

Abstract
Researchers seeking to improve the efficiency and cost effectiveness of the bioactive small-molecule discovery process have recently embraced selection-based approaches, which in principle offer much higher throughput and simpler infrastructure requirements compared with traditional small-molecule screening methods. Since selection methods benefit greatly from an information-encoding molecule that can be readily amplified and decoded, several academic and industrial groups have turned to DNA as the basis for library encoding and, in some cases, library synthesis. The resulting DNA-encoded synthetic small-molecule libraries, integrated with the high sensitivity of PCR and the recent development of ultra high-throughput DNA sequencing technology, can be evaluated very rapidly for binding or bond formation with a target of interest while consuming minimal quantities of material and requiring only modest investments of time and equipment. In this review we describe the development of two classes of approaches for encoding chemical structures and reactivity with DNA: DNA-recorded library synthesis, in which encoding and library synthesis take place separately, and DNA-directed library synthesis, in which DNA both encodes and templates library synthesis. We also describe in vitro selection methods used to evaluate DNA-encoded libraries and summarize successful applications of these approaches to the discovery of bioactive small molecules and novel chemical reactivity.
Introduction
The discovery of bioactive small molecules remains a major focus of both academic and industrial chemists. The global pharmaceutical industry spends ~$100 billion annually on research and development, reflecting the continued need for new therapies for the treatment of human disease.1 Bioactive molecules that do not meet all safety, efficacy, and marketability requirements of drugs have nonetheless proven valuable as probes to study a wide range of biological processes in the life sciences.2
The development of highly efficient methods to study the complete DNA, RNA, protein, or small-molecule content of cells has provided researchers with an enormous wealth of information on cellular targets implicated in human disease.3, 4 In order to find modulators of these disease-associated targets, researchers frequently use a drug discovery approach based on combinatorial or diversity-oriented synthesis5 coupled with high-throughput screening. This approach seeks to create libraries of hundreds or thousands of compounds using reactions capable of tolerating chemically and structurally diverse building blocks. Once a library has been synthesized, a variety of screening methods are used to interrogate the activity of each library member in a discrete biochemical or cell-based phenotypic assay. Hits that emerge from high-throughput screens then serve as lead compounds for further development towards probes or therapeutic agents.
While high-throughput screening has become widely adopted in the drug discovery industry, it can be time consuming and typically requires substantial investment in costly infrastructure such as liquid-handling robots and in consumable reagents such as pipette tips and multi-well plates. Moreover, due to the discrete nature of screening assays, screening time and cost scale approximately linearly with library size.
In contrast, selection-based approaches, in which all library members are simultaneously tested for their ability to interact with a target of interest in a single one-pot experiment, can be much more efficient than screening. Since the time and effort required to perform a selection are independent of library size, researchers have selected large (“high-complexity”, in evolution terminology) libraries containing up to 1015 members,6 while screening-based methods have not been applied to libraries with complexity greater than 108,7 and are typically applied to libraries in the range of 103–106 members. Moreover, selection methods regardless of the target typically require only modest, widely available infrastructure such as disposable vessels and filters or magnetic separators, in contrast with the more specialized and target-specific equipment used to perform screens on biological targets.
The ability to deconvolute and decode a complex mixture of chemical entities before and after selection is a critical requirement for the application of selections to small-molecule discovery. Since directed analysis based upon the physicochemical properties of the library members (such as mass or retention time) may not offer the sensitivity and resolution required to uniquely identify the components of a highly complex mixture, researchers have developed the use of molecular barcodes directly associated with each library member that can be decoded with high fidelity.8–11 The in vitro or in vivo selection of proteins and nucleic acids is made possible by their genetic encoding and by biosynthetic machinery that uniquely translates DNA sequences into corresponding RNA or protein molecules. Due to the association of genotype and phenotype, the identity of library members surviving selection can be analyzed by PCR amplification and DNA sequencing of their associated genes.
Nature does not provide corresponding encoding and translation machinery for synthetic molecules. In 1992, Brenner and Lerner12 described a theoretical framework for using DNA to encode synthetic peptides. In 2001, Liu and co-workers implemented the use of DNA-templated synthesis to generate DNA-encoded small molecules with arbitrary chemical structures.13 Additional methods for preparing and subjecting DNA-encoded small-molecule libraries to in vitro selection have been reported by several academic and industrial groups. This review summarizes the development of these approaches and their recent application to discover bioactive synthetic small molecules.
DNA as a Barcode for Library Synthesis
DNA offers several key features as a molecular barcode for synthetic libraries. Its information density is very high, such that a 20-mer can encode millions of library members with error-avoiding redundancy. Moreover, the ability of minute quantities (sub-fmol) of DNA to be routinely amplified by PCR enables library evaluation to take place with a sensitivity not achievable by most screening methods. Finally, the advent of highly efficient methods for the separation and comprehensive, statistical sequencing of complex mixtures of DNA offers crucial advantages for library evaluation. Because of these advances in ultra-high throughput DNA sequencing technologies, even small changes in the abundance of each member of a large DNA-encoded library upon exposure to a target-binding selection can be revealed in a single, cost-effective experiment. These features collectively explain the growing popularity of DNA as the basis for synthetic library encoding.
The theory of using DNA to encode a combinatorial chemistry library was first described in the literature by Brenner and Lerner in 1992.12 Their scheme used oligonucleotide synthesis to record the chemical steps of split-and-pool library synthesis performed on a common solid-phase substrate. Multiple rounds of small-molecule and DNA co-synthesis would result in a combinatorial library in which each library member is linked to a DNA sequence that encodes its identity.
Brenner and Lerner’s strategy was visionary but posed significant synthetic challenges since the co-synthesis of library members and oligonucleotides requires chemical compatibility between two combinatorial synthesis schemes. In 1993, two groups experimentally implemented the synthesis of DNA-encoded peptides. Early efforts by Brenner and Janda to synthesize DNA-encoded peptides (synthesis of a DNA-encoded library was never reported) were successful upon careful selection of a suitable linker conducive to both chemistries.14 Using a modified approach that was conceptually similar to that of Brenner and Lerner, Gallup and co-workers were able to construct a library of ~105 DNA-encoded peptides by synthesizing in parallel peptide and oligonucleotides on different sites of the same resin bead.15 In this strategy, the bead itself serves as the “linker” that associates genetic tag with its corresponding library structure. While Gallup and co-workers were able to isolate active peptide sequences from this library, they were forced to evaluate the library using a screening approach since the library was resin-bound. In vitro selections typically require solution-phase libraries since each library member must be able to freely access and compete for target binding (or for reaction with a target substrate). Notably, both of these early examples of DNA-encoded libraries required substantial modifications to standard oligonucleotide and peptide synthesis methodology such as acid-stable purine analogs that could survive TFA-mediated peptide side chain deprotection, methyl phosphate protecting groups instead of the standard 2-cyanoethyl phosphate protecting group, and careful optimization of reaction conditions.14, 15
In the following decade, researchers developed and implemented several distinct approaches to the synthesis of DNA-encoded libraries. Two classes of strategies were pursued: what we refer to as “DNA-recorded chemistry,” in which the oligonucleotide is added as an identification tag before or after a synthesis reaction, and what we term “DNA-directed chemistry,” in which the oligonucleotide both identifies and directs the synthesis of each library member. DNA-directed approaches allow iterated rounds of selection and, in principle, directed evolution of synthetic libraries since the library can be re-synthesized after selection in a process analogous to protein translation. DNA-recorded approaches, in contrast, lack the ability to undergo translation en masse from DNA sequences to corresponding synthetic molecules and thus do not support this capability. DNA-recorded strategies, however, offer simpler access to library members since oligonucleotide sequences do not direct chemical reactions during library synthesis and therefore library synthesis steps can precede and be performed independent of DNA polymerization events.
