.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zona L, Lupberger J, Sidahmed-Adrar N, Thumann C, Harris HJ, Barnes A, et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell host & microbe. 2013;13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 102.Pei R, Chen H, Lu L, Zhu W, Beckebaum S, Cicinnati V, et al. Hepatitis C virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. The Journal of general virology. 2011;92:2237–2248. doi: 10.1099/vir.0.032581-0. [DOI] [PubMed] [Google Scholar]
- 103.Nakamura H, Aoki H, Hino O, Moriyama M. HCV core protein promotes heparin binding EGF-like growth factor expression and activates Akt. Hepatol Res. 2011;41:455–462. doi: 10.1111/j.1872-034X.2011.00792.x. [DOI] [PubMed] [Google Scholar]
- 104.Perugorria MJ, Latasa MU, Nicou A, Cartagena-Lirola H, Castillo J, Goni S, et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology. 2008;48:1251–1261. doi: 10.1002/hep.22437. [DOI] [PubMed] [Google Scholar]
- 105.Huang G, Besner GE, Brigstock DR. Heparin-binding epidermal growth factor-like growth factor suppresses experimental liver fibrosis in mice. Lab Invest. 2012;92:703–712. doi: 10.1038/labinvest.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coriat R, Gouya H, Mir O, Ropert S, Vignaux O, Chaussade S, et al. Reversible decrease of portal venous flow in cirrhotic patients: a positive side effect of sorafenib. PLoS One. 2011;6:e16978. doi: 10.1371/journal.pone.0016978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Horwitz E, Stein I, Andreozzi M, Nemeth J, Shoham A, Pappo O, et al. Human and Mouse VEGFA-Amplified Hepatocellular Carcinomas Are Highly Sensitive to Sorafenib Treatment. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-13-0782. [DOI] [PubMed] [Google Scholar]
- 108.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 109.Fabris C, Falleti E, Cussigh A, Bitetto D, Fontanini E, Bignulin S, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol. 2011;54:716–722. doi: 10.1016/j.jhep.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 110.Fusco DN, Brisac C, John SP, Huang YW, Chin CR, Xie T, et al. A genetic screen identifies interferon-alpha effector genes required to suppress hepatitis C virus replication. Gastroenterology. 2013;144:1438–1449. doi: 10.1053/j.gastro.2013.02.026. 1449 e1431-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat Genet. 2011;43:455–458. doi: 10.1038/ng.809. [DOI] [PubMed] [Google Scholar]
- 112.Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J Immunol. 2003;171:5423–5429. doi: 10.4049/jimmunol.171.10.5423. [DOI] [PubMed] [Google Scholar]
- 113.Lo PH, Urabe Y, Kumar V, Tanikawa C, Koike K, Kato N, et al. Identification of a functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS One. 2013;8:e61279. doi: 10.1371/journal.pone.0061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoshida Y, Fuchs BC, Tanabe KK. Genomic risk of hepatitis C-related hepatocellular carcinoma. J Hepatol. 2012;56:729–730. doi: 10.1016/j.jhep.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 115.Lange CM, Bibert S, Dufour JF, Cellerai C, Cerny A, Heim MH, et al. Comparative genetic analyses point to HCP5 as susceptibility locus for HCV-associated hepatocellular carcinoma. J Hepatol. 2013;59:504–509. doi: 10.1016/j.jhep.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 116.Miki D, Ochi H, Hayes CN, Abe H, Yoshima T, Aikata H, et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat Genet. 2011 doi: 10.1038/ng.876. [DOI] [PubMed] [Google Scholar]
