

Abstract
Objective
To assess demographic, clinical, magnetic resonance imaging, and treatment exposure predictors of time to 3 or 12-month confirmed disability worsening in clinically isolated syndrome (CIS) and early multiple sclerosis (MS).
Methods
We utilized the MSBase Incident Study (MSBasis), a prospective cohort study of outcome after CIS. Predictors of time to first 3 and 12-month confirmed expanded disability status scale worsening were analyzed using Cox proportional hazards regression.
Results
About 1989 patients were analyzed, the largest seen-from-onset cohort reported to-date. A total of 391 patients had a first 3-month confirmed disability worsening event, of which 307 were sustained for 12 months. Older age at CIS onset (adjusted hazard ratio: aHR 1.17, 95% 1.06, 1.30), pyramidal (aHR 1.45, 95% CI 1.13, 1.89) and ambulation (HR 1.60, 95% CI 1.09, 2.34) system dysfunction, annualized relapse rate (aHR 1.20, 95% CI 1.18, 1.22), and lower proportion of observation time on treatment were associated with 3-month confirmed worsening. Predictors of time to 12-month sustained worsening included pyramidal system dysfunction (Hazard ratio: aHR 1.38, 95% CI 1.05, 1.83), and older age at CIS onset (aHR 1.17, 95% CI 1.04, 1.31). Greater proportion of follow-up time exposed to treatment was associated with greater reductions in the rate of worsening.
Interpretation
This study provides class IV evidence for a strong protective effect of disease-modifying treatment to reduce disability worsening events in patients with CIS and early MS, and confirms age and pyramidal dysfunction at onset as risk factors.
Introduction
Reduction in neurological disability is the most important goal of early treatment in multiple sclerosis (MS). Reflecting this, disability progression or “worsening” events have been used as the primary endpoint in several recent disease-modifying therapy (DMT) studies.1–3 The implicit assumption is that therapeutic intervention in the relapsing-remitting phase of MS reduces the incidence and rate of accumulation of irreversible disability.
The Expanded Disability Status Scale (EDSS) is an ordinal scale with 19 disease steps between 0 and 10. It is the accepted gold standard for assessing level of disability and worsening in MS clinical trials.4 In clinical trials, the identification of “confirmed disability worsening” is based on assessment of repeated EDSS measurements. An increase of 1 point on the EDSS above baseline subsequently confirmed at repeat assessment either 3 or 6 months later (3- or 6-month confirmed worsening) are commonly used.1,5–10 However, the duration of a confirmed disability worsening event is important, as some 3-month confirmed disability worsening events later remit.11
A first confirmed EDSS disability worsening in clinically isolated syndrome (CIS) and early MS heralds the onset of MS-related disability. Prior data suggest that DMT exposure in early MS reduces long-term disability worsening in comparison to later exposure to DMT,12 but these results require confirmation. As randomized clinical trials are typically of 2–3 years duration, their capacity to measure long-term confirmed progression or worsening rates are very limited. The aim of the current study was to assess the independent effect of DMT exposure on 3 month-confirmed (short-term) and 12-month (long-term) sustained MS disability worsening events in a large seen-from-onset cohort of patients, whilst controlling for putative demographic, clinical and magnetic resonance imaging (MRI) predictors of these outcomes.
Patients and Methods
Study population and design
MSBasis is an observational cohort study based on prospectively collected data, a substudy of the international MSBase Registry.13,14 Patients who experienced a CIS and were seen by a participating neurologist within 12 months of onset were recruited into this longitudinal study. MSBasis commenced in 2004 and, at date of data extraction, prospectively followed 3623 patients from MS specialist centers in 20 countries. Baseline data collected included demographics, date of CIS, Kurtzke Functional System (KFS) scores, and EDSS scores as well as cerebral MRI classification according to Barkhof-Tintore criteria. Follow-up data collected at a minimum annually included: date of visit, KFS/EDSS scores, relapses and treatment changes since the patient's last visit. Data entry was performed in or near real time at most centers as previously described.15 Human research ethics committee approval was obtained from the lead site (Melbourne Health HREC) and approvals or waivers, and written informed consent from patients were obtained at each participating site, as local laws required.
Patients with a primary progressive disease course were excluded from this study. A minimum of three visits per patient spanning a minimum 9 months, with full EDSS assessment was required to assess first confirmed 3-month disability worsening. Patients without a complete baseline KFS were excluded. Cerebral MRI was available for 88.0% of patients within 12 months of first clinic visit. Of the 3623 patients enrolled in MSBasis at the time of data extraction and compilation, 1989 patients were eligible for analysis following exclusions (largely inadequate follow-up time, as the study continues to recruit). Differences between included and excluded patients were not significant. For instance, 71.2% of eligible patients were female compared to 67.8% of excluded patients. Median age at onset was 32.9 years for eligible patients and 34.0 years in excluded patients.
Definitions
Disability worsening
Confirmed disability worsening events were defined as a minimum one-point increase in EDSS score above a baseline EDSS of between 1 and 5.5 confirmed at repeat assessment at least 3 months or 1 year later. Baseline EDSS scores of zero required a confirmed one and a half-point increase, and baseline EDSS scores equal to or above six required a half-point increase above baseline confirmed at least 3 months or 1 year later. EDSS scores recorded during relapses were excluded. Twelve-month sustained worsening was defined as that subset of the initial 3-month confirmed disability worsening who sustained this initial worsening for a minimum of 12 months following the date of the initial observed worsening.
Relapsing-remitting MS
As soon as MS is confirmed according to McDonald criteria, iMed software (Merck Serono, Geneva, Switzerland) classifies patients as relapsing-remitting MS (RRMS), this includes “CIS patients” fulfilling “McDonald MS” and those with clinically definite MS.
Baseline EDSS/KFS
Baseline EDSS and KFS here are derived from the EDSS visit immediately preceding a 3- or 12-month confirmed disability worsening event.
Baseline cerebral MRI
The T2 lesion load classifier was determined from the first MRI scan recorded within 12 months of CIS onset (median 33 days; inter-quartile range [IQR] 9, 110), using the Barkhof-Tintore categories of 0, 1–2, 3–8 or 9+ lesions.
Proportion of time on treatment
The proportion of time patients spent on treatment (PTT) was used to account for changing treatment status of patients over the observation period. It took into account the total time a patient spent on treatment, including any switches and gaps in treatment. All MS therapies were included in the calculation of the PTT. The PTT was censored at confirmed worsening, or most recent visit if a worsening event had not yet occurred. For the primary analysis, PTT was categorized according to the following ranges: no treatment (i.e., 0%), >0 to ≤50%, >50 to ≤80% and >80%.
No DMT
In the present study, no DMT refers to the patient group that did not receive DMT treatment (IFNβ, glatiramer acetate, fingolimod, natalizumab) prior to first disability worsening. This excludes the use of glucocorticoids to treat relapses.