Building on the original proposal of Brenner and Lerner, DNA-recorded library chemistry has been pioneered by Professor Dario Neri and co-workers (ETH Zurich, Switzerland) and commercialized by Philochem (Zurich, Switzerland) as well as researchers at Praecis Pharmaceuticals (Waltham, MA, USA) (now owned by GlaxoSmithKline) and NuEvolution (Copenhagen, Denmark).
Three distinct approaches to DNA-directed chemistry have been developed: DNA-templated synthesis (DTS) by Professor David Liu and co-workers (Harvard University, USA) and later commercialized by Ensemble Discovery (Cambridge, MA, USA), DNA-display or DNA-routing by Professor Pehr Harbury and co-workers (Stanford University, USA), and the YoctoReactor system developed by Vipergen (Copenhagen, Denmark). Professor Neri and coworkers have also developed an approach known as encoded self-assembling combinatorial (ESAC) libraries, in which DNA hybridization directly assembles each library member. In contrast to the DNA-directed methods described above, however, ESAC libraries cannot be subjected to iterated rounds of selection. These approaches will be described in further detail below.
Development of DNA-Directed Library Synthesis
In 2001, Gartner and Liu described DNA-templated organic synthesis (DTS), an approach to DNA-encoded chemistry in which a DNA oligonucleotide directs (or “templates”) bond-forming reactions by bringing DNA-linked reagents into proximity through Watson-Crick base pairing.13 Liu and co-workers were not the first to use nucleic acid hybridization to template chemical reactions. In 1966, Naylor and Gilham16 demonstrated that a polyA template could catalyze chemical phosphodiester bond formation between polyT hexamers, and pioneering work by Leslie Orgel and others17 has demonstrated the ability of an oligonucleotide template to catalyze the polymerization of activated nucleotide units, including those of DNA, RNA, and peptide nucleic acid (PNA). Liu and co-workers discovered that DTS is a general approach to increase the effective molarity of chemical reactants, enabling access to a wide variety of products with no structural similarity to nucleic acids.13 Because the products of DTS reactions are linked to DNA templates that encode and direct their synthesis, and because Watson-Crick base pairing between many oligonucleotides can take place in one solution in a highly sequence-specific manner, DTS can “translate” libraries of DNA into corresponding libraries of DNA-encoded synthetic molecules suitable for in vitro selection.
Demonstrating the versatility of the approach, Liu and co-workers have applied DTS to three distinct problems: 1) synthesis and selection of DNA-templated small-molecule libraries, 2) reaction discovery using DNA-encoded libraries of small-molecule substrates, and 3) a system for the in vitro translation, selection and amplification of sequence-defined synthetic polymers. In 2004, Gartner et al. described the first DNA-templated small-molecule library synthesis in which a pilot 65-membered macrocycle library was generated by three DNA-templated amine acylations and one Wittig macrocyclization reaction.18 Incorporation of a phenyl sulfonamide-containing building block into this library enabled a model selection against carbonic anhydrase to be performed successfully. Several years later, advances in DNA-templated library synthesis methodology enabled the rapid synthesis of a 13,000-membered DNA-templated macrocycle library suitable for in vitro selection and ligand discovery (Figure 1A).19 Bioactive macrocycles discovered from this library are described below.
Figure 1.


Approaches to DNA-directed library synthesis. A) Synthesis of a library of 13,824 macrocycles using DNA-templated synthesis. B) Synthesis of a DNA-encoded library using the “YoctoReactor” system. C) Synthesis of a library of 100 million peptoids using DNA-display. D) Library synthesis through the encoded self-assembling combinatorial (ESAC) approach.
DTS libraries have been applied to research programs beyond those seeking the discovery of functional DNA-encoded small-molecules. In 2004, Kanan et al. reported a DNA-encoded reaction discovery system20 that revealed a novel Pd(II)-catalyzed alkene-alkynamide coupling. In this system, two pools of DNA-linked substrates were hybridized, resulting in 168 pairwise substrate combinations, and subjected to in vitro selection for bond formation under a variety of reaction conditions. PCR amplification and DNA microarray analysis revealed bond-forming substrate combinations. Liu and co-workers developed a hybridization-independent DNA-encoded reaction discovery system that resulted in the discovery of an Au(III)- or acid-mediated hydroarylation reaction21 and a Ru(II)-catalyzed azide reduction22 that is triggered by visible light and is compatible with the functional groups present in proteins, nucleic acids, and carbohydrates.
