

Summary
By exploiting host cell machineries, viruses provide powerful tools for gaining insight into cellular pathways. Proteins from two unrelated viruses, human CMV (HCMV) and HCV, are documented to traffic sequentially from the ER into mitochondria, probably through the mitochondria-associated membrane (MAM) compartment. The MAM are sites of ER-mitochondrial contact enabling the direct transfer of membrane bound lipids and the generation of high calcium (Ca2+) microdomains for mitochondria signalling and responses to cellular stress. Both HCV core protein and HCMV UL37 proteins are associated with Ca2+ regulation and apoptotic signals. Trafficking of viral proteins to the MAM may allow viruses to manipulate a variety of fundamental cellular processes, which converge at the MAM, including Ca2+ signalling, lipid synthesis and transfer, bioenergetics, metabolic flow, and apoptosis. Because of their distinct topologies and targeted MAM sub-domains, mitochondrial trafficking (albeit it through the MAM) of the HCMV and HCV proteins predictably involves alternative pathways and, hence, distinct targeting signals. Indeed, we found that multiple cellular and viral proteins, which target the MAM, showed no apparent consensus primary targeting sequences. Nonetheless, these viral proteins provide us with valuable tools to access the poorly characterized MAM compartment, to define its cellular constituents and describe how virus infection alters these to its own end. Furthermore, because proper trafficking of viral proteins is necessary for their function, discovering the requirements for MAM to mitochondrial trafficking of essential viral proteins may provide novel targets for the rational design of anti-viral drugs.
Keywords: Cytomegalovirus, Hepatitis C virus, MAM, ER, mitochondria, protein trafficking
Introduction
Only two viral proteins are documented to traffic from the ER, possibly through the ER sub-domain called mitochondria-associated membrane (MAM), into mitochondria. We review literature characterizing the MAM, a key, yet poorly-understood ER sub-domain that directly contacts mitochondria and is involved in regulating lipid biosynthesis, calcium signalling, metabolism and cell survival (recently reviewed in [1]). Subsequently, we provide an overview of the viral proteins which target the MAM. Trafficking of viral proteins to the MAM is likely to place them at a nexus, where informed decisions about cell survival are made during times of stress. Furthermore, these two proteins are derived from two completely unrelated viruses; a human herpesvirus (HCMV) and a hepacivirus (HCV) suggesting the possibility that more examples of this phenomenon may be found in the future. These proteins will predictably include those encoded by conserved viral gene sequences, such as those from primate CMVs, as well as analogous viral proteins that target mitochondria-mediated apoptosis.
Historical perspective
By the 1960s, analyses of a wide variety of plant and animal tissues by EM had revealed close relationships between mitochondria and ER [2]. Morphologic extensions of the outer mitochondrial membrane (OMM) seemingly contiguous with adjacent ER membranes were repeatedly observed. These extensions were predominantly identified as smooth ER (SER), but ribosome-studded, rough ER (RER) were also observed in close proximity with mitochondria [2, 3]. The frequency and appearance of these contacts led some researchers to propose that the OMM was an extension of the ER [4]. Such proposals were controversial, considering the propensity for artefacts to be produced from EM fixation. Nonetheless, in 1973, Lewis and Tata [5] demonstrated the tight association between the ER and mitochondria by isolating a subcellular RER fraction with mitochondria in rat liver cells. These authors concluded that mitochondria are enmeshed or entangled by the ER and that this association might have a functional basis.
Connections between the ER and mitochondria are known as MAM
It is now well documented that mitochondria and the ER interact physically and functionally in order to integrate cellular responses and functions between these organelles. The ER controls trafficking and folding of membrane-anchored proteins and soluble proteins through the secretory pathway, the biosynthesis of lipids, and is the main store of intracellular calcium (Ca2+) [6, 7]. Mitochondrial functions include ATP generation, lipid oxidation, oxygen radical production, hormone metabolism and intrinsic cell death signaling [8-10]. Contacts between the ER and mitochondria are dynamic and transient [11-13], and regulate critical aspects of cellular physiology, including metabolic flow, Ca2+ signaling, bioenergetics and cell survival [14-19]. These close contacts characterise the MAM compartment which has been experimentally defined as ER sub-domains that co-sediment with mitochondria [5, 18, 20, 21]. The contacts between the ER and mitochondria [21, 22], and their quasi-synaptic organization [23], have been visualized using EM. The improved resolution of electron tomography has resolved physical links or tethers between the ER and mitochondria [22]. Associations (∼10 nm) between OMM and the SER are closer than associations (∼25 nm) of the RER with the OMM, which accommodate ribosomes [22]. Heterogeneity in the distances between the ER and OMM is consistent with the possibility that their contacts may be differentially regulated.
The MAM provides functional ER-mitochondrial junctions
The MAM appears to serve as a nexus, where pro-apoptotic (including Ca2+ signaling, ceramide transfer, HCV core protein) and anti-apoptotic (e.g., HCMV UL37 proteins) machineries as well as cellular stress sensors (sigma-1 receptor, Sig-1R) converge. The MAM associates with mitochondria at discrete junctions where the OMM and inner mitochondrial membranes (IMM) meet, to affect lipid synthesis and transfer, and ultimately cell fate decisions [24-26].
Lipid synthesis in the MAM
The MAM has lipid and protein compositions which can be distinguished from those of the bulk ER. Although the MAM was characterized by Meier and Meyer in 1981 [11], it was later ascribed a lipid synthetic function, when Jean Vance encountered it while studying phospholipid synthesis and distribution throughout subcellular organelles [18, 25]. The MAM, designated as Fraction X, was characterized in rat liver as either RER or SER, which remained tightly associated with mitochondria after homogenization, but which could be separated using Percoll gradients [5, 18, 27]. The MAM is enriched in lipid synthetic enzymes, including phosphatidylserine (PS) synthase types 1 (PSS-1) and 2 (PSS-2), phosphatidylethanolamine N-methyltransferase, fatty acid coenzyme A ligase 4 (FACL-4), ceramide synthase, and sphingolipid-specific glycotransferases [18, 25, 28-33]. In contrast, other enzymes targeted to the secretory pathway were lower in activities [18, 31, 34]. Based upon these findings, Vance proposed that Fraction X was a sub-domain of the ER, tightly associated with mitochondria, which allowed the generation of unique phospholipid pools, and their transport to and from mitochondria. An extensive review of the lipid synthetic capability of the MAM was recently published by Jean Vance [31]. Consistent with their high lipid composition, viruses that target this compartment affect lipid metabolism. HCV core protein associates with lipid droplets (LDs) and has a capacity to influence metabolic events involving lipid storage [35-38] and very low density lipoprotein assembly and secretion [39]. LD is a cytosolic organelle which stores neutral lipids, interacts with the ER, and facilitates transport of lipids between organelles. HCV core protein recruits replication complexes to the LD-associated membranes for virus particle assembly [40]. For its part, HCMV was found to dynamically regulate host bioactive sphingolipid levels during infection [41].
Cellular proteins that reside exclusively in the MAM have not been identified. Because the MAM coalesces within the existing framework of the secretory apparatus, it contains many ER-localized chaperones [14, 27, 34, 42]. Their broader distributions in multiple organelles make these unsuitable as MAM markers. Two lipid synthetic enzymes, PSS-1 and PSS-2, are largely absent from peripheral ER and mostly restricted to the MAM [25] and, therefore, serve as suitable organelle markers for identification of purified MAM fractions and display a broad reticular distribution in cells (Figure 1) [27, 42].
Figure 1. Multiple morphologies of MAM markers.
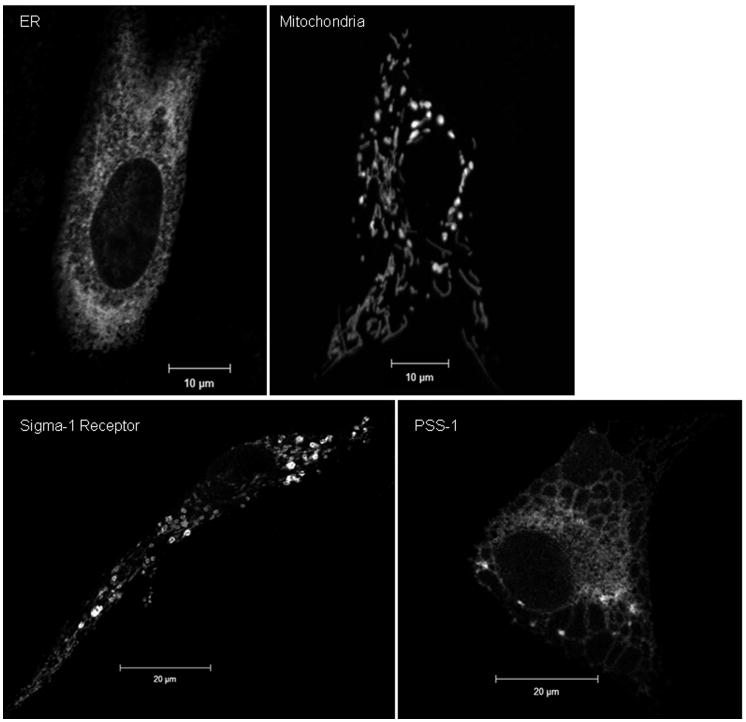
Representative morphologies of ER and mitochondrial organelles are shown in the top row. The characteristic reticular ER morphology was visualized using a commercially available, soluble ER lumen fusion protein, pECFP-ER (Clontech). Similarly, both the thread-like and punctuate morphologies associated with dynamic mitochondria are observed using a marker for the mitochondrial inner membrane, DsRed1-Mito (Clontech). Two different markers for the MAM are shown in the bottom row, Sig-1R (left panel) and PSS-1 (right panel), revealing multiple morphologies associated with this ER subdomain. Primary diploid fibroblasts were lipofected with either pECFP-ER, DsRed1-Mito, mEGFP-human PSS-1 [42], or with Sig1R-EYFP (a generous gift from Drs. Hayashi and Su) [14]. 24 hours after transfection, cells were fixed with ice cold methanol as described [42] and imaged using confocal microscopy with a Zeiss LSM510 and a 63× objective (NA 1.4). Panels show single optical sections (0.8 microns) of transfected cells.