Statistical analyses
Categorical variables were summarized using frequency and percentage and compared using Pearson's χ2. Continuous variables were assessed for normality using a Shapiro–Wilk test and described using mean and standard deviation (SD), or summarized using median and IQR as appropriate, and compared using one-way analysis of variance (ANOVA), or Kruskal–Wallis with Bonferroni's post hoc correction respectively. Predictors of first 3-month confirmed and 12-month sustained disability worsening were assessed using univariable and multivariable Cox proportional hazards regression. Hazard proportionality was assessed through analysis of scaled Schoenfeld residuals. In the absence of a worsening event, data were censored at the most recent EDSS clinic visit. Demographic, clinical, para-clinical, and treatment covariates that were associated with the worsening outcome on univariable analysis were then included in adjusted analyses. The multivariable modeling analysis was adjusted for clinic location (country) and sex. Interactions between model covariates were examined in the multivariable modeling. There were no significant interaction effects in the adjusted models. To assess whether subjects recording early worsening conferred an ascertainment bias upon the association between treatment and subsequent worsening, an additional model incorporating time from CIS onset to initiation of first DMT as a predictor variable was run as a sensitivity analysis. Kaplan–Meier survival curves were plotted for worsening-free survival. All reported P-values are exact and 2-tailed and for all analyses P < 0.05 was considered significant. All analyses were performed using Stata version 12.0 (StataCorp, College Station, TX).
Results
About 1989 CIS patients enrolled in MSBasis were included in this analysis (Fig.1). Table1 summarizes baseline demographic, clinical, para-clinical, and treatment characteristics of the cohort. A total of 6724 person-years of follow-up were analyzed, with median individual follow-up of 3.0 years (range 0.75–9.9). Mean age at CIS onset was 32.8 years (SD 10.0) and 71.3% of patients were female. Median EDSS at baseline was 1.5 (IQR: 1, 2), and 69% of patients with available MRI had fewer than nine T2 lesions at baseline.
Figure 1.
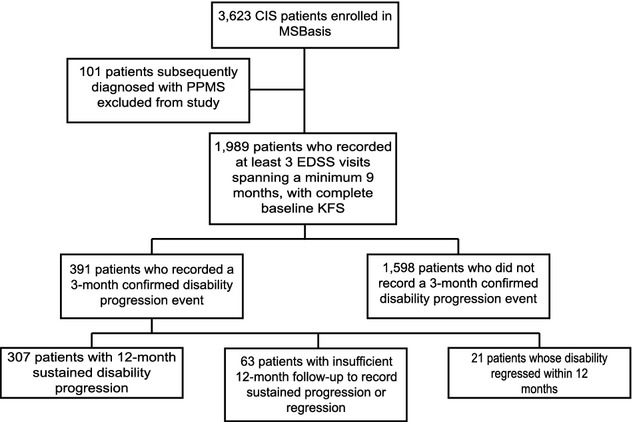
Summary of patients analyzed in the present analysis. Note the 12-month sustained disability worsening group is a subset of the 3-month confirmed disability worsening group.
Table 1.
Baseline demographic and clinical characteristics
| Variable | Level | All patients (n = 1989) |
|---|---|---|
| Visits assessed | Median (IQR) | 7 (4, 11) |
| Range | 3–66 | |
| Sex | Male | 571 (28.7%) |
| Female | 1418 (71.3%) | |
| Age at CIS | Mean (SD) | 32.9 (10.0) |
| Range | 6–68 | |
| EDSS | Median (IQR) | 1.5 (1, 2) |
| Range | 0–7.5 | |
| Affected KFS | ||
| (KFS score ≥1) | Ambulation | 208 (10.5%) |
| Pyramidal | 1134 (57.0%) | |
| Cerebellar | 528 (26.5%) | |
| Brainstem | 634 (31.9%) | |
| Sensory | 943 (47.4%) | |
| Bowel/Bladder | 304 (15.3%) | |
| Visual | 485 (24.4%) | |
| Mental | 285 (14.3%) | |
| T2 lesion load | ||
| 0 | 77 (3.9%) | |
| 1–2 | 106 (5.3%) | |
| 3–8 | 1027 (51.6%) | |
| ≥9 | 540 (27.1%) | |
| Not available | 239 (12.0%) | |
| Age at DMT start (years) | Mean (SD) | 33.2 (9.9) |
| Range | 6.4–67.6 | |
| First DMT | ||
| (prior to worsening) | No DMT | 650 (32.7%) |
| IFNβ-1a IM | 373 (18.8%) | |
| IFNβ-1a SC | 494 (24.8%) | |
| IFNβ-1b | 232 (11.7%) | |
| Glatiramer Acetate | 208 (10.5%) | |
| Fingolimod | 4 (0.2%) | |
| Natalizumab | 28 (1.4%) | |
| PTT | ||
| (censored at progression) | 0% | 666 (33.5%) |
| >0–50% | 349 (17.5%) | |
| >50–80% | 519 (26.1%) | |
| >80% | 455 (22.9%) | |
CIS, clinically isolated syndrome; EDSS, Expanded Disability Status Scale; KFS, Kurtzke Functional System; DMT, disease-modifying therapy; PTT, proportion of time on treatment; IQR, interquartile range; IM, intramuscular; SC, subcutaneous.
A total of 1339 (67.3%) of patients were exposed to at least one DMT during the observation period. These patients contributed 4895 person-years of follow-up of which 3237 person-years were on therapy. Mean (SD) follow-up was 3.4 years (2.2). Median time to first DMT from CIS onset was 0.7 years (IQR: 0.4, 1.2) whilst mean (SD) duration of first DMT (censoring at first worsening event) was 2.2 years (1.5). Of these, 19% (n = 252) were classified as CIS and 81% (n = 1087) of patients had RRMS at the time of treatment initiation.
About 391 (19.7%) patients had a first 3-month confirmed disability worsening event. Of these, 307 (78.5%) worsening events were sustained for at least 12 months. Twenty-one patients’ disability worsening regressed, and 63 had insufficient follow-up to demonstrate sustained worsening or regression, having been followed up for less than the 12 months required to assess sustained worsening at 12 months. Median (IQR) time to first 3-month confirmed disability worsening was 1.94 years (0.87, 3.39) whilst median time to 12-month sustained worsening was 2.85 years (1.82, 4.22). On adjusted modeling (Table2), annualized relapse rate (ARR) was independently predictive of increased rates of 3-month worsening (adjusted hazard ratio [aHR] 1.20, 95% CI 1.18, 1.22) but not of 12-month sustained worsening (aHR 1.01, 95% CI 0.98, 1.05). Additionally a baseline ambulation KFS score of ≥1 versus 0 (aHR 1.60; 95% CI 1.09, 2.34), and a baseline pyramidal KFS score of ≥2 versus 0–1 (aHR 1.45; 95% CI 1.13, 1.89) were predictive of 3-month confirmed disability worsening events. A baseline pyramidal score of ≥2 (vs. 0–1) remained predictive (aHR 1.38; 95% CI 1.05, 1.83) of 12-month sustained worsening. Older age at CIS onset was predictive of both 3-month (aHR 1.17 per 10 years, 95% CI 1.06, 1.30) and 12-month (HR 1.17 per 10 years, 95% CI 1.04, 1.31) disability worsening events. Sex was not predictive of disease worsening on adjusted or unadjusted analyses.
