

Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is the most abundant kinase within excitatory synapses in the mammalian brain. It interacts with and phosphorylates a large number of synaptic proteins, including major ionotropic glutamate receptors (iGluRs) and group I metabotropic glutamate receptors (mGluRs), to constitutively and/or activity-dependently regulate trafficking, subsynaptic localization, and function of the receptors. Among iGluRs, the N-methyl-D-aspartate receptor (NMDAR) is a direct target of CaMKII. By directly binding to an intracellular C-terminal (CT) region of NMDAR GluN2B subunits, CaMKII phosphorylates a serine residue (S1303) in the GluN2B CT. CaMKII also phosphorylates a serine site (S831) in the CT of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. This phosphorylation enhances channel conductance and is critical for synaptic plasticity. In addition to iGluRs, CaMKII binds to the proximal CT region of mGluR1a, which enables the kinase to phosphorylate threonine 871. Agonist stimulation of mGluR1a triggers a CaMKII-mediated negative feedback to facilitate endocytosis and desensitization of the receptor. CaMKII also binds to the mGluR5 CT. This binding seems to anchor and accumulate inactive CaMKII at synaptic sites. Active CaMKII dissociates from mGluR5 and may then bind to adjacent GluN2B to mediate the mGluR5-NMDAR coupling. Together, glutamate receptors serve as direct substrates of CaMKII. By phosphorylating these receptors, CaMKII plays a central role in controlling the number and activity of the modified receptors and determining the strength of excitatory synaptic transmission.
Keywords: NMDA, GluN2B, AMPA, GluA1, mGluR, PKC, calmodulin, synaptic plasticity
1 Introduction
L-glutamate is a major neurotransmitter in the mammalian brain. This transmitter interacts with two classes of receptors to regulate synaptic transmission: ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs). iGluRs are ligand-gated ion channels and are further classified into α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors (AMPAR), N-methyl-D-aspartate receptors (NMDAR), and kainate receptors [1]. All iGluRs form heteromers or homomers assembled from multiple subunits in order to gain full function. mGluRs, on the other hand, are a family of G protein-coupled receptors (GPCRs). Eight subtypes of mGluRs so far cloned are grouped into three functional groups (group I, II, and III). Among these groups, group I mGluRs (mGluR1 and mGluR5 subtypes) draw the most attention and have been extensively investigated in regulating various cellular and synaptic activities [2]. Stimulation of Gαq-coupled group I mGluRs activates phospholipase Cβ1 (PLCβ1) to hydrolyze phosphoinositide (PI) into inositol-1,4,5-triphosphate (IP3) and diacylglycerol. The former (IP3) releases Ca2+ from internal stores and the latter (diacylglycerol) activates protein kinase C (PKC). Released Ca2+ and activated PKC in turn modulate multiple downstream signaling targets.
iGluRs and group I mGluRs are mostly postsynaptic and are enriched in the postsynaptic density (PSD) microdomain. As surface membrane-bound receptors, their intracellular domains interact with a number of submembranous proteins, including various scaffolding proteins, signaling proteins, and protein kinases and phosphatases [1,2]. A notable protein kinase is Ca2+/calmodulin-dependent protein kinase II (CaMKII), which is a serine/threonine kinase abundant in the PSD [3]. CaMKII has many different isoforms, mainly including α and β isoforms in the central nervous system [4–6]. All isoforms structurally have three domains: an N-terminal catalytic domain, a central regulatory domain and a C-terminal association domain. The regulatory domain (residues 281–310) contains an autoinhibitory sequence, a calmodulin (CaM)-binding site and several autophosphorylation sites, i.e. threonine 286 (T286) and threonine 305/306. The catalytic domain transfers phosphate from ATP to serine or threonine residues in substrates. The regulatory domain governs activation of the kinase. At the inactive state, this domain binds to the catalytic domain and blocks the accessibility of the catalytic domain to substrates. When activated by Ca2+ and CaM, the regulatory domain dissociates from the catalytic domain and opens the catalytic domain to substrates. At the same time, active kinase undergoes autophosphorylation in the regulatory domain at an autophosphorylation site, i.e., threonine 286 (T286 in α isoform), which transforms the kinase to a Ca2+-independent (autonomous) state, a state with prolonged kinase activity even after the initial Ca2+ signals subside [4–6]. As a synapse-enriched kinase, CaMKII directly binds to NMDARs and mGluR1/5 (see below). Through closely interacting with glutamate receptors, CaMKII phosphorylates the receptors at specific serine or threonine sites [7,8]. The site-specific phosphorylation is either constitutively active or activity-dependently regulated by changing synaptic or intracellular inputs. Through the phosphorylation mechanism, CaMKII regulates expression of iGluRs and mGluRs at postsynaptic sites and controls glutamatergic synaptic transmission and synaptic plasticity. This review outlines the interaction between CaMKII and glutamate receptors and summarizes the role of CaMKII in phosphorylating and regulating iGluRs and group I mGluRs with a focus on recent progress.