The principles of DTS have also been applied to synthetic polymers capable of Watson-Crick base pairing with DNA templates. In 2009, Brudno et al.23 reported the synthesis and selection of a library of DNA-encoded synthetic PNA analogs. They demonstrated that PNA building blocks can be non-enzymatically “translated” from a DNA template sequence with high sequence fidelity, and that the resulting DNA-encoded synthetic polymer library could support iterated cycles of translation, selection, and amplification, enabling a single biotinylated library member to be successively enriched from a library of more than 108 PNAs through successive in vitro selection for streptavidin binding.23
In 2009, scientists at Vipergen applied the principles of DNA-templated synthesis to generate a 100-membered DNA-encoded peptide library.24 Their system, termed the “YoctoReactor”, relies on DNA-templated reactions occurring at the center of a DNA three-way junction (Figure 1B). Like other DTS-based systems, the YoctoReactor can support iterated rounds of selection, amplification, and translation. Indeed, in a model selection conducted over two rounds, Hansen et al. demonstrated >150,000-fold enrichment of a known ligand for [Leu]-enkephalin from a YoctoReactor-synthesized library.24
While synthetic small-molecule libraries made by split-and-pool synthesis typically use off-the-shelf chemical reagents for library synthesis, the DNA-templated approaches described above require the separate synthesis and purification of DNA-linked reagents for use in DTS steps. The approach to DNA-encoded library synthesis described by Harbury and co-workers, termed “DNA display,”25–27 circumvents this requirement by combining the ability of DNA to direct reactions in a sequence-specific manner with the ease of performing library synthesis using commercially available building blocks. In the DNA display method, immobilized DNA sequences complementary to individual library codons capture and segregate a subset of DNA-linked library members in a codon-dependent manner via Watson-Crick base pairing (Figure 1C). Spatial segregation of DNA sequences according to individual codon identity allows the small-molecule library to be generated by what is effectively DNA-directed split-and-pool chemical synthesis. The DNA display method, like other DNA-directed library synthesis approaches, allows resynthesis (“retranslation”) of active library members from DNA isolated after selection. One complication of the use of DNA to route attached small molecules during library synthesis is the apparent requirement for long coding sequences on the order of 300 nucleotides.25 Harbury and co-workers demonstrated the potential of the DNA display approach by synthesizing a 1-million-membered DNA-encoded synthetic peptide library and performing a model selection for binding a [Leu]-enkephalin antibody.26
Another DNA-directed library approach was reported by Neri and co-workers in 2004.28 Their approach, encoded self-assembling combinatorial (ESAC) libraries, uses DNA hybridization to combinatorially assemble independently constructed libraries of DNA-encoded small-molecule fragments in a sequence-programmed fashion. In this approach, two DNA-encoded libraries containing n unique elements can combine to generate a DNA-duplex library displaying n2 DNA-encoded non-covalent fragment combinations (Figure 1D). Neri and co-workers used ESAC to improve known ligands to albumin and carbonic anhydrase. In these initial efforts, one hybridization partner with known target affinity was kept constant, and the second strand consisting of a DNA-encoded library was hybridized to the first strand. Selection of the resulting DNA duplexes displaying two small molecules led Melkko et al. to identify and synthesize tighter binding derivatives of dansyl amide and 4-carboxybenzenesulfonamide.28 One inherent challenge in the ESAC approach is the lack of information from selection results on an appropriate structural scaffold for the selected bidentate ligand once combinations of fragments are identified.
Development of DNA-Recorded Library Synthesis
In contrast to DNA-directed libraries, DNA-recorded libraries cannot be generated from libraries of DNA and therefore cannot be subjected to iterated rounds of selection and translation. Nevertheless, DNA-recorded libraries offer advantages including their ease of synthesis and their ability to use encoding DNA sequences (codons) that do not necessarily support sequence-specific hybridization. These features have enabled researchers to construct DNA-recorded libraries containing up to 800 million individual library members.29 The general approach to DNA-recorded library synthesis involves alternating library synthesis steps with enzyme-catalyzed DNA polymerization or ligation of short DNA sequences used to encode each synthetic step.30 Library diversity is generated over repeated cycles of division, synthesis, and pooling, a well-established method in combinatorial chemistry known as “split-and-pool” synthesis.31
In 2008, the Neri lab reported the synthesis of two 4,000-membered DNA-recorded libraries created by the above strategy. Each library contained two diversity-generating building blocks coupled using either the Diels-Alder cycloaddition (Figure 2A)32 or amide bond formation.33 Synthesis of the encoding double-stranded DNA was achieved by Klenow fragment DNA polymerase-catalyzed primer extension templated by partially complementary coding oligonucleotides. Neri and co-workers used DNA-conjugated desthiobiotin as a positive control in the library and selected the resulting population against streptavidin. They used high-throughput DNA sequencing, the first reported application of this technology to a DNA-encoded library, to analyze the selection results and confirm enrichment of the positive control as well as several novel streptavidin-binding molecules.33
Figure 2.

Approaches to DNA-recorded library synthesis. A) Neri and co-workers synthesized a 4,000-membered DNA-encoded library using a bicyclic Diels-Alder-formed scaffold and DNA-tag synthesis using primer extension. A similar 4,000-membered DNA-encoded library was also synthesized using amide-bond-formation chemistry. B) Researchers at Praecis Pharmaceuticals assembled a 7-million-membered DNA-encoded library using alternating cycles of enzymatic DNA ligation and chemical synthesis on a triazine scaffold.
At Praecis Pharmaceuticals (Waltham, MA, USA), now owned by GlaxoSmithKline, researchers devised an encoding method that records each library synthesis reaction by prior enzymatic ligation of a double-stranded DNA tag. DNA ligation and chemical synthesis on a triazine scaffold were cycled in a split-and-pool manner over three rounds to create a DNA-encoded library of up to 7 million members (DEL-A) (Figure 2B), and over four rounds to create a library of up to 800 million members (DEL-B),29 the largest DNA-encoded small-molecule library reported to date. As a proof-of-principle demonstration, VX-680, a known Aurora A kinase inhibitor, was conjugated to DNA and spiked into DEL-A at the expected concentration of a single library member. Three rounds of panning (binding selection without amplification) of this library against Aurora A resulted in 100,000-fold enrichment of the positive control VX-680-encoding DNA as determined by high-throughput sequencing.29 Praecis scientists included a known p38 MAPK inhibitor pharmacophore, 3-amino-4-methyl-N-methoxybenzamide (AMMB), into DEL-A during a second proof-of-principle selection experiment. Selection of DEL-A against p38 MAPK resulted in substantial enrichment of library members containing the AMMB moiety.
Collectively, the results from the Neri group and Praecis/GSK validated the ability of split-and-pool synthesis to furnish large DNA-recorded libraries suitable for the discovery of protein-binding small molecules, and established high-throughput DNA sequencing as a critical component of molecular discovery from large DNA-encoded libraries.
Methods for In Vitro Selection of DNA-Encoded Libraries
In vitro selections for target affinity are a simple but very powerful tool for evaluating synthetic libraries.34–37 They enable the preferential enrichment of molecules with target-binding activity relative to non-binding molecules from a high-complexity library, facilitating the identification of rare library members with desirable properties. During a selection, the properties of each molecule are evaluated on the basis of that molecule’s individual properties at the same time in one pot, enabling extremely high throughput.6 In contrast, a screening approach by definition interrogates the properties of each library member discretely, a process that requires time, effort, and expense roughly proportional to library size, and that also typically requires the physical separation of each library member, or synthesis of each library member in a spatially segregated format.38
Binding selections are often performed using an immobilized target that chromatographically separates active library members bound to the target from non-binding library members still in solution (Fig. 3A). Washing removes inactive library members, and target-bound library members are then eluted by cleaving the target from solid support, by disrupting ligand-target interactions, or by treating with an excess of a competitive ligand. In the case of DNA-encoded libraries, the DNA encoding selection survivors can then be amplified by PCR and subjected to subsequent rounds of in vitro selection (perhaps under more stringent conditions) to further enrich the most active library members. Phage display first popularized this in vitro selection method,39 termed ‘panning’ in analogy to the process for removing contaminants from samples containing gold. Panning has been extensively used for the in vitro selection and evolution of nucleic acid aptamers long before its application to synthetic library evaluation.6
Figure 3.