Table 2.
Predictors of 3-month confirmed and 12-month sustained disability worsening events
| Predictor | Level | 3-month confirmed worsening events (n = 391) | 12-month sustained worsening events (n = 307) | ||
|---|---|---|---|---|---|
| n (% of level) | Adjusted HR (95% CI) P-value1 | n (% of level) | Adjusted HR (95% CI) P-value2 | ||
| Demographics | |||||
| Sex | Male | 110 (19.3%) | 1.05 (0.84, 1.31) 0.677 | 83 (14.5%) | 0.97 (0.75, 1.25) 0.823 |
| Female | 281 (19.8%) | 1.00 | 224 (15.8%) | 1.00 | |
| Age at CIS onset | Per 10 years | – | 1.17 (1.06, 1.30) 0.002 | – | 1.17 (1.04, 1.31) 0.007 |
| Clinical | |||||
| KFS ambulation | 0 | 318 (17.9%) | 1.00 | 254 (14.3%) | 1.00 |
| 1+ | 73 (35.1%) | 1.60 (1.09, 2.34) 0.015 | 53 (25.5%) | 1.45 (0.94, 2.22) 0.092 | |
| KFS pyramidal | 0–1 | 242 (16.5%) | 1.00 | 194 (13.2%) | 1.00 |
| 2+ | 149 (28.7%) | 1.45 (1.13, 1.89) 0.003 | 113 (21.7%) | 1.38 (1.05, 1.83) 0.023 | |
| KFS cerebellar | 0–1 | 306 (18.5%) | 1.00 | 243 (14.7%) | 1.00 |
| 2+ | 85 (25.6%) | 1.06 (0.78, 1.44) 0.710 | 64 (19.3%) | 1.33 (0.95, 1.86) 0.097 | |
| KFS bowel/bladder | 0–1 | 342 (18.5%) | 1.00 | 272 (14.8%) | 1.00 |
| 2+ | 49 (33.8%) | 0.78 (0.52, 1.18) 0.246 | 35 (24.1%) | 0.75 (0.47, 1.21) 0.241 | |
| Relapses | |||||
| ARR | – | 1.20 (1.18, 1.22) <0.001 | – | 1.01 (0.98, 1.05) 0.456 | |
| MRI | |||||
| T2 lesion load | 0 | 27 (35.1%) | 1.00 | 25 (32.5%) | 1.00 |
| 1–2 | 26 (24.5%) | 1.17 (0.67, 2.04) 0.578 | 22 (20.8%) | 1.06 (0.59, 1.92) 0.834 | |
| 3–8 | 195 (19.0%) | 0.91 (0.60, 1.38) 0.649 | 148 (14.4%) | 0.83 (0.53, 1.30) 0.424 | |
| ≥9 | 101 (18.7%) | 1.07 (0.69, 1.68) 0.753 | 78 (14.4%) | 0.98 (0.61, 1.58) 0.939 | |
| Not recorded | 42 (17.8%) | 0.88 (0.53, 1.46) 0.634 | 34 (14.2%) | 0.82 (0.48, 1.40) 0.472 | |
| Treatment | |||||
| Proportion follow-up years treated | 0 (no treatment) | 157 (23.6%) | 1.07 (0.81, 1.39) 0.645 | 142 (21.3%) | 0.75 (0.56, 1.02) 0.066 |
| >0–50% treated | 87 (24.9%) | 1.00 | 68 (19.5%) | 1.00 | |
| >50–80% treated | 95 (18.3%) | 0.64 (0.47, 0.86) 0.003 | 59 (11.4%) | 0.43 (0.31, 0.59) <0.001 | |
| >80–100% treated | 52 (11.4%) | 0.35 (0.25, 0.50) <0.001 | 38 (8.4%) | 0.24 (0.17, 0.35) <0.001 | |
Cox Proportional Hazards Regression.
Hazard proportionality test: p = 0.3025;
Hazard proportionality test: p = 0.2772
CIS, Clinically Isolated Syndrome; KFS, Kurtzke Functional System; ARR, annualized relapse Rate; DMT Disease Modifying Therapy; IM Intramuscular; SC Subcutaneous; CI Confidence Interval.
Unadjusted modeling further revealed that a baseline EDSS of 1–2.5 versus an EDSS of zero predicted an increased risk of 3-month (HR 2.34; 95% CI 1.72–3.28; P < 0.001) and 12-month (HR 2.01; 95% CI 1.44–2.80; P < 0.001) disability worsening events. Furthermore, an EDSS of 3–5.5 as compared to an EDSS of zero was predictive of a greater risk of 3-month (HR 3.3; 95% CI 2.29–4.89; P < 0.001) and 12-month (HR 2.88; 95% CI 1.91–4.36; P < 0.001) worsening. However, EDSS and KFS scores were highly correlated with each other and thus were unable to be combined in the adjusted model due to significant colinearity.
Although predictive on unadjusted analysis, the presence of nine or greater T2 lesions on baseline MRI was not independently associated with increased risk of either 3-month confirmed (aHR 1.07, 95% CI 0.69, 1.68) or 12-month sustained (aHR 0.98, 95% CI 0.61, 1.58) worsening in the multivariable models. A sensitivity analysis showed that those patients with higher lesion load were more likely to receive DMT earlier, and spent a greater proportion of the observation period on DMT treatment (Table3).
Table 3.
