

Abstract
Neural tube defects (NTDs), including anencephaly and spina bifida, arise from the failure of neurulation during early embryonic development. Neural tube defects are common birth defects with a heterogenous and multifactorial etiology with interacting genetic and environmental risk factors. Although the mechanisms resulting in failure of neural tube closure are unknown, up to 70% of NTDs can be prevented by maternal folic acid supplementation. However, the metabolic mechanisms underlying the association between folic acid and NTD pathogenesis have not been identified. This review summarizes our current understanding of the mechanisms by which impairments in folate metabolism might ultimately lead to failure of neural tube closure, with an emphasis on untangling the relative contributions of nutritional deficiency and genetic risk factors to NTD pathogenesis.
Keywords: folate, neural tube defect, metabolism, genetics, thymidylate
INTRODUCTION
Failure of the neural tube to fuse in its entirety during early embryogenesis results in a cluster of common developmental anomalies known as neural tube closure defects (NTDs). NTDs usually appear with herniation and exposure of nervous tissue in the cranial region (termed anencephaly) or the spinal region (termed spina bifida). These malformations are severe, irreversible, and debilitating. Anencephaly is incompatible with postnatal survival, whereas spina bifida results in a lifelong disability and often necessitates multiple surgical interventions. Although the morphogenic processes underlying both normal neurulation and its failure continue to be an area of active investigation, the ultimate causes and the associated biologic mechanisms of NTDs in mammals remain unknown. Over the past two decades, numerous genetic and environmental risk factors have been identified; however, the strongest association to date is between the B-vitamin folate and NTD risk. Early clinical observations in the 1960s led to an understanding that reduced maternal folate status was associated with elevated NTD risk (Hibbard, 1964, 1967; Smithells et al., 1976). Subsequent studies identified elevated maternal homocysteine, which is a biomarker of impaired folate status and/or metabolism, as a risk factor for NTDs (Steegers-Theunissen et al., 1991; Mills et al., 1995). Later, randomized control trials and population-wide fortification initiatives (Castilla et al., 2003; Mills and Signore, 2004; Sayed et al., 2008) verified the efficacy of folic acid supplementation in reducing both NTD occurrence (Czeizel and Dudas, 1992) and recurrence (MRC Vitamin Study Research Group, 1991) by up to 70%. Despite several decades of epidemiologic research indicating that folate is intimately linked to NTD risk, the metabolic mechanisms underlying the pathogenesis of folate-responsive NTDs have yet to be identified.
Although maternal folate status is linked to NTD risk, certain individuals within populations are at greater risk than others independent of folate status, indicating that folate deficiency alone is not sufficient to cause an NTD. A genetic component of NTDs has been long recognized; family history is one of the strongest risk factors for NTDs (Elwood et al., 1992). In addition, NTDs are more common among certain ethnic groups and in individuals with a previous NTD-affected pregnancy (Mitchell et al., 2004). Because folate status contributes to NTD risk, investigation of genetic risk factors in humans has focused primarily on variation within genes that encode proteins that bind, transport, process, or metabolize folate, with the assumption that genetically-induced alterations in folate status and/or metabolism are likely to contribute to NTD pathogenesis. Although polymorphisms have been identified in folate-related genes that contribute to risk of developing an NTD, the total genetic variation identified to date that contributes to NTD risk does not account for the overall genetic contribution to NTD incidence observed in human populations. Commonly, epidemiologic studies focus on synergistic gene-diet interactions and the identification of polymorphisms within folate-related genes that interact with low folate status to confer risk for developmental anomalies, including NTDs.
