

Abstract
Objective
Childhood absence epilepsy (CAE) is a childhood-onset generalized epilepsy. Recent fMRI studies have suggested that frontal cortex activity occurs before thalamic involvement in epileptic discharges suggesting that frontal cortex may play an important role in childhood absence seizures. Neurocognitive deficits can persist after resolution of the epilepsy. We investigate whether structural connectivity changes are present in the brains of CAE patients in young adulthood.
Methods
Cortical thickness measurements were obtained for 30 subjects with CAE (mean age 21 ± 2 years) and 56 healthy controls (mean age 24 ± 4) and regressed for age, sex, and total intracranial volume (TIV). Structural connectivity was evaluated by measuring the correlation between average cortical thicknesses in 915 regions over the brain. Maps of connectivity strength were then obtained for both groups.
Results
When compared to controls, the CAE group shows overall increased “connectivity” with focal increased connection strength in anterior regions including; the anterior cingulate and the insula and superior temporal gyrus bilaterally; the right orbito-frontal and supramarginal regions; and the left entorhinal cortex. Decreased connection strength in the CAE group was found in the left occipital lobe, with a similar trend in right occipital lobe.
Interpretation
Brains in young adults whose CAE was resolved had abnormal structural connectivity. Our findings suggest that frontal regions correlate most with cortical thickness throughout the brain in CAE patients, whereas occipital regions correlate most in well matched normal controls. We interpret this as evidence of a developmental difference in CAE that emphasizes these frontal lobe regions, perhaps driven by frontal lobe epileptiform activity.
Introduction
Childhood absence epilepsy (CAE) is a common form of childhood epilepsy characterized by frequent brief absence seizures (AS) and onset between the ages of 5 and 10 years in an “otherwise normal child”. Their EEG demonstrates rhythmic spike-and-wave activity, usually at 3 Hz, on an otherwise normal background.1 Although AS appear clinically bland, children often display significant neuro-psychological deficits suggesting a more pervasive effect of seizures, or the epilepsy syndrome, on cognition.2–4 CAE and the related epilepsy syndromes (the genetic generalized epilepsies [GGE]) are conceptualized as disorders of “bilaterally distributed networks” involving both cortical and subcortical structures.5 The structures in this network include, but are likely not limited to, the mesial and parietal cortex, thalamus, and striatum.6–8 Frontal cortical BOLD changes have been seen in a number of studies and the relative timing of the BOLD response indicates that they may also be important in seizure generation.9 This is also shown in resting state functional connectivity analysis.10 They are also hypothesized to be the substrate for the cognitive variations seen between cases with CAE and healthy controls.11,12 A number of studies have identified structural changes patients with GGE, including thalamic atrophy identified using voxel-based morphometry.13,14 This may arise as a consequence of the functional involvement of the thalamus in seizures. As yet, there has been no corresponding structural correlate observed for the functional activations in the cortex, although subtle changes in neocortical gray and white matter volumes have been observed on the lobar scale,15 and appear to be associated with EEG signals.16
Given the evidence of network disturbance in CAE, it is important to study the structural consequences of these changes at the network level. A powerful tool for the investigation of network disorders is connectivity analysis, which harnesses the formalism of graph theory to reveal large-scale changes in brain structure and dynamics.17 Brain networks constructed using these tools have revealed the importance of regions corresponding to the default mode network (DMN) and the occipital lobes in healthy controls.18 Structural networks can also be constructed using correlations between cortical thickness.19 Networks constructed in this fashion show maximal network activity in areas associated with the DMN in healthy controls,20 and have been applied to show altered structural organization in the brains of subjects with temporal lobe epilepsy21; a related seeded approach has already been used to investigate GGE and generalized tonic–clonic seizures only.22
In this study, our aim was to determine if there are changes in the structure of the cortex of CAE patients when compared to healthy controls. We examine absolute differences in cortical thickness and changes in cortical organization using unseeded cortical thickness-based structural network analysis in a well-characterized community-based cohort of young adults who were prospectively recruited as children and followed as part of a community-based cohort of newly diagnosed epilepsy.23,24
Methods
Subjects
A community-based cohort of subjects was recruited from the state of Connecticut, U.S.A., as part of a study on the long-term outcomes of childhood-onset epilepsy. Briefly, all children who were diagnosed with epilepsy for the first time in the state of Connecticut, U.S.A., between the years of 1992–1997 were eligible. They were recruited from 16 of 17 practicing pediatric neurologists in the state, as well as five adult neurologists and seven pediatricians.