Baseline demographic and clinical covariates by T2 lesion load
| Variable | Level | T2 lesion load | P-value | ||||
|---|---|---|---|---|---|---|---|
| 0 (n = 77) | 1–2 (n = 106) | 3–8 (n = 1027) | ≥9 (n = 540) | N/A (n = 239) | |||
| Demographics | |||||||
| Sex | Male | 14 | 30 | 306 | 161 | 60 | 0.1581 |
| Female | 63 | 76 | 721 | 379 | 179 | 0.1581 | |
| Age at onset | Mean (SD) | 32.3 (8.6) | 33.7 (9.8) | 32.5 (9.8) | 33.4 (10.4) | 33.3 (10.1) | 0.2262 |
| Clinical | |||||||
| EDSS | Median (IQR) | 1.5 (1, 2) | 1.5 (1, 2) | 1.5 (1, 2) | 1.5 (0, 2) | 1.5 (1, 2) | 0.40293 |
| Treatment | n = 18 (23%) | n = 43 (40.1%) | n = 705 (68.6%) | n = 407 (75.4%) | n = 166 (69.5%) | ||
| Time to first DMT from CIS onset | Median (IQR) | 2.5 (1.2, 3.7) | 0.9 (0.5, 1.5) | 0.8 (0.4, 1.3) | 0.6 (0.3, 1.1) | 0.7 (0.3, 1.0) | 0.00013 |
| Mean (SD) | 3.1 (2.5) | 1.2 (1.1) | 1.1 (1.2) | 0.8 (0.8) | 1.0 (1.2) | ||
| PTT (continuous) | Median (IQR) | 0.5 (0.3, 0.6) | 0.6 (0.3, 0.8) | 0.7 (0.5, 0.8) | 0.7 (0.5, 0.9) | 0.7 (0.4, 0.8) | 0.00033 |
| Mean (SD) | 0.4 (0.2) | 0.5 (0.3) | 0.6 (0.3) | 0.7 (0.3) | 0.6 (0.3) | ||
| Disease classification at treatment start | CIS | 1 (5.5%) | 6 (14.0%) | 124 (17.6%) | 96 (23.6%) | 25 (15.1%) | |
| RRMS | 17 (94.5%) | 37 (86.0%) | 581 (82.4%) | 311 (76.4%) | 141 (84.9%) | ||
Pearson's χ2.
One-way ANOVA, Bonferroni's post hoc test.
Kruskal–Wallis.
The effect of treatment on risk of experiencing a disability worsening event was also assessed in the adjusted regression model (Table2). Mean (SD) duration of treatment across the sample within the observation period was 1.62 years (1.91) and 1.73 years (1.92) for the 3-month confirmed and 12-month sustained worsening analyses, respectively (where treatment duration was censored at the time of the initial first worsening event). The median (IQR) number of DMT initiations per patient were 1 (0, 1) for both the 3-month and 12-month analyses. The cohort was sub-divided according to proportion of time treated (PTT). Increasing PTT resulted in a step-wise reduction in the rate of 3-month confirmed (Fig.2A) and 12-month sustained (Fig.2B) worsening events. Patients exposed to DMT for between 51% and 80% of their follow-up time had a 36% (aHR 0.64, 95% CI 0.47, 0.86) and a 57% (aHR 0.43, 95% CI 0.31, 0.59) reduction in the rate of 3-month confirmed and 12-month sustained disability worsening events, respectively, compared with patients treated for >0 but ≤50%. Longer DMT exposure was associated with even larger risk reductions, as patients treated for greater than 80% of their observation period had a 65% (aHR 0.35, 95% CI 0.25, 0.50) and a 76% (aHR 0.24, 95% CI 0.17, 0.35) reduction in the rate of 3-month and 12-month worsening events, respectively, compared to patients treated for >0 but ≤50% of their follow-up time. There was a weak effect of a PTT ≤50% relative to patients who were untreated (PTT of 0%) prior to a 12-month, but not a 3-month worsening event.
Figure 2.

Kaplan-Meier survival curves showing proportion of patients free of 3-month confirmed and 12-month sustained disability worsening by: proportion of time on treatment (PTT; A: 3-month disability worsening; B: 12-month disability worsening) and by disease-modifying therapy (DMT) product (C:3-month disability worsening; D: 12-month disability worsening).
The identity of the first DMT commenced during follow-up was co-linear with PTT and thus we ran an alternate adjusted model as a sensitivity analysis substituting PTT for DMT identity (Table4). Interferon beta-1a intramuscular (IM) (aHR 0.70, 95% CI 0.53, 0.94), Interferon beta-1b (aHR 0.58, 95% CI 0.41, 0.82), Interferon beta-1a subcutaneous (SC) (aHR 0.51, 95% CI 0.39, 0.68), and Glatiramer Acetate (aHR 0.59, 95% CI 0.41, 0.85) were all associated with decreased rates of first 3-month confirmed worsening compared with no exposure to DMT during follow-up (Fig.2C). Similarly, each of these DMT products were associated with comparable risk reduction in 12-month sustained worsening compared to no DMT (Table4, Fig.2D). Of those patients who experienced a 3-month confirmed disability worsening event, we found that 78.3% of worsening events were preceded by a relapse within 3 months of the worsening event in the no DMT group, whereas, 81.6% of worsening events were not preceded by a relapse in patients who were DMT treated.
Table 4.
Predictors of 3-month confirmed and 12-month sustained disability worsening events – sensitivity model substituting first DMT identity for PTT
| Predictor | Level | 3-month confirmed worsening events (n = 391) | 12-month sustained worsening events (n = 307) | ||
|---|---|---|---|---|---|
| n (% of level) | Adjusted HR (95% CI) P-value1 | n (% of level) | Adjusted HR (95% CI) P-value2 | ||
| Demographics | |||||
| Sex | Male | 110 (19.3%) | 1.02 (0.82, 1.28) 0.857 | 83 (14.5%) | 0.95 (0.73, 1.22) 0.669 |
| Female | 281 (19.8%) | 1.00 | 224 (15.8%) | 1.00 | |
| Age at CIS onset | Per 10 years | – | 1.16 (1.05, 1.29) 0.003 | – | 1.16 (1.04, 1.30) 0.010 |
| Clinical | |||||
| KFS Ambulation | 0 | 318 (17.9%) | 1.00 | 254 (14.3%) | 1.00 |
| 1+ | 73 (35.1%) | 1.60 (1.09, 2.33) 0.016 | 53 (25.5%) | 1.50 (0.97, 2.30) 0.066 | |
| KFS pyramidal | 0–1 | 242 (16.5%) | 1.00 | 194 (13.2%) | 1.00 |
| 2+ | 149 (28.7%) | 1.46 (1.14, 1.87) 0.003 | 113 (21.7%) | 1.39 (1.05, 1.84) 0.023 | |
| KFS cerebellar | 0–1 | 306 (18.5%) | 1.00 | 243 (14.7%) | 1.00 |
| 2+ | 85 (25.6%) | 1.06 (0.78, 1.44) 0.698 | 64 (19.3%) | 1.33 (0.95, 1.86) 0.092 | |
| KFS bowel/bladder | 0–1 | 342 (18.5%) | 1.00 | 272 (14.8%) | 1.00 |
| 2+ | 49 (33.8%) | 0.83 (0.55, 1.26) 0.388 | 35 (24.1%) | 0.80 (0.50, 1.28) 0.349 | |
| Relapses | |||||
| ARR | – | 1.20 (1.18, 1.21) <0.001 | – | 1.01 (0.97, 1.04) 0.820 | |
| MRI | |||||
| T2 lesion load | 0 | 27 (35.1%) | 1.00 | 25 (32.5%) | 1.00 |
| 1–2 | 26 (24.5%) | 1.12 (0.64, 1.95) 0.686 | 22 (20.8%) | 1.02 (0.57, 1.84) 0.940 | |
| 3–8 | 195 (19.0%) | 0.83 (0.55, 1.26) 0.379 | 148 (14.4%) | 0.78 (0.50, 1.21) 0.266 | |
| ≥9 | 101 (18.7%) | 0.94 (0.60, 1.47) 0.786 | 78 (14.4%) | 0.88 (0.55, 1.42) 0.602 | |
| Not recorded | 42 (17.8%) | 0.83 (0.50, 1.37) 0.466 | 34 (14.2%) | 0.80 (0.47, 1.36) 0.416 | |
| Treatment | |||||
| First DMT | No DMT | 153 (23.5%) | 1.00 | 139 (21.4%) | 1.00 |
| IFNβ-1a IM | 76 (20.4%) | 0.70 (0.53, 0.94) 0.015 | 56 (15.0%) | 0.53 (0.39, 0.74) <0.001 | |
| IFNβ-1b | 43 (18.5%) | 0.58 (0.41, 0.82) 0.002 | 33 (14.2%) | 0.47 (0.32, 0.69) <0.001 | |
| IFNβ-1a SC | 79 (16.0%) | 0.51 (0.39, 0.68) <0.001 | 57 (11.5%) | 0.41 (0.30, 0.57) <0.001 | |
| Glatiramer Acetate | 35 (16.8%) | 0.59 (0.41, 0.85) 0.005 | 20 (9.6%) | 0.33 (0.21, 0.54) <0.001 | |
Cox proportional hazards regression. DMT, disease-modifying therapy; PTT, proportion of time on treatment; CIS, clinically isolated syndrome; KFS, Kurtzke Functional System; ARR, annualized relapse rate; MRI, magnetic resonance imaging; IM, intramuscular; SC, subcutaneous.
