

Abstract
The anaplastic lymphoma kinase (ALK) receptor tyrosine kinase was initially discovered as a component of the fusion protein nucleophosmin (NPM)-ALK in anaplastic large-cell lymphoma (ALCL). Genomic alterations in ALK, including rearrangements, point mutations, and genomic amplification, have now been identified in several malignancies, including lymphoma, non-small cell lung cancer (NSCLC), neuroblastoma, inflammatory myofibroblastic tumor, and others. Importantly, ALK serves as a validated therapeutic target in these diseases. Several ALK tyrosine kinase inhibitors (TKIs), including crizotinib, ceritinib and alectinib, have been developed, and some of them have already been approved for clinical use. These ALK inhibitors have all shown remarkable clinical outcomes in ALK-rearranged NSCLC. Unfortunately, as is the case for other kinase inhibitors in clinical use, sensitive tumors inevitably relapse due to acquired resistance. This review focuses on the discovery, function, and therapeutic targeting of ALK, with a particular focus on ALK-rearranged NSCLC.
Introduction
Anaplastic lymphoma kinase (ALK) was initially discovered as the fusion gene nucleophosmin (NPM)-ALK in anaplastic large-cell non-Hodgkin's lymphoma (ALCL) (1). More than a decade later, the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion gene was first detected in lung cancer (2), and numerous other ALK fusions, differing by the N-terminal gene fusion partner, have since been identified in lung and other cancers. Additionally, ALK point mutations have been detected in neuroblastoma and thyroid cancers (3-6). Although many ALK mutations have been deposited in the COSMIC database following the development of next-generation sequencing based diagnostic techniques, the significance of these mutations have not yet been fully clarified. This review will focus on therapeutic strategies and acquired resistance in ALK-rearranged non-small cell lung cancer (NSCLC) and will briefly describe ALK alterations in other cancers.
Background: Function of ALK
ALK encodes a single transmembrane receptor tyrosine kinase that belongs to the insulin receptor superfamily. Although recent studies have identified pleiotrophin, midkine, and heparin as putative ALK ligands (7-9), a detailed understanding of ALK receptor activation and function remains to be determined. In mice, protein expression of ALK is observed in the thalamus, hypothalamus, mid-brain, and dorsal root ganglia 11 days post-coitum, and ALK expression is scarcely detectable in the adult mouse. ALK knockout mice grow without obvious abnormalities and achieve normal life spans. However, comparisons between wild-type (WT) and ALK knockout mice implicate ALK in the function of the frontal cortex and hippocampus in the adult brain. Behavioral response to ethanol is reduced in ALK knockout mice (10). Thus, normal ALK function in adult humans remains unclear.
ALK Alterations in Cancer
ALK point mutations in neuroblastoma (NB)
NB is the most common pediatric extra-cranial solid tumor and can occur in both infants and children. Point mutations in the ALK kinase domain have been detected in a subset of patients with both sporadic and hereditary NB (Fig. 1A). Mutated ALK serves as a “driver oncogene,” in these cases. A recent study of >1,500 NB patients identified ALK tyrosine kinase domain mutations in 8% of patient samples and reported transformation potential, affinity to ATP, and sensitivity to the ALK inhibitor crizotinib for each ALK mutant (11). Importantly, ALK has been validated as a therapeutic target in NB, however, only a minority of patients with neuroblastoma harboring ALK mutations actually respond to crizotonib (12). Since our understanding of the pathogenesis of ALK in NB is growing but still limited, further studies are needed to develop the most effective therapies for these patients.
Figure 1.

Genetic alterations of ALK. A, ALK point mutations identified in neuroblastoma. B, Schematic representations of ALK fusion proteins (top) and a list of various ALK fusion proteins described to date (bottom). Note that this list is not comprehensive.
ALK fusions
NPM-ALK, as a result of the t(2,5) chromosome rearrangement, was first identified in ALCL (1). Approximately 55% of ALCL patients harbor ALK gene rearrangements (13). To date, various other ALK fusions, differing by the N-terminal gene fusion partner, have been discovered in ALCL, including TPM3-ALK, TPM4-ALK, TFG-ALK, and others (14-16). In 2007, the EML4-ALK fusion was discovered in NSCLC (2), and analogous to ALCL, several other ALK fusions were reported thereafter (Fig. 1B) (2, 17-23). Common characteristics of ALK fusions include: 1) conserved breakpoints in the ALK gene which includes the entire ALK tyrosine kinase domain within each of the known ALK fusions; 2) a promoter derived from the N-terminal fusion partner which leads to constitutive expression of the ALK fusion protein; and 3) an oligomerization domain in the N-terminal fusion partner protein. In the case of full length ALK, ligand-mediated dimerization followed by trans-phosphorylation of the tyrosine kinase domain is believed to induce ALK activation (8). In the setting of ALK rearrangement, oligomerization of the ALK fusion protein mediated by the fusion partner's oligomerization domain induces constitutive activation of the kinase.
ALCL, the first tumor type in which ALK fusions were detected, infrequently affects pediatric patients. Approximately 50% of ALCLs harbor ALK fusions, with NPM-ALK being the most common fusion detected (1, 24). ALK-rearranged ALCL is highly responsive to combination chemotherapy regimens (e.g., CHOP); hence, chemotherapy remains the standard of care. However, once the tumor becomes chemotherapy-resistant, the ALK inhibitor crizotinib can result in dramatic and durable responses (12, 25, 26).