The profile of NTD pathogenesis is emerging as an interaction between predisposing genetic factors and primary or secondary nutrient deficiencies (Fig. 1). Although it was originally hypothesized that maternal folate supplementation lowered risk for NTDs by correcting a primary folate deficiency, it has become apparent that this explanation may not account for many cases of NTD prevention. Furthermore, folic acid supplementation may prevent NTDs even in the absence of overt maternal folate deficiency, because most women with an NTD-affected pregnancy are not folate-deficient (Molloy et al., 1985; Mills et al., 1992; Kirke et al., 1993). Increased folate intake, in the form of folic acid supplements or fortified food, may compensate for genetically-linked impairments in folate utilization and/or secondary nutrient deficiencies, not correct folate deficiency per se. Thus, investigations into gene-nutrient interactions that result in NTD pathogenesis now distinguish among: (1) single gene variants that affect folate status alone, (2) single gene variants that affect folate utilization and/or metabolism, and (3) single gene variants that affect both folate status and metabolism/utilization. Impairments in folate status can be the result of dietary folate deficiency, but can also result from genetic variation that effects cellular folate accumulation, including its absorption, cellular transport, processing, retention and degradation (Suh et al., 2001). Likewise, impairments in folate utilization and metabolism can result from genetic variation that affects the activity and/or stability of folate-dependent metabolic enzymes, but can also result from secondary deficiencies of nutrients intimately linked to folate metabolism, such as vitamin B12 and choline. Thus, both genetic risk factors and nutrient deficiencies contribute to impairments in folate status and/or impairments in folate utilization (Fig. 1). Untangling the relative contributions of the genetic and nutritional components of NTD risk will be required to identify the specific biologic pathways that lead to NTD pathogenesis, which will enable the design of better targeted and efficacious interventions for NTD prevention. In this review, our current understanding of the mechanisms underlying NTD pathogenesis in the context of folate-mediated one-carbon metabolism is summarized.
Figure 1.
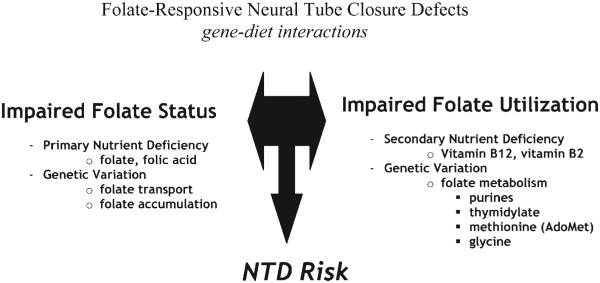
Gene nutrient interactions in neural tube closure defects. Low folate status interacts with impairments in one-carbon metabolism (folate utilization) to create risk for neural tube defects. Both folate status and folate utilization are compromised by interactions among genetic and environmental (nutritional) factors.
Folate-Mediated One-Carbon Metabolism
In the cell, folates function as a family of metabolic cofactors that carry and chemically activate single carbons, referred to as one-carbon units, for a variety of anabolic and catabolic reactions collectively known as folate-mediated one-carbon metabolism (OCM) (Fig. 2). Folate-activated one-carbons are carried by tetrahydrofolate (THF), the metabolically active form of folates. Tetrahydrofolate carries one-carbons at three different oxidation states, ranging from formaldehyde to methanol, and the one-carbon forms of folate can be interconverted enzymatically (Schirch and Strong, 1989; Appling, 1991; Wagner, 1995). Cellular folate cofactors also contain a poly-γ-glutamate peptide that varies in length in cells from three to nine glutamate residues. In the gut, the poly-γ-glutamate peptide is hydrolyzed, leaving monoglutamated folate derivatives that are transported across the intestinal mucosa. Folate monoglutamates, predominately in the form of 5-methyl-THF, are present in serum and transported into cells. Both retention of folate in the cell and the conversion to functional cofactors require the reestablishment of the poly-γ-glutamate peptide (Lin and Shane, 1994; Shane, 1995).
Figure 2.

Compartmentation of folate-mediated one-carbon metabolism in the cytoplasm and mitochondria. One-carbon metabolism in the cytoplasm is required for the de novo synthesis of purines and thymidylate and for the remethylation of homocysteine to methionine. One-carbon metabolism in mitochondria is required to generate formate for one-carbon metabolism in the cytoplasm, to generate the amino acid glycine, and to synthesize formylmethionyl-tRNA for protein synthesis in mitochondria. FTHFS, 10-formyltetrahydrofolate synthetase; MTHFC, methenyltetrahydrofolate cyclohydrolase; MTHFD, methylenetetrahydrofolate dehydrogenase; MTHFR, methylenetetrahydrofolate reductase; GARFT, phosphoribosylglycinamide formyltransferase; AICARFT, phosphoribosylaminoimidazolecarboxamide formyltransferase; cSHMT, cytoplasmic serine hydroxymethyltransferase; TS, thymidylate synthase; DHFR, dihydrofolate reductase; MS, methionine synthase; mSHMT, mitochondrial serine hydroxymethyltransferase; GCS, glycine cleavage system; SD, sarcosine dehydrogenase; DMGD, dimethylglycine dehydrogense; mMTHFD, mitochondrial methylenetetrahydrofolate dehydrogenase; mMTHFC, mitochondrial methenyltetrahydrofolate cyclohydrolase; MFT, methionyl-tRNA formyltransferase; mFTHFS, mitochondrial formyltetrahydrofolate synthetase.