2 Phosphorylation of NMDARs by CaMKII
NMDARs are assembled into a tetrameric structure by composing two obligatory GluN1 (formerly known as NR1) with two modulatory GluN2 subunits, mainly GluN2A (NR2A) or GluN2B (NR2B) [1]. All subunits share the similar membrane conformation: three membrane-spanning domains (M1, M3, and M4), a hydrophobic hairpin domain (M2), an extracellular N-terminus, and an intracellular C-terminus (CT). GluN2A and GluN2B CT regions are noticeably large, which together with the relatively shorter GluN1 CT constitute key zones harboring defined protein-protein interactions and phosphorylation [7,9].
Direct binding of CaMKII to GluN2A/B is noteworthy. In hippocampal and cortical neurons, CaMKII colocalized with NMDARs in confined subcellular compartments and CaMKII-NMDAR complexes were readily coimmunoprecipitated, while CaMKII did not coprecipitated with AMPARs [10–12]. In binding assays with purified recombinant proteins and/or peptides, CaMKII was found to directly bind to the GluN2A/B CT with high affinity [11–14]. The CaMKII binding to GluN2B CT has been most extensively characterized. It is now known that CaMKII binds to the membrane proximal residues 839–1120 and residues 1290–1310 in the GluN2B CT. Transient activity elicits Ca2+/CaM-dependent binding of GluN2B to a substrate-binding site (S-site) within the catalytic domain of CaMKII, whereas prolonged activity leads to persistent GluN2B binding to a T286-binding site (T-site) adjacent to the S-site [13–15]. The GluN2B binding to the T-site blocks the binding of the T-site to the regulatory region of CaMKII, thereby prolonging CaMKII activity after Ca2+ signals subside. Thus, activation of CaMKII by stimulating Ca2+-permeable NMDARs in forebrain neurons increased the CaMKII association with NMDARs [12,15]. This suggests a dynamic mechanism for the activity-dependent recruitment of CaMKII to the PSD to regulate a discrete set of local substrates.
Binding of CaMKII to GluN2B suggests potential phosphorylation of the receptor by the kinase. Indeed, the binding between the catalytic domain of CaMKII and GluN2B 1290–1309 represents an enzyme-substrate mode of interactions and results in phosphorylation of S1303 within the binding region [16]. The Km of S1303 phosphorylation was in a nanomolar range, much lower than that of other known substrates of CaMKII, indicating that the receptor is a high affinity substrate. As expected, phospho-S1303 (pS1303) is also subject to the dephosphorylation modulation by phosphatases. Both protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) dephosphorylated pS1303 in the GluN2B CT [17]. In the PSD, PP1 is a major phosphatase catalyzing S1303 dephosphorylation [18].