- 117.Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, et al. The Effect of PNPLA3 on Fibrosis Progression and Development of Hepatocellular Carcinoma: A Meta-analysis. Am J Gastroenterol. 2014 doi: 10.1038/ajg.2013.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ko C, Siddaiah N, Berger J, Gish R, Brandhagen D, Sterling RK, et al. Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: results from the National Hemochromatosis Transplant Registry. Liver Int. 2007;27:1394–1401. doi: 10.1111/j.1478-3231.2007.01596.x. [DOI] [PubMed] [Google Scholar]
- 119.Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, et al. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat. 2008;15:498–507. doi: 10.1111/j.1365-2893.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 120.Furutani T, Hino K, Okuda M, Gondo T, Nishina S, Kitase A, et al. Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology. 2006;130:2087–2098. doi: 10.1053/j.gastro.2006.02.060. [DOI] [PubMed] [Google Scholar]
- 121.Bonkovsky HL, Naishadham D, Lambrecht RW, Chung RT, Hoefs JC, Nash SR, et al. Roles of iron and HFE mutations on severity and response to therapy during retreatment of advanced chronic hepatitis C. Gastroenterology. 2006;131:1440–1451. doi: 10.1053/j.gastro.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 122.Varnholt H, Drebber U, Schulze F, Wedemeyer I, Schirmacher P, Dienes HP, et al. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology. 2008;47:1223–1232. doi: 10.1002/hep.22158. [DOI] [PubMed] [Google Scholar]
- 123.Ura S, Honda M, Yamashita T, Ueda T, Takatori H, Nishino R, et al. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology. 2009;49:1098–1112. doi: 10.1002/hep.22749. [DOI] [PubMed] [Google Scholar]
- 124.Toffanin S, Hoshida Y, Lachenmayer A, Villanueva A, Cabellos L, Minguez B, et al. MicroRNA-Based Classification of Hepatocellular Carcinoma and Oncogenic Role of miR-517a. Gastroenterology. 2011;140:1618–1628. doi: 10.1053/j.gastro.2011.02.009. e1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 126.Kojima K, Takata A, Vadnais C, Otsuka M, Yoshikawa T, Akanuma M, et al. MicroRNA122 is a key regulator of alpha-fetoprotein expression and influences the aggressiveness of hepatocellular carcinoma. Nat Commun. 2011;2:338. doi: 10.1038/ncomms1345. [DOI] [PubMed] [Google Scholar]
- 127.Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Banaudha K, Kaliszewski M, Korolnek T, Florea L, Yeung ML, Jeang KT, et al. MicroRNA silencing of tumor suppressor DLC-1 promotes efficient hepatitis C virus replication in primary human hepatocytes. Hepatology. 2011;53:53–61. doi: 10.1002/hep.24016. [DOI] [PubMed] [Google Scholar]
- 129.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Di Bisceglie AM, Shiffman ML, Everson GT, Lindsay KL, Everhart JE, Wright EC, et al. Prolonged Therapy of Advanced Chronic Hepatitis C with Low-Dose Peginterferon. N Engl J Med. 2008;359:2429–2441. doi: 10.1056/NEJMoa0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bruix J, Poynard T, Colombo M, Schiff E, Burak K, Heathcote EJ, et al. Maintenance therapy with peginterferon alfa-2b does not prevent hepatocellular carcinoma in cirrhotic patients with chronic hepatitis C. Gastroenterology. 2011;140:1990–1999. doi: 10.1053/j.gastro.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 132.Okita K, Izumi N, Matsui O, Tanaka K, Kaneko S, Moriwaki H, et al. Peretinoin after curative therapy of hepatitis C-related hepatocellular carcinoma: a randomized double-blind placebo-controlled study. J Gastroenterol. 2014 doi: 10.1007/s00535-014-0956-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) Jama. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Simon R. The use of genomics in clinical trial design. Clin Cancer Res. 2008;14:5984–5993. doi: 10.1158/1078-0432.CCR-07-4531. [DOI] [PubMed] [Google Scholar]