Folate-mediated OCM is a metabolic network of interdependent pathways that is compartmentalized in the mitochondria, the cytoplasm, and the nucleus. Folate metabolism in mitochondria is required for the production of formate, glycine and fmettRNA from the catabolism of choline, serine, and glycine (Shane, 1989; Appling, 1991). Once formed, formate traverses from the mitochondria to the cytoplasm, where it serves as a primary source of one-carbon units for cytoplasmic OCM. Folate-mediated OCM in the cytoplasm is essential for (1) de novo purine biosynthesis, (2) de novo thymidylate biosynthesis, and (3) the remethylation of homocysteine to form methionine. Methionine is required for the biosynthesis of S-adenosylmethionine (AdoMet), which is a cofactor that serves as the universal one-carbon donor for cellular methylation reactions including methylation of chromatin, proteins, lipids, and other small molecules (Shane, 1995; Wagner, 1995). During S-phase, the enzymes that constitute the de novo thymidylate biosynthesis cycle—serine hydroxymethyltransferase, thymidylate synthase and dihydrofolate reductase—are modified by the small ubiquitin-like modifier (SUMO) and transported to the nucleus for nuclear thymidylate biosynthesis (Anderson et al., 2007; Woeller et al., 2007). Impairments in folate-mediated OCM can result from diminished folate status, polymorphisms in genes that encode folate-metabolizing enzymes, or secondary micronutrient deficiencies that alter folate status, including other B vitamins (Bailey, 1995; Stover and Garza, 2002; Stover, 2004). Biomarkers of impaired OCM include diminished capacity to synthesize thymidylate de novo leading to increased uracil content into DNA (Blount et al., 1997), elevated serum homocysteine (Selhub, 1999), and DNA hypomethylation (Rampersaud et al., 2003; Bai et al., 2005).
Folate Transport
Sources of dietary folates include food folates, which contain a polyglutamate peptide, and folic acid, a synthetic dietary supplement and fortificant. Folic acid is a monoglutamic and oxidized form of folate that, unlike natural reduced folates, is chemically stable. Most folic acid is readily absorbed and converted to THF within the enterocytes where it becomes chemically indistinguishable from natural food folates (Gregory, 2001). Polyglutamated food folate derivatives must first be converted to monoglutamate derivatives by the enzyme folylpoly-γ-glutamate carboxypeptidase II (gene name GCPII) in the gut before absorption through the intestine (Tamura and Stokstad, 1973), which is accomplished through a recently identified intestinal folate receptor (PCFT) (Qiu et al., 2006). Serum folates are present in the form of monoglutamated 5-methyl-THF, which is taken up at the cell surface either by the reduced folate carrier (RFC), a facilitative anion-exchange carrier, or by an endocytotic process mediated by one of two folate receptors, FRα and FRβ (Kamen et al., 1988). The folate receptor is a membrane-anchored receptor with a glycosyl-phosphatidyl-inositolmoiety that has a high affinity for 5-methyl-THF (Kamen et al., 1988). Once inside the cell, polyglutamation of folate cofactors is catalyzed by the enzyme folylpoly-γ-glutamate synthetase, resulting in the sequestration of folates within the cell. The polyglutamate peptide also increases the affinity of THF cofactors for folate-dependent enzymes (Schirch and Strong, 1989; Wagner, 1995). Within the cell, 5-methyl-THF is the most abundant folate derivative.