Approximately 3%–7% of NSCLC cases harbor ALK rearrangements. Patients with ALK-rearranged NSCLC tend to be young, never or former light smokers with adenocarcinoma histology. The most common fusion variant detected in these patients is EML4-ALK, variant 1. The EML4 and ALK genes are both located on chromosome 2p, and the EML4-ALK fusion results from a chromosomal inversion at this genomic locus. Several different EML4-ALK fusions have been described, varying by the fusion breakpoint in the EML4 gene. The breakpoints in EML4 can occur at exons 2, 6, 13, 14, 15, 18 or 20, while the breakpoint in ALK is most commonly within exon 20. As described above, EML4 contains an oligomerization domain (in this case, a coiled–coil domain), which mediates oligomerization and constitutive activation of the ALK fusion protein. The oncogenic potential of EML4-ALK has been confirmed by tumor formation in NIH3T3 fibroblasts (2) and lung cancer development in an EML4-ALK transgenic mouse model (27). Kinase activity has been shown to be necessary for EML4-ALK tumor formation, as a mutant that abrogates ALK kinase activity (ALK K1150M) is no longer oncogenic.
Recently, a lung cancer mouse model was established by generating the EML4-ALK fusion gene using adenoviral Crispr-Cas9-mediated chromosomal inversion between the introns of the EML4 and ALK genes. This strategy was also utilized to generate other known oncogenic fusion kinases, including RET and ROS1 fusions in NSCLC (28).
Diagnostic Methods for Identifying ALK Fusions in Tumor Biopsy Samples
There are numerous methods whereby ALK can be detected in tumor biopsy samples. The method used depends, in part, on the tumor type. For example, ALK immunohistochemistry (IHC) is the standard method used to diagnose ALK-rearranged ALCL and IMT. However, the antibody used to determine ALK status in these tumor types did not appear to work well in ALK-rearranged NSCLC. ALK IHC can be used to diagnose ALK rearranged NSCLC, but a high affinity antibody is needed to detect expression of the ALK fusion (21). Fluorescence in situ hybridization (FISH) using ALK break-apart probes has been the standard way to detect ALK in NSCLC. In addition, there are other diagnostic methods as well, including RT-PCR and next-generation sequencing (29, 30). For a comprehensive overview of the pros and cons of each diagnostic modality, we would refer the reader to a recently published review (31).
ALK Inhibitors
Crizotinib
Crizotinib is an orally available ALK/MET/ROS1 tyrosine kinase inhibitor (TKI). Crizotinib was initially developed as a MET inhibitor, but during the dose escalation portion of the phase 1 trial (PROFILE 1001), ALK rearrangements were discovered in NSCLC. Based on this discovery, screening for ALK rearrangement was undertaken, and patients with ALK-rearranged NSCLC were quickly enrolled in this study. In the latest update of the phase 1 trial, the overall response rate (ORR) was 60.8% and progression-free survival (PFS) period was 9.7 months. Commonly observed AEs associated with crizotinib include mild visual and gastrointestinal (GI) tract disturbances and liver enzyme abnormalities. Based on high ORRs observed in both the phase 1 and phase 2 (PROFILE 1005) studies, crizotinib was granted accelerated approval by the US Food and Drug Administration (FDA) in 2011 for the treatment of advanced, ALK-rearranged NSCLC. Crizotinib was also approved in various countries, including Japan, based on these single-arm studies.
Two randomized phase 3 studies of crizotinib have now completed. In the second-line PROFILE 1007 study, crizotinib was compared with single-agent chemotherapy (pemetrexed or docetaxel) in patients with ALK-rearranged NSCLC who had disease progression after first line platinum-based chemotherapy. Crizotinib showed significantly longer PFS (7.7 vs. 3.0 months) and higher ORR (65% vs. 20%) compared with chemotherapy. There was no difference in OS between the groups (20.3 vs. 22.8 months, respectively), likely due to significant crossover of patients from the chemotherapy to the crizotinib group at the time of progression. In the recently published first-line study (PROFILE 1014), 343 patients with ALK-rearranged NSCLC who had not received prior systemic treatment were randomly assigned in a 1:1 ratio, to receive crizotinib or combination chemotherapy (pemetrexed plus either cisplatin or carboplatin). Similar to PROFILE 1007, PROFILE 1014 showed that PFS was significantly longer with crizotinib compared to chemotherapy (10.9 vs. 7.0 months, respectively) and that ORR was significantly higher (74% vs 45%, respectively) (32). The improvement in PFS seen with first-line crizotinib as compared to platinum-based chemotherapy in ALK-rearranged NSCLC is similar to that seen in multiple trials of first line EGFR inhibitors in EGFR-mutant NSCLC. In both randomized studies, patient reported outcomes also favored crizotinib (32, 33).
