

Abstract
Introduction
Multiple sclerosis (MS) is an enigmatic autoimmune-driven inflammatory/demyelinating disease of the human central nervous system (CNS), affecting brain, spinal cord, and optic nerves. The cause of the disease is not known and the number of effective treatments is limited. Despite some clear successes, translation of immunological discoveries in the mouse experimental autoimmune encephalomyelitis (EAE) model into effective therapies for MS patients has been difficult. This translation gap between MS and its elected EAE animal model reflects the phylogenetic distance between humans and their experimental counterpart, the inbred/specific pathogen free (SPF) laboratory mouse.
Objective
Here, we discuss that important new insights can be obtained into the mechanistic basis of the therapy paradox from the study of nonhuman primate EAE (NHP-EAE) models, the well-validated EAE model in common marmosets (Callithrix jacchus) in particular.
Interpretation
Data presented in this review demonstrate that due to a considerable immunological and pathological overlap with mouse EAE on one side and MS on the other, the NHP EAE model can help us bridge the translation gap.
Introduction
Multiple sclerosis (MS) is an enigmatic disease of the human central nervous system (CNS), affecting brain, spinal cord, and optic nerves. The cause of the disease is not known, but general consensus exists that autoimmune reactions against axon-enwrapping myelin sheaths and myelin-producing oligodendrocytes influence the disease course. The autoimmune concept is supported by genetic association of MS susceptibility with the immune system1 and the clinical effect of treatments modulating immunological processes.2 The mouse experimental autoimmune encephalomyelitis (EAE) model has shaped our understanding of immunopathogenic mechanisms. However, translation of scientific discoveries made in EAE into effective treatments for patients has been notoriously difficult. The frequent failure of new treatments to reproduce promising effects observed in the EAE model when they were tested in the clinic, indicate that essential elements of MS are missing in current EAE models. This raises the question which lessons can be learned from EAE in more human-like species, such as nonhuman primates (NHP).
Accumulating evidence demonstrates a decisive role of the environment on the manifestations of EAE in genetically susceptible animals. Mice bred and raised under germ-free conditions develop significantly attenuated EAE, indicating that CNS targeting autoreactive proinflammatory CD4+ T cells receive essential activation signals in the gut.3,4 In addition, we discovered a new pathogenic mechanism in a NHP model of MS, EAE in the common marmoset (Callithrix jacchus; CJ).5 This mechanism does not exist in mouse EAE and seems closely associated with the pathogen-educated nature of the primate immune system, which is shaped by chronic latent infections with herpesviruses such as Epstein–Barr virus (EBV) and cytomegalovirus (CMV). This new mechanism shows an MS-like response to treatment with clinically relevant monoclonal antibodies (mAb), illustrating its potential relevance for MS. Results obtained in a new model developed on the new mechanism shed light on the pathogenic role of CD8+ T cells and B cells and the elusive association of MS with EBV.
MS, a Human Disease of Unknown Cause(s)
The two prevailing concepts in the MS literature are that the primary pathogenic event in MS occurs either inside or outside the CNS.6 According to an “outside-in” paradigm, MS is caused by an exogenous factor, such as an infection, which in genetically susceptible individuals induces the activation of naïve autoreactive T and B cells present in the immune repertoire. The presence of autoreactive lymphocytes in the healthy human immune repertoire has been well established.7 Activation renders these autoreactive T cells capable of infiltrating the CNS. Interaction with local antigen presenting cells (APC) elicits a cascade of pathophysiological reactions that leads to CNS inflammation. An infectious cause of MS is supported by some epidemiological data.8
According to an “inside-out” paradigm, one or more pathogenic event(s) inside the CNS causes instability of myelin and release of myelin antigens,6 which may induce the activation of hyperreactive T and B cells in CNS draining lymph nodes.9 The initial pathogenic event is not necessarily unique to MS and may also differ between patients.
At this stage it remains unclear which of the two complementary views most closely approximates reality. It is interesting that spontaneous manifestation of MS pathology and symptoms has only been observed in the human population and is not found in animals, not even in our nearest kin, the hominoid primate (e.g., bonobo, chimpanzee). To our knowledge there is only one report documenting postinfectious inflammatory/demyelinating disease in a captive colony of Japanese macaques (Macaca fuscata).10 This may imply that the pathogenic event that triggers MS is specific for the human primate.
There is no single pathological aspect that is unique to this enigmatic disease. This may imply that there is no single cause of MS, but that multiple triggering factors may converge in causing instability of oligodendrocyte/myelin and neuro/axonal complexes. Whether myelin instability in MS is a direct consequence of autoimmune-driven inflammation, as observed in EAE models,11 or a secondary consequence of axon degeneration, as observed in Theiler's murine encephalomyelitis,12 has also not been formally established. It is thus well possible that MS is determined by a unique combination of immune responses to self-antigens released from unstable CNS regions.6
The Autoimmune Concept of MS
In an outside-in MS concept autoreactive T and B cells are activated by dynamic interactions between genetic and environmental (risk) factors. In addition, unknown stochastic factors may be involved. With regard to the genetic risk factors, genome-wide association studies have identified more than 100 genes contributing to MS susceptibility, although their individual contribution is usually modest.1 Although almost all identified genes have a function in cellular immunity, the major histocompatibility complex (MHC) exerts the strongest effect. MHC class II alleles, such as HLA-DRB1*1501/-DRB5*0101 and HLA-DQA1*0102/-DQB2*0602 and the MHC class I allele HLA-B7 were found associated with increased MS susceptibility, whereas other alleles seem to reduce the risk, such as HLA-DR1 and -DR8.13
Environmental risk factors can be broadly categorized as infectious, such as with EBV, or noninfectious, such as smoking and a low serum vitamin D level.14,15 The focus of this review is on infectious triggers of MS. Although a large number of bacteria and viruses have been proposed as candidate trigger, only few have survived rigorous testing. Seroepidemiological studies indicate age-at-risk (age ±15 years) exposure of genetically susceptible individuals to EBV as likely cause of MS (reviewed in 16).
EBV is a γ1-herpesvirus belonging to the large genus of primate lymphocryptoviruses (LCV). EBV infects about 90% of the adult human population. Although primary infection usually occurs in childhood without marked symptoms, exposure during adolescence can elicit infectious mononucleosis, a condition characterized by significant flu-like symptoms, such as fatigue and malaise, but also lymphadenopathy and often splenomegaly, reflecting the robust systemic immune response to the virus.17 EBV mainly infects B cells via binding CD21 and HLA-DR. LCV causing chronic latent infection of NHP resemble EBV in molecular and functional aspects.18 NHP are therefore potentially useful animal models of human EBV infection.