Studies to date have not provided conclusive evidence that common polymorphisms within genes that encode proteins that mediate folate transport and absorption affect folate status. There have not been any variants identified within the coding regions of the folate receptor genes, and investigations of polymorphisms in noncoding regions of FRα and FRβ have not yielded an association with NTD risk (Barber et al., 1998, 2000; O’Leary et al., 2003; Boyles et al., 2006). Coding variants within genes that impair folate transport and accumulation may not be compatible with life, resulting in embryonic lethality. It is also possible that upregulation of folate transporter expression in response to folate deficiency, if it occurs, could mask deleterious genetic variation. Nonetheless, genetic variation within the folate receptor genes does not confer risk for NTDs in humans. A common single nucleotide polymorphism (SNP) in the RFC1 gene has shown a moderate association with NTD risk under conditions of folate deficiency (Shaw et al., 2002; Morin et al., 2003a; Pei et al., 2005), although with low penetrance. In regard to genes encoding proteins that mediate folate processing, no polymorphisms have been identified within the gene encoding folylpoly-γ-glutamate synthetase that confer risk for NTDs in human populations. Similarly, a single variant identified in GCPII does not affect NTD risk in humans (Afman et al., 2003). Thus, there is little evidence that genetic variation disrupts folate transport, absorption, processing, and retention to a degree that independently contributes to NTD risk in human populations. However, given that NTDs are complex traits, genetic alterations of these processes might exacerbate nutritional deficiencies or metabolic impairments, and thereby sensitize population subgroups with genetically-induced impairments in OCM.
A definitive and causal role of embryonic folate deficiency in NTD pathogenesis has been demonstrated by the observation that genetic deletion of the gene encoding FRα, Folr1, results in NTDs in mice (Piedrahita et al., 1999). However, investigation of OCM in this mouse model has not provided mechanistic information on the identity of the one-carbon pathway component that underlies NTD pathogenesis. There are no differences observed in homocysteine levels in FolR1+/− dams maintained on a normal diet (Piedrahita et al., 1999), nor are there any differences in global DNA methylation in FolR1+/− embryos or FolR1−/− embryos rescued to gestation day 15.5 with folinic acid (Finnell et al., 2002). Furthermore, that NTDs in nullizygous FolR1 knockout embryos can be rescued by maternal supplementation with folic acid (Piedrahita et al., 1999) provides further support for the concept that genetic disruption of folate transport affects folate status in the absence of alterations in folate utilization, because reductions in transport capacity can be overcome by maternal vitamin supplementation. A similar finding has been observed with deletion of the gene encoding RFC in mice. Deletion of RFC results in early embryonic lethality; however, embryonic survival can be rescued up until gestation day 12 by maternal folic acid supplementation (Zhao et al., 2001). Collectively, these data suggest that adequate folate status mediated by cellular folate uptake is required for embryonic development and neural tube closure. However, in the absence of an association between human genes involved in folate uptakes and NTD risk, these models do not provide further insight into specific causes that underlie human folate-responsive NTDs.
Folate Metabolism in the Mitochondria
The primary role of folate metabolism in mitochondria is to generate formate and glycine from the enzymatic cleavage of serine. In certain tissues, glycine can also be catabolized to generate formate through mitochondrial folate metabolism (Christensen and MacKenzie, 2006). This pathway is initiated by the mitochondrial isoform of serine hydoxymethyltransferase (mSHMT; gene name Shmt2), which catalyzes the conversion of serine and THF to form glycine and methylene-THF. Methylene-THF can also be synthesized from glycine by the glycine cleavage system (Motokawa and Kikuchi, 1971), as well as from the catabolism of sarcosine and dimethylglycine (Wittwer and Wagner, 1981). Methylene-THF is oxidized to produce methenyl-THF in a reaction catalyzed by methylenetetrahydrofolate dehydrogenase (MTHFD), which is subsequently hydrolyzed to 10-formyl THF by the enzyme methenyltetrahydrofolate cyclohydrolase (MTHFC). The formyl group of 10-formyl-THF is hydrolyzed to generate free formate and THF to complete the cycle, in a reaction catalyzed by formyltetrahydrofolate synthetase (FTHFS) (Christensen et al., 2005). Formate then traverses into the cytoplasm and serves as a major source of one-carbon units for cytoplasmic OCM. Some of the genes encoding the enzymes that catalyze the generation of formate from 5,10-methylene-THF in the mitochondria have yet to be identified (Christensen and MacKenzie, 2006).