Syndromic classification was made independently by three experienced pediatric neurologists based on all clinical data, where disagreement existed consensus was reached in conference. Further details of the recruitment process are presented elsewhere.23–25 Of the 613 children recruited into the study, 74 (12.1%) were diagnosed with CAE. Criteria for identifying CAE were those commonly used at the time children were recruited.26
Data presented here were acquired when subjects of the study returned for a comprehensive assessment ∼15 years after having entered the study (2007–2013); this assessment included a structural MRI scan. Of the 74 children originally diagnosed with CAE, 30 remained in the study at this stage and consented to an MRI scan. These subjects comprised of 15 male subjects, 15 females, with a mean age of 21 years ± 2 SD. Also acquired were 56 control subjects (23 males, 33 females, mean age 21 years ± 2 SD). All controls were reported as neurologically normal, and independent review showed no abnormalities apparent on clinical MRI scans. Subjects with CAE had a mean time since last seizure of 9 years, with 22 (73%) having had complete remission of seizures for 2 years or more including cessation of antiepileptic drug (AED) use (20 (67%) having been in complete remission for 5 or more years). An additional three patients had experienced no seizures for 2 years or more but were still on medication at the time of scan, and only one patient has experienced a seizure subsequent to the scan. More detail on seizure remission in this cohort is reported elsewhere.27 CAE patients were initially treated either with valproic acid (11, 37%) or ethosuximide (18, 60%), or in one instance carbamazepine; 13 subjects were treated with more than two AEDs over the course of their epilepsy. Subjects had a median length of treatment (defined as the time from prescription of the first AED to total withdrawal of medication) of 6.4 years. Seven subjects (23%) were still using AEDs at the time of the scan. Subjects were scanned with a T1 MPRAGE sequence using a 1.5T Siemens Sonata (Siemens, Erlagen, Germany) scanner with the following image parameters: echo time 3.05 msec, repetition time 1730 msec, flip angle 15°, inversion time 1100 msec, and voxel size 0.94 × 0.94 × 1.6 mm. Research conducted was approved by the relevant review board human ethics committees at each site, and all subjects gave written, informed consent prior to the scan taking place.
Image analysis
Cortical thickness was measured using Freesurfer (http://surfer.nmr.mgh.harvard.edu/).28,29 Each scan was coregistered to a surface-based atlas.30 Standard vertex-wise statistical analysis of cortical thickness using a general linear model was performed to test for mean differences between groups. Age, sex, and estimated total intracranial volume (TIV) were used as covariates. Significance was tested using a vertex-wise false discovery rate corrected threshold of P < 0.05. A structural brain network was then constructed from the data. The native Freesurfer parcellation scheme was extended by subdividing each region into subregions of approximately equal area (mean 124 mm2, SD 30 mm2) using k-means clustering on the surface-based Euclidean distance, resulting in a total of 915 subregions over both hemispheres. Each subregion was treated as a node in the subsequent analysis.
The average cortical thickness in each node was calculated for each subject and regressed against age, sex, and TIV. Pearson's product-moment correlation coefficients R between each node and each other node was then estimated for both CAE and control groups. Negative values of R were excluded from subsequent analysis, so anticorrelations are ignored.31
We assume that the group correlation coefficient is indicative of some connection between different regions of cortex,32 and, therefore, we interpret the group correlation coefficient between the nodes i and j as the edge weight wij. The type I error rate of the correlation in the control population is used as a sparsity threshold; only those edges with a correlation significance level of P < 0.05 are considered as linked. The top 17.5% of edges in the control matrix pass this threshold; the same proportional threshold was applied to the CAE group to keep the number of edges constant in the two groups. This ensures that any topological differences between the two groups are not an artifact of unequal edge densities.33
Several network measures are employed here to identify differences in structural connectivity between the two groups; full definitions are described elsewhere.31 The Brain Connectivity Toolbox (http://www.brain-connectivity-toolbox.net)31 was used to calculate these measures on our graph. Briefly, we focus on degree, which is the total number of connections for a given node and gives an indication of the importance of the node to the network, strength, which is a weighted degree, and the clustering coefficient,34 which can imply functional segregation in the network. Here, we use it as a measure of “randomness” of a network, this is quantified using the small-worldness metric35 which compares clustering coefficient and a network's characteristic path length against random networks of the same degree. Networks are called small world if they have a small-world index greater than one. A small-world network lies between the two extremes of a completely ordered, lattice-like network, and a completely random network.34
Node-wise statistical analysis
The networks constructed based on correlations in cortical thickness have nodes that are nominally fixed at a position in template space, therefore statistical tests comparing the value of a network metric from one node to the other are justified. Variance was estimated using a permutation testing approach. The subjects were assigned a label, either CAE or control, and these were randomly permuted 500,000 times. For each permutation, the network was constructed using the method described above and a node-wise null distribution in strength was constructed. This distribution was used to estimate the T statistic for each node.