Methods to perform in vitro selections on DNA-encoded libraries. A) Traditional in vitro selection for target affinity. B) Traditional in vitro selection for bond formation. C) Selection for bond formation using reaction-dependent PCR (RDPCR). D) Selection for target binding using interaction-dependent PCR (IDPCR).
In 2003, Doyon et al.40 demonstrated the use of in vitro selection methodology with immobilized protein targets to enrich DNA-linked small-molecule ligands from mixtures containing predominantly non-binding small molecules. This approach has been widely used to process DNA-encoded chemical libraries. While target immobilization is the most straightforward approach to in vitro binding selection, it is not an absolute requirement and methods such as capillary electrophoresis enable the solution-phase separation of library member-target complexes from free non-binding library members.41
An in vitro binding selection can be adapted into a selection for bond formation or bond cleavage by linking changes in covalency among library members with the transfer of an affinity handle such as biotin, thereby enabling physical separation of reactive library members from unreactive ones (Fig. 3B). Researchers in the ribozyme and DNAzyme evolution field have used such in vitro bond-forming and bond-breaking selections to evolve RNA and DNA catalysts.6
While the traditional approaches to in vitro selection described above have proven extremely successful for the discovery of ligands and catalysts, they are subject to two fundamental limitations: the inability to select a library against multiple targets in one solution and the general reliance on physical separation of active and inactive library members (usually by target or ligand immobilization) to enable enrichment.42, 43 Since researchers are often interested in interrogating multiple targets to increase the likelihood of finding a potent and selective ligand, the first constraint restricts the throughput of the method. The second limitation requires purification of library member-target complexes from inactive library members and, when immobilized targets are used, introduces artifacts arising from matrix binding, imprecise control of target concentration, or loss of native target structure.44 In principle, a solution-phase in vitro selection approach capable of evaluating the covalent or non-covalent interactions between a library of ligands and a library of targets should be able to overcome the restrictions discussed above.
Towards this goal, Gorin et al. recently developed reaction-dependent PCR (RDPCR)45 as an in vitro selection method for interrogating solution-phase reactions between pairs of DNA-encoded small molecules (Fig. 3C). RDPCR relies on the difference in melting temperature between an intermolecular versus an intramolecular (known as a “hairpin”) DNA duplex. By linking a single-stranded oligonucleotide capable of initiating primer extension to one small molecule, covalent-bond formation between a DNA-encoded library member and oligo-linked small molecule results in an intramolecular duplex which can be selectively extended by a DNA polymerase and subsequently PCR amplified preferentially over the intermolecular duplex formed by non-reacting DNA-encoded library members (Fig. 3C). McGregor et al.46 expanded the principles of RDPCR to solution-phase non-covalent binding between a DNA-encoded library and an oligonucleotide-tagged protein or nucleic acid target in a method termed interaction-dependent PCR (IDPCR). IDPCR enables multiple selections to be performed in one solution by adding a DNA barcode to each target, thereby allowing all possible combinations of a library of small molecules and a library of macromolecular targets to be evaluated simultaneously in a single experiment (Fig. 3D). McGregor et al. used IDPCR to selectively enrich the DNA sequences encoding biotin+streptavidin, desthiobiotin+streptavidin, carboxybenzenesulfonamide+carbonic anhydrase, Gly-Leu-carboxybenzene sulfonamide+carbonic anhydrase, and antipain+trypsin small molecule-target pairs out of ~68,000 possible small molecule-target DNA barcode combinations in a single solution. The application of IDPCR and related methods could significantly facilitate multiplexed selection experiments that simultaneously identify ligand-receptor pairs, reveal ligand selectivity across multiple receptors, and profile receptor selectivity across many ligands.
Decoding Selection Outcomes
Every member of a DNA-encoded chemical library is linked to “genes” that encode their structures. Assuming the method to synthesize the library rigorously preserves the correspondence between gene sequence and library member structure (or, minimally, library member reaction history), the outcomes of in vitro selections can be revealed by comparing DNA sequences before and after selection. DNA sequences enriched upon selection suggest that the corresponding library members possess target affinity or reactivity. Rapid advances in DNA sequencing technologies over the past decade47, 48 have dramatically increased the resulting information content that results from decoding DNA-encoded chemical libraries while decreasing the cost by several orders of magnitude. As a result, it is currently possible to obtain detailed structure-activity relationships across large DNA-encoded libraries subjected to in vitro selection in less than a few weeks of time and at a cost of less than $100 per selection. Future next-generation DNA sequencing technologies will likely drive these time and cost requirements down by at least an additional order of magnitude within the next few years, such that the cost of evaluating many selections of DNA-encoded libraries containing thousands or millions of library members may become trivial.
During the initial growth of DNA-encoded library technology in the early 90s and the beginning of the millennium, automated Sanger “dideoxy” sequencing was the method of choice for determining DNA sequence identity and thus it saw application in many of the pilot studies mentioned above.18, 28, 49 While Sanger sequencing offers relatively long and accurate read lengths (a property that is not critical for decoding synthetic DNA tag sequences which are often short and designed to be maximally distinguishable), it is not a high-throughput technique, and is only useful and cost-effective when sampling a small number of sequences (<1,000) can give an accurate picture of the distribution of sequences in the entire population, as is the case for small DNA-encoded libraries or for multi-round selection efforts that have sufficiently enriched the population to converge to a small number of active sequences.