To date, there have been few reports or investigations into the role of mitochondrial folate metabolism in NTD pathogenesis; this includes investigations of human genetic susceptibility and genetically-manipulated mouse models. One limitation is the paucity of knowledge regarding the identity of the genes and enzymes that regulate OCM in the mitochondria and the degree to which the capacity of mitochondrial OCM, including formate production, affects cytoplasmic OCM. Furthermore, mitochondrial OCM plays different metabolic roles in different tissues and cell types. Whereas it has been shown in certain cell types that mitochondrial OCM is an essential source of glycine (Christensen and MacKenzie, 2006), it remains to be established definitively that formate derived from mitochondrial OCM is essential for cytoplasmic OCM. Recently, a mouse knockout model of the gene encoding the cytoplasmic SHMT isoform (cSHMT; Shmt1) was generated and was shown to be both viable and fertile (Macfarlane et al., 2008). The viability of this mouse model indicates that one-carbon units generated in the cytoplasm by cSHMT, through the expression of Shmt1, are not essential for growth and survival. This mouse model emphasizes the importance of mitochondrial OCM in the production of one-carbon units for folate-dependent anabolic reactions in the cytoplasm. Further investigation into the role of mitochondrial OCM in regulating folate-dependent anabolic pathways in the cytoplasm is warranted. In addition, exploration of human polymorphisms in mitochondrial folate-dependent enzymes and the creation of mouse models with disruptions in genes encoding the mitochondrial folate pathway will shed light on the potential contribution of mitochondrial folate metabolism to NTD pathogenesis.
Folate Metabolism in the Cytoplasm
Cytoplasmic OCM is essential for the de novo biosynthesis of nucleotides and AdoMet-dependent cellular methylation reactions (Fig. 2). The purine biosynthesis pathway utilizes the cofactor 10-formyl-THF as the one-carbon donor for carbons 2 and 8 in the purine ring. De novo thymidylate biosynthesis utilizes methylene-THF as a cofactor for the methylation of deoxyuridine monophosphate (dUMP) to form deoxythymidine monophosphate (dTMP), in a reaction catalyzed by the enzyme thymidylate synthase (TS). The one-carbons carried by 10-formyl-THF and methylene-THF can be generated in the cytoplasm from formate via the action of the adenosine triphosphate (ATP)–dependent and nicotinamide adenosine dinucleotide phosphate (NADPH)-dependent trifunctional enzyme, which contains FTHFS/MTHFC/MTHFD activities encoded by the MTHFD1 gene. Methylene-THF can also be synthesized from the pyridoxal phosphate (PLP)-dependent enzymatic conversion of serine and THF, catalyzed by the enzyme cytoplasmic SHMT (cSHMT, gene name Shmt1), a reaction that also generates glycine (Fig. 2). Methylene-THF can be irreversibly reduced by the enzyme methylene-THF reductase (MTHFR) to yield 5- methyl THF, which serves as the cofactor for the B12-dependent remethylation of homocysteine to form methionine, catalyzed by methionine synthase (MS; gene name MTR). Methionine can be adenosylated to form S-adenosylmethionine (AdoMet), which is the one-carbon donor for cellular methylation reactions. The transfer of the one-carbon from AdoMet yields the intermediate S-adenosylhomocysteine (AdoHcy), which is hydrolyzed to homocysteine and adenosine by the enzyme S-adenosylhomocysteine hydrolase.
Although much is known about OCM and its anabolic pathways in the cytoplasm, deciphering the causal metabolic pathway associated with NTD risk has been challenging. Within the cell, the concentration of folate-binding proteins and enzymes far exceeds the concentration of folate cofactors, and thus all cellular folate is protein bound (Suh et al., 2001). This indicates that folate-dependent anabolic reactions in the cytoplasm compete for a limiting pool of folate-derived one-carbon units and folate cofactors (Scott et al., 1981; Suh et al., 2001). This competition is greatest for the two anabolic reactions that utilize 5,10-methylene-THF, thymidylate biosynthesis, and homocysteine remethylation (leading to AdoMet biosynthesis). These two pathways are sensitive to folate deficiency; therefore, it is challenging to determine independently the effect of either thymidylate synthesis or homocysteine remethylation (and cellular methylation reactions) on NTD risk. It has been suggested by mathematical modeling (Green et al., 1988) and experimental data (Scott et al., 1981) that under normal cellular conditions, the biosynthesis of 5-methyl-THF is favored over the biosynthesis of thymidylate.