A concept connecting autoimmunity with infection is molecular mimicry,19 which implies that autoimmunity can be the consequence of an immune response against structurally related (mimicry) antigens shared between an infectious agent and host tissues. T cells and antibodies induced by the infection will therefore cross-react with host tissue. Activated T cells can infiltrate target organs, including the CNS in MS as illustrated by adoptive transfer EAE, which is induced by the transfer of myelin-sensitized T cells obtained from immunized mice to healthy MHC-matched recipient mice.20 Conceptually, engagement of transferred autoreactive T cells in cognate interactions inside the CNS with APC, such as microglia or coinfiltrated dendritic cells (DC), elicits EAE pathology. The APC present epitopes from CNS antigens that are recognized by T cells as if they were epitopes of the infecting pathogen. In MS T-cell cross-recognition of EBV and the CNS myelin antigen MBP has been documented.21 Another explanation for the association of EBV with MS might be that EBV-infected B cells are directly involved in the development of neuroinflammation.22 However, the validity of this finding was seriously criticized.23
Two lines of evidence obtained in NHP-EAE support an active role of EBV in MS (reviewed in 24). First, adoptive transfer of autologous LCV-infected B cells induced autoreactive T-cell activation and (mild) meningeal inflammation in rhesus monkeys25 and marmosets.26 Second, we observed that the clinical effect in the marmoset EAE (Cj-EAE) model of B-cell-depleting mAb was associated with their ability to deplete LCV-infected B cells.26
However, the prevalence of the HLA-DR2/-DQ6 haplotype in the human population is 10–25% and that of EBV infection in young adults is ±60%. As MS develops only in 1 per 1000 young adults, it is difficult to envisage how the interaction of two such common factors can be the cause of a relatively low prevalent disease. In addition, translation of pathogenic concepts developed in the EAE model into effective treatments for the MS patients has been notoriously difficult. We postulate that a refined and well-validated NHP model, such as the Cj-EAE model, can be useful in the search of missing links between EAE and MS.
The Common Marmoset EAE (Cj-EAE) Model
Cj is a small-bodied nonprotected Neotropical primate (weighing ±350 g at adult age) that breeds well in captivity. Marmosets share the outbred nature and a high degree of genetic, immunological, and microbiological similarity with humans. Marmosets and tamarins, another genus within the Callitrichidae family, are unique among mammalian species as they usually produce nonidentical twins that exchange hemopoietic stem cells in utero via the fused placental bloodstream. The ensuing chimerism not only implies mutual tolerance for alloantigens between fraternal siblings but also a high degree of immunological similarity. The complete Cj genome has now been sequenced and annotated.27
Hauser and colleagues first developed the Cj-EAE model using a mouse EAE protocol, that is, human myelin formulated with complete Freund's adjuvant (CFA) in combination with intravenous Bordetella pertussis particles.28,29 In our hands, these immunized marmosets developed acute EAE with severely destructive pathology.30 Slight modification of the protocol, usage of CFA containing less mycobacteria and omission of Bordetella injection, delivered an MS-like chronic disease model resembling relapsing-remitting MS (RRMS) in clinical and neuropathological presentation.30 The original concept was that, just like in classical rodent EAE models, Cj-EAE is driven by the synergistic action of T helper 1 (Th1) cells inducing inflammation and autoantibodies eliciting demyelination via macrophage- and complement-mediated cytotoxic injury to myelin sheaths as dominant pathogenic process (antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, respectively).5 For validation of this paradigm we used a fully human anti-IL-12p40 monoclonal antibody (mAb) ustekinumab, unexpectedly. The IL-12p40 subunit is shared by IL-12 and IL-23,31 which are the signature cytokines of two important autoimmune inflammatory pathways, respectively, driven by the activation of proinflammatory and encephalitogenic Th1 and Th17 cells.32 Although a convincing protective effect of the anti-IL-12p40 mAb was observed in the Cj-EAE model,33,34 the same mAb showed no detectable clinical effect in RRMS patients.35 The observation in mice that many of the pathogenic effects attributed to IL-12 are mediated by IL-23 via induction of Th17 cells,32 prompted us to test a novel anti-IL-17A antibody in Cj-EAE. We found only a trend toward delayed onset of clinical signs, suggesting that the treatment may affect events in late-stage disease.36
These paradoxical results do not imply that the Th1/Th17 paradigm is an artifact of the EAE model that is irrelevant for MS. It is well possible that the acute pathogenic mechanisms modeled in EAE operate early in the MS pathogenesis and may be extinguished when MS is diagnosed (see also: 37). The notion that different immunopathogenic processes may drive the initiation and progression of MS is strongly supported by the marmoset EAE model. The existence of distinct pathogenic mechanisms is well-illustrated by the observation in Cj38 as well as in Biozzi ABH mice39 that autoimmunity against MOG is dispensable in EAE initiation, but is essential in EAE progression. Fine mapping of the T-cell response against recombinant MOG (rMOG) revealed that initially Th1 cells are activated specific for the epitope MOG24–36.40 Progression of Cj-EAE involved activation of another autoimmune mechanism, mediated by cytotoxic effector memory T cells specific for the epitope MOG40–48.41 These EM-CTL have an important pathogenic role, as they seem to shift the focus of the autoimmune attack to the gray matter, where they induce severe demyelination.42
Two Faces of MOG in Autoimmunity
MOG, a unique constituent of the mammalian CNS, is expressed as a homodimeric complex in head-to-tail orientation on the surface of oligodendrocytes and the outer lamellae of the axon-enwrapping myelin sheaths within the CNS.43 MOG is encoded in the extended class I region of the MHC located in humans on chromosome 6p21.3-p22 at 60 kb telomeric to HLA-F. MOG transcripts are extensively spliced and splicing profiles are more complex in higher than in lower species.44 In the human brain at least 15 splice variants were found, but it remains unknown whether these have a different function and whether certain variants are associated with autoimmunity. Crystallization of the mouse43 and rat45 MOGED, elucidated the 3D configuration and provided insights into the antigenicity of the protein (see below). The surface-exposed Asn31Arg32Tyr33 motif encodes the N-glycosylation site.46 A refined analysis of the glycan attached to the Asn31 residue has recently been published and identified the C-type lectin DC-SIGN as a potential receptor for MOG within the CNS and draining lymph nodes of human and NHPs.47 We hypothesized that at the level of the interaction of MOG with DC-SIGN the decision on tolerance or immunity against MOG is made.48
Monomeric human MOG consists of an evolutionary conserved Ig-like extracellular domain (ED; residues 1–121) (Fig.1) that contains the EAE-associated B- and T-cell epitopes, two hydrophobic segments domains (residues 121–151 and 174–201), of which only the first is transmembrane spanning and the second is partially buried in the lipid bilayer, and a short intracytoplasmic tail that may be connected to the oligodendrocyte cytoskeleton.49
Figure 1.

Alignment of mammalian MOGED sequences. The depicted sequences were downloaded from the Swissprot database (http://www.uniprot.org/uniprot/?query=myelin+oligodendrocyte+glycoprotein&sort=score). (A) Highlighted in yellow is the highly conserved domain MOG21–52, which contains the only N-glycosylation site at Asn31 (marked with #). (B) The conserved sequence 21–50 contains the two most relevant epitopes, namely residues 24–36 (Th1 epitope; yellow) and residues 40–48 (NK-CTL epitope; green). *M. fascicularis = cynomolgus macaque; M. Mulatta = rhesus macaque; S. sciureus = Saimiri sciureus.