Hazard proportionality test: P = 0.2981.
Hazard proportionality test: P = 0.1973.
Patients experiencing comparatively early disability worsening following CIS could have less opportunity and thus lower probability of initiating treatment prior to the worsening event relative to patients recording a confirmed worsening later on in post-CIS follow-up. To assess whether this biased, or otherwise influenced, the associations of treatment with worsening, we performed a sensitivity analysis for time to either first 3- or 12-month sustained worsening including time to first DMT as an additional model covariate. Every 1-year increase in the time to first post-CIS treatment initiation was associated with a reduction in the risk of subsequent 3-month (HR 0.75, 95% CI 0.65, 0.87) and 12-month sustained worsening (HR 0.73, 95% CI 0.61, 0.87). However, an increasing proportion of follow-up time on treatment remained associated with a reduction in the risk of worsening. In a further sensitivity analysis we showed that increasing time to second attack was not associated with time to either 3-month (HR 0.95, 95% CI 0.88, 1.02) or 12-month sustained worsening (HR 0.94, 95% CI 0.87, 1.02).
Discussion
Early clinical and para-clinical factors that could predict disability outcome are of great interest in MS. Previous studies in CIS and RRMS cohorts have proposed various prognostic indicators.16–23 However, factors contributing to individual disability worsening rates after CIS have not previously been examined in a contemporary cohort.
We found that rates of 3-month confirmed and 12-month sustained disability worsening events after CIS onset were significantly and substantially reduced by DMT treatment. In our study, we assessed DMT use in two ways. Firstly, we used a contemporaneous cohort of patients who were not treated prior to a first disability worsening event. Secondly, to account for the fact that clinically stable patients tend to remain untreated, and to inherently control for treatment effect within individuals, we assessed the cumulative proportion of time individuals spent on treatment over the observation period. We found a significant benefit of treatment if patients were treated for greater than 50% of the observation period, versus less than 50% of the time, with a further benefit if patients were treated for greater than 80% of the observation period. This effect was more pronounced for 12-month sustained disability worsening events. Similar results have previously been reported in a post hoc analysis of an earlier trial24; where IFNβ-1a IM had a more potent effect on 12-month confirmed worsening events than 6-month worsening events.8 Interestingly, our study found little difference in the rate of disability accumulation between those patients with a PTT of ≤50% and those who were untreated. Two recent long-term follow-up studies25,26 of pivotal trials also aimed to determine the effect of cumulative treatment dose or duration on long-term disability measures. Whilst both studies had relatively small numbers of patients and were likely underpowered, they did reported trends in delay to disability milestones in higher dose/duration groups.25,26 In our cohort, median disease duration at treatment initiation was less than 1 year. Our results are consistent with those of a previous study which found that patients treated within a year of RRMS onset had a 37% reduced risk of experiencing a one-point EDSS worsening relative to those treated more than a year after RRMS onset.12
Here, we also report that the two strongest independent clinical predictors of individual first 3-month confirmed worsening events are baseline dysfunction within the pyramidal system and ARR. The effect of pyramidal system dysfunction is consistent with previous reports from RRMS cohorts that have identified initial motor symptoms as a poor prognostic indicator.17,19,20,23 Our results extend previous analyses27 by demonstrating that, ARR is independently predictive of 3-month individual worsening events in early MS. However, this relationship between ARR and disability worsening requires further examination, and will be interrogated further as our data set matures.
Past CIS and RRMS studies have reported that older age at onset is a poor prognostic indicator.18,20–23 Consistent with these studies, we found that older age at CIS diagnosis was an independent predictor of both 3- and 12-month sustained disability worsening events. Our analysis shows no independent adverse effect of male sex, consistent with previous studies.18,20,22,23
Natural history CIS and RRMS studies have consistently found that high T2 lesion load and volume are associated with poor prognostic outcomes.16,18,28–31 In our contemporary cohort, we report that T2 lesion load is, at best, a weak predictor of the rate of disability accumulation. We observed that patients with a high T2 lesion load were, on average, treated earlier and longer with DMT than those with a low lesion load. Our data therefore suggest that DMT intervention in individuals with a high T2 load may in part offset the adverse effect of high T2 lesion load at onset on disability accumulation. However, this is a correlation only and will need to be examined further with a larger sample possessing greater follow-up.