Lipid transfer from the MAM to the OMM
Membrane continuities between the MAM and mitochondria permit direct delivery of membrane-bound lipids to the OMM. The MAM associates with the OMM at sites of IMM contacts [21] that may facilitate the transfer of integral membrane products to the IMM. PSS has been detected at these contact sites [21]. Vance and her colleagues demonstrated the transfer of PS in rat liver from MAM to mitochondria using a reconstituted system in vitro [43]. Translocation of PS to mitochondria was ATP-dependent, required a mitochondrial membrane protein, and was stimulated by magnesium and Ca2+ [43]. PS is a relatively minor constituent of cellular membranes but plays important roles in signalling and apoptosis [44, 45]. During the early phases of apoptosis, PS on the plasma membrane becomes externalized to the outer leaflet [44, 46], which is widely used as an early marker for apoptosis.
MAM de novo ceramide synthesis and transfer to mitochondria
De novo synthesis is important for generating the ceramide that induces mitochondrial mediated apoptosis [47]. Increased levels of ceramide and ganglioside GD3 in the OMM play roles in initiation and propagation of mitochondrial mediated apoptotic death [24, 47, 48]. Ceramide, synthesized in the MAM, can directly move into the OMM [24] with random collision kinetics. Transfer appears to be catalyzed, although the protein involved in the transfer has not yet been identified. Transferred ceramide can reach critical levels and result in ceramide channel formation in the OMM. These channels can initiate the execution phase of apoptosis by releasing pro-apoptotic proteins, including cytochrome c, from mitochondria [24].
Calcium microdomains
Another critical function of the MAM is the control of Ca2+ transfer between the ER and mitochondria [10, 17, 49, 50]. The release of Ca2+ from the ER is closely coupled with its uptake by mitochondria, suggesting a privileged Ca2+ transfer between the organelles [23]. Ca2+ release from ER stores via inositol 1,4,5-triphosphate receptors (IP3Rs), which are highly compartmentalized in the MAM, generates microdomains of high Ca2+ concentrations >20-fold higher than levels in the cytosol [23, 26, 49, 51]. The functional significance of MAM for Ca2+ transfer was demonstrated by the reduction of Ca2+ transfer following weakening of the physical coupling between ER and mitochondria [22]. Conversely, tightening of the connections improved mitochondrial Ca2+ uptake.
IP3R, cytosolic glucose regulated protein 75 (Grp75), and voltage dependent anion channel 1 (VDAC1), an OMM channel, form a macromolecular complex at the ER-mitochondrial interface [15]. The complex functionally controls Ca2+ movement from ER stores through the cytosol into mitochondria. Unlike Ca2+ coupling between the dihydropyridine Ca2+ channel and the ryanodine receptor, synchronized activation of multiple IP3Rs results in optimal activation of mitochondrial Ca2+ uptake [23]. These microdomains provide sufficient Ca2+ concentrations to activate the low-affinity mitochondrial Ca2+ uniporter [10, 23, 52].
Chaperones that localize to the MAM include calnexin, BiP, and Sig-1Rs [14]. Sig-1R localizes to globular, lipid enriched compartments (Figure 1) directly apposed to mitochondria [14, 53]. Within the MAM, Sig-1Rs and BiP form a complex that is sensitive to decreasing levels of ER Ca2+. Declining ER Ca2+ levels promote the rapid dissociation of Sig-1R from BiP and activate its chaperone function. Sig-1R then associates with and stabilizes IP3R. This selectively affects Ca2+ mobilization from the MAM to mitochondria but not Ca2+ influx from the bulk cytosol into mitochondria [14]. Prolonged ER stress can provoke Sig-1R relocalization to the ER periphery [14, 53]. Thus, some MAM constituents, such as Sig-1R, are responsive to Ca2+ levels and ER stress.
Regulation and stabilization of ER-mitochondrial contacts
The functional association of ER and mitochondria is transient and dynamic [51]. These contacts appear to be stabilized, in part, by the IP3R-Grp75-VDAC protein complex [15]. Indeed, siRNA knockdown of Grp75 reduced mitochondrial Ca2+ uptake through the ER-mitochondrial complex [15]. Further, association of ER to mitochondria, at least in some sub-domains, is regulated by Ca2+ [54]. Agonist induced Ca2+ oscillations suppress mitochondrial motility [55]. It has been proposed that mitochondrial motility and subcellular distribution are controlled by cytosolic Ca2+ and ATP levels, thereby ensuring generation of energy where and when it is needed [56].
In addition to the roles of the IP3R-Grp75-VDAC complex and Ca2+ in regulating ER-mitochondrial connections, MAM association with mitochondria is stabilized by PACS-2, a multifunctional sorting protein [32]. Reduction of PACS-2 levels by siRNA knockdown caused extensive mitochondrial fragmentation and uncoupling of the ER from mitochondria [32]. It has recently been found that mitofusin 2 (Mfn2), a mitochondrial dynamin-related protein, which alters mitochondrial morphology, directly tethers the MAM to mitochondria, thereby increasing IP3R Ca2+ signalling to mitochondria [57].
Human Cytomegalovirus
Medical importance
HCMV, a medically significant beta-herpesvirus containing a linear dsDNA genome, is the leading viral cause of congenital birth defects and the leading non-genetic cause of neurosensory hearing loss in developed countries [58-60]. Microcephaly and polymicrogyria are the most prominent features of the brain abnormalities in infants congenitally infected with HCMV [61, 62]. HCMV is also a significant pathogen in immunosuppressed individuals, particularly in transplant recipients [60, 63]. An association between HCMV infection and cardiac allograft rejection and restenosis has been suggested [64, 65]. HCMV seropositive persons are at a greater risk than others of developing restenosis after coronary atherectomy [65]. High HCMV antibody titers are associated with coronary artery disease and may predict post-coronary balloon angioplasty restenosis [66].
HCMV UL37 proteins
During lytic growth in cultured cells, HCMV expresses its gene products in a temporally regulated manner [60]. The immediate early (IE) proteins, produced first, include several anti-apoptotic proteins, which protect the infected cell from programmed cell death. Several UL37 isoforms (Figure 2A), the UL37 exon 1 (UL37x1) protein (pUL37x1), full-length UL37 glycoprotein (gpUL37), and UL37 medium protein (pUL37M) as well as the UL36 protein (pUL36), are encoded by the HCMV UL36-38 IE locus [67-71]. pUL37x1, also known as viral mitochondria-localized inhibitor of apoptosis (vMIA), is the predominant UL37 product during permissive HCMV infection [71, 72]. Although gpUL37 shares its N-terminal sequences with pUL37x1\vMIA, its expression is tightly regulated at the level of RNA [67, 73, 74] and the protein is produced at very low levels during HCMV infection of cultured fibroblasts [70, 73, 75, 76]. pUL37M has not yet been detected in infected cells but its encoding transcript has [68, 74, 77]. gpUL37 and pUL37M also display anti-apoptotic activity in transfected cells [68].
Figure 2. HCMV UL37 proteins.
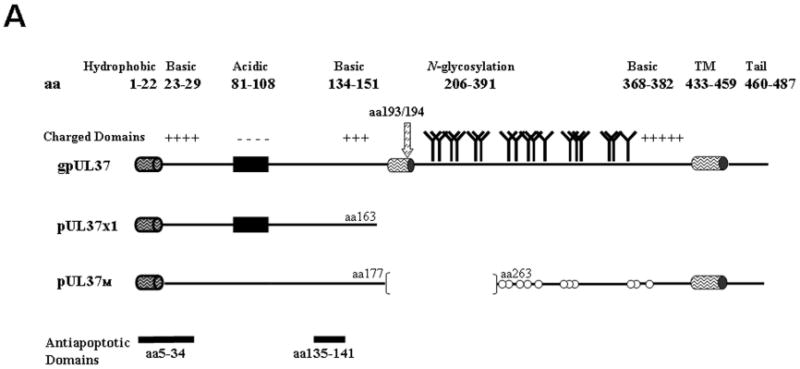
A. Schematic representation of the predominant UL37 protein isoforms. The bipartite leader sequence is shared by all UL37 isoforms and is comprised of a hydrophobic domain (cylinder, aa 1 to 22) and downstream residues (aa 23 to 34). Internal TM domain (cylinder, aa 178 to 196), internal signal peptidase I cleavage site (arrow, aa 193/194), consensus N-glycosylation sites, and C-terminal TM domain (aa 433 to 459) are indicated for gpUL37. Elements common to multiple UL37 isoforms are repeated in each protein. Consensus N-glycosylation sites for pUL37M are marked by open circles to denote that they are not used, as this isoform lacks an internal TM domain needed to direct downstream sequences into the ER lumen. The two anti-apoptotic domains (aa 5 to 34 and 135 to 141), present in all three isoforms, are highlighted at the bottom of the figure.