Autoimmunity against MOG appears a crucial factor in the development of chronic Cj-EAE, as was demonstrated by the observation that the normal chronic EAE course is impaired when MOG-deficient mouse myelin was used for the immunization.38 MOG is best known for its capacity to induce in the EAE model50 autoantibodies that opsonize myelin and elicit demyelination via cytotoxicity of complement (CDC) and macrophages (ADCC).51
From an evolutionary point of view it is difficult to understand that expression of a potentially harmful protein without a clear beneficial function is permitted in a vital organ as the brain. We therefore examined whether MOG may have a beneficial role in the healthy brain. A physiological role of MOG in the CNS has not been clearly established thus far, although several functions were proposed. These include an adhesive function in maintaining the structural integrity of CNS myelin, involvement in the microtubule integrity of oligodendrocytes and binding of complement factors.43,45 The developmental pattern of MOG expression in the CNS coinciding with late stages of myelination52 hints at a role in the formation of compact CNS myelin. However, mice lacking MOG expression in their CNS develop normally without detectable myelination defects.53
Studies in wild type and MOG-deficient mice showed that potentially pathogenic anti-MOG T cells are not completely cleared from the immune repertoire during thymic selection.54 Interestingly, T cells from MOG-deficient mice displayed higher reactivity to recombinant MOG, measured by proliferation and cytokine production, and were more encephalitogenic, as assessed by adoptive transfer.55 These observations hint at a dual function of MOG, namely a homeostatic role inside the CNS and an antigenic role outside the CNS in the lymphoid organs.
Innate Immune Recognition of Myelin
Myeloid dendritic cells (mDC) have a central regulatory role in tolerance and immunity.48 For this task mDC are equipped with pattern-recognition receptors (PRR) binding conserved pathogen-associated molecular patterns (PAMPS), which inform the cell about the presence of friend or foe.48 Danger-sensing receptors inducing DC activation include toll-like and nod-like receptor families (TLR and NLR).56,57 C-type lectin receptors (CLR) are PRR with a role in the sensing of nondanger signals, which antagonize DC activation.58 In contrast to TLR, CLR can mediate antigen internalization. CLR also recruit signaling molecules that modulate activation signals relayed via danger receptors. It can thus be envisaged that via the integration of activation signals received via TLR/NLR and inhibitory signals received via CLR, DC are instructed whether they should adopt an immunogenic or a tolerogenic function59 (Fig.2).
Figure 2.
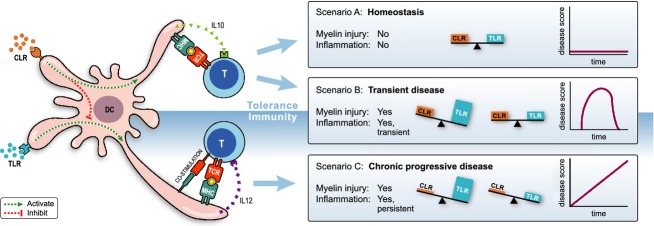
Yin-Yang regulation of myeloid APC. Depicted is a myeloid APC, for example, a microglia or dendritic cell, which expresses C-type lectin (CLR) and toll-like receptors (TLR). Via the integration of inhibitory input signals via CLR and stimulatory input signals via TLR the APC is instructed whether it needs to display tolerogenic or immunogenic activity. In a healthy brain APC are in a tolerogenic state as danger signals received via TLR are counterbalanced by inhibitory signals via CLR, for example, binding of normally glycosylated MOG to DC-SIGN (scenario A). CNS infection or tissue damage induces increase in danger signals, resulting in a, CLR/TLR dysbalance and maturation of APC to an immunogenic state. When the danger signals are cleared, the CLR/TLR balance that maintains homeostasis is restored (scenario B). When scenario B occurs in inflamed tissue where normal glycosylation is disturbed resulting in impaired CLR signaling, return to homeostasis after clearance of the danger does not occur. APS, antigen presenting cell; CNS, central nervous system; DC, dendritic cell.
DC-SIGN is a transmembrane receptor expressed on immature mDC in peripheral tissues, mature mDC in lymphoid organs and on subsets of macrophages. DC-SIGN has broad physiological functions.60 DC-SIGN expression on mDC is upregulated by IL-4 and downregulated by inflammatory factors.61 DC-SIGN contains a Ca2+-dependent mannose-binding carbohydrate recognition domain with dual specificity for high-mannose and fucose-type sugars, such as Lewis X. Various self-antigens are decorated with glycans that mediate binding to DC-SIGN, such as the adhesion molecules ICAM-2 and ICAM-3, the MOG-related milk protein butyrophilin and tumor antigens, such as the carcinoembryonic antigens CEA and CEACAM1. Moreover, a range of pathogens express carbohydrate epitopes that bind DC-SIGN (see references in review 58). Accumulating evidence indicates that these pathogens use DC-SIGN binding for avoiding a neutralizing immune response.62
We analyzed the interaction of myelin purified from healthy human brain white matter with human mDC.47 Lipid-rich myelin emulsified in aqueous buffers forms micellar structures of variable sizes, which can be separated by flow cytometry. We observed that DC-SIGN binding is confined to larger myelin particles, whereas smaller particles did not bind. A possible explanation is that for binding to DC-SIGN MOG needs to multimerize in lipid rafts, which because of their size can only be formed in large myelin particles.63 The functional consequence of MOG interaction with DC-SIGN is modulation of the mDC response to TLR triggering. The MOG-mediated binding of DC-SIGN to myelin converted mDC activation signals from LPS-TLR4 interaction into tolerogenic signals. This resulted in inhibition of mDC maturation and NALP3 inflammasome formation as well as the secretion of anti-inflammatory cytokines, such as IL-10. Small myelin particles that lack MOG multimers or myelin produced under inflammatory conditions, inducing altered glycosylation of MOG, did not bind DC-SIGN. In this circumstance TLR4 stimulation by LPS induced strong proinflammatory signals, expressed by NALP3 inflammasome activation and secretion of IL-17A.
DC-SIGN-expressing APC relevant for Cj-EAE are microglia located within the CNS47 and phagocytic cells within the CNS draining cervical and lumbar lymph nodes.64 We hypothesize the following scenarios: Injury to myelin without simultaneous inflammation, a situation present in noninflammatory brain injury, does not (necessarily) induce autoimmunity as tolerogenic mechanisms are activated (scenario B in Fig.2). In contrast, myelin injury together with inflammation, a condition present in inflammatory-demyelinating MS lesions, induces autoimmune mechanisms that amplify injury and perpetuate inflammation (scenario C in Fig.2).
Adaptive Immune Recognition of MOG
Most EAE studies focused on the MOGED, which is used as a nonglycosylated recombinant protein. Despite the clear pathogenic role of MOG in EAE, there is controversy about the relevance of anti-MOG T cells and antibodies in MS. Part of the controversy may be due to the assays utilized in different studies. Tests of cells and sera from MS patients for reactivity with recombinant human MOGED, often gave contradictory results. However, assays based on cells expressing conformationally intact, glycosylated MOG on their surface detected different serum IgG reactivity between healthy controls and pediatric MS patients with clinically isolated syndrome or an acute disseminated encephalomyelitis ADEM-like disease presentation. The same assays only rarely detected differences between adult MS patients and healthy controls.65
Figure1A shows MOGED amino acid sequences from a variety of mammalian species are aligned. Noteworthy is the remarkable sequence homology (>90%) between two species widely separated in evolution, that is, the naked mole rat (Heterocephalus glaber) and man (Homo sapiens). The MOGED contains a domain of 30 amino acids length (21–50) that is almost completely conserved in evolution (Fig.1B). The only difference is a nonsynonymous substitution of proline and serine at position 42. The Ser42Pro substitution alters the protein structure,66 the strength of antibody binding67 and the autoantibody response in B6 mice.68 The two key pathogenic T-cell epitopes of Cj-EAE model (24–36 and 40–48) are juxta-positioned within this conserved 21–50 region.