- 135.EASL-EORTC. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943. doi: 10.1016/j.jhep.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 136.Davila JA, Henderson L, Kramer JR, Kanwal F, Richardson PA, Duan Z, et al. Utilization of surveillance for hepatocellular carcinoma among hepatitis C virus-infected veterans in the United States. Ann Intern Med. 2011;154:85–93. doi: 10.7326/0003-4819-154-2-201101180-00006. [DOI] [PubMed] [Google Scholar]
- 137.Smith JO, Sterling RK. Systematic review: non-invasive methods of fibrosis analysis in chronic hepatitis C. Aliment Pharmacol Ther. 2009;30:557–576. doi: 10.1111/j.1365-2036.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 138.Singal AG, Mukherjee A, Elmunzer BJ, Higgins PD, Lok AS, Zhu J, et al. Machine learning algorithms outperform conventional regression models in predicting development of hepatocellular carcinoma. Am J Gastroenterol. 2013;108:1723–1730. doi: 10.1038/ajg.2013.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.El-Serag HB, Kanwal F, Davila JA, Kramer J, Richardson P. A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology. 2014;146:1249–1255. doi: 10.1053/j.gastro.2014.01.045. e1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nahon P, Zucman-Rossi J. Single nucleotide polymorphisms and risk of hepatocellular carcinoma in cirrhosis. J Hepatol. 2012;57:663–674. doi: 10.1016/j.jhep.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 141.Tanabe KK, Lemoine A, Finkelstein DM, Kawasaki H, Fujii T, Chung RT, et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. Jama. 2008;299:53–60. doi: 10.1001/jama.2007.65. [DOI] [PubMed] [Google Scholar]
- 142.Abu Dayyeh BK, Yang M, Fuchs BC, Karl DL, Yamada S, Sninsky JJ, et al. A functional polymorphism in the epidermal growth factor gene is associated with risk for hepatocellular carcinoma. Gastroenterology. 2011;141:141–149. doi: 10.1053/j.gastro.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhong JH, You XM, Gong WF, Ma L, Zhang Y, Mo QG, et al. Epidermal growth factor gene polymorphism and risk of hepatocellular carcinoma: a meta-analysis. PLoS One. 2012;7:e32159. doi: 10.1371/journal.pone.0032159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nahon P, Sutton A, Rufat P, Charnaux N, Mansouri A, Moreau R, et al. A variant in myeloperoxidase promoter hastens the emergence of hepatocellular carcinoma in patients with HCV-related cirrhosis. J Hepatol. 2012;56:426–432. doi: 10.1016/j.jhep.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 145.Marcolongo M, Young B, Dal Pero F, Fattovich G, Peraro L, Guido M, et al. A seven-gene signature (cirrhosis risk score) predicts liver fibrosis progression in patients with initially mild chronic hepatitis C. Hepatology. 2009;50:1038–1044. doi: 10.1002/hep.23111. [DOI] [PubMed] [Google Scholar]
- 146.Hoshida Y, Villanueva A, Sangiovanni A, Sole M, Hur C, Andersson KL, et al. Prognostic Gene-Expression Signature for Patients with Hepatitis C-Related Early-Stage Cirrhosis. Gastroenterology. 2013;144:1024–1030. doi: 10.1053/j.gastro.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wong KF, Xu Z, Chen J, Lee NP, Luk JM. Circulating markers for prognosis of hepatocellular carcinoma. Expert opinion on medical diagnostics. 2013;7:319–329. doi: 10.1517/17530059.2013.795146. [DOI] [PubMed] [Google Scholar]
- 148.Fuchs BC, Wang H, Yang Y, Wei L, Polasek M, Schuhle DT, et al. Molecular MRI of collagen to diagnose and stage liver fibrosis. J Hepatol. 2013;59:992–998. doi: 10.1016/j.jhep.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mazzaferro V, Romito R, Schiavo M, Mariani L, Camerini T, Bhoori S, et al. Prevention of hepatocellular carcinoma recurrence with alpha-interferon after liver resection in HCV cirrhosis. Hepatology. 2006;44:1543–1554. doi: 10.1002/hep.21415. [DOI] [PubMed] [Google Scholar]