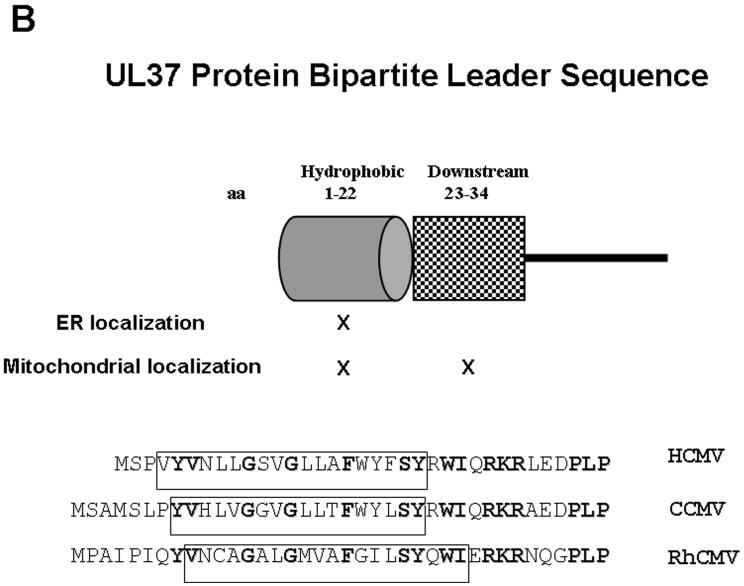
B. Cartoon representation of the pUL37x1\vMIA N-terminal bipartite leader sequence, highlighting the sequences needed for ER and mitochondrial importation [71]. The lower panel displays the sequence similarities in pUL37x1\vMIA leader sequences of HCMV (Accession number=ABV71569), CCMV (Accession number=AAM00687), and rhesus monkey CMV (Accession number=AAZ80560). Bolded residues are strictly conserved, while those boxed show regions of alpha helical structure (predicted by HMMTOP).
The UL37x1 ORF is highly invariant in primary HCMV strains [78]. pUL37x1\vMIA is required for the growth of primary HCMV strains [79] and of some lab strains (AD169) [72, 80-82] but not others (TownevarATCC) [83] in cultured human diploid fibroblasts.
Sequential trafficking of HCMV UL37 proteins
ER to mitochondrial trafficking is common to all studied UL37 isoforms [71, 84] and suggests important functions during HCMV infection. The potent pUL37x1\vMIA anti-apoptotic activity requires its trafficking into the OMM. UL37 proteins are inserted into the ER membrane by their N-terminal hydrophobic leader, which remains uncleaved [70, 71, 84]. gpUL37 has two additional downstream transmembrane (TM) domains spanning aa 178-196 and aa 433-459. The second TM introduces downstream UL37 residues into ER lumen, where its consensus N-glycosylation sites are modified [70, 75, 84]. By internal signal peptidase cleavage, two stable fragments, pUL37NH2 and gpUL37COOH, are produced. pUL37x1\vMIA, pUL37NH2 and gpUL37COOH traffic into the MAM [84]. The MAM serves as a sorting compartment from where gpUL37COOH traffics to the secretory apparatus whereas pUL37x1\vMIA and pUL37NH2, traffic to the OMM [75, 84].
During their trafficking, pUL37x1\vMIA and pUL37NH2 remain membrane anchored by their uncleaved leader with downstream sequences (aa 23-163 and aa 23-193, respectively) in the cytosol [84]. This anchoring and topology have implications for its activities, allowing recruitment and retention of pro-apoptotic proteins, such as Bax and Bak, on the OMM during mitochondrial mediated apoptosis. Although pUL37x1\vMIA interacts tightly with membrane inserted Bax, it fails to interact with soluble Bax [85]. Surprisingly, pUL37x1\vMIA is also documented to interact with cellular proteins located in the IMM, such as adenine nucleotide translocase (ANT) and the mitochondrial phosphate carrier (PiC) [68, 86]. These interactions may reflect pUL37x1\vMIA trafficking from the MAM whose contacts with OMM occur at sites of contact with the IMM.
The sequential trafficking of UL37 proteins was initially suggested by the trafficking of the ER signal peptidase cleavage products into the MAM and mitochondria as well as by the requirement for the pUL37x1\vMIA hydrophobic leader for its trafficking into mitochondria [71, 75]. This trafficking pathway was verified using a full length gpUL37 cleavage mutant [84]. Abrogation of its internal cleavage site generated a triply membrane-anchored, gpUL37 cleavage site mutant. Mitochondrial importation of this N-glycosylated full-length mutant unequivocally established the precursor/product relationship of the ER to mitochondrial pUL37 species [75, 84].
The pUL37x1\vMIA hydrophobic leader (aa 1-22) targets the nascent protein to the ER membrane; whereas aa 2-34 are required for mitochondrial importation (Figure 2B). pUL37x1\vMIA mutants (Δ2-23) did not traffic efficiently to the ER, MAM or mitochondria, whereas pUL37x1Δ23-34 was defective in mitochondrial import [71, 78]. The likely importance of these sequences is supported by their conservation. Forty percent of the primary residues in these HCMV domains are conserved in both chimpanzee CMV (CCMV) and rhesus monkey CMV (RhCMV) pUL37x1\vMIA; while 70% are conserved between HCMV and CCMV.
UL37 protein functions
Anti-apoptosis
Most HCMV mutants defective in pUL37x1\vMIA anti-apoptotic functions are also defective in growth [72, 81]. The UL37 NH2-terminal bipartite mitochondrial targeting signal [71] constitutes the first UL37x1\vMIA anti-apoptotic domain, which, when combined with a downstream domain (aa 118-147), is sufficient to confer potent anti-apoptotic activity (Figure 2A) [68, 78, 85, 87-90]. Even though it lacks primary sequence homology with Bcl-2 family members, pUL37x1\vMIA is known to interact with Bax, Bak, and growth arrest and DNA damage 45 protein (GADD45) [68, 85, 87, 90] (Table 1). In addition, there is evidence that pUL37x1\vMIA interacts with ANT and the mitochondrial PiC, reducing ATP generation in transfected cells [68, 86, 91, 92].
Table 1. HCMV pUL37x1\vMIA functional interactions*.
| Interacting Protein | Sub-cellular Location | Activity | References | |
|---|---|---|---|---|
| Adenine nucleotide translocase (ANT) | Inner mitochondrial membrane | anti-apoptosis |
Goldmacher et al., 1999 Vieira et al., 2001 Poncet et al., 2006 |
|
| Bcl-2 family members | ||||
| BAX | Outer mitochondrial Membrane (OMM) | anti-apoptosis |
Poncet et al., 2004; 2006 Arnoult et al., 2004 Pauleau et al., 2007 Norris and Youle, 2008 |
|
| BAK | OMM | anti-apoptosis |
Karbowski et al., 2006 Norris and Youle, 2008 |
|
| Growth arrest and DNA damage 45 (GADD45) | Cytosol | anti-apoptosis | Smith and Mocarski 2005 | |
| Inorganic phosphate carrier (PiC) | Inner mitochondrial membrane | decrease ATP synthesis | Poncet et al., 2006 | |
| PiC | Inner mitochondrial membrane | mitochondrial fragmentation | Pauleau et al., 2008 | |
| Unknown | ER membrane | Ca2+ release from ER | Sharon-Friling et al., 2006 | |
| Unknown | Intramitochondrial | inhibition of HtrA2/Omi-dependent, cmvPCD† | McCormick et al., 2008 | |
Proteins that have been documented to interact with pUL37x1\vMIA are listed. Also included are the probable site of interaction, the resulting activity and the reference for each interacting protein.
Serine protease-dependent CMV infected cell specific programmed cell death
HCMV UL37 proteins recruit Bax to the OMM through residues (aa 135-141) in its second anti-apoptotic domain, trigger Bax oligomerization, and thereby block formation of the permeabilization complex [85, 87, 90]. Only primate cytomegaloviruses have conserved sequence homologues of pUL37x1\vMIA [93]; although murine CMV encodes a functional analogue, m38.5, which binds to Bax and has anti-apoptotic activity [83, 94, 95]. Another IE product of the HCMV UL36-38 locus, pUL36, targets caspase 8 and is defective in some HCMV strains [69]. An early product encoded by the same gene locus, the UL38 protein (pUL38), blocks the action of tuberous sclerosis tumor suppressor complex, a modulator of mTORC1 and protects the infected cell from apoptosis during late times [96, 97]. Of the HCMV UL36-38 anti-apoptotic proteins, only the UL37 proteins target mitochondria localized, Bax -mediated initiation and execution of apoptosis at the OMM and display this unconventional ER to mitochondria protein trafficking.
Inhibition of serine protease-dependent CMV infected cell specific programmed cell death (cmvPCD)
Late events in the HCMV infected cell result in cell fragmentation through the action of serine proteases and independent of caspases [98]. HCMV pUL37x1\vMIA has evolved to also suppress this cmvPCD effected by mitochondrial localized serine protease HtrA2/Omi, although it is not clear whether this suppression results from direct or indirect effects of pUL37x1\vMIA on HtrA2/Omi [98].