B-cell epitopes
Studies with the mAb 8-18C5, which was a potent inducer of CNS demyelination in marmosets69 showed that the dominant B-cell epitope is conformation-dependent. The mAb binds to an epitope formed by three loops at the membrane-distal end of the molecule.
For the studies in Cj-EAE recombinant proteins were used representing the MOGED fragment from rat (rrMOGED) or human origin (rhMOGED).28,70 Marmosets immunized with rrMOGED (Ser42) or rhMOGED(Pro42) in CFA developed a similar polyclonal IgG response.40,71 In our hands the reaction of immune sera with ELISA plate-bound rhMOGED was reduced >90% when the immune sera were preincubated with synthetic MOG54–76 peptide.72 This observation complies with the identification of peptide 65–75 of rrMOGED as part of a marmoset B-cell epitope.73
Marta et al. have reported the remarkable observation that C57/BL6 mice immunized with rhMOG, but not those immunized with rrMOG, produce IgG antibodies against the N-linked carbohydrate epitope.68 This finding was attributed to the P42 residue as in mice immunized with rhMOG (Ser42), IgG antibody induction against glycosylated myelin was markedly reduced.68 Theoretically, such glycan-binding antibodies may promote EAE development by blocking the homeostatic interaction of MOG with DC-SIGN on DC and microglia and thus abrogate the maintenance of homeostasis. This remains to be proven.
T-cell epitopes
The human MOGED domains containing dominant T-cell epitopes in rhMOG-induced Cj-EAE overlap in part with those identified for MS (residues 14–46, 34–56, 64–78).40 With two peptides Cj-EAE could be elicited, MOG14–36 and MOG34–56. These two peptides induced two different pathogenic pathways leading both to clinical EAE (Fig.3).5 One pathway is mediated by Th1 cells specific for MOG epitope 24–36. Immunization with MOG peptide 14–36 in CFA40 or adoptive transfer of a Th1 clone specific for MOG peptide 21–4074 elicited mild clinical EAE with modest inflammatory pathology in the white matter without demyelination.74 The second pathway is activated by immunization with MOG peptide 34–56 in CFA or even incomplete Freund's adjuvant (IFA) resulting in severe clinical EAE with MS-like inflammation and demyelination within the white and gray matter of the brain (Fig.4).50
Figure 3.
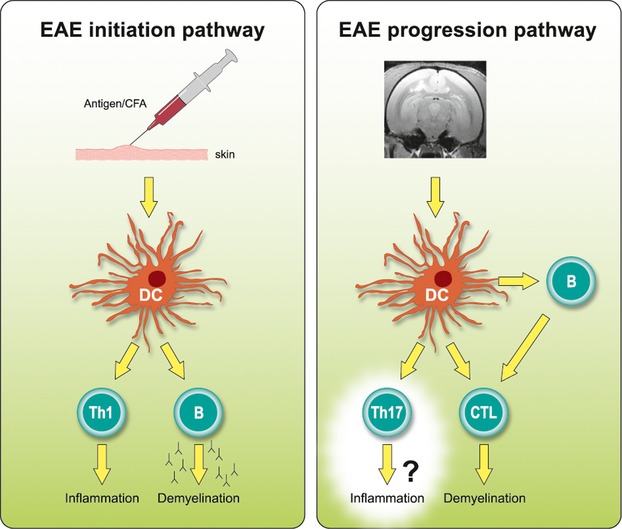
EAE development in marmosets involves two consecutively activated pathways. Injection of rhMOG/CFA emulsion into the skin induces local activation of APC, which induce activation of MHC class II/Caja-DRB1*W1201-restricted Th1 cells specific for MOG24–36 and B cells against conformational epitopes. The combined autoimmune attack initiates the inflammation and demyelination of white matter. Antigens released from such primary lesions can be retrieved within DC-SIGN+ APC within the cervical lymph nodes.64 Autoimmune factors induced at this location, in particular MHC-E restricted CTL, are critical mediators of chronic EAE. Virus-infected B cells have an important role in the progression pathway as requisite APC of the CTL. EAE, experimental autoimmune encephalomyelitis; CFA, complete Freund's adjuvant; APS, antigen presenting cell; MHC, major histocompatibility complex; DC, dendritic cell.
Figure 4.
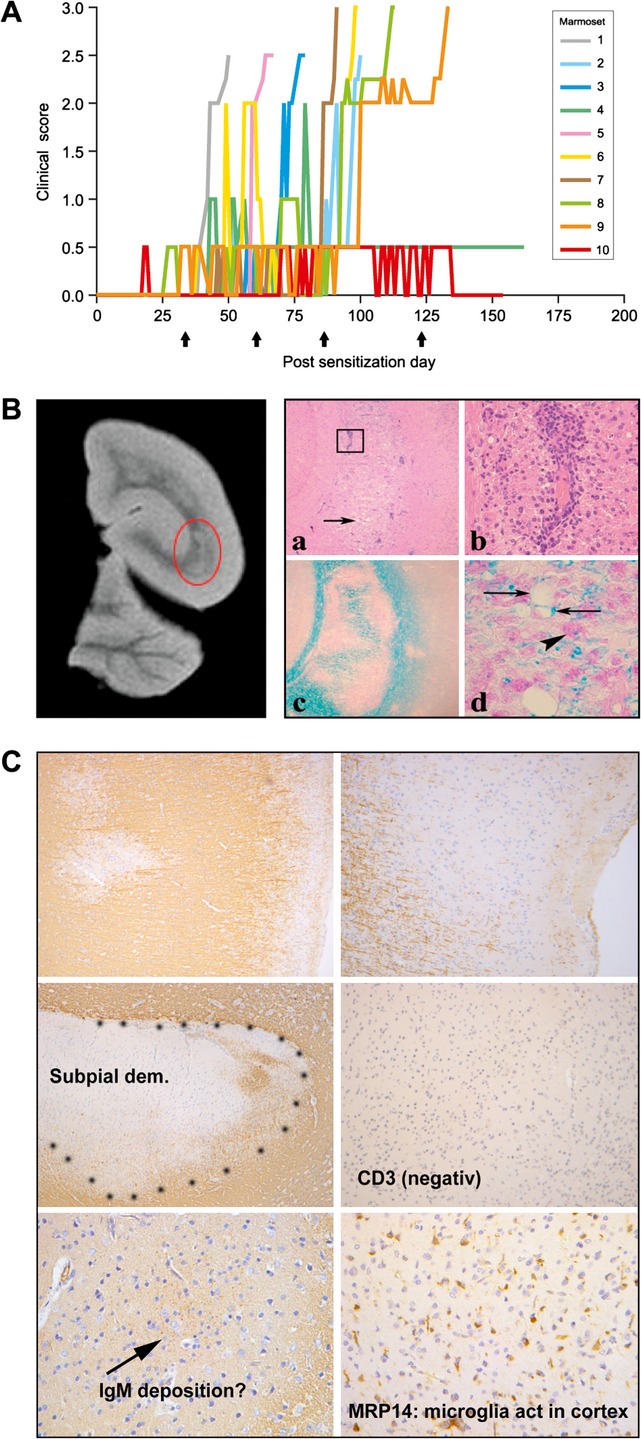
Clinical and pathological presentation of the EAE model induced with MOG34–56/IFA. A total of 10 marmosets were immunized with MOG34–56/IFA on days 0, 28, 56, 84, and 112 (arrows). (A) Nine monkeys developed clinically evident progressive EAE; one monkey went into remission after short lasting neurological impairment Characteristic pathological changes in cerebral white matter (B) and cortical gray matter (C) are shown. EAE, experimental autoimmune encephalomyelitis; IFA, incomplete Freund's adjuvant.