Protein phosphorylation is a key mechanism regulating localization and function of modified proteins. Thus, CaMKII-mediated S1303 phosphorylation is assumed to have a significant impact on receptor activity, although direct evidence seems lacking. CaMKII has been shown to enhance desensitization of GluN2B-but not GluN2A-containg NMDARs in HEK293 cells [19]. However, the direct role of the CaMKII-mediated S1303 phosphorylation in this event is unclear. Interestingly, S1303 is also a substrate site for other kinases. Both PKC and death-associated protein kinase 1 (DAPK1) phosphorylated this site [20,21]. Through phosphorylating S1303, PKC potentiated GluN1/GluN2B-mediated currents [20] and DAPK1 enhanced GluN1/GluN2B channel conductance [21]. Thus, S1303 may act as a point of convergence for these kinases to modulate GluN2B-containing NMDARs. An additional layer of crosstalk involving CaMKII is that CaMKII coupled GluN2B and casein kinase 2 (CK2) to form a tri-molecular complex, through which CaMKII increased CK2-mediated phosphorylation of GluN2B at S1480 [22].
Phosphorylation status at GluN2B S1303 also regulates its interactions with CaMKII. An early study showed that phosphorylation of S1303 by CaMKII inhibited CaMKII-GluN2B binding and promoted slow dissociation of preformed CaMKII/GluN2B complexes [13]. A further study found that phosphorylation of S1303 allowed the receptor to recognize and accept the specific form of CaMKII: S1303 phosphorylation permits the binding of Ca2+/CaM-activated CaMKIIα but not Ca2+/CaM-independent T286-autophosphorylated CaMKIIα [17].
3 Phosphorylation of AMPARs by CaMKII
Like NMDARs, AMPARs become functional when four subunits (GluA1–4, previously named GluR1–4) form a homomeric or heteromeric dimer-of-dimers structure [1]. Moreover, membrane-bound GluA1–4 subunits have the similar membrane-spanning domains as described above for NMDARs, although their CT regions are much shorter as compared to GluN2A/B. Despite the smaller size, CT regions of GluA1–4 are sufficient to harbor robust phosphorylation by various kinases, including CaMKII [7,8]. CaMKII is positioned close to AMPARs by interacting with adjacent GluN2B [12]. The specific site in GluA1 sensitive to CaMKII is S831, which is also phosphorylated by PKC [23–25].
Reversible phosphorylation of GluA1 S831 by CaMKII generally potentiates the strength of AMPAR channels. In an early study, an S831D mutation (phosphomimetic mutation) increased single channel conductance of recombinant GluA1 AMPARs in HEK293 cells [26]. A recent study showed that CaMKII phosphorylation of S831 enhanced the coupling efficiency of glutamate binding to channel gating [27], suggesting a mechanism to underlie the CaMKII potentiation of AMPAR conductance. Of note, the CaMKII potentiation via S831 was restricted to homomeric GluA1 and was lacking in a GluA1/GluA2 heteromeric complex [28]. Thus, the role of the CaMKII-S831 pathway in augmenting channel conductance may be limited in brain regions where GluA1/GluA2 heteromers are predominant. The S831-directed increase in conductance was also observed following activation of PKC, and an S831A mutation (phosphodeficient mutation) blocked the PKC effect [29]. Apparently, either CaMKII or PKC can phosphorylate S831 to potentiate AMPARs. A complex interplay among CaMKII, PKC and their associated scaffolding proteins may exist to determine the sensitivity of S831 to either kinase under different conditions [29]. In addition, the S831 regulation of the channel property relies on coexpression of GluA1 with transmembrane AMPA receptor regulatory proteins (TARPs) [27].
The role of S831 phosphorylation in common forms of synaptic plasticity, long-term potentiation (LTP) and long-term depression (LTD), has been investigated in details [8,30]. Increased activity of CaMKII drove GluA1-containing AMPARs into synapses, which may form a metaplastic basis for LTP [31,32]. However, the CaMKII effect was not diminished by mutating S831 to S831A. Thus, even though active CaMKII increased synaptic receptor delivery, the importance of S831 phosphorylation to this particular process seems to be limited. Nevertheless, S831 phosphorylation may contribute to LTP through a different mechanism, e.g., augmenting channel conductance. While direct evidence is lacking, this role of S831 is implied by the findings that 1) S831 phosphorylation was consistently increased during LTP [25,33,34], 2) S831/845A mutant mice, i.e., double phosphomutants lacking both S831 and S845 (PKA-sensitive) phosphorylation sites, showed a faster decaying LTP and a deficit in LTD [35], and 3) S831/845D double phosphomimetic gene knockin sufficiently lowered the threshold for LTP induction, increasing the probability of synaptic plasticity [36]. Surprisingly, mice with single S831 or S845 mutation (i.e., S831A or S845A mutants) displayed normal LTP [37]. This suggests that either S831 or S845 alone is not necessary for LTP. In other words, either site alone can support LTP. The two sites may substitute each other for supporting LTP when either site is lacking. Only when both sites are mutated, is LTP blunted. As expected, the CaMKII binding to GluN2B is critical for anchoring the kinase to phosphorylate GluA1 and contributes to LTP since mutant mice with the impaired CaMKII binding to GluN2B exhibited overall decreases in GluA1 phosphorylation at the CaMKII site, LTP, and learning behavior [38].