ALK fusions have been found in various cancers in addition to ALCL and NSCLC. Approximately 50% of inflammatory myofibroblastic tumors (IMTs), which are more common in children, are positive for ALK gene rearrangements (34), with some cases showing marked sensitivity to crizotinib (35). Additionally, in NBs, crizotinib showed some activity against the ALK mutation R1275Q, but no activity against the F1174L mutation (10). The latter observation is consistent with the finding that an acquired F1174L mutation can mediate crizotinib resistance in ALK-rearranged IMTs (36). Several ongoing studies are aimed at determining the efficacy of crizotinib in ALK-rearranged malignancies other than ALCL and NSCLC. In addition, one arm of the NCI-MATCH (Molecular Analysis for Therapy Choice Program) trial, will test the efficacy of crizotinib in ALK-rearranged cancers other than NSCLC.
Resistance to crizotinib
Although most patients with ALK-rearranged NSCLC respond to crizotinib, tumors inevitably relapse, often after only 1–2 years of treatment. This type of resistance is referred to as acquired resistance. The central nervous system (CNS) is a frequent site of relapse on crizotinib. However, failure in the CNS likely represents a pharmacokinetic issue rather than biologic resistance, since penetration of crizotinib into the CNS is limited. In one patient, the cerebrospinal fluid (CSF) concentration of crizotinib was 0.26% of that in plasma (37). Despite its limited CNS penetration, crizotinib does have some intracranial activity, as shown in a recent retrospective analysis of patients who enrolled into PROFILE 1005 or 1007 with untreated brain metastases (38). The intracranial response rate among patients with measurable, untreated brain lesions was 18% and intracranial disease control rate was 56% at 12 weeks. Nevertheless, the brain was the most common site of relapse in patients with and without brain metastases at baseline. The limited CNS penetration of crizotinib may be due in part to ABCB1-mediated export of the drug (38-40). Consistent with this notion, an ABCB1 (abcb1a and abcb1b)-knockout mouse model demonstrated higher crizotinib concentrations in the CSF than WT mice (41).
A variety of different mechanisms mediating acquired resistance to crizotinib have been described. Mutations within the ALK kinase domain have been identified in approximately 1/3 of crizotinib-resistant tumor specimens (42). As shown in Fig. 2, the most frequently observed mutations are L1196M and G1269A. The L1196M mutation is the gatekeeper mutation in the ALK kinase domain, analogous to the T790M epidermal growth factor receptor (EGFR)-TKI-resistant mutation. Many small-molecule kinase inhibitors have exploited a conserved hydrophobic residue within the ATP binding site for binding specificity. Since the gatekeeper controls access of the inhibitors to a hydrophobic pocket deep in the active site that is not contacted by ATP, the substitution of the gatekeeper residue with bulky side chains is a common mechanism of resistance to pharmacological ATP-competitive kinase inhibitors (43). The G1269 residue resides next to the DFG motif within the kinase activation loop – a portion of the kinase domain necessary for kinase activity. Substitution of glycine at 1269, which has the smallest side chain, to alanine causes steric hindrance, resulting in decreased affinity for crizotinib (44, 45). Additionally, multiple mutations in the ALK kinase domain, such as L1152R, 1151 T-ins, C1156Y, I1171T, F1174L, G1202R, and S1206Y, are associated with crizotinib resistance, even though the frequencies of these mutations are low (36, 46-49). The G1202R mutation leads to a large basic residue that would be predicted to cause steric interference with ALK inhibitor binding (47).
Figure 2.
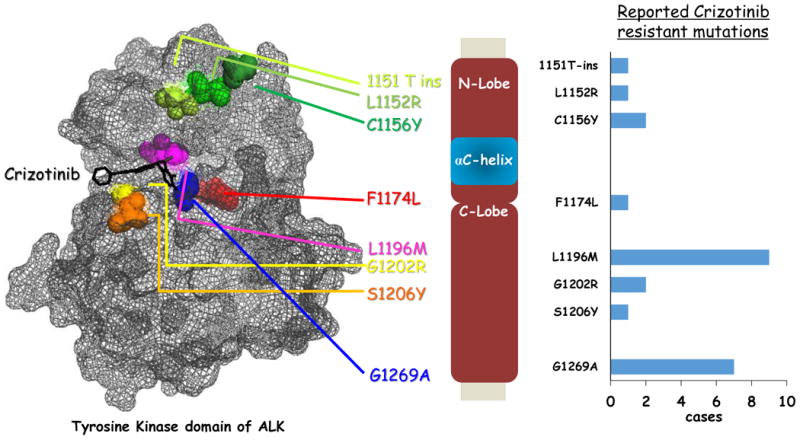
Crizotinib resistant mutations mapped on ALK kinase domain. Reported crizotinib resistance mutations (point mutations and insertion mutation) in the ALK kinase domain were mapped on the structure data of ALK kinase domain (PDB: 2XP2) The bar graph on the right shows the frequency (reported cases) of each mutation detected directly from the crizotinib resistant patients.
Amplification of the ALK fusion gene is also observed in about 9% of crizotinib-resistant cases (42, 47). ALK amplification was first noted as a potential resistance mechanism in cell line models of crizotinib resistance. The ALK-rearranged NSCLC cell line, H3122, was grown in increasing concentrations of crizotinib until resistant cells emerged. Cells grown in 600 nM of crizotinib had intermediate levels of resistance, while those cells cultured in 1 μM of crizotinib had high level resistance. Those cells with intermediate resistance exhibited significant EML4-ALK gene amplification but no ALK mutation, whereas fully resistant cells harbored both ALK gene amplification and the L1196M gatekeeper mutation (50).