MOG, a Critical Autoantigen in Chronic EAE
Marmosets given a single immunization with rhMOGED in the strong bacterial adjuvant CFA, developed a severe neurological disease, characterized by MS-like CNS pathology.75,76 Thus far, almost all immunized marmosets (>95%; N > 100) developed clinically evident MS-like disease irrespective of their MHC differences. In the few exceptions without detectable neurological symptoms MS-like neuropathology was nevertheless found. Serial magnetic resonance imaging showed that the development of brain lesions in this model starts within the white matter and spreads to the cortical gray matter at later stages of the disease (Fig.5). The frequent detection of MS-like demyelination75 and diffuse atrophy77 in the cerebral gray matter is an important, albeit not unique, aspect of the Cj-EAE model.
Figure 5.
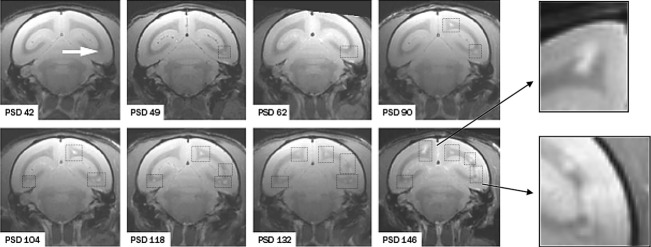
Brain lesion development in the rhMOG/CFA EAE model disseminated in time and space. Depicted is a series of T2-weighted MRI scans of the same coronal brain section recorded at periodic intervals (psd = postsensitization day). The arrow in the first picture points to the first abnormality recorded at 6 weeks after EAE induction. At 7 days later the first lesion has been enlarged and again 40 days later a second lesion can be observed. Squares with dotted lines are placed around lesions for better visualization. The inserts show an enhancement of lesions expanded into the cortical gray matter, which is a late event. CFA, complete Freund's adjuvant; EAE, experimental autoimmune encephalomyelitis.
The 100% incidence of induced EAE is higher than expected for an outbred species but could be explained by the observation that EAE susceptibility maps to the ubiquitously expressed monomorphic MHC class II allele, Caja-DRB1*W1201.78 Presentation of MOG24–36 by Caja-DR12 molecules induced Th1 cells40 and marmosets immunized with MOG14–36 peptide in CFA developed only mild clinical EAE with small-sized inflammatory lesions.40 With the data reported by Genain et al.69 and Menge et al.79 in mind, one might speculate that the absence of demyelination is due to a lack of autoantibody production against conformational epitopes.
In a significant proportion of the cases, those with a rapid disease progression in particular, we found activation of T cells specific for other parts of the rhMOG molecule, including the peptides 4–11, 11–21, 34–56, 64–86, 74–96, and 94–110.41 The pathogenic potential of the N-terminal 4–21 peptide or the C-terminal 94–110 peptide, which is encephalitogenic in DA rats,80 still needs to be confirmed for marmosets. Cj sensitized against MOG74–96 peptide, being encompassed within the dominant MOG64–96 T-cell epitope in MS,81 failed to develop EAE but developed severe neurological defects following booster-immunization with MOG34–56 in IFA. It was already observed in rhesus monkeys that T cells sensitized against MOG34–56 cross-react with a peptide derived from the UL86 ORF encoded major capsid protein of human CMV and its macaque equivalent in rhesus monkeys.82
The synthetic peptide-in-oil formulation (MOG34–56/IFA) lacked detectable innate stimulatory activity and therefore did not elicit EAE in Biozzi ABH and C57BL6 mice.50 Nevertheless, >90% of marmosets immunized with MOG34–56/IFA (N > 30) developed full-blown clinical EAE, associated with MS-like pathology in the CNS white and gray matter (Fig.4).50 We therefore speculated that the immunization with MOG34–56/IFA might induce the activation of effector memory T cells from the preexisting anti-CMV repertoire. However, formal proof awaits purification and molecular characterization of the marmoset CMV, that is, CalHV2.
Anti-B-Cell Antibody Trials Highlight a Pathogenic Role of LCV-Infected B Cells
Anti-CD20 mAbs, such as rituximab, ocrelizumab, or ofatumumab, have a remarkable clinical effect in RRMS that could not be attributed to the abrogation of autoantibody production.83 We speculated that detailed analysis of the immune reactions in Cj-EAE that are modified by B-cell depletion might help gaining insight into essential pathogenic mechanisms. For the studies in Cj-EAE we used HuMab7D8, a clonal variant of the fully human anti-CD20 mAb ofatumumab with equivalent specificity and efficacy.84 The mAb was evaluated in the EAE models induced with rhMOG/CFA, in which anti-rhMOG antibodies mediating demyelination are formed, or with MOG34–56/IFA, where anti-rhMOG antibodies are not formed. In both Cj-EAE models we observed profound suppression of neurological deficits together with modulation of T- and B-cell functions by anti-CD20 mAb treatment.76,85,86 Intriguingly, in the rhMOG/CFA model indirect peripheral B-cell depletion via treatment with fully human mAbs against two major stimulatory factors of B-cell development and survival, BLyS/BAFF (belimumab) or APRIL, exerted a disappointing clinical effect although serum levels of anti-MOG antibodies were reduced.87 The marginal clinical effect of anti-BlyS mAb compared with the robust treatment effect of anti-CD20 mAb the treatment with anti-CD20 mAb reminds to the ineffectivity of atacicept, a soluble version of the joint TACI receptor of BLyS and APRIL, in clinical trials. An unexpected increase in inflammatory disease activity observed in one trial led to suspension of all atacicept trials in MS.88
We chose to investigate in the marmoset EAE model why the three mAbs (anti-CD20, anti-BlyS and anti-APRIL), which eventually all induced depletion of peripheral CD20+ B cells albeit with different kinetics, had such a remarkably different clinical effect. We observed that treatment with anti-CD20 mAb induced a sharp reduction in CalHV3 virus DNA copy numbers in lymphoid organs. CalHV3 is a Cj γ-herpesvirus with comparable B-cell transforming activity as its human counterpart EBV.89 In monkeys treated with anti-BLyS or anti-APRIL mAbs we did not observe depletion of CalHV3 DNA, rather an increment of CalHV3 DNA copy numbers.26 On the basis of these findings, we postulated that the subset of CalHV3-infected marmoset B cells may have a central role in Cj-EAE development, namely as APC for the subset of EM-CTL, which can induce demyelination of gray matter independent of autoantibody. The observation that infusion of EBV-induced B lymphoblastoid cells prepulsed with MOG34–56 induced the in vivo activation of MOG34–56-specific marmoset T cells, albeit without clinical signs, supports this hypothesis.26
We examined the secondary lymphoid organs (SLO) of EAE marmosets from the placebo, anti-CD20, anti-BlyS and anti-APRIL treatment groups for histological changes that might explain the discrepant clinical effects of these mAbs. We observed that the anti-CD20 mAb induced profound changes in the immunogenic milieu inside SLO resulting in impairment of T-cell activation, which was not observed in monkeys treated with anti-BlyS or anti-APRIL mAbs.90 In the anti-CD20-treated Cj-EAE cases the T cells retained high expression of CCR7 and might therefore not be released into the circulation. We thus propose that the brisk and profound beneficial effect of rituximab in RRMS might be explained by retention of pathogenic T cells within SLO. Note that the clinical effect of the S1P receptor antagonist fingolimod in RRMS is based on the same principles.91
Concluding Remarks
Despite obvious similarities between the EAE model and MS, there are several discrepancies, which require clarification. The main discrepancy is in the pathogenic role of CD4+ proinflammatory T cells. Th1 and Th17 have an important pathogenic role in mouse and marmoset EAE models, as illustrated by the efficacy of therapies targeting this EAE initiation mechanism. However, difficulties encountered in the translation of these findings to the MS patient have raised questions on the relevance of the EAE initiation pathway for MS. A second discrepancy is the absence of neurodegeneration in the EAE model, indicating that there may be no causal relation between this pathological aspect of MS and the autoimmune process. Advantage can be taken from the availability of well-characterized NHP-EAE models, which can be used to test whether these paradoxes are due to technical shortcomings of the models or to inconsistencies in the current concept of MS pathogenesis.92
The most important message of this publications is that NHP-EAE models are unique because, similar to humans, autoimmunity and the ensuing autoimmune neurological disease develop in a mature immune system. Maturity is induced by the daily combat with environmental pathogens and with chronic latent infection by endogenous pathogens. As reviewed elsewhere, the need to suppress exacerbation of the endogenous infections creates a repertoire of highly reactive effector memory, which are potentially autoreactive. Absence of a pathogen-educated immune repertoire in inbred/SPF (specific pathogen free) rodents may be the missing link between MS and the EAE model. NHP model can therefore provide relevant information on the still poorly understood relation between infection and autoimmunity.