There is compelling evidence from randomized controlled trials (RCTs) that DMT reduce relapse rates and T2 lesion burden in RRMS patients. However, the effect of DMT on reduction in disability worsening, particularly long-term confirmed worsening, is less certain. Randomized controlled trials of first-line therapies in CIS cohorts have demonstrated that early treatment delays conversion to CDMS16,32–34 and the BENEFIT extension study has provided evidence for 3-year reductions in the risk of disability worsening in early versus late treatment groups, however, this was not sustained at 5 years.35,36 Similarly, long-term follow-up studies of RRMS RCTs have demonstrated that, in general, patients in the “early” treatment arms of these trials exhibit slower rates of disability worsening compared to patients initially randomized to the placebo arms of the respective trials.37 Studies in real-world clinical settings have generally found that DMT treatment results in slower EDSS worsening.38,39 However, a recent study failed to find a significant effect of DMT on delay to disability milestones relative to a contemporary or to a historical untreated cohort.22 Patients in that study had a mean age of 38 years and a median disease duration of 3.0 years at treatment start, a comparatively long delay in treatment start relative to disease duration. In contrast, examining the hazard of 12-month sustained worsening events in CIS and early MS, we clearly show a marked reduction in these worsening events due to DMT treatment. One possible explanation for these discordant results is that treatment, when initiated in our cohort, commenced much sooner (8 months) and at a younger age (33.2 years) after CIS onset. Our data suggest that the principal mechanism of action by which DMT treatment slows the rate of disability worsening in the early phase of MS is via the reduction in relapse-associated persistent disability, as even in early relapsing-remitting MS, relapse-independent worsening events were not prevented by injectable DMT.
Strengths and limitations
The size, and cumulative follow-up available for our cohort allowed us to analyze the independent effect of significant variables in our multivariable models. Our capacity to assign hazard ratios to the predictive variables in our multivariable models now opens the possibility for providing individualized risk prediction.
One limitation of our study is that treatment adherence was not assessed. It is possible that treatment effects in highly adherent patient groups could be larger than those reported in our study. A further limitation of our study is selection bias. As the participating centers were specialized MS clinics, the data set contains few patients with normal cerebral MRI scans, who are known to be at low risk of MS conversion and disability accumulation, and the results are not applicable to this population. However, at the participating centers, all patients presenting within 12 months of CIS symptoms were recruited if consent was obtained. Given the observational nature of this study, no specific intervention was prescribed to patients, and physicians together with their patients made treatment decisions. Unlike an RCT, this study lacks randomization and may therefore suffer from indication bias. However, the greatest treatment effect we found was that of cumulative time on treatment. Furthermore, in our adjusted analyses, we found that all first-line therapies performed well, reducing rates of 12-month disability accumulation by 47–67%, with no significant differences between individual preparations. This is in line with a previous report that found no difference between individual IFNβ preparations in the cumulative probability of RRMS patients remaining progression-free at 12 and 24 months.40 The intrinsic bias of RCT is that they recruit patients with highly active disease and are therefore likely to find significant effects of treatment in treated versus placebo arms. In our large, perhaps more representative patient cohort, we were nonetheless able to demonstrate a potent treatment effect.
Clinical, pathological, and MRI findings have demonstrated that inflammation and axonal transection occur early in the disease course, potentially leading to irreversible disability.31,41–43 Our study provides evidence that in a real-world clinical setting, DMT is very effective in reducing the hazard of individual disability worsening events if used early and consistently over an extended period, supporting the hypothesis that treatment efficacy is greatest in early MS. An analysis of the long-term effects of cumulative treatment duration on 5 and 10-year disability outcomes will be warranted in this cohort once a sufficient follow-up period has elapsed.
Acknowledgments
MSBase Study Group Co-investigators: Thor Petersen, MD (Kommunehospitalet, Arhus C, Denmark, Site PI); Eva Havrdova, MD, PhD (Charles University, Prague, Czech Republic, Site PI); Orla Gray, MD (Craigavon Area Hospital, Portadown, UK, Site PI); Mark Paine, MBBS (St Vincent's Hospital, Melbourne, Australia, Site PI); Carolyn Young, MD (The Walton Centre, Liverpool, UK, Site PI); Ludwig Kappos (Universitätsspital Basel, Basel, Switzerland, Site PI). We thank the following MSBase study group members for aiding in the collection of clinical outcomes data: From the Royal Melbourne Hospital, Australia, Anneke van der Walt (PhD), Mark Marriott (PhD), Trevor Kilpatrick (PhD), John King (MD); From Box Hill Hospital, Monash University, Australia, Olga Skibina (MD); From the University of Bari, Italy, Pietro Iaffaldano (MD); From Department of Neuroscience and Imaging, University “G. d'Annunzio”, Italy, Giovanna De Luca (MD), Valeria Di Tommaso (MD), Daniela Travaglini (MD), Erika Pietrolongo (MD), Maria di Ioia (MD) and Deborah Farina (MD); From Ospedale di Macerata, Italy, Elisabetta Cartechini (MD), Eugenio Pucci (MD) and Matteo Diamanti; From John Hunter Hospital, Australia, David Williams (MD), Lisa Dark (MD) and Karen Ribbons (PhD); From FLENI, Argentina, Jorge Correale (MD) and Celica Ysrraelit (MD); From Hospital Italiano, Argentina, Juan Ignacio Rojas (MD) and Liliana Patrucco (MD); From Flinders University and Medical Centre, Sharon Barlow (MN); From New York University Langone Medical Center, U.S.A., Ilya Kister (MD). We thank the MSBase operations team (Royal Melbourne Hospital, Australia): Jill Byron, Eloise Hinson and Lisa Morgan and the MSBase technical team (Rodanotech, Switzerland): Samir Merchati, Eric Bianchi, Alexandru Bulla and Matthieu Corageoud.
Study funding: This study was supported by a project grant from the NHMRC (1032484) as well as funding support from Serono (Geneva), which provided a nonconditional grant to the MSBase Foundation to cover investigator payments for the first 2000 patients recruited. Serono (later Merck Serono), did not participate in the analyses or have access to the manuscript.
Data access, responsibility, and analysis: “Jokubaitis and Spelman had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.”
Author Contributions
Study conceptualization and design: V. G. J., T. S., H. B., M. T. Data analysis and interpretation: V. G. J., T. S., T. K., H. B., M. T. Manuscript drafting: V. G. J. Manuscript review and approval: V. G. J, T. S., T. K., G. I., F. G. M., P. D., M. G., A. L., P. G., R. H., J. A. C. G., C. O. G., C. B., G. G., R. F. B., G. I., J. L. S., F. V., V. vP., T. P. B., M. F., F. M., E. C., R. A., R. B., M. B., M. S., N. V., J. H., C. S., M. L. S., M. P. A., D. L., D. P., H. B., M. T. Study funding: V. G. J., D. L., H. B. Data acquisition and study supervision: G. I., F. G. M., P. D., M. G., A. L., P. G., R. H., J. A. C. G., C. O. G., C. B., G. G., R. F. B., G. I., J. L. S., F. V., V. vP., T. P. B., M. F., F. M., E. C., R. A., R. B., M. B., M. S., N. V., J. H., C. S., M. L. S., M. P. A., D. L., D. P., H. B., M. T.