In another approximately 40% of patients with crizotinib-resistant tumors, alternative pathway-mediated survival signal activation, so-called “bypass pathway” activation, has been observed (Fig. 3). For example, in some patients and in H3122-derived crizotinib-resistant cells, EGFR activation was observed (without EGFR mutation or amplification on the genomic level), and EGFR inhibitor co-treatment resensitized the cells to crizotinib. In addition, cKIT gene amplification was identified in 2 cases (47). A concomitant EGFR activating mutation (L858R) or KRAS mutation has also been reported in crizotinib-resistant patients (46, 48). Recently, activation of insulin-like growth factor 1 receptor (IGF1R) was identified as a crizotinib-resistance mechanism in vitro and in vivo, and treatment with crizotinib and an IGF1R inhibitor resensitized crizotinib-resistant cells (51). In ALK-rearranged ALCL cells, IGF1R binds to and activates NPM-ALK, while IGF1R inhibition suppresses growth of ALK-rearranged ALCL cells (52). Furthermore, the Y644F and Y664F mutations in NPM-ALK were found to inhibit binding with IGF1R, resulting in loss of oncogenicity of NPM-ALK (53). Recently, activation of Src was also found to confer resistance to crizotinib (and other ALK-TKIs) in a cell line-derived model and resistant cancer cell lines derived from ALK-TKI-resistant patients (54).
Figure 3.

Mechanisms of acquired resistance to ALK inhibitor therapy in ALK-rearranged NSCLC. Reported crizotinib, alectinib or ceritinib resistant mechanisms are shown. Bold characters indicate those resistance mechanisms which have been detected in patient tumor samples.
Next-generation ALK inhibitors
A number of new ALK inhibitors are currently under development. Among them, ceritinib is the most advanced in the clinic and was granted accelerated approval by the US FDA in April 2014. Alectinib has also been approved in Japan, and both alectinib and AP26113 were granted FDA breakthrough therapy designation. These drugs have demonstrated robust clinical activity in patients who developed resistance to crizotinib, and they are now being investigated in the front-line setting.
Ceritinib
Ceritinib is a highly potent oral ALK inhibitor. At the enzymatic level, the half maximal inhibitory concentration (IC50) of ceritinib is approximately 150 pM, which is 20-fold lower than that of crizotinib (44). In addition to ALK, ceritinib potently inhibits several other kinases, including IGF1R (8 nM), insulin receptor (7 nM), and testis-specific kinase substrate (23 nM) (55). Moreover, ceritinib has documented efficacy against crizotinib resistant ALK mutations, including L1196M, G1269A, I1171T, and S1206Y crizotinib-resistant mutations (44).
In a phase I clinical trial of 114 patients with ALK-rearranged NSCLC, the ORR among patients treated with ceritinib at doses of 400 to 750 mg daily was 58% and PFS was 7.0 months (56). In the subgroup of patients who had previously received crizotinib, the ORR was 56% (45/80) and the PFS was 6.9 months. Interestingly, approximately 20 patients underwent rebiopsy after relapsing on crizotinib and prior to starting on ceritinib. Responses were observed in patients with and without identifiable resistance mutations, suggesting that most crizotinib-resistant tumors remain ALK-dependent. Importantly, ceritinib also has documented efficacy in patients with CNS disease, as intracranial responses were observed in patients with untreated brain metastases. The most common ceritinib-associated AEs were nausea (82%), diarrhea (75%), vomiting (65%), fatigue (47%), and elevated transaminase levels, all reversible and resolved by ceritinib discontinuation (56).
While ceritinib was able to re-induce responses in the majority of crizotinib-resistant patients, acquired resistance to ceritinib ultimately developed. Four of 10 patients who initially received crizotinib and then were treated with ceritinib developed a secondary G1202R or F1174C/V mutation. In 1 case, after ceritinib treatment, a F1174V and G1202R mutation was found in malignant fluid collected from the left and right lung, respectively. The ability of these mutations to confer ceritinib resistance was confirmed in Ba/F3 cells (44). In addition, a MAP2K1 K57N activating mutation was recently detected in a ceritinib-resistant tumor sample (54). Src activation may also mediate ceritinib resistance. Ongoing studies are directed at better understanding mechanisms of acquired resistance to ceritinib.
Alectinib
Alectinib (CH5424802/RO5424802) is a potent and selective ALK TKI (IC50, 2 nM in vitro kinase assay). Analogous to ceritinib, alectinib can inhibit multiple crizotinib-resistant ALK mutations (e.g., L1196M, G1269A, C1156Y, and F1174L) (45, 57). The efficacy of alectinib was first evaluated in ALK-TKI naïve patients in a multicenter Japanese phase 1/2 clinical trial (AF-001JP). In contrast to the crizotinib and ceritinib studies which used ALK FISH as the diagnostic test, AF-001JP required that tumors be positive for ALK gene rearrangement by both immunohistochemistry and FISH. Of 46 patients receiving 300 mg of alectinib twice a day (the phase 2 recommended dose in Japan), the ORR was 93.5% and median PFS exceeded 27 months (58). In a phase I/II clinical trial of US patients previously treated with crizotinib (AF-002JG), the ORR was 54% at the time of data cut-off, and 5 of 9 patients with untreated CNS disease responded to alectinib (59). In a separate series, among 4 patients who had received prior crizotinib and ceritinib and then developed leptomeningeal disease, 3 achieved a significant partial response (PR) while the fourth experienced disease stabilization (60). Unlike crizotinib and ceritinib, alectinib is not transported by ABCB1 and thus can achieve high levels in the CNS (61). Recently, two randomized phase 3 trials (ALEX (NCT02075840) and J-ALEX) were launched, comparing alectinib vs. crizotinib as first-line therapy for patients with advanced ALK-rearranged NSCLC.