Mitochondrial fragmentation
Through the action of pUL37x1\vMIA HCMV infection causes fragmentation of mitochondria that results from decreased fusion [88, 99, 100]. Because Ca2+ waves can result in mitochondrial Ca2+ overload with its resulting loss of membrane potential and release of pro-apoptotic factors, mitochondrial fragmentation can serve as protective mechanism to suppress apoptotic cell death [99]. Mitochondrial fragmentation induced by pUL37x1\vMIA can be reversed by increased fusion through ectopic expression of Bax [100]. The anti-apoptotic and mitochondrial fragmentation activities of pUL37x1\vMIA are genetically separable, because mutants defective in anti-apoptosis still cause mitochondrial fragmentation [85]. Rather, pUL37x1\vMIA mediated mitochondrial fragmentation results from inhibition of mitochondrial PiC [92].
Ca2+ efflux and F-actin rearrangement
HCMV pUL37x1\vMIA co-localizes with SERCA, a sarcoplasmic reticulum Ca2+ ATPase, induces Ca2+ release from the ER, and causes cytoskeletal changes during infection [72, 86]. Ca2+ release reorganises the cellular F-actin network and produces the characteristic early cytopathology of HCMV-infected cells [72, 86, 87]. The defect in actin polymerization is not secondary to a defect in Rho-GTPase function [86] nor does pUL37x1\vMIA have a direct catabolic effect on actin [86].
Transactivation
The HCMV UL36-38 locus is required for its oriLyt DNA replication [101-104]. Expression of nuclear genes, including induction of the human heat shock protein 70 (hsp70) and selected HCMV early genes, can be regulated by pUL37x1\vMIA and gpUL37 [78, 105, 106]. The pUL37x1\vMIA acidic domain (aa 81-108) plays a role in transactivation of HCMV early gene promoters required for viral DNA replication [107] while the gpU37COOH TM/cytosolic domains transactivate cellular promoters [105]. pUL37x1\vMIA also negatively regulates the HCMV US3 IE promoter [108]. Because of their predominant ER/MAM/mitochondrial localization, the mechanism of transactivation by UL37 IE proteins is predictably indirect, possibly by Ca2+ signalling, rather than direct interactions with transcription machinery as observed with nuclear regulatory factors.
Hepatitis C Virus
Medical importance
HCV is the major causative agent of non-A, non-B hepatitis [109] and is estimated to chronically infect 3-5% of the world's population [110-112]. Importantly, 10-20% of chronically infected individuals develop liver cirrhosis, while 1-5% develop hepatocellular carcinoma [113].
HCV is an enveloped virus of the Flaviviridae family, with an uncapped positive-sense, single-stranded RNA genome of ∼9.6 kb, which encodes a single ORF [114-116]. The highly mutable genome gives rise to different viral subpopulations found in serum, liver, and peripheral blood mononuclear cells; and has lead to the classification of HCV into 6 major genotypes, which differ in genetic sequence by >30% [111].
HCV core protein
Translation of the HCV ORF initiates from an internal ribosome entry site within the 5′ untranslated region and produces a polyprotein (∼3000 aa) which is post-translationally cleaved by cellular and viral proteases to generate ten viral proteins. The most N-terminal of these is the structural capsid protein, the core or C protein. Cleavage of the large HCV polyprotein at aa191/192 by a host signal peptidase creates an immature membrane-anchored form of the core protein, p23 (Figure 3). The immature core protein has three domains [36, 117]. The N-terminal Domain I (aa1-118) is hydrophilic and high in basic residues, has potential phosphorylation sites [118], and functions in protein stability and non-specific RNA binding. Domain II (aa119-174) consists of a hydrophobic stretch of residues, which are necessary for proper protein folding, multimerization, and association of the mature core protein with membranes and LDs. Domain III (aa175-191) represents the signal sequence of the HCV E1 protein. Subsequently, the C-terminal membrane anchor (Domain III) is thought to be cleaved by an intramembrane presenilin-type signal peptide peptidase [36, 119, 120] to release mature core protein, p21.
Figure 3. HCV core protein domains.
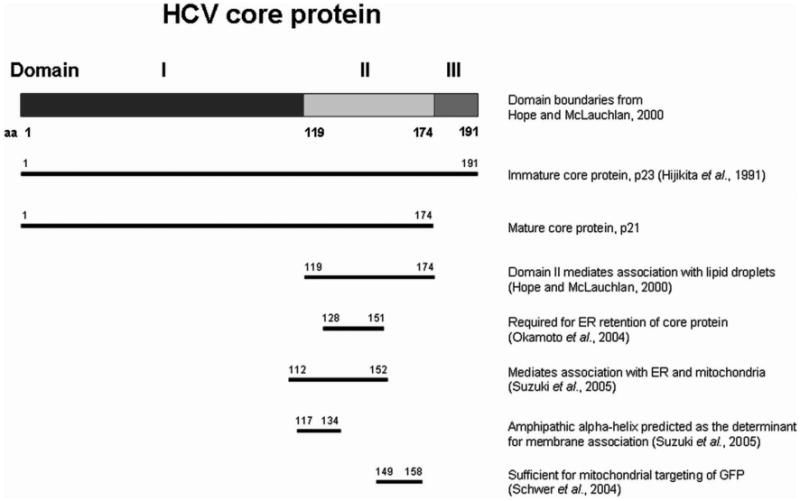
The domains of HCMV core protein important for membrane association and proper subcellular protein trafficking are shown. On the right are descriptions of the amino acid segment as well as the references which characterized them.
Localization and targeting of the HCV core protein
Mature HCV core protein is mostly cytosolic and associates with ER membranes and LDs [35, 121, 122]. HCV core protein recruits nonstructural proteins, HCV RNAs, and the replication complex to LD-associated membranes [40]. This recruitment appears to be important for the assembly of infectious progeny. The HCV core protein is also detected in the nucleus [123-126], associated with the medial-Golgi compartment [127], on the RER in close proximity to mitochondria [121], as well as in direct contact with OMM [125, 128, 129].
HCV core protein targeting sequences
Nuclear localization of the core protein uses multiple, redundant primary sequence domains scattered throughout the protein [130]. In contrast, a short sequence within Domain II suffices to target the HCV core protein to its multiple cytosolic locations. McLauchlan and Hope first identified aa 119-174 in Domain II as necessary for LD association and protein stability of the core protein [117]. Subsequently, others found aa 128-151 sufficient for ER retention of the precursor core protein (containing the C-terminal membrane anchor) [131] and aa112-152 of the mature core protein, lacking the C-terminal membrane anchor, sufficient to mediate membrane association with both ER and mitochondrial membranes [130]. The amphipathic alpha-helix therein (aa117-134) was predicted to direct membrane association [130]. Conversely, aa 149-158 were sufficient to target a recombinant green fluorescent protein (GFP) to the OMM [128]. Whether mitochondrial targeting of the core-GFP recombinant protein (which shares aa 149-152 but lacks the amphipathic alpha-helix) occurs by a similar mechanism as the previously characterized sequences has yet to be determined.
Targeting of HCV core protein to different subcellular locations also depends on factors beyond its primary sequence. Protein expression levels affected its subcellular trafficking. In transiently transfected HeLa and Huh-7 cells, high expression levels targeted the core protein predominantly to ER and LDs. However, low core protein levels resulted in mitochondrial localization [128]. High expression levels may saturate trafficking machinery and result in targeting to alternate or secondary pathways. Alternatively, over-expression of some viral proteins may alter the physiological state of the cell and, thereby affect protein trafficking. Interestingly, recent studies link regulation of HCV core protein expression levels with the levels of cellular vimentin, an intermediate filament protein, which attaches to the ER, mitochondria and nucleus [132]. Additionally, it was reported that cell-specific variations in LD composition may affect the subcellular localization of HCV core protein [117].
HCV core protein activities
The HCV core protein is rich in proline and basic residues and binds circularized HCV genomes to form viral nucleocapsids [133]. Besides playing a role in virion assembly, the HCV core protein has been implicated in the alteration of cellular signalling pathways involved in lipid metabolism [35, 39], apoptosis [134], transcription [135, 136], and transformation [137-140]. These alterations can lead to enhancement of de novo fatty acid biosynthesis [141], increased production of cellular reactive oxygen species [142], and impairment of the mitochondrial electron transfer system [142].
Induction of apoptosis
ER stress
Over-expression of a transfected HCV genotype 1b core gene or of the full length HCV replicon reproducibly causes ER stress and induction of Grp78/BiP, Grp94, calreticulin, and SERCA expression levels [143]. HCV core protein overexpression also increases CHOP/GADD153 expression, causes Bax translocation to the OMM, and induces apoptosis [143].
ER Ca2+ depletion
Over-expression of the HCV core protein also causes ER store Ca2+ depletion. Although ER stress occurred prior to ER Ca2+ depletion, no association between the two events has been verified. In liver-derived cells, ER Ca2+ depletion appeared to result from inhibition in Ca2+ absorption into the ER, conflicting with similar studies in transfected Jurkat (lymphoid) cells, which attributed ER Ca2+ depletion to induction of Ca2+ leakage from the ER [144].
Both ER Ca2+ depletion and CHOP/GADD153 induction are known to activate Bax and trigger its translocation to OMM [145, 146]. In cells expressing core protein, mitochondrial membrane depolarization, resulting from Bax translocation, could be inhibited either by treatment of cells with a Ca2+ chelator or by treatment with the pancaspase inhibitor, z-VAD-fmk. Thus, HCV core protein may redundantly encode pro-apoptotic functions via ER stress activation as well as ER Ca2+ depletion.