4 CaMKII and group I mGluRs
Group I mGluRs are distributed in broad brain regions [39,40] and are mostly postsynaptic [41,42]. As such, these receptors are actively involved in the regulation of various normal cellular and synaptic activities and are linked to the pathogenesis of different neurological and neuropsychiatric disorders [2]. Like typical GPCRs, group I mGluRs are anchored in plasma membranes by seven transmembrane helices, which give rise to three intracellular loops and an intracellular CT. The CT regions of the long-form splice variants (mGluR1a, mGluR5a, and mGluR5b) are particularly large. This renders these variants accessibility by various submembranous binding partners. A number of mGluR1/5-interacting proteins have been identified, which actively modulate subcellular and subsynaptic distribution and function of the receptors [43,44].
Among interacting partners are a group of protein kinases, including CaMKII. Multiple CaMKII binding and phosphorylation consensus motifs, RXXS/T [45], exist in the mGluR1/5 CT [46], suggesting potential of CaMKII-mGluR1/5 interactions. In fact, a recent study demonstrated a direct interaction between CaMKII and mGluR1a [47]. It was found that the purified catalytic domain of CaMKIIα bound to a membrane-proximal region of recombinant mGluR1a CT. The interaction also occurred between native CaMKIIα and mGluR1a in the striatum. Ca2+ signals profoundly enhanced the interaction in striatal neurons. Since activation of mGluR1/5 with an agonist (DHPG) induced a transient interaction between the two proteins, CaMKII is believed to be activity-dependently activated and recruited to the receptor to form a feedback loop. The direct interaction resulted in phosphorylation of mGluR1a at a specific threonine site (T871). This site is consistent with the consensus phosphorylation motif and lies within the center of the CaMKIIα binding motif in mGluR1a-CT.
Like most GPCRs, mGluR1a undergoes feedback endocytosis and desensitization after prolonged and repeated agonist stimulation [48–51]. This process involves either second messenger-dependent protein kinases or GPCR kinases (GRKs) [52]. Through a phosphorylation-sensitive mechanism, these kinases trigger an agonist-induced internalization of receptors, leading to desensitization. The CaMKII inhibitor KN93 blocked the internalization and homologous desensitization of mGluR1a induced by glutamate in HEK293 cells [53]. KN93 also reduced the internalization and heterologous desensitization of mGluR1a following muscarinic M1 receptor stimulation [53,54]. Thus, CaMKII in addition to PKC [52] contributes to mGluR1a desensitization, although underlying mechanisms are poorly understood. In a recent attempt to unravel the mechanism, KN93 was found to block the binding of CaMKIIα to mGluR1a CT in striatal neurons, which presumably reduced the CaMKIIα-mediated phosphorylation of mGluR1a, and thereby resensitized the responsivity of the receptor to subsequent agonist stimulation [47]. Disrupting the CaMKIIα-mGluR1a association with an interaction-dead peptide produced the same effect [47]. Thus, CaMKII serves as an important element in a feedback loop promoting mGluR1 desensitization. The observation that DHPG readily activated CaMKII in striatal and hippocampal neurons [55,56] supports the CaMKII-regulated feedback model. The fact that the CaMKII binding and phosphorylation site is immediately adjacent to the mGluR1a region harboring G proteins is noteworthy [52].