The major AEs of alectinib therapy are dysgeusia, photosensitivity, decreased neutrophil count, elevated liver function tests, increased creatinine, and increased creatine phosphokinase, although almost all AEs have been mild (grade 1-2). Notably, GI toxicity is observed at a relatively high frequency in crizotinib- and ceritinib-treated patients, but not in those receiving alectinib (59, 62).
Patients treated with alectinib invariably relapse, as they do on crizotinib and ceritinib. The V1180L and I1171T ALK kinase domain mutations were identified in H3122 cells made resistant to alectinib in vitro. I1171 mutations have subsequently been identified in patients treated with alectinib (23, 49, 63). The V1180L mutation is located sterically next to the L1196 gatekeeper residue. Both V1180L and I1171T confer resistance to both crizotinib and alectinib, but not ceritinib. Interestingly, in a patient with alectinib resistance due to an acquired I1171T mutation, the resistant tumor responded to ceritinib therapy (63). Additionally, the G1202R mutation has also been identified in an alectinib-resistant specimen. The G1202R mutation appears to be a highly refractory mutation, conferring high-level resistance to crizotinib, alectinib, and ceritinib (49, 64). Finally, MET gene amplification (65) has been identified from a patient-derived sample as a potential mechanism of resistance to alectinib. This patient subsequently responded to crizotinib, which can effectively inhibit both ALK and MET.
AP26113
AP26113 is a potent ALK inhibitor (IC50, 0.62 nM in vitro kinase assay). Like ceritinib and alectinib, AP26113 can inhibit multiple crizotinib-resistant ALK mutants, including the L1196M gatekeeper mutation (50). In addition to ALK, AP26113 has some activity against the T790M gatekeeper-mutated EGFR in vitro. An ongoing phase I/II clinical trial reported an ORR of 72% and median PFS of 56 weeks among 72 patients with advanced ALK-rearranged NSCLC at the time of data cut-off. Of 65 patients pretreated with crizotinib, the ORR was 69% with a median PFS of 47.3 weeks (66). Commonly observed AEs were nausea, diarrhea, and fatigue. Because serious early-onset pulmonary symptoms (such as dyspnea) have occurred in 10% of patients receiving AP26113, particularly within the first week, the design of the phase I/II study was modified so that patients would start at a low dose of 90 mg daily and step up to 180 mg daily after one week if no pulmonary symptoms.
X-396
X396 is a potent ALK inhibitor with a similar chemical structure as crizotinib, but with a 10-fold higher potency. X-396 has documented efficacy against crizotinib-resistant mutations, such as L1196M and C1156Y (67). In an ongoing phase I clinical trial of X-396, 10/17 (59%) of ALK-rearranged NSCLC patients achieved a PR, and SD was observed in 2 (12%). Additionally, of 13 patients pretreated with crizotinib, 7 responded to X-396. Major AEs were rash, nausea, vomiting, fatigue, and edema (68). The phase I trial of this agent is ongoing (NCT01625234).
PF-06463922
PF-06463922 shares a structural basis with crizotinib but has an approximately 60-fold higher ALK inhibitory activity in NIH3T3 cells and >10-fold in vitro (69). This drug has been optimized to overcome the limitations of crizotinib with regards to selectivity, potency and CNS penetration. Thus, PF-06463922 is effective against all the crizotinib-resistant mutations identified in patients thus far. The IC50 of PF-06463922 for Ba/F3 cells expressing the G1202R mutant was 77 nM, the same as that of crizotinib for wild-type EML4-ALK (80 nM). Additionally, PF-06463922 is not an ABCB1 substrate and effectively penetrates into the CNS. In preclinical studies, PF-06463922 demonstrated potent intracranial activity in vivo (69-71). A phase I/II trial was launched in early 2014 and is ongoing (NCT01970865).
Other Potential Therapeutic Strategies to Overcome ALK TKI Resistance
To date, the majority of studies aimed at ALK TKI resistance have focused on next-generation ALK inhibitors, which have greater potency against ALK than crizotinib, and can overcome at least some of the known crizotinib-resistance mutations within the ALK tyrosine kinase domain. However, in addition to the use of next-generation ALK inhibitors, several other potential strategies to combat ALK TKI resistance have been proposed and some are actively being investigated, including:
ALK TKI with other kinase inhibitor
Combination strategies that target both ALK and a second kinase may be needed to overcome any of a number of different “bypass pathways” mediating ALK-TKI resistance. Potential combinations include ALK TKI with EGFR inhibitor, cKIT inhibitor, MEK inhibitor, or SRC inhibitor. The selection of the appropriate combination strategy may need to be individualized based on the resistance mechanism(s) identified in resistant tumor specimens.