Recent advances in the field have allowed studies using HCV infection in vitro. Intriguingly, infection of liver derived cells with an HCV genotype 2a virus did not result in induction of Grp78 or ER stress [147]. However, similar to the transfection studies, HCV infection did cause increased Bax activation as well as induction of apoptosis through the intrinsic pathway. ER Ca2+ depletion was not assessed in these infection studies. Whether this downplays the role of ER stress during HCV infection or simply reflects a genotype specific phenomenon [148] has yet to be determined.
Targeting of viral proteins to the MAM and mitochondria
Little is known about how MAM domain boundaries are demarcated within the broader context of the ER, the extent of continuity of ER-mitochondrial bridges formed by MAM, or how limited cargo exchange between ER and mitochondria is accomplished without extensive amalgamation of the two organelles. Understanding how proteins are targeted to, and through, the MAM will certainly yield valuable insights into its structure and function.
Both soluble and integral membrane cellular proteins have been found associated with or within the MAM. Some MAM proteins target high lipid density domains or lipid rafts. However, to our knowledge, there is no published evidence of cellular proteins trafficking from the MAM into OMM, although their lipid products and Ca2+ are readily transferred between these compartments. In contrast, viral proteins, including HCMV UL37 proteins and HCV core protein, appear to traffic from the MAM into mitochondria. Predictably, we will discover differences in how they use this trafficking pathway.
Although HCV core protein can exist as an integrally-associated membrane precursor protein, its predominant form is a mature, soluble and cytosolic protein, which associates peripherally with cellular membranes. In contrast, HCMV UL37 proteins remain membrane anchored during their sequential trafficking. Because of their distinct topologies and targeted MAM domains, these proteins use different targeting signals to traffic to the MAM (Figure 2 and 3). We found that comparison of multiple cellular and viral proteins that target the MAM show no apparent consensus targeting sequences (data not shown).
We propose that HCMV UL37 proteins may traffic through the MAM into the OMM by one of a few mechanisms (Figure 4). Based upon the precedent of lipid exchange at the MAM and OMM at membrane bridges and the presence of visible tethers between the MAM and OMM [22, 43], pUL37x1\vMIA could traffic by membrane bridges used for lipid exchange. There is some evidence that HCMV pUL37x1\vMIA is located in close proximity in the MAM to mitochondria, potentially sites of their transfer [42, 128]. However, the resolution of the confocal microscopy is not sufficient to confirm direct transfer of viral proteins. Alternatively, trafficking could be mediated by vesicles as in the secretory pathway or by transfer of MAM lipid-rich domains, possibly rafts, between closely apposed MAM and OMM membranes. This may reflect the high density of lipid synthetic enzymes and of their products in the MAM. We have indications that HCMV pUL37x1\vMIA is in high lipid domains in close proximity to Sig-1R (Figure 5). We are investigating whether direct or indirect mechanisms underlie the transfer of HCMV UL37 proteins.
Figure 4. Models of translocation of HCMV pUL37x1\vMIA protein from the MAM to the OMM.
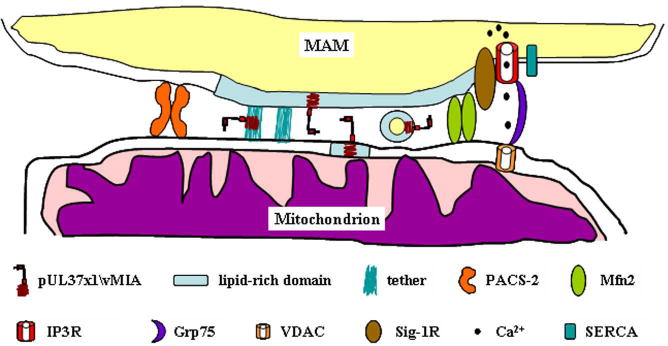
Detail of the proximity between the MAM and mitochondria is shown. The lipid enriched membrane of the MAM is represented (blue region) in the membrane. Complexes stabilizing the ER-mitochondrial contacts (PACS-2, Mfn2, and IP3R-Grp75-VDAC) are represented as well. Tethers observed by Csordas et al. [22] may serve as sites for direct transfer of pUL37x1\vMIA. Alternatively, lipid rich membrane domains may be distorted allowing close proximity between the membranes and the generation of vesicles/microvesicles or as lipid rafts that may traffic to the OMM. The key of the symbols is at the bottom of the figure.
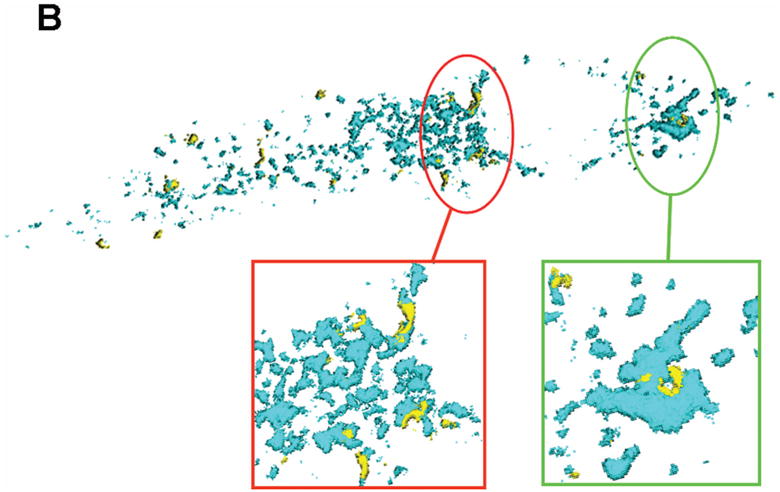
Figure 5. Physical proximity of HCMV pUL37x1 to lipid enriched MAM domains.
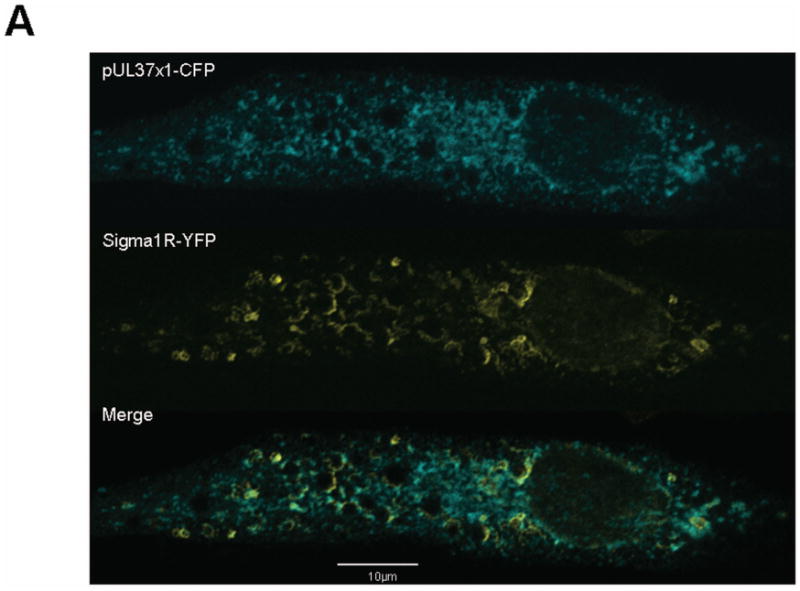
A. Shown is a single optical section of human fibroblasts expressing HCMV pUL37x1-Cerulean fluorescent protein (CFP) and Sig-1R-enhanced yellow fluorescent protein (EYFP). 24 hours after transfection, cells were fixed and imaged as in Figure 1.
B. Surface-rendered 3D recreation of a Z-stack series through the transfected cell in Panel A. Regions where pUL37x1 and Sig-1R are in close proximity are circled and enlarged on the bottom of the figure. Similar morphology and curvatures are seen with both proteins in these regions, suggesting that they occupy the same compartment.
Summary
Both RNA (cytoplasmic) and DNA (nuclear) viruses tap into the MAM, indicating convergent evolution has targeted this crucial ER-subdomain. The MAM monitors in both ER and mitochondria and responsively regulates metabolism, protein and lipid trafficking, calcium signalling, and cell survival. Trafficking of viral proteins to the MAM places them at a nexus where ER and mitochondrial functions are integrated. Machineries that regulate apoptosis, Ca2+ signalling, lipid synthesis and transport and metabolism can be usurped at this site for viral replication. Gaining the ability to manipulate diversified cellular machineries, by accessing MAM sub-domains, predictably affords viruses the flexibility to usurp control from a wider variety of cell types under a variety of physiological states. Manipulation of these cellular pathways will dictate, in large measure, the ability of the infectious progeny to be produced in the infected cell. Particularly in terms of medically pertinent HCMV infection, it is easy to conceive a role of the “quasi-synaptic” MAM for viral persistence in neuronal and cardiac cells, as well as in disease phenotype.
Acknowledgments
The authors are grateful to Drs. Teruo Hayashi and Tsung-Ping Su for the generous gift of the Sigma-1-EYFP expression vector.
Our studies were funded, in part, by NIAID, NIH (R01 AI057906), and by Discovery and Research Advisory Council funds from Children's Research Institute and the CNMC Board of Visitors. The confocal microscopy imaging was supported by a core grant (1P30HD40677) to the Children's Mental Retardation and Developmental Disabilities Research Center.