Conflict of Interest
Vilija Jokubaitis’ salary is supported by NHMRC project grant #1032484 and has received conference travel support from Novartis. Tim Spelman received compensation for travel and speaker honoraria from Biogen Idec. Tomas Kalincik received compensation for travel from Novartis, Biogen Idec, Sanofi Aventis, Teva and Merck Serono. Guillermo Izquierdo received speaking honoraria from Biogen Idec, Novartis, Sanofi, Merck Serono and Teva. Francois Grand'Maison received honoraria from Biogen Idec, Genzyme, Novartis, and Roche. Pierre Duquette did not declare any competing interests. Marc Girard received consulting fees from Teva Canada Innovation, Biogen Idec, Novartis and Genzyme Sanofi; lecture payments from Teva Canada Innovation, Novartis and EMD Serono. Dr. Girard has also received a research grant from Canadian Institutes of Health Research. Alessandra Lugaresi is a Bayer Schering, Biogen Idec, Genzyme, Merck Serono Advisory Board Member. She received travel grants and honoraria from Bayer Schering, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis, and Teva, research grants from Bayer Schering, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis, and Teva, travel and research grants from the Associazione Italiana Sclerosi Multipla and was a Consultant of “Fondazione Cesare Serono”. Pierre Grammond is a Novartis, Teva-neuroscience, Biogen Idec advisory board member, consultant for Merck Serono, received payments for lectures by Merck Serono, Teva-Neuroscience and Canadian Multiple sclerosis society, and received grants for travel from Teva-Neuroscience and Novartis. Raymond Hupperts received honoraria as consultant on scientific advisory boards from Merck Serono, Biogen Idec, Sanofi-Genzyme, and Teva, research funding from Merck Serono and Biogen Idec, and speaker honoraria from Sanofi-Genzyme. Jose Antonio Cabrera-Gomez did not declare any competing interests. Celia Oreja-Guevara received honoraria as consultant on scientific advisory boards from Biogen Idec, Bayer Schering, Merck Serono, Teva, and Novartis; has participated in clinical trials/other research projects by Biogen Idec, GSK, Teva, and Novartis. Cavit Boz has received travel grants from Merck Serono, Biogen Idec, Novartis, Bayer Schering, Merck Serono, and Teva; has participated in clinical trials by Sanofi Aventis, Roche and Novartis. Giorgio Giuliani did not declare any competing interests. Ricardo Fernández-Bolaños did not declare any competing interests. Gerardo Iuliano had travel/accommodations/meeting expenses funded by Bayer Schering, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis, and Teva. Jeannette Lechner-Scott has accepted travel compensation from Novartis, Biogen, and Merck Serono. Her institution receives the honoraria for talks and advisory board commitment and also clinic support from Bayer Health Care, Biogen Idec, CSL, Genzyme-Sanofi, Merck Serono, and Novartis. Freek Verheul is an advisory board member for Teva Biogen Merck Serono, and Novartis. Vincent van Pesch has served on advisory boards for Biogen Idec and Genzyme; has received travel grants from Biogen Idec, Bayer Schering, Sanofi Aventis, Merck Serono, and Novartis Pharma; has received consultancy fees from Biogen Idec, Teva, and Novartis Pharma; has received research grants from Bayer Schering. Tatjana Petkovska-Boskova did not declare any competing interests. Marcela Fiol received honoraria from Merck Serono and Bayer. Fraser Moore has participated in clinical trials sponsored by EMD Serono and Novartis. Edgardo Cristiano received honoraria as consultant on scientific advisory boards by Biogen Idec, Bayer Schering, Merck Serono, Genzyme, and Novartis; has participated in clinical trials/other research projects by Merck Serono, Roche, and Novartis. Raed Alroughani received honororia from Biologix, Bayer, Merck Sorono, GSK, and Novartis, and served on advisory board for Biologix, Novartis, and Merck Sorono. Roberto Bergamaschi received speaker honoraria from Bayer Schering, Biogen, Novartis, Sanofi-Aventis, Teva; research grants from Bayer Schering, Biogen, Novartis, Sanofi-Aventis, Teva; congress and travel expense compensations from Bayer Schering, Biogen, Novartis, Sanofi-Aventis, Teva. Michael Barnett has served on scientific advisory boards for Biogen Idec, Novartis, and Genzyme and has received conference travel support from Biogen Idec and Novartis. He serves on steering committees for trials conducted by Novartis. His institution has received research support from Biogen Idec, Merck Serono and Novartis. Mark Slee has participated in, but not received honoraria for, advisory board activity for Biogen Idec, Merck Serono, Bayer Schering, Sanofi Aventis, and Novartis. Norbert Vella received compensation for travel and honoraria from Novartis, Biogen Idec, Glaxo-Smith-Kline. Joseph Herbert did not declare any competing interests. Cameron Shaw did not declare any competing interests. Maria Laura Saladino did not declare any competing interests. Maria Pia Amato received honoraria as consultant on scientific advisory boards by Biogen Idec, Bayer Schering, Merck Serono, Teva, and Sanofi-Aventis; has received research grants by Biogen Idec, Bayer Schering, Merck Serono, Teva, and Novartis. Danny Liew did not declare any competing interests. Damiano Paolicelli received honoraria for consultancy and/or speaking from Biogen Idec, Merck Serono, Bayer Schering, Novartis, and TEVA. Helmut Butzkueven has served on scientific advisory boards for Biogen Idec, Novartis, and Sanofi-Aventis and has received conference travel support from Novartis, Biogen Idec, and Sanofi Aventis. He serves on steering committees for trials conducted by Biogen Idec and Novartis, and has received research support from Merck Serono, Novartis, and Biogen Idec. Maria Trojano has served on scientific advisory boards for Biogen Idec, Novartis, and Merck Serono, received speaking honoraria from Biogen Idec, Bayer Schering, Sanofi Aventis, Merck Serono, Teva, and Novartis; has received research grants from Biogen Idec, Merck Serono, and Novartis.