Obviously, there are many outstanding questions waiting for an answer, such as:
Which events trigger the formation of primary lesions in MS?
How might the marmoset EBV homolog CalHV3 alter the pathogenic function of B cells?
How do the NK-CTL induce demyelination of cortical gray matter?
Is it possible to selectively deplete the EBV-infected B-cell subset and is this sufficient for the prevention of ongoing disease?
We believe that the unique marmoset EAE model provides an excellent experimental platform to help with finding an answer for these burning questions.
Acknowledgments
We thank Michel Vierboom (BPRC) for critical review of the manuscript and Henk van Westbroek (BPRC) for the artwork.
Conflict of Interest
Dr. Bruno Gan Support for the salary of a Research Registrar; fees as Consultant; support for the attendance of scientific conferences for Teva UK. Support for laboratory based MS research; support for the attendance of scientific conferences; fees as Consultant for Biogen Idec. Support for laboratory based MS research; support for the attendance of scientific conferences for Novartis. Support for the salary of a Research Registrar; support for the attendance of scientific conferences; fees as Consultant for Merck Serono. Support for laboratory based MS research from Genzyme. Support for laboratory based MS research; support for the attendance of scientific conferences for Bayer Schering. Received personal fees as advisory board member for Roche. Dr. 't Hart reports fee for service test of anti-B cell mAb from Glaxo Smith Kline, Human Genome Sciences. Fee for service test of anti-IL-17A mAb, Celltech-UCB. Fee for service test of anti-IL-12p40 mAb, Centocor outside the submitted work.
References
- Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi N, Gran B, Constantinescu CS. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J Neurochem. 2010;115:829–844. doi: 10.1111/j.1471-4159.2010.06982.x. [DOI] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, Mues M, Koutrolos M, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- ‘t Hart BA, Gran B, Weissert R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med. 2011;17:119–125. doi: 10.1016/j.molmed.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13:507–514. doi: 10.1038/nrn3275. [DOI] [PubMed] [Google Scholar]
- Pette M, Fujita K, Wilkinson D, et al. Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc Natl Acad Sci USA. 1990;87:7968–7972. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ‘t Hart BA, Hintzen RQ, Laman JD. Multiple sclerosis – a response-to-damage model. Trends Mol Med. 2009;15:235–244. doi: 10.1016/j.molmed.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Axthelm MK, Bourdette DN, Marracci GH, et al. Japanese macaque encephalomyelitis: a spontaneous multiple sclerosis-like disease in a nonhuman primate. Ann Neurol. 2011;70:362–373. doi: 10.1002/ana.22449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine CS, Cannella B, Hauser SL, Genain CP. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Ann Neurol. 1999;46:144–160. doi: 10.1002/1531-8249(199908)46:2<144::aid-ana3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Tanaka T, Saijoh Y, Fujinami RS. Targeting inflammatory demyelinating lesions to sites of Wallerian degeneration. Am J Pathol. 2007;171:1563–1575. doi: 10.2353/ajpath.2007.070147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T, Hillert J. The genetics of multiple sclerosis and its experimental models. Curr Opin Neurol. 2008;21:255–260. doi: 10.1097/WCO.0b013e3282fd10cc. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann Neurol. 2007;61:504–513. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- Pender MP. CD8+ T-cell deficiency, Epstein-Barr virus infection, vitamin D deficiency, and steps to autoimmunity: a unifying hypothesis. Autoimmune Dis. 2012;2012:1–16. doi: 10.1155/2012/189096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- Wang F, Rivailler P, Rao P, Cho Y. Simian homologues of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci. 2001;356:489–497. doi: 10.1098/rstb.2000.0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinami RS, Oldstone MB, Wroblewska Z, et al. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci USA. 1983;80:2346–2350. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- Lang HL, Jacobsen H, Ikemizu S, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM NeuroproMiSe EBVWG. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue–report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134(Pt 9):2772–2786. doi: 10.1093/brain/awr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ‘t Hart BA, Jagessar SA, Haanstra K, et al. The primate EAE model points at EBV-infected B cells as a preferential therapy target in multiple sclerosis. Front Immunol. 2013;4:145. doi: 10.3389/fimmu.2013.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haanstra KG, Wubben JA, Jonker M, ‘t Hart BA. Induction of encephalitis in rhesus monkeys infused with lymphocryptovirus-infected B-cells presenting MOG34-56 peptide. PLoS One. 2013;8:e71549. doi: 10.1371/journal.pone.0071549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagessar SA, Fagrouch Z, Heijmans N, et al. The different clinical effects of anti-BLyS, anti-APRIL and anti-CD20 antibodies point at a critical pathogenic role of gamma-herpesvirus infected B cells in the marmoset EAE model. J Neuroimmune Pharmacol. 2013;8:727–738. doi: 10.1007/s11481-013-9448-6. [DOI] [PubMed] [Google Scholar]
- Marmoset Genome S, Analysis C, Marmoset Genome S, Analysis C. The common marmoset genome provides insight into primate biology and evolution. Nat Genet. 2014;46:850–857. doi: 10.1038/ng.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genain CP, Hauser SL. Experimental allergic encephalomyelitis in the New World monkey Callithrix jacchus. Immunol Rev. 2001;183:159–172. doi: 10.1034/j.1600-065x.2001.1830113.x. [DOI] [PubMed] [Google Scholar]
- Massacesi L, Genain CP, Lee-Parritz D, et al. Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: a new model for multiple sclerosis. Ann Neurol. 1995;37:519–530. doi: 10.1002/ana.410370415. [DOI] [PubMed] [Google Scholar]
- ‘t Hart BA, Bauer J, Muller HJ, et al. Histopathological characterization of magnetic resonance imaging- detectable brain white matter lesions in a primate model of multiple sclerosis: a correlative study in the experimental autoimmune encephalomyelitis model in common marmosets (Callithrix jacchus. Am J Pathol. 1998;153:649–663. doi: 10.1016/s0002-9440(10)65606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig BM. Drug evaluation: CNTO-1275, a mAb against IL-12/IL-23p40 for the potential treatment of inflammatory diseases. Curr Opin Investig Drugs. 2007;8:947–954. [PubMed] [Google Scholar]