Results
Cortical thickness differences
No significant differences between CAE and controls were observed using this analysis. Subsequent connectivity results are not, therefore, driven by absolute differences in surface-based cortical thickness measurements.
Nodal distribution
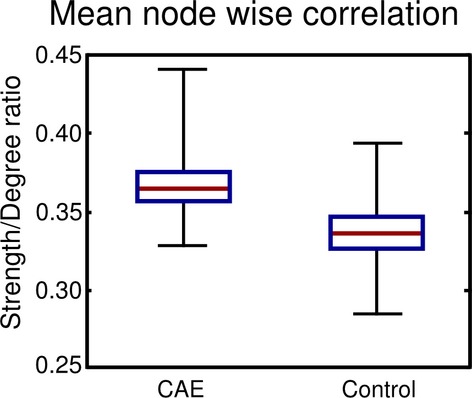
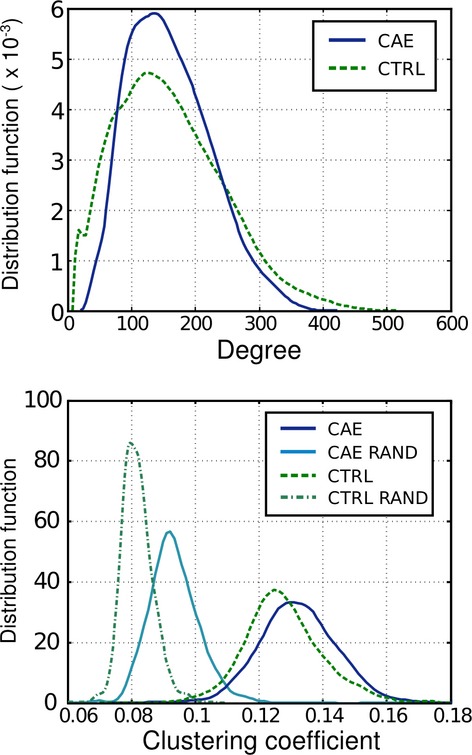
The node degree, strength, clustering coefficient, and network small-worldness index were calculated for the CAE and control group adjacency matrices. The strength and degree correlated linearly for both control and CAE groups but with different gradients. These differing ratios are plotted in Figure1. The ratio of strength to degree showed a significant difference between the subjects; the CAE subjects’ ratio was increased by 0.0297 (P = 0.025). This means that, on average, the CAE brain regions are more “connected”, that is, more highly correlated, than the control brain. Increased strength-to-degree ratio implies a given number of links will have a higher strength in CAE than in controls. The degree distribution for both subject groups is plotted in Figure2. It is evident from Figure2 that the control distribution has a longer high-degree tail, implying that there are a small number of nodes that have a greater number of connections with the rest of the brain in comparison with CAE.
Figure 1.
Node-based degree and clustering coefficient distributions for childhood absence epilepsy (solid lines) and control (dashed lines) groups. The clustering coefficient distributions are plotted alongside those for random graphs of similar degree distribution. A nonnormal degree distribution is observed for both groups indicating nonrandom network organization. The clustering coefficient is also significantly larger than that of a random distribution. These distributions are normalized so that the area under the curve is unity.
Figure 2.
The ratio of strength to degree for all nodes for both childhood absence epilepsy (CAE) and control groups networks. This is a measure of average edge strength. The red line denotes the median value, the box the 25th and 75th quartiles, and the whiskers denote the maximum and minimum, respectively. We see CAE has significantly larger average edge strength. This is driven by overall increases in the magnitude of the correlation coefficient for the CAE group compared to control. The CAE group evidently has more homogeneous cortical thickness across the group.
The clustering coefficient distributions for both CAE and controls are also plotted in Figure2. The clustering coefficient for two random networks with the same degree profile as each subject group is plotted alongside in the clustering plot. There is a clear difference in clustering between the random networks and the two subject groups; this implies the networks are nonrandom. The CAE group displays greater clustering than controls. Similar results have been seen in functional connectivity studies.36 We observed small-worldness in both CAE and control groups with values of 1.70, (95% confidence intervals, family-wise error corrected, 1.67–1.73) for the CAE group and 1.45 (1.41–1.49) for controls. All values are significantly greater than 1, which indicates that these graphs represent “small-world” networks. This suggests that the CAE group is more clustered than controls; indeed, such a difference in the small-worldness metric between groups can indicate that the CAE group has a more lattice-like structure, indicative of network more vulnerable to target attack.37
Surface distributions
The node strength is overlaid on the inflated template surface in Figure3A for the left hemisphere, and Figure3B for the right hemisphere. In order to get a sense of the anatomical distribution, the strength is displayed for values greater than the 50th percentiles of the combined strength distribution for both groups. The T statistic is displayed in Figure4, for (a) thresholded at P < 0.05 family-wise error (FWE) corrected, and (b) thresholded at P < 0.05 uncorrected. As the Bonferroni correction is quite conservative, the uncorrected T scores are displayed in Figure4B to present the shape of the data underlying the threshold. Those nodes which passed the corrected significance threshold are listed in Table1. It is known that across-subject variance in cortical thickness is somewhat heterogeneous throughout the cortex.38 To ensure our results were not confounded by sensitivity variation, we determined which of the 915 nodes would have been sensitive to detect the minimum reported effect size. The minimum significant effect size was −5.43; given the variance at each node, this effect size would have been detectable in 88% of nodes.