References
- Kappos L, Traboulsee A, Constantinescu C, et al. Long-term subcutaneous interferon beta-1a therapy in patients with relapsing-remitting MS. Neurology. 2006;67:944–953. doi: 10.1212/01.wnl.0000237994.95410.ce. [DOI] [PubMed] [Google Scholar]
- Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- Group PS. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352:1498–1504. [PubMed] [Google Scholar]
- Jacobs L, Rudick R, Simon J. Extended observations on MS patients treated with IM interferon-beta1a (Avonex): implications for modern MS trials and therapeutics. J Neuroimmunol. 2000;107:167–173. doi: 10.1016/s0165-5728(00)00232-0. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, O'Brien P, Erickson BJ, et al. The Mayo Clinic-Canadian Cooperative trial of sulfasalazine in active multiple sclerosis. Neurology. 1998;51:1342–1352. doi: 10.1212/wnl.51.5.1342. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Goodkin DE, Jacobs LD, et al. Impact of interferon beta-1a on neurologic disability in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG) Neurology. 1997;49:358–363. doi: 10.1212/wnl.49.2.358. [DOI] [PubMed] [Google Scholar]
- Wolinsky JS, Narayana PA, O'Connor P, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann Neurol. 2007;61:14–24. doi: 10.1002/ana.21079. [DOI] [PubMed] [Google Scholar]
- Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. 1995;45:1277–1285. [PubMed] [Google Scholar]
- Liu C, Blumhardt LD. Disability outcome measures in therapeutic trials of relapsing-remitting multiple sclerosis: effects of heterogeneity of disease course in placebo cohorts. J Neurol Neurosurg Psychiatry. 2000;68:450–457. doi: 10.1136/jnnp.68.4.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojano M, Pellegrini F, Paolicelli D, et al. Real-life impact of early interferon beta therapy in relapsing multiple sclerosis. Ann Neurol. 2009;66:513–520. doi: 10.1002/ana.21757. [DOI] [PubMed] [Google Scholar]
- MSBase . Available at: http://www.msbase.org.
- Butzkueven H, Chapman J, Cristiano E, et al. MSBase: an international, online registry and platform for collaborative outcomes research in multiple sclerosis. Mult Scler. 2006;12:769–774. doi: 10.1177/1352458506070775. [DOI] [PubMed] [Google Scholar]
- Meyniel C, Spelman T, Jokubaitis VG, et al. Country, sex, EDSS change and therapy choice independently predict treatment discontinuation in multiple sclerosis and clinically isolated syndrome. PLoS One. 2012;7:e38661. doi: 10.1371/journal.pone.0038661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357:1576–1582. doi: 10.1016/s0140-6736(00)04725-5. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain. 2003;126:770–782. doi: 10.1093/brain/awg081. [DOI] [PubMed] [Google Scholar]
- D'Alessandro R, Vignatelli L, Lugaresi A, et al. Risk of multiple sclerosis following clinically isolated syndrome: a 4-year prospective study. J Neurol. 2013;260:1583–1593. doi: 10.1007/s00415-013-6838-x. [DOI] [PubMed] [Google Scholar]
- Damasceno A, Von Glehn F, Brandao CO, et al. Prognostic indicators for long-term disability in multiple sclerosis patients. J Neurol Sci. 2013;324:29–33. doi: 10.1016/j.jns.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Healy BC, Engler D, Gholipour T, et al. Accounting for disease modifying therapy in models of clinical progression in multiple sclerosis. J Neurol Sci. 2011;303:109–113. doi: 10.1016/j.jns.2010.12.024. [DOI] [PubMed] [Google Scholar]
- Runmarker B, Andersen O. Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow-up. Brain. 1993;116(Pt 1):117–134. doi: 10.1093/brain/116.1.117. [DOI] [PubMed] [Google Scholar]
- Shirani A, Zhao Y, Karim ME, et al. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA. 2012;308:247–256. doi: 10.1001/jama.2012.7625. [DOI] [PubMed] [Google Scholar]
- Trojano M, Avolio C, Manzari C, et al. Multivariate analysis of predictive factors of multiple sclerosis course with a validated method to assess clinical events. J Neurol Neurosurg Psychiatry. 1995;58:300–306. doi: 10.1136/jnnp.58.3.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG) Ann Neurol. 1996;39:285–294. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- Ebers GC, Traboulsee A, Li D, et al. Analysis of clinical outcomes according to original treatment groups 16 years after the pivotal IFNB-1b trial. J Neurol Neurosurg Psychiatry. 2010;81:907–912. doi: 10.1136/jnnp.2009.204123. [DOI] [PubMed] [Google Scholar]
- Uitdehaag B, Constantinescu C, Cornelisse P, et al. Impact of exposure to interferon beta-1a on outcomes in patients with relapsing-remitting multiple sclerosis: exploratory analyses from the PRISMS long-term follow-up study. Ther Adv Neurol Disord. 2011;4:3–14. doi: 10.1177/1756285610391693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremlett H, Yousefi M, Devonshire V, et al. Impact of multiple sclerosis relapses on progression diminishes with time. Neurology. 2009;73:1616–1623. doi: 10.1212/WNL.0b013e3181c1e44f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brex PA, Ciccarelli O, O'Riordan JI, et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346:158–164. doi: 10.1056/NEJMoa011341. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131:808–817. doi: 10.1093/brain/awm329. [DOI] [PubMed] [Google Scholar]
- O'Riordan JI, Thompson AJ, Kingsley DP, et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS. A 10-year follow-up. Brain. 1998;121(Pt 3):495–503. doi: 10.1093/brain/121.3.495. [DOI] [PubMed] [Google Scholar]
- Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology. 2006;67:968–972. doi: 10.1212/01.wnl.0000237354.10144.ec. [DOI] [PubMed] [Google Scholar]
- Comi G, Martinelli V, Rodegher M, et al. Effects of early treatment with glatiramer acetate in patients with clinically isolated syndrome. Mult Scler. 2013;19:1074–1083. doi: 10.1177/1352458512469695. [DOI] [PubMed] [Google Scholar]
- Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006;67:1242–1249. doi: 10.1212/01.wnl.0000237641.33768.8d. [DOI] [PubMed] [Google Scholar]
- Kinkel RP, Kollman C, O'Connor P, et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology. 2006;66:678–684. doi: 10.1212/01.wnl.0000200778.65597.ae. [DOI] [PubMed] [Google Scholar]
- Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet. 2007;370:389–397. doi: 10.1016/S0140-6736(07)61194-5. [DOI] [PubMed] [Google Scholar]
- Kappos L, Freedman MS, Polman CH, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009;8:987–997. doi: 10.1016/S1474-4422(09)70237-6. [DOI] [PubMed] [Google Scholar]
- Trojano M, Paolicelli D, Tortorella C, et al. Natural history of multiple sclerosis: have available therapies impacted long-term prognosis? Neurol Clin. 2011;29:309–321. doi: 10.1016/j.ncl.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Brown MG, Kirby S, Skedgel C, et al. How effective are disease-modifying drugs in delaying progression in relapsing-onset MS? Neurology. 2007;69:1498–1507. doi: 10.1212/01.wnl.0000271884.11129.f3. [DOI] [PubMed] [Google Scholar]
- Trojano M, Pellegrini F, Fuiani A, et al. New natural history of interferon-beta-treated relapsing multiple sclerosis. Ann Neurol. 2007;61:300–306. doi: 10.1002/ana.21102. [DOI] [PubMed] [Google Scholar]
- Trojano M, Liguori M, Paolicelli D, et al. Interferon beta in relapsing-remitting multiple sclerosis: an independent postmarketing study in southern Italy. Mult Scler. 2003;9:451–457. doi: 10.1191/1352458503ms948oa. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, et al. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Popescu BF, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]