ALK TKI with heat shock protein (HSP90) inhibitor
ALK fusion proteins bind to Hsp90 and are thought to depend on Hsp90 as a chaperone protein to form tertiary structure and stabilize the protein. Hsp90 inhibitor treatment induced degradation and reduction of ALK fusion proteins, regardless of ALK-TKI-resistant mutations, and induced cell death in ALK-rearranged cancers (50, 72). Hsp90 inhibitors have shown clinical activity in primarily TKI-naïve, ALK-rearranged lung cancer (73), although developing Hsp90 inhibitors for the treatment of ALK-rearranged cancer could be challenging because of the lower ORR and greater toxicity compared with ALK-TKIs. Notably, different EML4-ALK fusion variants may have different sensitivities to Hsp90 inhibitor therapy, with the EML4-ALK (E6; A20) (variant 3) being the least sensitive in vitro (74). The combination of ALK- and Hsp90 inhibitors is currently being investigated in the clinic (72).
ALK TKI with immunotherapy
Checkpoint inhibitors, specifically PD1 and PDL1 inhibitors, have shown promising activity in advanced NSCLC with response rates ranging from 15-20% and remarkably prolonged durations of response among those patients with response. Interestingly, response rates seem to be lower among never smokers compared to smokers, raising the question of whether patients with targetable genetic alterations like ALK and EGFR, may be less responsive to immunotherapies, at least when given as monotherapies. A recent report suggests that EGFR-mutant lung cancers inhibit antitumor immunity by activating the PD-1/PD-L1 pathway to suppress T-cell function (75, 76). This result, along with others, has generated tremendous interest in combining checkpoint inhibitors with TKIs in the treatment of oncogene-addicted NSCLC, including ALK-rearranged NSCLC. To date, one study combining ceritinib with nivolumab, a PD1 antibody, is about to launch (NCT02393625), and several others are being planned. The ceritinib/nivolumab combination study will enroll both TKI-sensitive and TKI-resistant patients. This ALK-TKI with immunotherapy is also discussed in this CCR Focus (77).
ALK TKI with chemotherapy
Currently, pemetrexed with or without crizotinib is being studied in patients with advanced, ALK-rearranged NSCLC that has progressed after crizotinib. Retrospective analyses first suggested that patients with ALK-rearranged lung cancer may be particularly responsive to pemetrexed-based chemotherapy (78, 79). The randomized trials of crizotinib compared to chemotherapy (either first line platinum/pemetrexed or second-line pemetrexed) also suggest a modest improvement in response and progression-free survival in ALK patients compared to historical controls (32, 33). Based on these data, an ongoing study is evaluating pemetrexed with or without crizotinib for patients with advanced ALK-rearranged NSCLC that has progressed after crizotinib (NCT02134912).
Conclusion and Future Directions
ALK represents a validated therapeutic target in numerous malignancies, including NSCLC, ALCL, and IMT. Numerous ALK inhibitors are in clinical development, and these agents have already shown remarkable efficacy in cohorts of patients with ALK mutant tumors, particularly ALK-rearranged NSCLC. Despite these remarkable results, therapeutic resistance is common and represents a significant barrier to the successful treatment of patients with ALK-rearranged tumors. ALK inhibitor resistance has proven to be a complex and heterogeneous process, with multiple mechanisms at play, including ALK amplification, ALK kinase domain mutation, and activation of a variety of different bypass signaling pathways. Moreover, to add to the complexity of this disease, numerous resistance mechanisms co-existing within the same patient have been documented. To overcome resistance, multiple promising ‘next-generation’ ALK kinase inhibitors and rational combinatorial strategies are under development. Since the spectrum of activity of each ALK inhibitor against different resistance mutations varies, repeat-biopsy to identify the resistance mechanisms can be critical for making therapeutic decisions, particularly after failure of a second ALK TKI.
Furthermore, since ALK alterations are only detected in small cohorts of patients, NCI and cooperative group studies, such as the NCI MATCH trial and the emerging NCI ALK Master Protocol, are being conceived to systematically study treatment responses in patients with tumors harboring ALK alterations. The NCI MATCH trial will include multiple phase 2 studies that will enroll patients with advanced solid tumors and lymphomas. After assessing tumors for the presence of targetable genetic abnormalities, patients will be assigned to one of the phase 2 studies based not on their type of cancer but on the identified genetic target. The NCI ALK Master protocol will test the potential role of multiple different next generation ALK inhibitors in the first-line setting, as compared with crizotinib. This will be an important study to evaluate how best to sequence ALK inhibitors in managing patients with advanced, ALK-rearranged NSCLC. The current treatment strategy is to use crizotinib as first-line therapy, based on PROFILE 1014, and to consider a next generation ALK inhibitor at the time of relapse. As mentioned previously, the ALEX trials will compare alectinib with crizotinib as first-line therapy for ALK-rearranged NSCLC, with PFS as the primary endpoint. However, the trial design for the ALEX studies does not include crossover of patients at progression, precluding comparison of first-line alectinib with sequential crizotinib followed by alectinib. In contrast, the ALK Master protocol will incorporate crossover for each study arm at the time of progression, and this will enable evaluation of sequential therapy with crizotinib followed by next generation inhibitor, as well as next generation inhibitor followed by crizotinib.