CaMKII also binds to the mGluR5a CT [57]. The CT region which CaMKIIα binds to is also open to CaM [58]. This forms a basis for CaM and CaMKIIα to compete with each other for binding to mGluR5. However, CaMKII binds to a different site in mGluR5 as compared to the site in mGluR1a [57] and CaM did not bind to mGluR1a [58]. More notably, the CaMKII binding to mGluR5a was differentially regulated by Ca2+. While Ca2+ enhanced the binding of active CaMKII to mGluR1a, Ca2+ reduced the association of active CaMKII with mGluR5. A further study in striatal neurons [57] seems to suggest an interesting model which may process an mGluR5 subtype-specific role in potentiating NMDARs [59]. A stepwise scenario is that inactive CaMKII binds to mGluR5 at a high level. As such, the kinase is anchored and accumulated at the perisynaptic site. When neuronal activity is enhanced together with a cytoplasmic Ca2+ rise, CaMKII becomes activated and thereby loses its affinity for mGluR5, leading to dissociations of the kinase from the receptor. After CaMKIIα dissociations, Ca2+-activated CaM occupies the same binding site and prevents the further accessibility of CaMKIIα to the site. Meanwhile, the dissociated CaMKIIα provides a source of the kinase to interact with adjacent substrates, such as GluN2B, to phosphorylate and regulate NMDARs.
5 Conclusions
The phosphorylation-dependent posttranslational modification of glutamate receptors has been a central topic for more than a decade. Among multiple protein kinases that readily phosphorylate iGluRs and mGluRs, CaMKII plays a significant role. This synapse-enriched serine and threonine kinase directly binds to the NMDAR subunit GluN2B and group I mGluR CT regions. As a Ca2+-sensitive kinase, CaMKII binds to these receptors in a Ca2+-dependent fashion. Interestingly, while the CaMKII binding to GluN2B and mGluR1a is upregulated by Ca2+, the binding to mGuR5 is down-regulated. Accordingly, CaMKII Ca2+- and activity-dependently phosphorylates GluN2B (S1303), mGluR1a (T871) and GluA1 (S831), and mGluR5 mainly serves to anchor and accumulate the kinase at synaptic sites (Fig. 1). CaMKII phosphorylation at a specific site (either constitutive or activity-dependent) distinctively regulates the biochemical, biophysical and functional properties of modified receptors. For instance, CaMKII-mediated GluA1 S831 phosphorylation enhances GluA1/AMPAR channel conductance and contributes to LTP. T871 phosphorylation of mGluR1a by CaMKII forms a negative feedback loop controlling agonist-induced endocytosis and desensitization. The CaMKII-mGluR5 interaction may enable the kinase to play a bridge role in coupling mGluR5 to GluN2B/NMDARs. In sum, CaMKII, as the most abundant kinase within excitatory synapses, is intimately involved in the phosphorylation and regulation of both iGluRs and mGluRs.
Fig. 1.
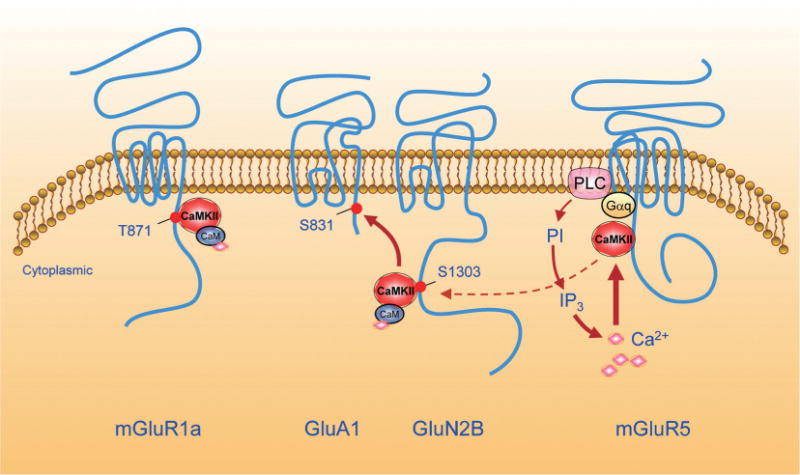
A schematic diagram illustrating CaMKII interactions with glutamate receptors at a postsynaptic site. Active CaMKII binds to the proximal region of the mGluR1a CT and phosphorylates T871. This subsequently promotes the agonist-induced endocytosis and desensitization of the receptor. Active CaMKII also binds to the GluN2B CT and phosphorylates S1303. Binding to the GluN2B CT places the kinase nearby GluA1 and enables the kinase to phosphorylate GluA1 at S831. Inactive CaMKII binds to the mGluR5 CT under basal conditions. mGluR5 activation activates the downstream PLC-IP3 pathway to release Ca2+. Released Ca2+ in turn activates CaMKII and dissociates the preformed kinase from mGluR5. Released CaMKII may then relocate to the adjacent GluN2B CT to phosphorylate and regulate NMDARs. See text for abbreviations.