DNA sequencing by hybridization to a microarray of complementary DNA oligonucleotides is a technique that provides a global profile of a DNA population and is not subject to the stochasticity present in methods that sample individual population members. Neri and co-workers successfully used DNA microarrays to analyze the results of affinity selections performed with a DNA-encoded library.28 The application of microarray-based methods for decoding complex populations of DNA sequences is unfortunately limited by the inherent specificity of DNA hybridization and the physical limitation on the number of probes that can be arrayed on a chip; consequently, this method is not suitable for analyzing very large DNA libraries containing many highly-similar encoding sequences. Microarray-based sequencing is an efficient method for analyzing modestly-sized populations of DNA sequences and has been repeatedly used by Liu and co-workers for decoding selection results from DNA-encoded reaction discovery experiments.20–22
Over the past six years, “next-generation” DNA sequencing technology has become readily available to the scientific community. One of the first such technologies to be commercialized, “454 sequencing”50 (454 Life Sciences,Branford, CT, USA, now owned by Roche), was initially able to provide on the order of ~10,000–100,000 sequence reads per sample, and was quickly adopted by the DNA-encoded library community, seeing application in selection analysis performed by Praecis Pharmaceuticals29 and by Neri and co-workers.33 More recently, Illumina’s (San Diego, CA, USA) Solexa sequencing technology51 has become the industry standard for providing very large numbers of sequences from a single experiment (~1–10 million reads per sample) with read lengths (~30–100 bases) that are sufficient to decode most types of DNA-encoded libraries. Kleiner et al.52 used Solexa sequencing to interrogate the selection of a DNA-templated macrocycle library against 36 protein targets. To maximize the capabilities of the technology, PCR barcoding was used to facilitate the parallel sequencing of all 36 selections as one sample. Similarly, Neri and co-workers have reported the use of Illumina technology to interrogate DNA-encoded libraries.53, 54
As DNA-encoded libraries have increased in size, the unprecedented sample coverage provided by high-throughput sequencing technology has become a crucial tool for the analysis of DNA-encoded library selections since it is the only method capable of detecting very rare library members or modest enrichments in a library member’s abundance upon selection. Continued advances in these rapidly developing technologies will make selection-based approaches increasingly efficient and cost-effective.
Bioactive Synthetic Small Molecules Discovered from DNA-Encoded Libraries
Researchers have recently integrated the developments summarized above to discover dozens of de novo synthetic small molecules with the ability to bind, inhibit, or activate biomedically relevant targets. The first example of such a discovery was reported by Harbury and colleagues in 2007.49 They generated a 100-million-membered peptoid library using the DNA display method and subjected this library to affinity selection against the SH3 domain of the proto-oncogene Crk. To favor the selection of Crk-SH3 ligands, the building blocks of the library were chosen based on a Crk-SH3-peptide co-crystal structure. After five rounds of selection, Sanger sequencing of 960 library clones revealed that the library sequences converged on ten families of structurally related compounds. Synthesis of one molecule per family gave rise to six Crk-SH3-binding poly-peptoids with dissociation constants ranging from 16 – 97 µM. These affinities are consistent with those observed for natural Crk-SH3 ligands, and the 16 µM peptoid binder (Table 1) was the highest affinity wholly unnatural SH3 ligand known at the time.
Table 1.
Bioactive synthetic small molecules discovered from DNA-encoded chemical libraries and in vitro selection.
| Target | Structure | Kd / IC50 |
|---|---|---|
| Crk-SH3 | 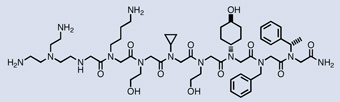 |
16 µM |
| HSA | 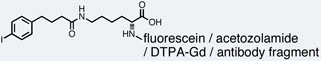 |
3.2 µM |
| MMP3 |  |
9.9 µM |
| Bcl-xL | 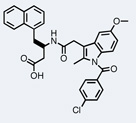 |
930 nM |
| TNF | 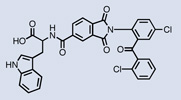 |
15 µM |
| CA IX | 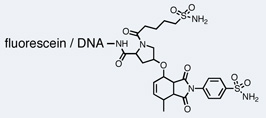 |
260 nM |
| Aurora A | 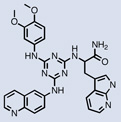 |
270 nM |
| p38 MAPK |  |
7 nM |
| Src | 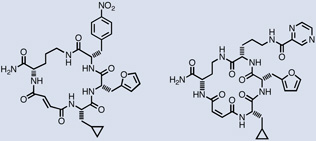 |
680 nM / 960 nM |
| p38α-MK2 | 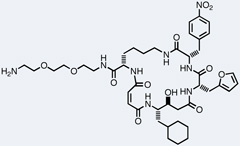 |
3.4 µM |
Several molecules discovered from DNA-encoded chemical libraries have shown activity in biological settings. One well-characterized bioactive molecule used for in vivo applications was reported by Neri and co-workers. They isolated a novel class of structurally-related human serum albumin-binding small molecules from a 619-membered DNA-encoded chemical library with affinities ranging from 3.2 µM to 55 µM.55 The highest-affinity ligand was applied as an albumin affinity tag by replacing the encoding DNA with other molecules of interest (Table 1).56, 57 In mouse studies, conjugates of the albumin binder and the commonly used blood pool contrast agents, fluorescein and Gd-DTPA, displayed increased circulatory half-lives and consequently improved performance as imaging agents over their unmodified counterpart. Further studies in mice demonstrated the benefits of endowing acetazolamide,56 a general carbonic anhydrase inhibitor routinely used for the treatment of a number of diseases including glaucoma, and a tumor-targeting antibody fragment57 with albumin-binding properties using the albumin affinity tag. In the first example, conjugation of the albumin binder to acetazolamide restricted the low molecular weight drug to the extracellular space and allowed specific inhibition of extracellular tumor-associated enzymes carbonic anhydrase IX and XII, resulting in tumor growth inhibition in a combination therapy with sunitinib in mice.56 In the second example, conjugation of the albumin binder to the antibody fragment increased the delivery of the protein to the tumor and resulted in superior tumor-to-organ ratios in comparison to the unmodified antibody fragment.57
ESAC libraries led to the discovery of matrix metalloproteinase 3 (MMP3) inhibitors, representing the first enzyme inhibitors isolated from a DNA-encoded chemical library.58 Initially, panning a 550-membered DNA-encoded library against MMP3 identified a starting compound which was then used as a lead structure for the assembly of an ESAC library. Selection of the dual fragment library against MMP3 resulted in several combinations of synergistically binding molecules. Suitable conjugation of these binding moieties resulted in an MMP3 inhibitor with an IC50 of 9.9 µM (Table 1). In contrast to most MMP inhibitors, the discovered molecule lacks common Zn-chelating groups.
The emergence of high-throughput DNA sequencing technology has facilitated the isolation of active molecules from increasingly larger libraries. In a collaboration between Philochem and Cambridge Antibody Technology (Cambridge, UK) (now MedImmune), a 4,000-membered amide-bond synthesized library was deployed for the identification of molecules binding to the anti-apoptotic protein Bcl-xL.59 High-throughput sequencing after affinity selection identified 34 molecules, of which 31 bound to the target with dissociation constants of 60 µM or better. The highest affinity binder (Kd = 930 nM) bound competitively with the Bak peptide, a natural ligand of Bcl-xL (Table 1), and induced cell death in Raji cells with an EC50 value of 77 µM. Interestingly, the molecule is a conjugate between indomethacin and a β-amino acid derivative. Indomethacin was originally discovered as an anti-inflammatory drug but has also been reported to be cytotoxic and to induce apoptosis in several cancer cell lines. In another example from the Neri lab, three binders of tumor necrosis factor (TNF) were isolated from a 4,000-membered Diels-Alder based library. Among these compounds, the most potent molecule displayed a dissociation constant of 20 µM.60 Further modification resulted in a small molecule capable of completely inhibiting TNF-induced apoptosis in cells, when applied at high concentrations (Table 1).