Abbreviations used
- ANT
adenine nucleotide translocase
- Ca2+
calcium
- CFP
Cerulean fluorescent protein
- CCMV
chimpanzee cytomegalovirus
- cmvPCD
serine protease-dependent CMV infected cell specific programmed cell death
- DPM-1
dolichyl phosphate mannose synthase 1
- EGFP
enhanced green fluorescent protein
- EYFP
enhanced yellow fluorescent protein
- FACL-4
fatty acid coenzyme A ligase 4
- GADD45
Growth arrest and DNA damage 45
- GFP
green fluorescent protein
- gpUL37
UL37 glycoprotein
- gpUL37COOH
carboxyl-terminal fragment of gpUL37
- Grp75
glucose regulated protein 75
- Grp78\BiP
glucose regulated protein 78
- HCMV
human cytomegalovirus
- hsp70
heat shock protein 70
- IE
immediate early
- IMM
inner mitochondrial membrane
- IP3R
inositol 1,4,5-triphosphate receptor
- LD
lipid droplet
- MAM
mitochondria-associated membrane
- mEGFP
monomeric enhanced green fluorescent protein
- Mfn2
mitofusin 2
- MT
microtubule
- OMM
outer mitochondrial membrane
- PiC
inorganic phosphate carrier
- PS
phosphatidylserine
- PSS-1
phosphatidylserine synthase type 1
- PSS-2
phosphatidylserine synthase type 2
- pUL37x1
UL37 exon 1 protein
- pUL38
UL38 protein
- RER
rough ER
- RhCMV
rhesus monkey cytomegalovirus
- SERCA
a sarcoplasmic reticulum Ca2+ ATPase
- SER
smooth ER
- siRNA
small interfering RNA
- Sig-1R
sigma-1 receptor
- TM
transmembrane
- UL37x1
UL37 exon 1
- pUL37NH2
UL37 amino-terminal fragment
- VDAC
voltage dependent anion channel
- vMIA
viral mitochondria-localized inhibitor of apoptosis
References
- 1.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruby JR, Dyer RF, Skalko RG. Continuities between mitochondria and endoplasmic reticulum in the mammalian ovary. Z Zellforsch Mikrosk Anat. 1969;97:30–37. doi: 10.1007/BF00331868. [DOI] [PubMed] [Google Scholar]
- 3.Spacek J, Lieberman AR. Relationships between mitochondrial outer membranes and agranular reticulum in nervous tissue: ultrastructural observations and a new interpretation. J Cell Sci. 1980;46:129–147. doi: 10.1242/jcs.46.1.129. [DOI] [PubMed] [Google Scholar]
- 4.Bracker CE, Grove SN. Continuity between cytoplasmic endomembranes and outer mitochondrial membranes in fungi. Protoplasma. 1971;73:15–34. doi: 10.1007/BF01286408. [DOI] [PubMed] [Google Scholar]
- 5.Lewis JA, Tata JR. A rapidly sedimenting fraction of rat liver endoplasmic reticulum. J Cell Sci. 1973;13:447–459. doi: 10.1242/jcs.13.2.447. [DOI] [PubMed] [Google Scholar]
- 6.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 7.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 8.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls DG. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int J Biochem Cell Biol. 2002;34:1372–1381. doi: 10.1016/s1357-2725(02)00077-8. [DOI] [PubMed] [Google Scholar]
- 10.Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004:re1. doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- 11.Meier PJ, Spycher MA, Meyer UA. Isolation and characterization of rough endoplasmic reticulum associated with mitochondria from normal rat liver. Biochim Biophys Acta. 1981;646:283–297. doi: 10.1016/0005-2736(81)90335-7. [DOI] [PubMed] [Google Scholar]
- 12.Marsh BJ, Mastronarde DN, Buttle KF, Howell KE, McIntosh JR. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc Natl Acad Sci U S A. 2001;98:2399–2406. doi: 10.1073/pnas.051631998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickett CB, Montisano D, Eisner D, Cascarano J. The physical association between rat liver mitochondria and rough endoplasmic reticulum. I. Isolation, electron microscopic examination and sedimentation equilibrium centrifugation analyses of rough endoplasmic reticulum-mitochondrial complexes. Exp Cell Res. 1980;128:343–352. doi: 10.1016/0014-4827(80)90070-1. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 15.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabadkai G, Rizzuto R. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett. 2004;567:111–115. doi: 10.1016/j.febslet.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 17.Pizzo P, Pozzan T. Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol. 2007;17:511–517. doi: 10.1016/j.tcb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- 19.Walter L, Hajnoczky G. Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J Bioenerg Biomembr. 2005;37:191–206. doi: 10.1007/s10863-005-6600-x. [DOI] [PubMed] [Google Scholar]
- 20.Shore GC, Tata JR. Two fractions of rough endoplasmic reticulum from rat liver. I. Recovery of rapidly sedimenting endoplasmic reticulum in association with mitochondria. J Cell Biol. 1977;72:714–725. doi: 10.1083/jcb.72.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ardail D, Gasnier F, Lerme F, Simonot C, Louisot P, Gateau-Roesch O. Involvement of mitochondrial contact sites in the subcellular compartmentalization of phospholipid biosynthetic enzymes. J Biol Chem. 1993;268:25985–25992. [PubMed] [Google Scholar]
- 22.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. Embo J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stiban J, Caputo L, Colombini M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J Lipid Res. 2008;49:625–634. doi: 10.1194/jlr.M700480-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- 26.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 27.Bozidis P, Williamson CD, Colberg-Poley AM. Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Current Protocols in Cell Biology. 2007;3.27:1–23. doi: 10.1002/0471143030.cb0327s37. [DOI] [PubMed] [Google Scholar]
- 28.Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ardail D, Popa I, Bodennec J, Louisot P, Schmitt D, Portoukalian J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem J. 2003;371:1013–1019. doi: 10.1042/BJ20021834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voelker DR. Bridging gaps in phospholipid transport. Trends Biochem Sci. 2005;30:396–404. doi: 10.1016/j.tibs.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Vance JE. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: two metabolically related aminophospholipids. J Lipid Res. 2008;49:1377–1387. doi: 10.1194/jlr.R700020-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. Embo J. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, Zollo M, Borsani G, Ballabio A, Renieri A. FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics. 1998;47:350–358. doi: 10.1006/geno.1997.5104. [DOI] [PubMed] [Google Scholar]
- 34.Rusiñol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- 35.Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, Eder G, Schaff Z, Chapman MJ, Miyamura T, Brechot C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci U S A. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo J. 2002;21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997;78(Pt 7):1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 38.Sabile A, Perlemuter G, Bono F, Kohara K, Demaugre F, Kohara M, Matsuura Y, Miyamura T, Brechot C, Barba G. Hepatitis C virus core protein binds to apolipoprotein AII and its secretion is modulated by fibrates. Hepatology. 1999;30:1064–1076. doi: 10.1002/hep.510300429. [DOI] [PubMed] [Google Scholar]
- 39.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, Koike K, Pessayre D, Chapman J, Barba G, Brechot C. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. Faseb J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 40.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 41.Machesky NJ, Zhang G, Raghavan B, Zimmerman P, Kelly SL, Merrill AH, Jr, Waldman WJ, Van Brocklyn JR, Trgovcich J. Human cytomegalovirus regulates bioactive sphingolipids. J Biol Chem. 2008;283:26148–26160. doi: 10.1074/jbc.M710181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bozidis P, Williamson CD, Colberg-Poley AM. Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J Virol. 2008;82:2715–2726. doi: 10.1128/JVI.02456-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiao YJ, Balcerzak B, Vance JE. A mitochondrial membrane protein is required for translocation of phosphatidylserine from mitochondria-associated membranes to mitochondria. Biochem J. 1998;331(Pt 1):217–223. doi: 10.1042/bj3310217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 45.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 46.Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–18364. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- 47.Perry DK. Serine palmitoyltransferase: role in apoptotic de novo ceramide synthesis and other stress responses. Biochim Biophys Acta. 2002;1585:146–152. doi: 10.1016/s1388-1981(02)00335-9. [DOI] [PubMed] [Google Scholar]
- 48.Garofalo T, Giammarioli AM, Misasi R, Tinari A, Manganelli V, Gambardella L, Pavan A, Malorni W, Sorice M. Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 2005;12:1378–1389. doi: 10.1038/sj.cdd.4401672. [DOI] [PubMed] [Google Scholar]
- 49.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- 50.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 51.Filippin L, Magalhaes PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem. 2003;278:39224–39234. doi: 10.1074/jbc.M302301200. [DOI] [PubMed] [Google Scholar]
- 52.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 53.Hayashi T, Su TP. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J Pharmacol Exp Ther. 2003;306:718–725. doi: 10.1124/jpet.103.051284. [DOI] [PubMed] [Google Scholar]