While early studies establish the solid relationship between CaMKII and glutamate receptors, further studies are needed to answer some more insightful questions. First, biochemical mechanisms underlying the phosphorylation-mediated regulation are unclear. Further studies on protein chemistry are needed to elucidate stepwise protein reactions to a site-specific phosphorylation. Second, structural interplay between CaMKII and a given glutamate receptor may take place either constitutively or in an activity-dependent manner. Structure biology studies are thus useful to define a structural basis required for accommodating interactions between CaMKII and glutamate receptors and to reveal the role of phosphorylation at a specific site in the configuration of protein structures. Third, tremendous crosstalk among synaptic proteins including CaMKII and glutamate receptors occurs given the facts that 1) CaMKII phosphorylates not only NMDARs, AMPARs and group I mGluRs, bus also kainate receptors [60] and a large number of other synaptic targets [61,62], and 2) a same receptor is phosphorylated by different kinases and is also subject to dephosphorylation by various PPs. Additionally, glutamate receptors are also subject to other posttranslational modifications, such as palmitoylation, ubiquitination and sumoylation [7]. Thus, crosstalk in the local synaptic network at different levels will be an interesting topic in the future. Finally, phosphorylation of glutamate receptors is linked to a variety of neurological disorders [7]. Phospho- and site-specific antibodies and loss-of-function mutation mice (by replacing serine and threonine by alanine) are direct and useful tools to evaluate the importance of a specific phosphorylation site or a set of defined phosphorylation sites in the pathogenesis or progression of neurological illnesses.
Acknowledgments
This review was supported by NIH R01 DA010355 and R01 MH061469.
References
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niswend CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly PT, McGuinness TL, Greengard P. Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci U S A. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudmon A, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 5.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Griffith LC. Regulation of calcium/calmodulin-dependent protein kinase II activation by intramolecular and intermolecular interactions. J Neurosci. 2004;24:8394–8398. doi: 10.1523/JNEUROSCI.3604-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mao LM, Guo ML, Jin DZ, Fibuch EE, Choe ES, Wang JQ. Post-translational modification biology of glutamate receptors and drug addiction. Front Neuroanat. 2011;5:19. doi: 10.3389/fnana.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol. 2012;22:470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang JQ, Guo ML, Jin DZ, Xue B, Fibuch EE, Mao LM. Roles of subunit phosphorylation in regulating glutamate receptor function. Eur J Pharmacol. 2014;728:183–187. doi: 10.1016/j.ejphar.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 1998;71:1733–1741. doi: 10.1046/j.1471-4159.1998.71041733.x. [DOI] [PubMed] [Google Scholar]
- 11.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 12.Leonard AS, Lim IA, Hemsworth DE, Home MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 14.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 15.Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omkumar RV, Melinda O, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- 17.Raveendran R, Devi Suma Priya S, Mayadevi M, Steephan M, Santhoshkumar TR, Cheriyan J, Sanalkumar R, Pradeep KK, James J, Omkumar RV. Phosphorylation status of the NR2B subunit of NMDA receptor regulates its interaction with calcium/calmodulin-dependent protein kinase II. J Neurochem. 2009;110:92–105. doi: 10.1111/j.1471-4159.2009.06108.x. [DOI] [PubMed] [Google Scholar]