Figure 3.
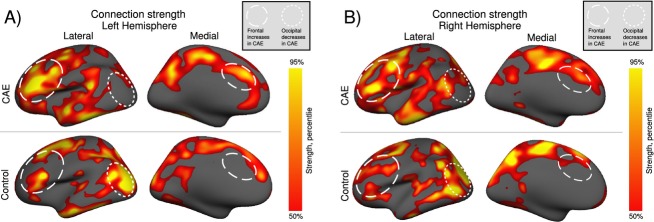
Weighted connection strength overlaid on an inflated cortical surface for the (A) left, and (B) right hemispheres. Only connection strengths greater than the median are displayed. Of note is the bright occipital/parietal region in controls that is not seen in childhood absence epilepsy (CAE). This implies that the cortical thickness of the occipital lobe in controls is most highly correlated with the rest of the brain, whereas this is not the case in the CAE group. Also evident is a large increase in the anterior cingulate cortex in the disease group. These changes are apparently part of an overall trend for posterior dominance in controls and anterior dominance in CAE. The activation pattern is broadly similar in both left and right hemispheres, with increased medial and lateral frontal and decreased posterior connectivity in the CAE group.
Figure 4.
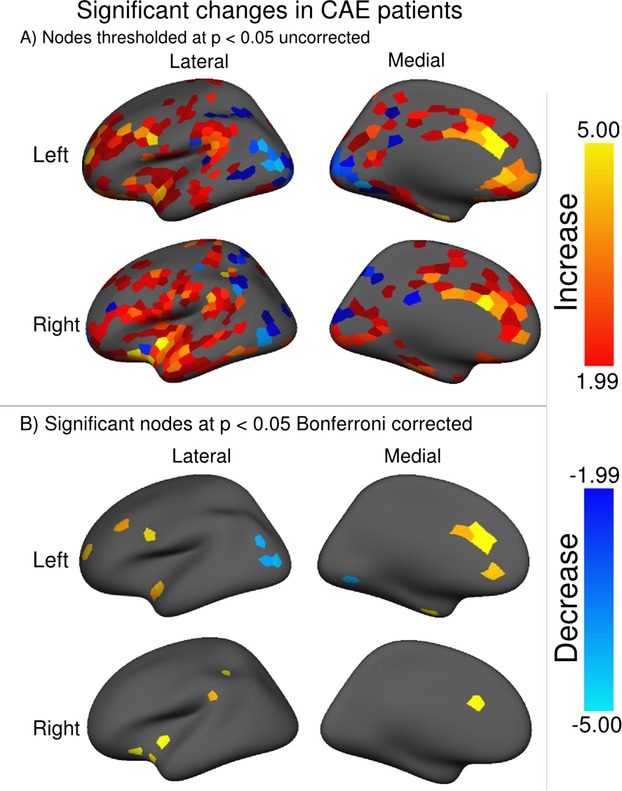
Node-wise differences between the childhood absence epilepsy and control groups. Difference is measured using a node-wise one sample T-test between the measured strength differences and the mean of a null distribution created using random permutations over the subject group. The T statistic is displayed thresholded at (A) P < 0.05 uncorrected to give a sense of the overall distribution, and (B) P < 0.05 FWE corrected to display those nodes with significant difference between the two groups.
Table 1.