Purine Biosynthesis
Rapid cell proliferation is essential for neural tube closure, thereby requiring increased rates and dependence on de novo nucleotide biosynthesis. Although the thymidylate biosynthesis and homocysteine remethylation pathways are highly sensitive to folate status, the purine biosynthetic pathway appears to be less sensitive to conditions of folate deficiency (Field et al., 2006). The “higher priority” conferred to purine biosynthesis within the OCM network may be due to the dependency of most organisms on maintenance of de novo purine biosynthesis for development and survival. The purine biosynthetic machinery utilizes exclusively 10-formyl-THF as the folate cofactor for the biosynthesis of the purine ring. In addition to 10-formyl-THF, there are two other folate derivatives found in cells at the level of oxidation of formate: 5-formyl-THF and 5,10-methenyl-THF. These folate derivatives participate in a futile cycle that involves the SHMT-catalyzed synthesis of 5-formyl-THF from methenyl-THF, and the ATP-dependent conversion of 5-formyl-THF back to methenyl-THF by the enzyme methenyl-THF synthetase (MTHFS) (Bertrand and Jolivet, 1989; Stover and Schirch, 1990). Although neither of these folate derivatives serve as substrates for folate-dependent anabolic reactions, recent evidence indicates that regulation of MTHFS and 5-formyl levels may impact cellular folate accumulation and regulate purine biosynthesis. Anguera et al. (2003) demonstrated that increased MTHFS expression depleted cellular folate concentration, and Field et al. (2006) showed that MTHFS expression enhanced purine biosynthesis through the sequestration and potential shunting of 10-formyl-THF into the purine biosynthetic pathway. Furthermore, the binding of 10-formyl-THF to MTHFS inhibits 5-formyl-THF metabolism, enabling its accumulation. In this regard, 5-formyl-THF may function as a storage form of folate in the cell that can be quickly mobilized for purine biosynthesis (Field et al., 2006). Thus, ongoing investigations of de novo purine biosynthesis and its regulation are elucidating mechanisms by which purine biosynthesis is protected within the OCM network even during periods of folate deficiency. Disruptions in synthesis, utilization, or distribution of the formyl folate cofactors by MTHFD1 and MTHFS could therefore potentially alter purine biosynthesis and/or folate status.
Information regarding the potential contribution of folate-dependent enzymes that regulate or provide substrates for purine biosynthesis to NTD risk is limited because few gene variants within these enzymes have been investigated in relation to NTDs in humans. There have been no polymorphisms identified within the gene encoding MTHFS that have been investigated in relation to NTDs, although the data described previously warrant such investigation. The most studied gene variant associated with purine biosynthesis is the R653Q transition in MTHFD1. A strong association has been found between MTHFD1 R653Q and an increased maternal risk for NTDs (Brody et al., 2002; De Marco et al., 2006; Parle-McDermott et al., 2006), although negative results have also been reported (Hol et al., 1998; van der Linden et al., 2007a). Despite this strong association, however, the metabolic basis for increased risk associated with the SNP has not been elucidated. The R653Q SNP does not appear to affect total folate levels (Brody et al., 2002), arguing against an effect mediated by changes in folate status. Furthermore, the SNP has not been found to be associated with elevated plasma homocysteine (Brody et al., 2002; Konrad et al., 2004), suggesting that metabolic disruption is likely located at the level of purine and/or thymidylate biosynthesis. Recently, the biochemical impact of the R653Q transition on MTHFD1 stability and function was explored. The R653Q variant is thermolabile in vitro, resulting in a 36% reduction in enzymatic activity. Furthermore, the variant was also associated with impaired de novo purine biosynthesis, as determined by a 26% reduction in the incorporation of 14C-labeled formate into genomic DNA in murine cells (Christensen et al., 2008). In addition to uncovering a metabolic disruption associated with the MTHFD1 R653Q polymorphism, these data provide preliminary evidence to suggest that impairments in de novo purine biosynthesis may underlie NTD pathogenesis. Additional investigation of the biochemical effect of the SNP on enzyme function and cellular folate utilization may provide further insight into the mechanism by which this polymorphism confers NTD risk. In addition, the use of animal models to elucidate the effects of MTHFD1 deficiency on folate status and utilization will be useful in ascertaining the role of this potential genetic component in NTD pathogenesis.