- 18.Prabhu Ramya R, Suma Priya S, Mayadevi M, Omkumar RV. Regulation of phosphorylation at Ser(1303) of GluN2B receptor in the postsynaptic density. Neurochem Int. 2012;61:981–985. doi: 10.1016/j.neuint.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 19.Sessoms-Sikes S, Honse Y, Lovinger DM, Colbran RJ. CaMKIIalpha enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol Cell Neurosci. 2005;29:139–147. doi: 10.1016/j.mcn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Liao GY, Wagner DA, Hsu MH, Leonard JP. Evidence for direct protein kinase-C mediated modulation of N-methyl-D-aspartate receptor current. Mol Pharmacol. 2001;59:960–964. doi: 10.1124/mol.59.5.960. [DOI] [PubMed] [Google Scholar]
- 21.Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, Roche KW. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 2013;3:607–614. doi: 10.1016/j.celrep.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 24.Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- 25.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 26.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir RL, Traynelis SF. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh MC, Derkach VA. Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat Neurosci. 2005;8:853–854. doi: 10.1038/nn1476. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins MA, Travnelis SF. PKC phosphorylates GluA1-Ser831 to enhance AMPA receptor conductance. Channels (Austin) 2012;6:60–64. doi: 10.4161/chan.18648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HK. Synaptic plasticity and phosphorylation. Pharmacol Ther. 2006;112:810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 32.Estaban JA, Shi H, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor sub-units controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 33.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 34.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 35.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 36.Makino Y, Johnson RC, Yu Y, Takamiya K, Huganir RL. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc Natl Acad Sci U S A. 2011;108:8450–8455. doi: 10.1073/pnas.1105261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Borse N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tallaksen-Greene SJ, Kaatz KW, Romano C, Albin RL. Localization of mGluR1a-like immunoreactivity and mGluR5a-like immunoreactivity in identified population of striatal neurons. Brain Res. 1998;780:210–217. doi: 10.1016/s0006-8993(97)01141-4. [DOI] [PubMed] [Google Scholar]
- 41.Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. 1997;13:219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 42.Takumi Y, Matsubara A, Rinvik E, Ottersen OP. The arrangement of glutamate receptors in excitatory synapses. Annu NY Acad Sci. 1999;868:474–482. doi: 10.1111/j.1749-6632.1999.tb11316.x. [DOI] [PubMed] [Google Scholar]
- 43.Enz R. Metabotropic glutamate receptors and interacting proteins: evolving drug targets. Curr Drug Targets. 2012;13:145–156. doi: 10.2174/138945012798868452. [DOI] [PubMed] [Google Scholar]
- 44.Fagni L. Diversity of metabotropic glutamate receptor-interacting proteins and pathophysiological functions. Adv Exp Med Biol. 2012;970:63–79. doi: 10.1007/978-3-7091-0932-8_3. [DOI] [PubMed] [Google Scholar]
- 45.White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem. 1998;273:3166–3179. doi: 10.1074/jbc.273.6.3166. [DOI] [PubMed] [Google Scholar]
- 46.Mao LM, Liu XY, Zhang GC, Chu XP, Fibuch EE, Wang LS, Liu Z, Wang JQ. Phosphorylation of group I metabotropic glutamate receptors (mGluR1/5) in vitro and in vivo. Neuropharmacology. 2008;55:403–408. doi: 10.1016/j.neuropharm.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin DZ, Guo ML, Xue B, Fibuch EE, Choe EE, Mao LM, Wang JQ. Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2013;33:3402–3412. doi: 10.1523/JNEUROSCI.3192-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schoepp DD, Johnson BG. Selective inhibition of excitatory amino acid-stimulated phosphoinositide hydrolysis in the rat hippocampus by activation of protein kinase C. Biochem Pharmacol. 1988;37:4299–4305. doi: 10.1016/0006-2952(88)90610-7. [DOI] [PubMed] [Google Scholar]