Neri and co-workers recently reported the discovery of inhibitors towards carbonic anhydrase IX (CA IX).54 Six inhibitors were isolated from a 1-millon-membered DNA-recorded library with inhibition activities as potent as IC50 = 240 nM (Table 1). All inhibitors included one or two sulfonamide-containing building blocks, functional groups known to bind enzymes containing zinc in their active site. A fluorescently labeled derivative of one of the bis(sulfonamide) compounds was shown to specifically localize at tumors overexpressing CA IX in two mouse models.54
Two groups have reported inhibitors for several disease-related kinases discovered using DNA-encoded libraries. Researchers at Praecis Pharmaceuticals/GSK discovered two families of Aurora A inhibitors from a 7-million-membered DNA-recorded library synthesized with a triazine chemical scaffold as described above.29 The most potent of these compounds inhibited Aurora A with an IC50 of 270 nM (Table 1). While the compounds were unrelated to previously described Aurora A inhibitors, they contained structural features common to kinase inhibitors. A co-crystal structure of one of the inhibitors showed the molecule binding in the ATP-binding pocket of the kinase and the former point of attachment to DNA exposed to solvent. Encouraged by these results, a DNA-recorded library of up to 800 million compounds was synthesized using the same triazine scaffold and was used to isolate molecules that bind p38 MAP kinase.29 In vitro selection revealed a family of enriched molecules sharing three out of the four library building blocks. Since the selection profile did not indicate any structural preference at the fourth building block, compounds corresponding to hydrolysis products of that synthetic step were included in the evaluation of hits. Indeed, one of these molecules lacking a fourth building block proved to be the most potent p38 MAP kinase inhibitor with an IC50 of 7 nM (Table 1). Crystallization experiments demonstrated binding of this molecule in the ATP pocket with the DNA attachment site pointing towards solvent.
Liu and co-workers described the first de novo discovery of active molecules from a DNA-templated library. Parallel selections of a 13,824-membered macrocycle library against 36 proteins resulted in the discovery of several novel classes of kinase inhibitors and activators.52 The most potent compounds isolated inhibited Src kinase with IC50 values of 680 and 960 nM (Table 1). Even though the identified inhibitors contained no building blocks strongly resembling ATP, they bound to Src in an ATP-competitive manner and with unusually high target specificity. The second family of kinase-binding macrocycles inhibited the kinases Akt3, MAPKAPK2, and Pim1 with low micromolar IC50 values (Table 1). Interestingly, one of the macrocycles in this family enhanced VEGFR2 activity significantly, suggesting that these macrocycles can also bind kinases in a manner that increases their activity. A third family of macrocycles inhibited their target kinases with modest activity, the best IC50value being 11 µM for the p38α-MAPKAP2 cascade.
Although industrial drug-discovery activities are often not immediately reported in the literature, the number of companies successfully harnessing the power of DNA-encoded chemical libraries and in vitro selection for drug discovery is further suggestive of the recent significant impact of these technologies. Indeed, interest in DNA-encoded chemical libraries has grown continuously since their initial reports (Table 2). This interest is reflected both by the growing number of new companies entering the field, and by the recent establishment of collaborative efforts (or acquisitions) involving large pharmaceutical companies and companies with expertise in DNA-encoded library synthesis and selection.
Table 2.
Companies and industrial collaborations engaged in small-molecule discovery using DNA-encoded libraries.
| Biotech | Founded | Collaboration/Acquisition | Initiation |
|---|---|---|---|
| Ensemble Therapeutics | 2004 | Bristol-Myers Squibb Pfizer |
2009 2010 |
| NuEvolution | 2001 | Merck Lexicon Pharmaceuticals Novartis |
2008 2008 2009 |
| Philochem | 2006 | Cambridge Antibody Technology | 2007 |
| Praecis Pharmaceuticals | 1993 | GlaxoSmithKline (acquisition) | 2007 |
| Vipergen | 2005 |
Conclusion/Discussion
Over the past decade, DNA-encoded chemical libraries have offered a valuable and versatile alternative to labor- and cost-intensive high-throughput screening campaigns. Several independent groups in academia and industry have reported the synthesis of many diverse DNA-encoded libraries using the methods summarized in this article. Selection of these libraries has yielded dozens of novel ligands for biomedically relevant targets. Although the chemistry that can be performed in the presence of DNA represents only a subset of known chemical reactions, the variety of synthetic schemes used to create DNA-encoded libraries to date have resulted in large, diverse collections of small molecules including libraries based on triazines, bicyclic Diels-Alder products, macrocycles, oxazolidines, peptoids, and PNA. The ligands, inhibitors, and activators isolated from DNA-encoded chemical libraries are likewise chemically diverse (Table 1). Moreover, the nature of the technology readily allows independently synthesized libraries to be combined so long as their template sequences are distinguishable, thereby increasing the complexity of libraries that can still be simultaneously assayed in each selection.
Selection methods require minimal infrastructure, have enormous throughput, and are versatile in target scope. While target-binding selections do not explicitly require binding to a specific site on the target, the majority of compounds surviving selection generally bind at predisposed locations, including enzyme active sites, enabling the isolation of novel inhibitors. Since minimal assay optimization is required for each individual target, selections against multiple targets are easily performed in parallel. This concept of rapidly assaying members of a large library for interaction with many targets has been extended by the development of IDPCR, in which all members of a library of DNA-encoded small molecules are simultaneously evaluated for binding to all members of a library of DNA-tagged macromolecules in a single experiment.46
The selection-based methods enabled by DNA-encoded chemical libraries can be tailored to yield more sophisticated insights beyond simple target affinity. For example, Wrenn et al.49 incorporated an elution step using an excess of the natural ligand to favor the selection of active-site binders. Alternatively, selections can be designed for the opposite outcome. Performing selections for kinase binding in the presence of high ATP or substrate peptide concentrations, for example, should preferentially enrich allosteric ligands. A target can likewise be locked in a catalytically inactive state to facilitate the selection of binders that stabilize the inactive conformation. Finally, free off-target macromolecules, such as related enzymes for which inhibition is undesirable, can be added to selections to remove library members with undesired specificities. Therefore, in vitro selections can be performed in replicates under slightly varied conditions to isolate multifunctional library members and to gain insights into corresponding complex structure-activity relationships.