A list of brain regions that show a significant difference in strength between CAE and controls
| Regions with significant differences in weighted connectivity (Bonferroni corrected P-values) | |||
|---|---|---|---|
| Region name | Left | Right | |
| (A) CAE increase | |||
| Frontal | Anterior cingulate (caudal) | 0.0003* | 0.0003* |
| Anterior cingulate (rostral) | 0.015* | >0.5 | |
| Middle frontal gyrus | 0.010* | 0.392 | |
| Supramarginal | 0.076 | 0.004* | |
| Lateral orbito-frontal | 0.109 | 0.004* | |
| Precentral | 0.006 | 0.212 | |
| Insula | 0.397 | 0.00005* | |
| Temporal | Superior temporal | 0.018* | 0.063 |
| Entorhinal | 0.005* | >0.5 | |
| Fusiform | >0.5 | 0.00007* | |
| (B) CAE decreases | |||
| Posterior | Lateral occipital | 0.016* | 0.058 |
| Inferior parietal | 0.031* | 0.075 | |
| Lingual | 0.033* | >0.5 | |
All increases were in fronto-temporal regions, all decreases in parieto-occipital regions. Nodes are considered significant if the difference passed a two-tailed P < 0.05 threshold. P-values were Bonferroni corrected for the number of nodes; values passing significance at this threshold are marked with asterisk (*). We see the caudal anterior cingulate is maximally affected bilaterally. Lateral and medial occipital decreases pass the significance threshold on the left side only, although a general trend toward occipital decrease is observed, see Figure3A. CAE, childhood absence epilepsy.
Sparsity threshold
In order to determine if the observed changes between the two groups are an artifact attributable to our choice of sparsity thresholds, which is a well-known confound when comparing two different networks where the observed effect may be driven by the graph density,33 we repeated our analysis for a range of proportional thresholds ranging from 0% to 100% (that is, from completely sparse to entirely unthresholded). This analysis clearly demonstrates that the results presented in Figures3A and B, 4 are not driven by the choice of sparsity threshold. We present these results as Figure S1.
Discussion
We have demonstrated statistically significant differences in connectivity patterns between CAE subjects and healthy controls. In subjects with CAE, cortical thickness connectivity is greatest in fronto-temporal regions, whereas in control subjects the greatest connectivity is in occipital regions. This closely mirrors resting state functional connectivity increases observed in the lateral and medial frontal cortex in patients with CAE,10 and agrees qualitatively with EEG-fMRI studies where the characteristic 3 Hz spike-and-wave is associated in activity in the frontal cortex.11 These are regions associated with impaired attention in these patients.12 In addition, the overall “connectedness” of cortical regions is greater in subjects with CAE. These results are not driven by absolute changes in the thickness of the cortex, but rather reflect differences in correlations across cortical networks. The connectivity findings of the control group, with highest correlation with the occipital regions, are consistent with the early activity of the visual cortex during development, the high metabolic rate of this area, and the finding of occipital emphasis in many structural and function connectivity measures in controls.18,20,39 The CAE group, however, is dominated by anterior cortical regions.
There are a number of possible explanations for increased overall connectivity and the changed emphasis from occipital to frontal regions that we have demonstrated in CAE subjects. During development, processes such as synaptic pruning, use-dependent plasticity, apoptosis and myelination, as well as neuronal firing patterns, can influence the structural development of the cortex in subtle ways. In this particular CAE cohort 67% of subjects have been free from seizures for at least 5 years by the time of scan, yet the subtle structural connectivity changes are still evident. This suggests that the findings are not simply the acute effect of seizures, but may reflect neurodevelopmental factors involved in these epilepsies. The normal pattern of correlation of brain regions with the occipital cortex may relate to the extensive functional and structural network connections of the occipital cortex. As a “visual” animal the normal firing pattern of the visual cortex in humans may influence the development of a range of other cortical regions (i.e., regions that fire together may be better correlated in cortical thickness than regions that are not driven in this way) and lead to stronger correlations with the cortical thickness measure throughout the brain. In CAE the altered pattern of cortical thickness connectivity suggests a fundamental change in the development of connectivity patterns perhaps influenced by differences in neuronal firing due to the underlying genetic factors which lead to the development of this disorder.
There is a strong case that a correlation exists between thickness of the neocortex and the axonal connections between them, and, ultimately, the individual genome.40,41 Hence, changes to structural cortical connectivity in CAE may indicate cortical reaction to abnormal firing, or may be some structural marker of the genetic precursor to absence epilepsy reflected in the large-scale structural organization of the cortex. Some authors have suggested that subcortical and fronto-cortical abnormalities may relate both to seizures and neurobehavioral comorbidities in GGE.42,43 Importantly, cognitive impairment has been identified prior to seizure onset.3,44 This can be interpreted as evidence of an underlying genetically determined neurodevelopmental basis for both the epilepsy and the cognitive problems. These changes may impact on cognition and cortical organization without necessarily being expressed as clinical seizures. Further studies examining anatomical correlations in children with CAE from onset to remission, including first-degree relatives not suffering from epilepsy, may explore these issues. A study of this sort could also examine the impact of AED use, and gender effects, on anatomical covariance in CAE.