- 49.Thomsen C, Mulvihill ER, Haldeman B, Pickering DS, Hampson DR, Suzdak PD. A pharmacological characterization of the mGluR1α subtype of the metabotropic glutamate receptor expressed in a cloned baby hamster kidney cell line. Brain Res. 1993;619:22–28. doi: 10.1016/0006-8993(93)91592-g. [DOI] [PubMed] [Google Scholar]
- 50.Alaluf S, Mulvihill ER, McIlhinney RA. Rapid agonist mediated phosphorylation of the metabotropic glutamate receptor 1-alpha by protein kinase C in permanently transfected BHK cells. FEBS Lett. 1995;367:301–305. doi: 10.1016/0014-5793(95)00575-t. [DOI] [PubMed] [Google Scholar]
- 51.Sato M, Tabata T, Hashimoto K, Nakamura K, Nakao K, Katsuki M, Kitano J, Moriyoshi K, Kano M, Nakanishi S. Altered agonist sensitivity and desensitization of neuronal mGluR1 responses in knock-in mice by a single amino acid substitution at the PKC phosphorylation site. Eur J Neurosci. 2004;20:947–955. doi: 10.1111/j.1460-9568.2004.03552.x. [DOI] [PubMed] [Google Scholar]
- 52.Dhami GK, Ferguson SS. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther. 2006;111:260–271. doi: 10.1016/j.pharmthera.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 53.Mundell SJ, Pula G, McIlhinney RA, Roberts PJ, Kelly E. Desensitization and internalization of metabotropic glutamate receptor 1a following activation of heterologous Gq/11-coupled receptors. Biochemistry. 2004;43:7541–7551. doi: 10.1021/bi0359022. [DOI] [PubMed] [Google Scholar]
- 54.Mundell SJ, Matharu AL, Pula G, Holman D, Roberts PJ, Kelly E. Metabotropic glutamate receptor 1 internalization induced by muscarinic acetylcholine receptor activation: differential dependency of internalization of splice variants on nonvisual arrestins. Mol Pharmacol. 2002;61:1114–1123. doi: 10.1124/mol.61.5.1114. [DOI] [PubMed] [Google Scholar]
- 55.Choe ES, Wang JQ. Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci Lett. 2001;313:129–132. doi: 10.1016/s0304-3940(01)02258-3. [DOI] [PubMed] [Google Scholar]
- 56.Mockett BG, Guevremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J Neurosci. 2011;31:7380–7391. doi: 10.1523/JNEUROSCI.6656-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin DZ, Guo ML, Xue B, Mao LM, Wang JQ. A role of CaMKIIα in linking mGluR5 to NMDA receptor phosphorylation in neurons. J Neurochem. 2013;127:620–631. doi: 10.1111/jnc.12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi KY, Chung S, Roche KW. Differential binding of calmodulin to group I metabotropic glutamate receptors regulates receptor trafficking and signaling. J Neurosci. 2011;31:5921–5930. doi: 10.1523/JNEUROSCI.6253-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonsi P, Platania P, Martella G, Madeo G, Vita D, Tassone A, Bernardi G, Pisani A. Distinct roles of group I mGlu receptors in striatal function. Neuropharmacology. 2008;55:392–395. doi: 10.1016/j.neuropharm.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 60.Carta M, Opazo P, Veran J, Athane A, Choquet D, Coussen F, Mulle C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013;32:496–510. doi: 10.1038/emboj.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshimura Y, Shinkawa T, Taoka M, Kobayashi K, Isobe T, Yamauchi T. Identification of protein substrates of Ca2+/calmodulin-dependent protein kinase II in the postsynaptic density by protein sequencing and mass spectrometry. Biochem Biophys Res Commun. 2002;290:948–954. doi: 10.1006/bbrc.2001.6320. [DOI] [PubMed] [Google Scholar]
- 62.Colbran RJ. Targeting of calcium/calmodulin-dependent protein kinase II. Biochem J. 2004;378:1–16. doi: 10.1042/BJ20031547. [DOI] [PMC free article] [PubMed] [Google Scholar]