- 150.Shiratori Y, Shiina S, Teratani T, Imamura M, Obi S, Sato S, et al. Interferon therapy after tumor ablation improves prognosis in patients with hepatocellular carcinoma associated with hepatitis C virus. Ann Intern Med. 2003;138:299–306. doi: 10.7326/0003-4819-138-4-200302180-00008. [DOI] [PubMed] [Google Scholar]
- 151.Printz C. Clinical trials of note. Sorafenib as adjuvant treatment in the prevention of disease recurrence in patients with hepatocellular carcinoma (HCC) (STORM) Cancer. 2009;115:4646. doi: 10.1002/cncr.24673. [DOI] [PubMed] [Google Scholar]
- 152.Ikeda K, Arase Y, Kobayashi M, Saitoh S, Someya T, Hosaka T, et al. Significance of multicentric cancer recurrence after potentially curative ablation of hepatocellular carcinoma: a longterm cohort study of 892 patients with viral cirrhosis. J Gastroenterol. 2003;38:865–876. doi: 10.1007/s00535-003-1163-2. [DOI] [PubMed] [Google Scholar]
- 153.Schmidt WN, Nelson DR, Pawlotsky JM, Sherman KE, Thomas DL, Chung RT. Direct Acting Antiviral Agents and the Path to Interferon Independence. Clin Gastroenterol Hepatol. 2013 doi: 10.1016/j.cgh.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370:1973–1982. doi: 10.1056/NEJMoa1402869. [DOI] [PubMed] [Google Scholar]
- 155.Shiratori Y, Imazeki F, Moriyama M, Yano M, Arakawa Y, Yokosuka O, et al. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann Intern Med. 2000;132:517–524. doi: 10.7326/0003-4819-132-7-200004040-00002. [DOI] [PubMed] [Google Scholar]
- 156.Lok AS, Everhart JE, Wright EC, Di Bisceglie AM, Kim HY, Sterling RK, et al. Maintenance Peginterferon Therapy and Other Factors Associated With Hepatocellular Carcinoma in Patients With Advanced Hepatitis C. Gastroenterology. 2011 doi: 10.1053/j.gastro.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Menon KV, Hakeem AR, Heaton ND. Meta-analysis: recurrence and survival following the use of sirolimus in liver transplantation for hepatocellular carcinoma. Aliment Pharmacol Ther. 2013;37:411–419. doi: 10.1111/apt.12185. [DOI] [PubMed] [Google Scholar]
- 158.Sahasrabuddhe VV, Gunja MZ, Graubard BI, Trabert B, Schwartz LM, Park Y, et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:1808–1814. doi: 10.1093/jnci/djs452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Emberson JR, Kearney PM, Blackwell L, Newman C, Reith C, Bhala N, et al. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One. 2012;7:e29849. doi: 10.1371/journal.pone.0029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108:881–891. doi: 10.1038/ajg.2013.5. quiz 892. [DOI] [PubMed] [Google Scholar]
- 161.Bravi F, Bosetti C, Tavani A, Gallus S, La Vecchia C. Coffee reduces risk for hepatocellular carcinoma: an updated meta-analysis. Clin Gastroenterol Hepatol. 2013;11:1413–1421. doi: 10.1016/j.cgh.2013.04.039. e1411. [DOI] [PubMed] [Google Scholar]
- 162.Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology. 2011;54:1947–1955. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- 163.Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072–2079. doi: 10.1158/1078-0432.CCR-13-0547. [DOI] [PubMed] [Google Scholar]
- 164.Boehm JS, Hahn WC. Towards systematic functional characterization of cancer genomes. Nat Rev Genet. 2011;12:487–498. doi: 10.1038/nrg3013. [DOI] [PubMed] [Google Scholar]
- 165.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, et al. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell. 2013;153:389–401. doi: 10.1016/j.cell.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 166.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 167.Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Molecular systems biology. 2014;10:721. doi: 10.1002/msb.145122. [DOI] [PMC free article] [PubMed] [Google Scholar]