Longitudinal structural imaging studies in children with idiopathic epilepsy suggest that altered patterns of brain development may occur in areas that include the prefrontal cortex.43 In that study, no association between these altered patterns and AED use could be found. The effect of medication in childhood cannot be excluded as having contributed to this finding, but it is more likely that processes associated with the epilepsy or seizures themselves would have the major effect on focal areas of cortex. In fact, longitudinal structural imaging studies in children with idiopathic epilepsy suggest that altered patterns of brain development may occur in areas that include the prefrontal cortex.43 Thickening or thinning of gray matter during childhood and adolescence is a core process of cortical maturation that can differ according to the area of the brain involved. It is likely that patients diagnosed with CAE would have a different cortical developmental trajectory than healthy controls. Gray matter thinning is associated with synaptic pruning, myelination, and apoptosis, and these processes have been suggested to underpin cognitive and behavioral development.45 Cortical thinning during development is known and involves primary sensorimotor cortices first, then multimodal cortices such as the frontal and temporal cortex, which continues during late adolescence.46,47 The absence of any absolute cortical thickness changes observed in this cohort may be due to any changes being so subtle as to escape detection, yet the development of these changes is reflected in the organization of the cortical thickness network. Indeed, if we accept that changes in cortical thickness morphology arise due to cellular hypertrophy-induced by seizures,48 then we would not expect to find absolute changes in a cohort of patients who have largely remitted from their seizures. Abnormal thickness of cortex has been demonstrated in development-related epilepsy syndromes such as BECTS.49,50 These processes of cortical brain maturation and ongoing functional refinement of cortical networks may be critical processes where abnormalities of network function or abnormalities within gene expression pathways may lead to epilepsy or its comorbidities.51
Conclusion
Extensive changes in brain networks are observed when comparing CAE to healthy controls, particularly a marked relative increase in covariance of the medial frontal lobe bilaterally, and a relative decrease in covariance in the occipital and parietal lobes. Our findings provide evidence of a structural network basis for what is typically considered a purely functional disease and demonstrates abnormal cortical organization that persists after the resolution of the seizures which may be a reflection of the etiology of CAE.
Acknowledgments
This study was supported by the National Institutes of Health – NINDS (NS-R37-31146; Principal Investigator, A. T. Berg). The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. This research was supported by a Victorian Life Sciences Computation Initiative (VLSCI) grant number VR0056 on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, Australia. M. Pedersen supported by a University of Melbourne Research Scholarship.
Conflict of Interest
Dr. Berg reports grants from NINDS, during the conduct of the study; other from Eisai, grants from Pediatric Epilepsy Research Foundation, personal fees and other from Korean Epilepsy Society, personal fees from AAN Continuum, outside the submitted work; and Editorial Board member for Neurology and Epilepsy & Behavior. Dr. Jackson has received honoraria from UCB. The remaining authors have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Mean connection strength in selected significant regions over a range of sparsity thresholds. Sparsity refers to the fraction of top connections allowed in the graph; increasing the sparsity threshold increases the allowed density of the graph. The differences between CAE and control are consistent over the complete range of sparsity thresholds, demonstrating that the significant differences between the groups are not artifacts of connection density.
References
- Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- Caplan R, Siddarth P, Stahl L, et al. Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities. Epilepsia. 2008;49:1838–1846. doi: 10.1111/j.1528-1167.2008.01680.x. [DOI] [PubMed] [Google Scholar]
- Masur D, Shinnar S, Cnaan A, et al. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology. 2013;81:1572–1580. doi: 10.1212/WNL.0b013e3182a9f3ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega C, Guo J, Killory B, et al. Symptoms of anxiety and depression in childhood absence epilepsy. Epilepsia. 2011;52:e70–e74. doi: 10.1111/j.1528-1167.2011.03119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Archer JS, Abbott DF, Waites AB, Jackson GD. fMRI “deactivation” of the posterior cingulate during generalized spike and wave. Neuroimage. 2003;20:1915–1922. doi: 10.1016/s1053-8119(03)00294-5. [DOI] [PubMed] [Google Scholar]