DNA-encoded chemical libraries have benefitted greatly from advances in high-throughput sequencing technology. Although developed for rapid and cost-effective genome sequencing, next-generation DNA sequencing technologies have been highly enabling in the evaluation of selections on DNA-encoded chemical libraries. The ability to obtain millions of sequence counts per selection has obviated the need for post-selection libraries to converge on a small number of surviving sequences since DNA-encoded libraries containing up to ~106 members can now be sequenced with a high-degree of coverage. DNA-encoded library evaluation will continue to benefit from the ongoing development of DNA sequencing methods that offer longer read lengths and even greater throughput.
The fact that most DNA-encoded libraries contain thousands to millions of molecules, rather than billions or trillions, reflects a tradeoff between library size (complexity) and the degree to which library size and quality have been rigorously characterized. Larger libraries are typically achieved by adding additional steps to library synthesis, increasing the frequency among library members that an unplanned reaction has taken place or an anticipated reaction has failed to occur, potentially complicating the interpretation and recapitulation of selection data. As the success of the Praecis/GSK efforts demonstrate, however, even libraries containing non-productive reactions can yield the discovery of potent and selective ligands when coupled with sufficient downstream characterization and deconvolution. Collectively, the rapid development and successful application of DNA-encoded library synthesis and evaluation methods over the past ten years suggests that these platforms will continue to reshape academic and industrial small molecule-discovery efforts.
Acknowledgements
We thank Dr. Mary M. Rozenman for help in preparing Figure 1 and Dr. David J. Gorin and Lynn M. McGregor for assistance in preparing Figure 3.
Biographies

Ralph E. Kleiner obtained his AB in Chemistry (2005) from Princeton University, where he studied de novo protein design with Prof. Michael Hecht. He received his PhD in Chemistry (2011) under the supervision of Prof. David Liu at Harvard University, where he developed and applied methods for the genetic encoding and in vitro selection of synthetic small molecules and polymers. His interests lie on the interface of chemistry and biology, particularly in the areas of molecular evolution and chemical genetics.

Christoph Dumelin received his MSc in biochemistry (2003) and his PhD (2007) from the ETH Zurich. During his doctoral studies with Prof. Dario Neri he developed and applied methodology for the identification of bioactive small molecules from DNA-encoded chemical libraries. After working at Philochem (2007–2008) on the preclinical development of a blood pool contrast agent arising from his graduate studies, he joined the group of Prof. David Liu at Harvard University as a postdoctoral fellow. His research interests lie in drug discovery and the application of chemical tools to probe biological systems.

David R. Liu performed research on sterol biosynthesis under Professor E. J. Corey's guidance throughout his undergraduate years at Harvard. Liu graduated in 1994 with a bachelor's degree in chemistry before entering the Ph.D. program at U. C. Berkeley. In the group of Professor Peter Schultz, Liu initiated the first general effort to expand the genetic code in living cells. He earned his Ph.D. in 1999 and became Assistant Professor of Chemistry and Chemical Biology at Harvard University in the same year. Currently, Liu is Professor of Chemistry and Chemical Biology at Harvard University, a Howard Hughes Medical Institute Investigator, and a Senior Associate Member of the Broad Institute of Harvard and MIT. Professor Liu’s research applies evolutionary principles to the study and manipulation of biological and synthetic molecules. His major research interests include (i) the evolution, delivery, and characterization of synthetic regulatory elements, macromolecules that precisely manipulate information flow in human cells; (ii) the discovery of new structures and functions among cellular nucleic acids; and (iii) the discovery of bioactive synthetic small molecules, functional synthetic polymers, and new chemical reactions through DNA-templated synthesis and in vitro selection.
Footnotes
Part of a themed issue on the advances in DNA-based nanotechnology.
References
- 1.The Pharmaceutical Industry in Figures. European Federation of Pharmaceutical Industries and Associations; 2010. [Google Scholar]
- 2.Stockwell BR. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaddurah-Daouk R, Kristal BS, Weinshilboum RM. Annu. Rev. Pharmacol. Toxicol. 2008;48:653–683. doi: 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- 4.Kramer R, Cohen D. Nat Rev Drug Discov. 2004;3:965–972. doi: 10.1038/nrd1552. [DOI] [PubMed] [Google Scholar]
- 5.Tan DS. Nat. Chem. Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 6.Wilson DS, Szostak JW. Annu. Rev. Biochem. 1999;68:611–648. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 7.Agresti JJ, Antipov E, Abate AR, Ahn K, Rowat AC, Baret JC, Marquez M, Klibanov AM, Griffiths AD, Weitz DA. Proc Natl Acad Sci U S A. 2010;107:4004–4009. doi: 10.1073/pnas.0910781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Affleck RL. Curr Opin Chem Biol. 2001;5:257–263. doi: 10.1016/s1367-5931(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 9.Barnes C, Balasubramanian S. Curr Opin Chem Biol. 2000;4:346–350. doi: 10.1016/s1367-5931(00)00098-3. [DOI] [PubMed] [Google Scholar]
- 10.Czarnik AW. Curr. Opin. Chem. Biol. 1997;1:60–66. doi: 10.1016/s1367-5931(97)80109-3. [DOI] [PubMed] [Google Scholar]
- 11.Ede NJ, Wu Z. Curr Opin Chem Biol. 2003;7:374–379. doi: 10.1016/s1367-5931(03)00061-9. [DOI] [PubMed] [Google Scholar]
- 12.Brenner S, Lerner RA. Proc. Natl. Acad. Sci. USA. 1992;89:5381–5383. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gartner ZJ, Liu DR. J. Am. Chem. Soc. 2001;123:6961–6963. doi: 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen J, Brenner S, Janda KD. J. Am. Chem. Soc. 1993;115:9812–9813. [Google Scholar]