- 54.Wang HJ, Guay G, Pogan L, Sauve R, Nabi IR. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J Cell Biol. 2000;150:1489–1498. doi: 10.1083/jcb.150.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brough D, Schell MJ, Irvine RF. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem J. 2005;392:291–297. doi: 10.1042/BJ20050738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–672. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 58.Nagy A, Endreffy E, Streitman K, Pinter S, Pusztai R. Incidence and outcome of congenital cytomegalovirus infection in selected groups of preterm and full-term neonates under intensive care. In Vivo. 2004;18:819–823. [PubMed] [Google Scholar]
- 59.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93–99. doi: 10.1097/00006454-199202000-00007. [DOI] [PubMed] [Google Scholar]
- 60.Mocarski ES, Jr, Shenk T, Pass RF. Cytomegaloviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Vol. 2. Philadelphia: Wolters Kluwer Health, Lippincott Williams & Wilkins; 2007. pp. 2701–2772. [Google Scholar]
- 61.Becroft DM. Prenatal cytomegalovirus infection: epidemiology, pathology and pathogenesis. Perspect Pediatr Pathol. 1981;6:203–241. [PubMed] [Google Scholar]
- 62.Perlman JM, Argyle C. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol. 1992;31:64–68. doi: 10.1002/ana.410310112. [DOI] [PubMed] [Google Scholar]
- 63.Boeckh M, Nichols WG. The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood. 2004;103:2003–2008. doi: 10.1182/blood-2003-10-3616. [DOI] [PubMed] [Google Scholar]
- 64.Valantine H, Zuckermann A. From clinical trials to clinical practice: an overview of Certican (everolimus) in heart transplantation. J Heart Lung Transplant. 2005;24:S185–190. doi: 10.1016/j.healun.2005.01.013. discussion S210-181. [DOI] [PubMed] [Google Scholar]
- 65.Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Epstein SE. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med. 1996;335:624–630. doi: 10.1056/NEJM199608293350903. [DOI] [PubMed] [Google Scholar]
- 66.Blum A, Giladi M, Weinberg M, Kaplan G, Pasternack H, Laniado S, Miller H. High anti-cytomegalovirus (CMV) IgG antibody titer is associated with coronary artery disease and may predict post-coronary balloon angioplasty restenosis. Am J Cardiol. 1998;81:866–868. doi: 10.1016/s0002-9149(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 67.Kouzarides T, Bankier AT, Satchwell SC, Preddy E, Barrell BG. An immediate early gene of human cytomegalovirus encodes a potential membrane glycoprotein. Virology. 1988;165:151–164. doi: 10.1016/0042-6822(88)90668-x. [DOI] [PubMed] [Google Scholar]
- 68.Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han JW, Lutz RJ, Watanabe S, Cahir McFarland ED, et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci U S A. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci U S A. 2001;98:7829–7834. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Al-Barazi HO, Colberg-Poley AM. The human cytomegalovirus UL37 immediate-early regulatory protein is an integral membrane N-glycoprotein which traffics through the endoplasmic reticulum and Golgi apparatus. J Virol. 1996;70:7198–7208. doi: 10.1128/jvi.70.10.7198-7208.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mavinakere MS, Colberg-Poley AM. Dual targeting of the human cytomegalovirus UL37 exon 1 protein during permissive infection. J Gen Virol. 2004;85:323–329. doi: 10.1099/vir.0.19589-0. [DOI] [PubMed] [Google Scholar]
- 72.Sharon-Friling R, Goodhouse J, Colberg-Poley AM, Shenk T. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci U S A. 2006;103:19117–19122. doi: 10.1073/pnas.0609353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tenney DJ, Colberg-Poley AM. Expression of the human cytomegalovirus UL36-38 immediate early region during permissive infection. Virology. 1991;182:199–210. doi: 10.1016/0042-6822(91)90663-v. [DOI] [PubMed] [Google Scholar]
- 74.Adair R, Liebisch GW, Colberg-Poley AM. Complex alternative processing of human cytomegalovirus UL37 pre-mRNA. J Gen Virol. 2003;84:3353–3358. doi: 10.1099/vir.0.19404-0. [DOI] [PubMed] [Google Scholar]
- 75.Mavinakere MS, Colberg-Poley AM. Internal cleavage of the human cytomegalovirus UL37 immediate-early glycoprotein and divergent trafficking of its proteolytic fragments. J Gen Virol. 2004;85:1989–1994. doi: 10.1099/vir.0.80094-0. [DOI] [PubMed] [Google Scholar]
- 76.Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA, 3rd, Kouzarides T, Martignetti JA, et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990;154:125–169. doi: 10.1007/978-3-642-74980-3_6. [DOI] [PubMed] [Google Scholar]
- 77.Adair R, Liebisch GW, Lerman BJ, Colberg-Poley AM. Human cytomegalovirus temporally regulated gene expression in differentiated, immortalized retinal pigment epithelial cells. J Clin Virol. 2006;35:478–484. doi: 10.1016/j.jcv.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 78.Hayajneh WA, Colberg-Poley AM, Skaletskaya A, Bartle LM, Lesperance MM, Contopoulos-Ioannidis DG, Kedersha NL, Goldmacher VS. The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology. 2001;279:233–240. doi: 10.1006/viro.2000.0726. [DOI] [PubMed] [Google Scholar]
- 79.Jurak I, Brune W. Induction of apoptosis limits cytomegalovirus cross-species infection. EMBO J. 2006;25:2634–2642. doi: 10.1038/sj.emboj.7601133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu D, Silva MC, Shenk T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A. 2003;100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reboredo M, Greaves RF, Hahn G. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J Gen Virol. 2004;85:3555–3567. doi: 10.1099/vir.0.80379-0. [DOI] [PubMed] [Google Scholar]
- 82.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A. 2003;100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCormick AL, Meiering CD, Smith GB, Mocarski ES. Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J Virol. 2005;79:12205–12217. doi: 10.1128/JVI.79.19.12205-12217.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mavinakere MS, Williamson CD, Goldmacher VS, Colberg-Poley AM. Processing of human cytomegalovirus UL37 mutant glycoproteins in the endoplasmic reticulum lumen prior to mitochondrial importation. J Virol. 2006;80:6771–6783. doi: 10.1128/JVI.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pauleau AL, Larochette N, Giordanetto F, Scholz SR, Poncet D, Zamzami N, Golmacher VS, Koemer G. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene. 2007;26:7067–7080. doi: 10.1038/sj.onc.1210511. [DOI] [PubMed] [Google Scholar]
- 86.Poncet D, Pauleau AL, Szabadkai G, Vozza A, Scholz SR, Le Bras M, Briere JJ, Jalil A, Le Moigne R, Brenner C, et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J Cell Biol. 2006;174:985–996. doi: 10.1083/jcb.200604069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem. 2004;279:22605–22614. doi: 10.1074/jbc.M308408200. [DOI] [PubMed] [Google Scholar]
- 88.McCormick AL, Smith VL, Chow D, Mocarski ES. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol. 2003;77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith G, Mocarski ES. Contribution of GADD45 family members to cell death suppression by cellular bcl-xL and cytomegalovirus vMIA. J Virol. 2005;79:14923–14932. doi: 10.1128/JVI.79.23.14923-14932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci U S A. 2004;101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vieira HL, Belzacq AS, Haouzi D, Bernassola F, Cohen I, Jacotot E, Ferri KF, El Hamel C, Bartle LM, Melino G, et al. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene. 2001;20:4305–4316. doi: 10.1038/sj.onc.1204575. [DOI] [PubMed] [Google Scholar]
- 92.Pauleau AL, Galluzzi L, Scholz SR, Larochette N, Kepp O, Kroemer G. Unexpected role of the phosphate carrier in mitochondrial fragmentation. Cell Death Differ. 2008;15:616–618. doi: 10.1038/sj.cdd.4402295. [DOI] [PubMed] [Google Scholar]
- 93.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 2003;316:221–233. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 94.Jurak I, Schumacher U, Simic H, Voigt S, Brune W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol. 2008;82:4812–4822. doi: 10.1128/JVI.02570-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Norris KL, Youle RJ. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol. 2008;82:6232–6243. doi: 10.1128/JVI.02710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol. 2007;81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe. 2008;3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCormick AL, Roback L, Mocarski ES. HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA) PLoS Pathog. 2008;4:e1000063. doi: 10.1371/journal.ppat.1000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roumier T, Szabadkai G, Simoni AM, Perfettini JL, Paulau AL, Castedo M, Metivier D, Badley A, Rizzuto R, Kroemer G. HIV-1 protease inhibitors and cytomegalovirus vMIA induce mitochondrial fragmentation without triggering apoptosis. Cell Death Differ. 2006;13:348–351. doi: 10.1038/sj.cdd.4401750. [DOI] [PubMed] [Google Scholar]