- Gotman J, Grova C, Bagshaw A, et al. Generalized epileptic discharges show thalamocortical activation and suspension of the default state of the brain. Proc Natl Acad Sci USA. 2005;102:15236–15240. doi: 10.1073/pnas.0504935102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller F, Siebner HR, Wolff S, et al. Simultaneous EEG-fMRI in drug-naive children with newly diagnosed absence epilepsy. Epilepsia. 2008;49:1510–1519. doi: 10.1111/j.1528-1167.2008.01626.x. [DOI] [PubMed] [Google Scholar]
- Carney PW, Masterton RAJ, Harvey AS, et al. The core network in absence epilepsy. Differences in cortical and thalamic BOLD response. Neurology. 2010;75:904–911. doi: 10.1212/WNL.0b013e3181f11c06. [DOI] [PubMed] [Google Scholar]
- Bai X, Guo J, Killory B, et al. Resting functional connectivity between the hemispheres in childhood absence epilepsy. Neurology. 2011;76:1960–1967. doi: 10.1212/WNL.0b013e31821e54de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney PW, Masterton RAJ, Flanagan D, et al. The frontal lobe in absence epilepsy EEG-fMRI findings. Neurology. 2012;78:1157–1165. doi: 10.1212/WNL.0b013e31824f801d. [DOI] [PubMed] [Google Scholar]
- Killory BD, Bai X, Negishi M, et al. Impaired attention and network connectivity in childhood absence epilepsy. Neuroimage. 2011;56:2209–2217. doi: 10.1016/j.neuroimage.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Briellmann RS, Pell GS, et al. Thalamic atrophy in childhood absence epilepsy. Epilepsia. 2006;47:399–405. doi: 10.1111/j.1528-1167.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- Pardoe H, Pell GS, Abbott DF, et al. Multi-site voxel-based morphometry: methods and a feasibility demonstration with childhood absence epilepsy. Neuroimage. 2008;42:611–616. doi: 10.1016/j.neuroimage.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan R, Levitt J, Siddarth P, et al. Frontal and temporal volumes in childhood absence epilepsy. Epilepsia. 2009;50:2466–2472. doi: 10.1111/j.1528-1167.2009.02198.x. [DOI] [PubMed] [Google Scholar]
- Betting LE, Li LM, Lopes-Cendes I, et al. Correlation between quantitative EEG and MRI in idiopathic generalized epilepsy. Hum Brain Mapp. 2010;31:1327–1338. doi: 10.1002/hbm.20944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore E, Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci. 2009;10:186–198. doi: 10.1038/nrn2575. [DOI] [PubMed] [Google Scholar]
- Hagmann P, Cammoun L, Gigandet X, et al. Mapping the structural core of human cerebral cortex. PLoS Biol. 2008;6:e159. doi: 10.1371/journal.pbio.0060159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AC. Networks of anatomical covariance. Neuroimage. 2013;80:489–504. doi: 10.1016/j.neuroimage.2013.05.054. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, He Y, Rosa-Neto P, et al. Revealing modular architecture of human brain structural networks by using cortical thickness from MRI. Cereb Cortex. 2008;18:2374–2381. doi: 10.1093/cercor/bhn003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt BC, Worsley KJ, Besson P, et al. Mapping limbic network organization in temporal lobe epilepsy using morphometric correlations: insights on the relation between mesiotemporal connectivity and cortical atrophy. Neuroimage. 2008;42:515–524. doi: 10.1016/j.neuroimage.2008.04.261. [DOI] [PubMed] [Google Scholar]
- Bernhardt BC, Rozen DA, Worsley KJ, et al. Thalamo–cortical network pathology in idiopathic generalized epilepsy: insights from MRI-based morphometric correlation analysis. Neuroimage. 2009;46:373–381. doi: 10.1016/j.neuroimage.2009.01.055. [DOI] [PubMed] [Google Scholar]
- Berg AT, Levy SR, Testa FM, Shinnar S. Classification of childhood epilepsy syndromes in newly diagnosed epilepsy: interrater agreement and reasons for d. Epilepsia. 1999;40:439–444. doi: 10.1111/j.1528-1157.1999.tb00738.x. [DOI] [PubMed] [Google Scholar]
- Berg AT, Shinnar S, Levy SR, Testa FM. Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia. 1999;40:445–452. doi: 10.1111/j.1528-1157.1999.tb00739.x. [DOI] [PubMed] [Google Scholar]
- Berg AT, Mathern GW, Bronen RA, et al. Frequency, prognosis and surgical treatment of structural abnormalities seen with magnetic resonance imaging in childhood epilepsy. Brain. 2009;132:2785–2797. doi: 10.1093/brain/awp187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger J, Bureau M, Dravet C, Genton P. Epileptic syndromes in infancy, childhood, and adolescence. 2nd ed. Montrouge: John Libbey Eurotext; 1992. [Google Scholar]