Finally, as next-generation sequencing based techniques increasingly become the standard of care in oncology practice, ALK alterations (amplification, mutations, and fusions) will be increasingly detected. Overall, an increased knowledge of the molecular biology of ALK as well as a more thorough study of ALK as a therapeutic target in the clinic has the potential to improve the treatment of many cancer patients.
Acknowledgments
Grant Support: R. Katayama was supported by JSPS KAKENHI grant number 25710015 and the Ministry of Health Labour and Welfare. C.M. Lovly was supported by the NIH under award numbers R01CA121210 and P01CA129243. A.T. Shaw was supported by the NIH under award number R01CA164273.
C.M. Lovly reports receiving a commercial research grant from AstraZeneca and Novartis; speakers bureau honoraria from Harrison and Star; and is a consultant/advisory board member for Novartis. A.T. Shaw is a consultant/advisory board member for ARIAD Pharmaceuticals, Chugai Pharmaceutical, Daiichi Sankyo, Genentech, Ignyta, Novartis, Pfizer, and Roche.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed by the other author.
References
- 1.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 2.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 4.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011;71:4403–11. doi: 10.1158/0008-5472.CAN-10-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 7.Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, et al. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem. 2002;277:35990–8. doi: 10.1074/jbc.M205749200. [DOI] [PubMed] [Google Scholar]
- 8.Murray PB, Lax I, Reshetnyak A, Ligon GF, Lillquist JS, Natoli EJ, Jr, et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci Signal. 2015;8:ra6. doi: 10.1126/scisignal.2005916. [DOI] [PubMed] [Google Scholar]
- 9.Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malerczyk C, et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem. 2001;276:16772–9. doi: 10.1074/jbc.M010660200. [DOI] [PubMed] [Google Scholar]
- 10.Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. 2008;33:685–700. doi: 10.1038/sj.npp.1301446. [DOI] [PubMed] [Google Scholar]
- 11.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–94. doi: 10.1016/j.ccell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–80. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111:5496–504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood. 1999;94:3265–8. [PubMed] [Google Scholar]
- 15.Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93:3088–95. [PubMed] [Google Scholar]
- 16.Siebert R, Gesk S, Harder L, Steinemann D, Grote W, Schlegelberger B, et al. Complex variant translocation t(1;2) with TPM3-ALK fusion due to cryptic ALK gene rearrangement in anaplastic large-cell lymphoma. Blood. 1999;94:3614–7. [PubMed] [Google Scholar]
- 17.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 19.Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R, et al. KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012;7:e31323. doi: 10.1371/journal.pone.0031323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong DW, Leung EL, Wong SK, Tin VP, Sihoe AD, Cheng LC, et al. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer. 2011;117:2709–18. doi: 10.1002/cncr.25843. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143–9. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi K, Soda M, Togashi Y, Ota Y, Sekiguchi Y, Hatano S, et al. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica. 2011;96:464–7. doi: 10.3324/haematol.2010.033514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, et al. Identification of a novel HIP1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to alectinib. J Thorac Oncol. 2014;9:1821–5. doi: 10.1097/JTO.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 24.Morris SW, Xue L, Ma Z, Kinney MC. Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of non-Hodgkin's lymphoma. Br J Haematol. 2001;113:275–95. doi: 10.1046/j.1365-2141.2001.02574.x. [DOI] [PubMed] [Google Scholar]
- 25.Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Institute. 2014;106:djt378. doi: 10.1093/jnci/djt378. [DOI] [PubMed] [Google Scholar]
- 26.Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364:775–6. doi: 10.1056/NEJMc1013224. [DOI] [PubMed] [Google Scholar]
- 27.Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105:19893–7. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–7. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–24. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 30.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 31.Rogers TM, Russell PA, Wright G, Wainer Z, Pang JM, Henricksen LA, et al. Comparison of Methods in the Detection of ALK and ROS1 Rearrangements in Lung Cancer. J Thorac Oncol. 2015;10:611–8. doi: 10.1097/JTO.0000000000000465. [DOI] [PubMed] [Google Scholar]
- 32.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–77. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 33.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 34.Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–95. doi: 10.1158/2159-8290.CD-14-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–33. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–43. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29:e443–5. doi: 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 38.Costa DB, Shaw AT, Ou SH, Solomon BJ, Riely GJ, Ahn MJ, et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015 Jan 26; doi: 10.1200/JCO.2014.59.0539. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camidge DR. Taking aim at ALK across the blood-brain barrier. J Thorac Oncol. 2013;8:389–90. doi: 10.1097/JTO.0b013e3182864e7c. [DOI] [PubMed] [Google Scholar]
- 40.Takeda M, Okamoto I, Nakagawa K. Clinical impact of continued crizotinib administration after isolated central nervous system progression in patients with lung cancer positive for ALK rearrangement. J Thorac Oncol. 2013;8:654–7. doi: 10.1097/JTO.0b013e31828c28e7. [DOI] [PubMed] [Google Scholar]