- Gran B, Zhang GX, Rostami A. Role of the IL-12/IL-23 system in the regulation of T-cell responses in central nervous system inflammatory demyelination. Crit Rev Immunol. 2004;24:111–128. doi: 10.1615/critrevimmunol.v24.i2.20. [DOI] [PubMed] [Google Scholar]
- Brok HP, Van Meurs M, Blezer E, et al. Prevention of experimental autoimmune encephalomyelitis in common marmosets using an anti-IL-12p40 monoclonal antibody. J Immunol. 2002;169:6554–6563. doi: 10.4049/jimmunol.169.11.6554. [DOI] [PubMed] [Google Scholar]
- ‘t Hart BA, Brok HP, Remarque E, et al. Suppression of ongoing disease in a nonhuman primate model of multiple sclerosis by a human-anti-human IL-12p40 antibody. J Immunol. 2005;175:4761–4768. doi: 10.4049/jimmunol.175.7.4761. [DOI] [PubMed] [Google Scholar]
- Segal BM, Constantinescu CS, Raychaudhuri A, et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- Kap YS, Jagessar SA, van Driel N, et al. Effects of early IL-17A neutralization on disease induction in a primate model of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2011;6:341–353. doi: 10.1007/s11481-010-9238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mix E, Meyer-Rienecker H, Hartung HP, Zettl UK. Animal models of multiple sclerosis–potentials and limitations. Prog Neurobiol. 2010;92:386–404. doi: 10.1016/j.pneurobio.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagessar SA, Smith PA, Blezer E, et al. Autoimmunity against myelin oligodendrocyte glycoprotein is dispensable for the initiation although essential for the progression of chronic encephalomyelitis in common marmosets. J Neuropathol Exp Neurol. 2008;67:326–340. doi: 10.1097/NEN.0b013e31816a6851. [DOI] [PubMed] [Google Scholar]
- Smith PA, Heijmans N, Ouwerling B, et al. Native myelin oligodendrocyte glycoprotein promotes severe chronic neurological disease and demyelination in Biozzi ABH mice. Eur J Immunol. 2005;35:1311–1319. doi: 10.1002/eji.200425842. [DOI] [PubMed] [Google Scholar]
- Brok HP, Uccelli A, Kerlero De Rosbo N, et al. Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets: the encephalitogenic T cell epitope pMOG24-36 is presented by a monomorphic MHC class II molecule. J Immunol. 2000;165:1093–1101. doi: 10.4049/jimmunol.165.2.1093. [DOI] [PubMed] [Google Scholar]
- Kap YS, Smith P, Jagessar SA, et al. Fast progression of recombinant human myelin/oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis in marmosets is associated with the activation of MOG34-56-specific cytotoxic T cells. J Immunol. 2008;180:1326–1337. doi: 10.4049/jimmunol.180.3.1326. [DOI] [PubMed] [Google Scholar]
- Jagessar SA, Heijmans N, Blezer EL, et al. Unravelling the T-cell-mediated autoimmune attack on CNS myelin in a new primate EAE model induced with MOG34-56 peptide in incomplete adjuvant. Eur J Immunol. 2012;42:217–227. doi: 10.1002/eji.201141863. [DOI] [PubMed] [Google Scholar]
- Clements CS, Reid HH, Beddoe T, et al. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:11059–11064. doi: 10.1073/pnas.1833158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarasse C, Della Gaspera B, Lu CW, et al. Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem. 2006;98:1707–1717. doi: 10.1111/j.1471-4159.2006.04053.x. [DOI] [PubMed] [Google Scholar]
- Breithaupt C, Schubart A, Zander H, et al. Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc Natl Acad Sci USA. 2003;100:9446–9451. doi: 10.1073/pnas.1133443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd PM, Dwek RA. Glycosylation: heterogeneity and the 3D structure of proteins. Crit Rev Biochem Mol Biol. 1997;32:1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallejo JJ, Ilarregui JM, Kalay H, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med. 2014;211:1465–83. doi: 10.1084/jem.20122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Van Vliet SJ, Engering A, et al. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- della Gaspera B, Pham-Dinh D, Roussel G, et al. Membrane topology of the myelin/oligodendrocyte glycoprotein. Eur J Biochem. 1998;258:478–484. doi: 10.1046/j.1432-1327.1998.2580478.x. [DOI] [PubMed] [Google Scholar]
- Jagessar SA, Kap YS, Heijmans N, et al. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J Neuropathol Exp Neurol. 2010;69:372–385. doi: 10.1097/NEN.0b013e3181d5d053. [DOI] [PubMed] [Google Scholar]
- Linington C, Lassmann H. Antibody responses in chronic relapsing experimental allergic encephalomyelitis: correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG) J Neuroimmunol. 1987;17:61–69. doi: 10.1016/0165-5728(87)90031-2. [DOI] [PubMed] [Google Scholar]
- Pham-Dinh D, Mattei MG, Nussbaum JL, et al. Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex. Proc Natl Acad Sci USA. 1993;90:7990–7994. doi: 10.1073/pnas.90.17.7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarasse C, Daubas P, Mars LT, et al. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin Invest. 2003;112:544–553. doi: 10.1172/JCI15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazilleau N, Delarasse C, Sweenie CH, et al. Persistence of autoreactive myelin oligodendrocyte glycoprotein (MOG)-specific T cell repertoires in MOG-expressing mice. Eur J Immunol. 2006;36:533–543. doi: 10.1002/eji.200535021. [DOI] [PubMed] [Google Scholar]
- Linares D, Mana P, Goodyear M, et al. The magnitude and encephalogenic potential of autoimmune response to MOG is enhanced in MOG deficient mice. J Autoimmun. 2003;21:339–351. doi: 10.1016/j.jaut.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8:372–379. doi: 10.1038/nri2296. [DOI] [PubMed] [Google Scholar]
- Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11:807–822. doi: 10.1038/nri3095. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallejo JJ, van Kooyk Y. Endogenous ligands for C-type lectin receptors: the true regulators of immune homeostasis. Immunol Rev. 2009;230:22–37. doi: 10.1111/j.1600-065X.2009.00786.x. [DOI] [PubMed] [Google Scholar]
- ‘t Hart BA, van Kooyk Y. Yin-Yang regulation of autoimmunity by DCs. Trends Immunol. 2004;25:353–359. doi: 10.1016/j.it.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallejo JJ, van Kooyk Y. The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol. 2013;34:482–486. doi: 10.1016/j.it.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Relloso M, Puig-Kroger A, Pello OM, et al. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-beta, and anti-inflammatory agents. J Immunol. 2002;168:2634–2643. doi: 10.4049/jimmunol.168.6.2634. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- Kim T, Pfeiffer SE. Myelin glycosphingolipid/cholesterol-enriched microdomains selectively sequester the non-compact myelin proteins CNP and MOG. J Neurocytol. 1999;28:281–293. doi: 10.1023/a:1007001427597. [DOI] [PubMed] [Google Scholar]