- Berg AT, Rychlik K. The course of childhood-onset epilepsy over the first two decades: a prospective, longitudinal study. Epilepsia. 2014;56:40–48. doi: 10.1111/epi.12862. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis, I: segmentation and surface reconstruction. Neuroimage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis, II: inflation, flattening and a surface-based coordinate system. Neuroimage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Rubinov M, Sporns O. Complex network measures of brain connectivity: uses and interpretations. Neuroimage. 2010;52:1059–1069. doi: 10.1016/j.neuroimage.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Lerch JP, Worsley K, Shaw WP, et al. Mapping anatomical correlations across cerebral cortex (MACACC) using cortical thickness from MRI. Neuroimage. 2006;31:993–1003. doi: 10.1016/j.neuroimage.2006.01.042. [DOI] [PubMed] [Google Scholar]
- Achard S, Bullmore E. Efficiency and cost of economical brain functional networks. PLoS Comput Biol. 2007;3:e17. doi: 10.1371/journal.pcbi.0030017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH. Collective dynamics of “small-world” networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Humphries MD, Gurney K. Network, “small-world-ness”: a quantitative method for determining canonical network equivalence. PLoS One. 2008;3:e0002051. doi: 10.1371/journal.pone.0002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten SC, Douw L, Bartolomei F, et al. Indications for network regularization during absence seizures: weighted and unweighted graph theoretical analyses. Exp Neurol. 2009;217:197–204. doi: 10.1016/j.expneurol.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Bernhardt BC, Chen Z, He Y, et al. Graph-theoretical analysis reveals disrupted small-world organization of cortical thickness correlation networks in temporal lobe epilepsy. Cereb Cortex. 2011;21:2147–2157. doi: 10.1093/cercor/bhq291. [DOI] [PubMed] [Google Scholar]
- Pardoe HR, Abbott DF, Jackson GD The Alzheimer's Disease Neuroimaging Initiative. Sample size estimates for well-powered cross-sectional cortical thickness studies. Hum Brain Mapp. 2013;34:3000–3009. doi: 10.1002/hbm.22120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-H, Fiecas M, Gutiérrez ED, et al. Genetic topography of brain morphology. Proc Natl Acad Sci USA. 2013;110:17089–17094. doi: 10.1073/pnas.1308091110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci USA. 2000;97:5621–5626. doi: 10.1073/pnas.090504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsipher DT, Seidenberg M, Guidotti L, et al. Thalamofrontal circuitry and executive dysfunction in recent-onset juvenile myoclonic epilepsy. Epilepsia. 2009;50:1210–1219. doi: 10.1111/j.1528-1167.2008.01952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Siddarth P, Toga AW, et al. Effects of childhood absence epilepsy on associations between regional cortical morphometry and aging and cognitive abilities. Hum Brain Mapp. 2011;32:580–591. doi: 10.1002/hbm.21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AT, Hesdorffer DC, Zelko FAJ. Special education participation in children with epilepsy: what does it reflect? Epilepsy Behav. 2011;22:336–341. doi: 10.1016/j.yebeh.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan TL, Baaré WFC, Stiles J, Madsen KS. Chapter 5 – postnatal brain development: structural imaging of dynamic neurodevelopmental processes. In: Braddick O, Atkinson J, Innocenti GM, editors. Progress in brain research. Amsterdam: Elsevier; 2011. pp. 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio A, Watkins KE, Chadwick M, et al. Longitudinal changes in grey and white matter during adolescence. Neuroimage. 2010;49:94–103. doi: 10.1016/j.neuroimage.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Shaw P, Kabani NJ, Lerch JP, et al. Neurodevelopmental trajectories of the human cerebral cortex. J Neurosci. 2008;28:3586–3594. doi: 10.1523/JNEUROSCI.5309-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothwell S, Meredith GE, Phillips J, et al. Neuronal hypertrophy in the neocortex of patients with temporal lobe epilepsy. J Neurosci. 2001;21:4789–4800. doi: 10.1523/JNEUROSCI.21-13-04789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overvliet GM, Besseling RMH, Jansen JFA, et al. Early onset of cortical thinning in children with rolandic epilepsy. Neuroimage Clin. 2013;2:434–439. doi: 10.1016/j.nicl.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoe HR, Berg AT, Archer JS, et al. A neurodevelopmental basis for BECTS: evidence from structural MRI. Epilepsy Res. 2013;105:133–139. doi: 10.1016/j.eplepsyres.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nijs L, Wolkoff N, Grisar T, Lakaye B. Juvenile myoclonic epilepsy as a possible neurodevelopmental disease: role of EFHC1 or Myoclonin1. Epilepsy Behav. 2013;28(suppl 1):S58–S60. doi: 10.1016/j.yebeh.2012.06.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mean connection strength in selected significant regions over a range of sparsity thresholds. Sparsity refers to the fraction of top connections allowed in the graph; increasing the sparsity threshold increases the allowed density of the graph. The differences between CAE and control are consistent over the complete range of sparsity thresholds, demonstrating that the significant differences between the groups are not artifacts of connection density.