- 100.Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 101.Pari GS, Kacica MA, Anders DG. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J Virol. 1993;67:2575–2582. doi: 10.1128/jvi.67.5.2575-2582.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith JA, Pari GS. Expression of human cytomegalovirus UL36 and UL37 genes is required for viral DNA replication. J Virol. 1995;69:1925–1931. doi: 10.1128/jvi.69.3.1925-1931.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iskenderian AC, Huang L, Reilly A, Stenberg RM, Anders DG. Four of eleven loci required for transient complementation of human cytomegalovirus DNA replication cooperate to activate expression of replication genes. J Virol. 1996;70:383–392. doi: 10.1128/jvi.70.1.383-392.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reid GG, Ellsmore V, Stow ND. An analysis of the requirements for human cytomegalovirus oriLyt-dependent DNA synthesis in the presence of the herpes simplex virus type 1 replication fork proteins. Virology. 2003;308:303–316. doi: 10.1016/s0042-6822(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 105.Hayajneh WA, Contopoulos-Ioannidis DG, Lesperance MM, Venegas AM, Colberg-Poley AM. The carboxyl terminus of the human cytomegalovirus UL37 immediate-early glycoprotein is conserved in primary strains and is important for transactivation. J Gen Virol. 2001;82:1569–1579. doi: 10.1099/0022-1317-82-7-1569. [DOI] [PubMed] [Google Scholar]
- 106.Yee LF. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology. 2007;363:174–188. doi: 10.1016/j.virol.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 107.Colberg-Poley AM, Huang L, Soltero VE, Iskenderian AC, Schumacher RF, Anders DG. The acidic domain of pUL37x1 and gpUL37 plays a key role in transactivation of HCMV DNA replication gene promoter constructions. Virology. 1998;246:400–408. doi: 10.1006/viro.1998.9212. [DOI] [PubMed] [Google Scholar]
- 108.Biegalke BJ. Human cytomegalovirus US3 gene expression is regulated by a complex network of positive and negative regulators. Virology. 1999;261:155–164. doi: 10.1006/viro.1999.9881. [DOI] [PubMed] [Google Scholar]
- 109.Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 110.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 111.Giannini C, Brechot C. Hepatitis C virus biology. Cell Death Differ. 2003;10(Suppl 1):S27–38. doi: 10.1038/sj.cdd.4401121. [DOI] [PubMed] [Google Scholar]
- 112.Lemon S, Walker C, Alter M, Yi M. Hepatitis C Virus. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Vol. 2. Philadelphia: Wolters Kluwer Health, Lippincott Williams & Wilkins; 2007. pp. 1253–1304. [Google Scholar]
- 113.Di Bisceglie AM. Hepatitis C. Lancet. 1998;351:351–355. doi: 10.1016/S0140-6736(97)07361-3. [DOI] [PubMed] [Google Scholar]
- 114.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, et al. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J Virol. 1991;65:1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gubler D, Kuno G, Markoff L. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Vol. 2. Philadelphia: Wolters Kluwer Health, Lippincott Williams & Wilkins; 2007. pp. 1153–1252. [Google Scholar]
- 117.Hope RG, McLauchlan J. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J Gen Virol. 2000;81:1913–1925. doi: 10.1099/0022-1317-81-8-1913. [DOI] [PubMed] [Google Scholar]
- 118.Lu W, Ou JH. Phosphorylation of hepatitis C virus core protein by protein kinase A and protein kinase C. Virology. 2002;300:20–30. doi: 10.1006/viro.2002.1524. [DOI] [PubMed] [Google Scholar]
- 119.Lemberg MK, Martoglio B. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol Cell. 2002;10:735–744. doi: 10.1016/s1097-2765(02)00655-x. [DOI] [PubMed] [Google Scholar]
- 120.Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science. 2002;296:2215–2218. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- 121.Moradpour D, Englert C, Wakita T, Wands JR. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology. 1996;222:51–63. doi: 10.1006/viro.1996.0397. [DOI] [PubMed] [Google Scholar]
- 122.Yasui K, Wakita T, Tsukiyama-Kohara K, Funahashi SI, Ichikawa M, Kajita T, Moradpour D, Wands JR, Kohara M. The native form and maturation process of hepatitis C virus core protein. J Virol. 1998;72:6048–6055. doi: 10.1128/jvi.72.7.6048-6055.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lo SY, Masiarz F, Hwang SB, Lai MM, Ou JH. Differential subcellular localization of hepatitis C virus core gene products. Virology. 1995;213:455–461. doi: 10.1006/viro.1995.0018. [DOI] [PubMed] [Google Scholar]
- 124.Matsuura Y, Harada T, Makimura M, Sato M, Aizaki H, Suzuki T, Miyamura T. Characterization of HCV structural proteins expressed in various animal cells. Intervirology. 1994;37:114–118. doi: 10.1159/000150365. [DOI] [PubMed] [Google Scholar]
- 125.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 126.Ravaggi A, Natoli G, Primi D, Albertini A, Levrero M, Cariani E. Intracellular localization of full-length and truncated hepatitis C virus core protein expressed in mammalian cells. J Hepatol. 1994;20:833–836. doi: 10.1016/s0168-8278(05)80157-6. [DOI] [PubMed] [Google Scholar]
- 127.Martire G, Viola A, Iodice L, Lotti LV, Gradini R, Bonatti S. Hepatitis C virus structural proteins reside in the endoplasmic reticulum as well as in the intermediate compartment/cis-Golgi complex region of stably transfected cells. Virology. 2001;280:176–182. doi: 10.1006/viro.2000.0733. [DOI] [PubMed] [Google Scholar]
- 128.Schwer B, Ren S, Pietschmann T, Kartenbeck J, Kaehlcke K, Bartenschlager R, Yen TS, Ott M. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J Virol. 2004;78:7958–7968. doi: 10.1128/JVI.78.15.7958-7968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 130.Suzuki R, Sakamoto S, Tsutsumi T, Rikimaru A, Tanaka K, Shimoike T, Moriishi K, Iwasaki T, Mizumoto K, Matsuura Y, et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J Virol. 2005;79:1271–1281. doi: 10.1128/JVI.79.2.1271-1281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Okamoto K, Moriishi K, Miyamura T, Matsuura Y. Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J Virol. 2004;78:6370–6380. doi: 10.1128/JVI.78.12.6370-6380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nitahara-Kasahara Y, Fukasawa M, Shinkai-Ouchi F, Sato S, Suzuki T, Murakami K, Wakita T, Hanada K, Miyamura T, Nishijima M. Cellular vimentin content regulates the protein level of hepatitis C virus core protein and the hepatitis C virus production in cultured cells. Virology. 2008 doi: 10.1016/j.virol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 133.Santolini E, Migliaccio G, La Monica N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J Virol. 1994;68:3631–3641. doi: 10.1128/jvi.68.6.3631-3641.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C, Lai MM. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J Virol. 1998;72:3691–3697. doi: 10.1128/jvi.72.5.3691-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hsieh TY, Matsumoto M, Chou HC, Schneider R, Hwang SB, Lee AS, Lai MM. Hepatitis C virus core protein interacts with heterogeneous nuclear ribonucleoprotein K. J Biol Chem. 1998;273:17651–17659. doi: 10.1074/jbc.273.28.17651. [DOI] [PubMed] [Google Scholar]
- 136.You LR, Chen CM, Yeh TS, Tsai TY, Mai RT, Lin CH, Lee YH. Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol. 1999;73:2841–2853. doi: 10.1128/jvi.73.4.2841-2853.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ray RB, Lagging LM, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. 1996;70:4438–4443. doi: 10.1128/jvi.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jin DY, Wang HL, Zhou Y, Chun AC, Kibler KV, Hou YD, Kung H, Jeang KT. Hepatitis C virus core protein-induced loss of LZIP function correlates with cellular transformation. Embo J. 2000;19:729–740. doi: 10.1093/emboj/19.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yoshida T, Hanada T, Tokuhisa T, Kosai K, Sata M, Kohara M, Yoshimura A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J Exp Med. 2002;196:641–653. doi: 10.1084/jem.20012127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chang J, Yang SH, Cho YG, Hwang SB, Hahn YS, Sung YC. Hepatitis C virus core from two different genotypes has an oncogenic potential but is not sufficient for transforming primary rat embryo fibroblasts in cooperation with the H-ras oncogene. J Virol. 1998;72:3060–3065. doi: 10.1128/jvi.72.4.3060-3065.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fukasawa M, Tanaka Y, Sato S, Ono Y, Nitahara-Kasahara Y, Suzuki T, Miyamura T, Hanada K, Nishijima M. Enhancement of de novo fatty acid biosynthesis in hepatic cell line Huh7 expressing hepatitis C virus core protein. Biol Pharm Bull. 2006;29:1958–1961. doi: 10.1248/bpb.29.1958. [DOI] [PubMed] [Google Scholar]
- 142.Koike K, Moriya K. Metabolic aspects of hepatitis C viral infection: steatohepatitis resembling but distinct from NASH. J Gastroenterol. 2005;40:329–336. doi: 10.1007/s00535-005-1586-z. [DOI] [PubMed] [Google Scholar]
- 143.Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, Paterlini-Brechot P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- 144.Bergqvist A, Sundstrom S, Dimberg LY, Gylfe E, Masucci MG. The hepatitis C virus core protein modulates T cell responses by inducing spontaneous and altering T-cell receptor-triggered Ca2+ oscillations. J Biol Chem. 2003;278:18877–18883. doi: 10.1074/jbc.M300185200. [DOI] [PubMed] [Google Scholar]
- 145.Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- 146.Pan Z, Bhat MB, Nieminen AL, Ma J. Synergistic movements of Ca(2+) and Bax in cells undergoing apoptosis. J Biol Chem. 2001;276:32257–32263. doi: 10.1074/jbc.M100178200. [DOI] [PubMed] [Google Scholar]
- 147.Deng L, Adachi T, Kitayama K, Bungyoku Y, Kitazawa S, Ishido S, Shoji I, Hotta H. Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J Virol. 2008;82:10375–10385. doi: 10.1128/JVI.00395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pazienza V, Clement S, Pugnale P, Conzelmann S, Pascarella S, Mangia A, Negro F. Gene expression profile of Huh-7 cells expressing hepatitis C virus genotype 1b or 3a core proteins. Liver Int. 2008 doi: 10.1111/j.1478-3231.2008.01866.x. [DOI] [PubMed] [Google Scholar]