- de Vos AF, van Meurs M, Brok HP, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002;169:5415–5423. doi: 10.4049/jimmunol.169.10.5415. [DOI] [PubMed] [Google Scholar]
- Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013;9:455–461. doi: 10.1038/nrneurol.2013.118. [DOI] [PubMed] [Google Scholar]
- Albouz-Abo S, Wilson JC, Bernard CC, von Itzstein M. A conformational study of the human and rat encephalitogenic myelin oligodendrocyte glycoprotein peptides 35-55. Eur J Biochem. 1997;246:59–70. doi: 10.1111/j.1432-1033.1997.t01-2-00059.x. [DOI] [PubMed] [Google Scholar]
- Breithaupt C, Schafer B, Pellkofer H, et al. Demyelinating myelin oligodendrocyte glycoprotein-specific autoantibody response is focused on one dominant conformational epitope region in rodents. J Immunol. 2008;181:1255–1263. doi: 10.4049/jimmunol.181.2.1255. [DOI] [PubMed] [Google Scholar]
- Marta CB, Oliver AR, Sweet RA, et al. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci USA. 2005;102:13992–13997. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genain CP, Nguyen MH, Letvin NL, et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest. 1995;96:2966–2974. doi: 10.1172/JCI118368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brok HP, Bauer J, Jonker M, et al. Non-human primate models of multiple sclerosis. Immunol Rev. 2001;183:173–185. doi: 10.1034/j.1600-065x.2001.1830114.x. [DOI] [PubMed] [Google Scholar]
- von Budingen HC, Hauser SL, Fuhrmann A, et al. Molecular characterization of antibody specificities against myelin/oligodendrocyte glycoprotein in autoimmune demyelination. Proc Natl Acad Sci USA. 2002;99:8207–8212. doi: 10.1073/pnas.122092499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ‘t Hart BA, Massacesi L. Clinical, pathological, and immunologic aspects of the multiple sclerosis model in common marmosets (Callithrix jacchus. J Neuropathol Exp Neurol. 2009;68:341–355. doi: 10.1097/NEN.0b013e31819f1d24. [DOI] [PubMed] [Google Scholar]
- Mesleh MF, Belmar N, Lu CW, et al. Marmoset fine B cell and T cell epitope specificities mapped onto a homology model of the extracellular domain of human myelin oligodendrocyte glycoprotein. Neurobiol Dis. 2002;9:160–172. doi: 10.1006/nbdi.2001.0474. [DOI] [PubMed] [Google Scholar]
- Villoslada P, Abel K, Heald N, et al. Frequency, heterogeneity and encephalitogenicity of T cells specific for myelin oligodendrocyte glycoprotein in naive outbred primates. Eur J Immunol. 2001;31:2942–2950. doi: 10.1002/1521-4141(2001010)31:10<2942::aid-immu2942>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Pomeroy IM, Matthews PM, Frank JA, et al. Demyelinated neocortical lesions in marmoset autoimmune encephalomyelitis mimic those in multiple sclerosis. Brain. 2005;128(Pt 11):2713–2721. doi: 10.1093/brain/awh626. [DOI] [PubMed] [Google Scholar]
- Kap YS, Bauer J, Driel NV, et al. B-cell depletion attenuates white and gray matter pathology in marmoset experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2011;70:992–1005. doi: 10.1097/NEN.0b013e318234d421. [DOI] [PubMed] [Google Scholar]
- Pomeroy IM, Jordan EK, Frank JA, et al. Diffuse cortical atrophy in a marmoset model of multiple sclerosis. Neurosci Lett. 2008;437:121–124. doi: 10.1016/j.neulet.2008.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxiadis GG, van der Wiel MK, Brok HP, et al. Reactivation by exon shuffling of a conserved HLA-DR3-like pseudogene segment in a New World primate species. Proc Natl Acad Sci USA. 2006;103:5864–5868. doi: 10.1073/pnas.0600643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge T, von Budingen HC, Lalive PH, Genain CP. Relevant antibody subsets against MOG recognize conformational epitopes exclusively exposed in solid-phase ELISA. Eur J Immunol. 2007;37:3229–3239. doi: 10.1002/eji.200737249. [DOI] [PubMed] [Google Scholar]
- de Graaf KL, Albert M, Weissert R. Autoantigen conformation influences both B- and T-cell responses and encephalitogenicity. J Biol Chem. 2012;287:17206–17213. doi: 10.1074/jbc.M111.304246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerlero de Rosbo N, Hoffman M, Mendel I, et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol. 1997;27:3059–3069. doi: 10.1002/eji.1830271144. [DOI] [PubMed] [Google Scholar]
- Brok HP, Boven L, van Meurs M, et al. The human CMV-UL86 peptide 981-1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34-56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol. 2007;182:135–152. doi: 10.1016/j.jneuroim.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Barun B, Bar-Or A. Treatment of multiple sclerosis with Anti-CD20 antibodies. Clin Immunol. 2012;142:31–37. doi: 10.1016/j.clim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Bleeker WK, Munk ME, Mackus WJ, et al. Estimation of dose requirements for sustained in vivo activity of a therapeutic human anti-CD20 antibody. Br J Haematol. 2008;140:303–312. doi: 10.1111/j.1365-2141.2007.06916.x. [DOI] [PubMed] [Google Scholar]
- Kap YS, van Driel N, Blezer E, et al. Late B cell depletion with a human anti-human CD20 IgG1kappa monoclonal antibody halts the development of experimental autoimmune encephalomyelitis in marmosets. J Immunol. 2010;185:3990–4003. doi: 10.4049/jimmunol.1001393. [DOI] [PubMed] [Google Scholar]
- Jagessar SA, Heijmans N, Bauer J, et al. B-cell depletion abrogates T cell-mediated demyelination in an antibody-nondependent common marmoset experimental autoimmune encephalomyelitis model. J Neuropathol Exp Neurol. 2012;71:716–728. doi: 10.1097/NEN.0b013e3182622691. [DOI] [PubMed] [Google Scholar]
- Jagessar SA, Heijmans N, Bauer J, et al. Antibodies against human BLyS and APRIL attenuate EAE development in marmoset monkeys. J Neuroimmune Pharmacol. 2012;7:557–570. doi: 10.1007/s11481-012-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Hartung HP, Freedman MS, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014;13:353–363. doi: 10.1016/S1474-4422(14)70028-6. [DOI] [PubMed] [Google Scholar]
- Cho Y, Ramer J, Rivailler P, et al. An Epstein-Barr-related herpesvirus from marmoset lymphomas. Proc Natl Acad Sci USA. 2001;98:1224–1229. doi: 10.1073/pnas.98.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kap YS, van Driel N, Laman JD, et al. CD20+ B cell depletion alters T cell homing. J Immunol. 2014;192:4242–4253. doi: 10.4049/jimmunol.1303125. [DOI] [PubMed] [Google Scholar]
- Chiba K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther. 2005;108:308–319. doi: 10.1016/j.pharmthera.2005.05.002. [DOI] [PubMed] [Google Scholar]
- ‘t Hart BA, Jagessar SA, Kap YS, et al. Improvement of preclinical animal models for autoimmune-mediated disorders via reverse translation of failed therapies. Drug Discov Today. 2014;19:1394–1401. doi: 10.1016/j.drudis.2014.03.023. [DOI] [PubMed] [Google Scholar]