- 15.Needels MC, Jones DG, Tate EH, Heinkel GL, Kochersperger LM, Dower WJ, Barrett RW, Gallop MA. Proc. Natl. Acad. Sci. USA. 1993;90:10700–10704. doi: 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naylor R, Gilham PT. Biochemistry. 1966;5:2722–2728. doi: 10.1021/bi00872a032. [DOI] [PubMed] [Google Scholar]
- 17.Joyce GF. Cold Spring Harbor Symposium on Quantitative Biology. Plainview, NY: 1987. [Google Scholar]
- 18.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tse BN, Snyder TM, Shen YS, Liu DR. J. Am. Chem. Soc. 2008;130:15611–15626. doi: 10.1021/ja805649f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozenman MM, Kanan MW, Liu DR. J. Am. Chem. Soc. 2007;129:14933–14938. doi: 10.1021/ja074155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Kamlet AS, Steinman JB, Liu DR. Nature Chemistry. 2011;3:146–153. doi: 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brudno Y, Birnbaum ME, Kleiner RE, Liu DR. Nat. Chem. Bio. 2010;6:148–155. doi: 10.1038/nchembio.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen MH, Blakskjaer P, Petersen LK, Hansen TH, Hojfeldt JW, Gothelf KV, Hansen NJV. J. Am. Chem. Soc. 2009;131:1322–1327. doi: 10.1021/ja808558a. [DOI] [PubMed] [Google Scholar]
- 25.Halpin DR, Harbury PB. PLOS Biol. 2004;2:1015–1021. doi: 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halpin DR, Harbury PB. PLOS Biol. 2004;2:1022–1030. doi: 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halpin DR, Lee JA, Wrenn SJ, Harbury PB. PLOS Biol. 2004;2:1031–1038. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melkko S, Scheuermann J, Dumelin CE, Neri D. Nat Biotechnol. 2004;22:568–574. doi: 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- 29.Clark MA, Acharya RA, Arico-Muendel CC, Belyanskaya SL, Benjamin DR, Carlson NR, Centrella PA, Chiu CH, Creaser SP, Cuozzo JW, Davie CP, Ding Y, Franklin GJ, Franzen KD, Gefter ML, Hale SP, Hansen NJV, Israel DI, Jiang J, Kavarana MJ, Kelley MS, Kollmann CS, Li F, Lind K, Mataruse S, Medeiros PF, Messer JA, Myers P, O'Keefe H, Oliff MC, Rise CE, Satz AL, Skinner SR, Svendsen JL, Tang L, Vloten Kv, Wagner RW, Yao G, Zhao B, Morgan BA. Nat. Chem. Biol. 2009;5:647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- 30.Kinoshita Y, Nishigaki K. Nucleic Acids Symp. Series. 1995;34:201–202. [PubMed] [Google Scholar]
- 31.Lam KS, Lebl M, Krchnak V. Chem. Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 32.Buller F, Mannocci L, Zhang Y, Dumelin CE, Scheuermann J, Neri D. Bioorg. Med. Chem. Lett. 2008;18:5926–5931. doi: 10.1016/j.bmcl.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 33.Mannocci L, Zhang Y, Dumelin CE, Scheuermann J, Neri D. Proc. Natl. Acad. Sci. USA. 2008;105:17670–17675. doi: 10.1073/pnas.0805130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buller F, Mannocci L, Scheuermann J, Neri D. Bioconjug Chem. 2010;21:1571–1580. doi: 10.1021/bc1001483. [DOI] [PubMed] [Google Scholar]
- 35.Clark MA. Curr Opin Chem Biol. 2010;14:396–403. doi: 10.1016/j.cbpa.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Liu DR. Angew. Chem. Int. Ed. 2004;43:4848–4870. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- 37.Scheuermann J, Neri D. Chembiochem. 2010;11:931–937. doi: 10.1002/cbic.201000066. [DOI] [PubMed] [Google Scholar]
- 38.Broach JR, Thorner J. Nature. 1996;384:14–16. [PubMed] [Google Scholar]
- 39.Smith GP, Petrenko VA. Chem. Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 40.Doyon JB, Snyder TM, Liu DR. J. Am. Chem. Soc. 2003;125:12372–12373. doi: 10.1021/ja036065u. [DOI] [PubMed] [Google Scholar]
- 41.Krylov SN. In: Aptamers in Bioanalysis. Editon edn. Mascini M, editor. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2008. [Google Scholar]
- 42.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Z, Cuozzo J. J Biomol Screen. 2009;14:1157–1164. doi: 10.1177/1087057109350114. [DOI] [PubMed] [Google Scholar]
- 44.Vijayendran RA, Leckband DE. Anal Chem. 2001;73:471–480. doi: 10.1021/ac000523p. [DOI] [PubMed] [Google Scholar]
- 45.Gorin DJ, Kamlet AS, Liu DR. J Am Chem Soc. 2009;131:9189–9191. doi: 10.1021/ja903084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGregor LM, Gorin DJ, Dumelin CE, Liu DR. J. Am. Chem. Soc. 2010;132:15522–15524. doi: 10.1021/ja107677q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pettersson E, Lundeberg J, Ahmadian A. Genomics. 2009;93:105–111. doi: 10.1016/j.ygeno.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Mardis ER. Annu. Rev. Genomics Hum. Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 49.Wrenn SJ, Weisinger RM, Halpin DR, Harbury PB. J. Am. Chem. Soc. 2007;129:13137–13143. doi: 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jano SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleiner RE, Dumelin CE, Tiu GC, Sakurai K, Liu DR. J. Am. Chem. Soc. 2010;132:11779–11791. doi: 10.1021/ja104903x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buller F, Steiner M, Scheuermann J, Mannocci L, Nissen I, Kohler M, Beisel C, Neri D. Bioorg. Med. Chem. Lett. 2010;20:4188–4192. doi: 10.1016/j.bmcl.2010.05.053. [DOI] [PubMed] [Google Scholar]
- 54.Buller F, Steiner M, Frey K, Mircsof D, Scheuermann J, Kalisch M, Buhlmann P, Supuran CT, Neri D. ACS Chem Biol. 2011 doi: 10.1021/cb1003477. [DOI] [PubMed] [Google Scholar]
- 55.Dumelin CE, Trussel S, Buller F, Trachsel E, Bootz F, Zhang Y, Mannocci L, Beck SC, Brumea-Mirancea M, Seeliger MW, Baltes C, Muggler T, Kranz F, Rudin M, Melkko S, Scheurermann J, Neri D. Angew. Chem. Int. Ed. 2008;47:3196–3201. doi: 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]
- 56.Ahlskog JK, Dumelin CE, Trussel S, Marlind J, Neri D. Bioorg. Med. Chem. Lett. 2009;19:4851–4856. doi: 10.1016/j.bmcl.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 57.Trussel S, Dumelin CE, Frey K, Villa A, Buller F, Neri D. Bioconjug. Chem. 2009;20:2286–2292. doi: 10.1021/bc9002772. [DOI] [PubMed] [Google Scholar]
- 58.Scheuermann J, Dumelin CE, Melkko S, Zhang Y, Mannocci L, Jaggi M, Sobek J, Neri D. Bioconjug. Chem. 2008;19:778–785. doi: 10.1021/bc7004347. [DOI] [PubMed] [Google Scholar]
- 59.Melkko S, Mannocci L, Dumelin CE, Villa A, Sommavilla R, Zhang Y, Grutter MG, Keller N, Jermutus L, Jackson RH, Scheuermann J, Neri D. ChemMedChem. 2010;5:584–590. doi: 10.1002/cmdc.200900520. [DOI] [PubMed] [Google Scholar]
- 60.Buller F, Zhang Y, Scheuermann J, Schafer J, Buhlmann P, Neri D. Chem. Biol. 2009;16:1075–1086. doi: 10.1016/j.chembiol.2009.09.011. [DOI] [PubMed] [Google Scholar]