- 41.Chuan Tang S, Nguyen LN, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer. 2014;134:1484–94. doi: 10.1002/ijc.28475. [DOI] [PubMed] [Google Scholar]
- 42.Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19:4273–81. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–18. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–73. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kodama TT, Tsukaguchi T, Hasegawa M, Yoshida M, Takanashi K, Kondoh O, et al. Selective ALK inhibitor alectinib (CH5424802/RO5424802) with potent antitumor activity in models of crizotinib resistance, including intracranial metastases [abstract]. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 2014 Apr 5-9; San Diego, CA. [Google Scholar]; Cancer Res. 19 Suppl. Vol. 74. Philadelphia (PA): AACR; 2014. Abstract nr 754. [Google Scholar]
- 46.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toyokawa G, Hirai F, Inamasu E, Yoshida T, Nosaki K, Takenaka T, et al. Secondary mutations at I1171 in the ALK gene confer resistance to both crizotinib and alectinib. J Thorac Oncol. 2014;9:e86–7. doi: 10.1097/JTO.0000000000000358. [DOI] [PubMed] [Google Scholar]
- 50.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20:1027–34. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi P, Lai R, Lin Q, Iqbal AS, Young LC, Kwak LW, et al. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood. 2009;114:360–70. doi: 10.1182/blood-2007-11-125658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi B, Vishwamitra D, Granda JG, Whitton T, Shi P, Amin HM. Molecular and functional characterizations of the association and interactions between nucleophosmin-anaplastic lymphoma kinase and type I insulin-like growth factor receptor. Neoplasia. 2013;15:669–83. doi: 10.1593/neo.122012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–6. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56:5675–90. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 56.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–90. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013;14:590–8. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 59.Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–28. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- 60.Gainor JF, Sherman CA, Willoughby K, Logan J, Kennedy E, Brastianos PK, et al. Alectinib salvages CNS metastases in ALK-positive lung cancer patients previously treated with crizotinib and ceritinib. J Thorac Oncol. 2015;10:232–6. doi: 10.1097/JTO.0000000000000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol. 2014;74:1023–8. doi: 10.1007/s00280-014-2578-6. [DOI] [PubMed] [Google Scholar]
- 62.Sato W, Fukazawa N, Nakanishi O, Baba M, Suzuki T, Yano O, et al. Reversal of multidrug resistance by a novel quinoline derivative, MS-209. Cancer Chemother Pharmacol. 1995;35:271–7. doi: 10.1007/BF00689444. [DOI] [PubMed] [Google Scholar]
- 63.Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, et al. Two Novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20:5686–96. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014;9:549–53. doi: 10.1097/JTO.0000000000000094. [DOI] [PubMed] [Google Scholar]
- 65.Toyokawa G, Seto T, Takenoyama M, Ichinose Y. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. 2014;9:e27–8. doi: 10.1097/JTO.0000000000000113. [DOI] [PubMed] [Google Scholar]
- 66.Gettinger S, Bazhenova L, Salgia R, Langer C, Gold K, Rosell R, et al. ALK Inhibitor AP26113 in patients with advanced malignancies, including ALK+ non-small cell lung cancer (NSCLC): updated efficacy and safety data [abstract]. Proceedings of the ESMO 2014 Congress; 2014 Sep 26-30; Madrid, Spain. [Google Scholar]; European Society for Medical Oncology. Lugano (Switzerland): 2014. Abstract nr 1292P. [Google Scholar]
- 67.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71:4920–31. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horn L, Infante JR, Blumenshcein G, Wakelee HA, Arkenau HT, Dukart G, et al. A phase I trial of X-396, a novel ALK inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014;32:5s(suppl) abstr 8030ˆ. [Google Scholar]
- 69.Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57:4720–44. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- 70.Yamazaki S, Lam JL, Zou HY, Wang H, Smeal T, Vicini P. Translational pharmacokinetic-pharmacodynamic modeling for an orally available novel inhibitor of anaplastic lymphoma kinase and c-Ros oncogene 1. J Pharmacol Exp Ther. 2014;351:67–76. doi: 10.1124/jpet.114.217141. [DOI] [PubMed] [Google Scholar]
- 71.Yamazaki S, Lam JL, Zou HY, Wang H, Smeal T, Vicini P. Mechanistic understanding of translational pharmacokinetic-pharmacodynamic relationships in nonclinical tumor models: a case study of orally available novel inhibitors of anaplastic lymphoma kinase. Drug Metab Dispos. 2015;43:54–62. doi: 10.1124/dmd.114.061143. [DOI] [PubMed] [Google Scholar]
- 72.Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3:430–43. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28:4953–60. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Richards MW, Law EW, Rennalls LP, Busacca S, O'Regan L, Fry AM, et al. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc Natl Acad Sci U S A. 2014;111:5195–200. doi: 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25:1935–40. doi: 10.1093/annonc/mdu242. [DOI] [PubMed] [Google Scholar]
- 77.Soria JC, Marabelle A, Brahmer JR, Gettinger S. Immune checkpoint modulation for non–small cell lung cancer. Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2959. [DOI] [PubMed] [Google Scholar]
- 78.Camidge DR, Kono SA, Lu X, Okuyama S, Baron AE, Oton AB, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6:774–80. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JO, Kim TM, Lee SH, Kim DW, Kim S, Jeon YK, et al. Anaplastic lymphoma kinase translocation: a predictive biomarker of pemetrexed in patients with non-small cell lung cancer. J Thorac Oncol. 2011;6:1474–80. doi: 10.1097/JTO.0b013e3182208fc2. [DOI] [PubMed] [Google Scholar]