Thymidylate Biosynthesis
Thymidylate synthase catalyzes the transfer of a one-carbon moiety from 5,10-methylene-THF to uridylate (dUMP) to form thymidylate (dTMP) de novo. In this reaction, folate serves both as a source of an activated one-carbon and as a source of reducing equivalents. The substrate 5,10-methylene-THF is synthesized from the MTHFD-catalyzed reduction of methenyl-THF and by the PLP-dependent conversion of serine and THF to form glycine and methylene-THF, catalyzed by cSHMT (Fig. 2). The transfer of the one-carbon from methylene-THF to dUMP also involves the oxidation of THF to dihydrofolate, which is recycled back into folate pools by the enzyme dihydrofolate reductase (DHFR). Decreased capacity to synthesize thymidylate de novo because of folate deficiency is associated with increased rates of uracil misincorporation into DNA (Blount et al., 1997). Uracil misincorporation can result in increased genomic instability (Reidy, 1987; Andersen et al., 2005). Other instances of genomic instability, including telomeric and centromeric instability, have been reported to cause NTDs in mice (Herrera et al., 1999; Hollander et al., 1999; Wang et al., 2004). In addition, impairments in de novo thymidylate biosynthesis affect DNA replication, reducing proliferative capacity essential for neurulation.
Recent evidence suggests that the partitioning of one-carbon units toward de novo thymidylate biosynthesis represents a point of sensitive regulation within cytoplasmic OCM. The thymidylate biosynthesis machinery, including cSHMT, TS, and DHFR, was shown to translocate to the nucleus at S-phase following modification of the enzymes with the small ubiquitin-like modifier (SUMO) (Anderson et al., 2007; Woeller et al., 2007). Compartmentation of de novo thymidylate biosynthesis in the nucleus enables the preferential shunting of cSHMT-derived one-carbon units into the thymidylate biosynthesis pathway. Other studies have demonstrated that cSHMT activity is rate limiting for de novo thymidylate biosynthesis (Herbig et al., 2002). In addition to stimulating thymidylate biosynthesis, cSHMT also regulates 5-methyl-THF utilization in the cytoplasm (Herbig et al., 2002). 5-Methyl-THF is bound tightly by cSHMT, preventing its use for the remethylation of homocysteine to methionine, which impairs AdoMet biosynthesis and ultimately cellular methylation reactions (Fig. 2). Therefore, cSHMT mediates the competition between thymidylate biosynthesis and homocysteine remethylation for folate-activated one-carbons. In cells that do not express cSHMT, data indicate that the partitioning of one-carbon units in the form of methylene-THF is favored in the direction of the generation of 5-methyl-THF at the expense of thymidylate biosynthesis (Scott et al., 1981; Green et al., 1988). However, increased cSHMT expression favors one-carbon flux in the direction of thymidylate biosynthesis at the expense of cellular methylation. Recently, our laboratory generated a mouse model with a null cSHMT allele that confirms the metabolic role of cSHMT (Macfarlane et al., 2008). Loss of cSHMT expression does not affect folate status, but does result in an enhanced methylation potential (as indicated by the hepatic AdoMet/AdoHcy ratio) and increased uracil levels in nuclear DNA. Together, these data indicate that cSHMT regulates folate utilization by balancing the partitioning of one-carbon units between thymidylate biosynthesis and cellular methylation, in the absence of effects on folate status.
Investigation of the thymidylate biosynthesis pathway as a genetic risk factor for NTDs in humans has been inconclusive. The only human polymorphisms in the TS gene that have been investigated in relation to human NTD risk are present in noncoding regions, and only two studies have yielded conflicting findings (Volcik et al., 2003b; Wilding et al., 2004). The effect of a common polymorphism in the promoter/enhancer region of the TS gene has yet to be clearly delineated, and the effect of altered TS expression on OCM and folate status has yet to be investigated in a mouse model. As with genes involved in purine biosynthesis, it is likely that mutations within the TS gene that markedly affect function are associated with gestational lethality. Recently, a noncoding deletion allele in the gene encoding DHFR was investigated in relation to NTDs; however, results from three studies have been inconclusive in determining a role for DHFR in mediating NTD risk (Johnson et al., 2004; Parle-McDermott et al., 2007; van der Linden et al., 2007b). A handful of studies have investigated the effect of the common L474F polymorphism within the gene encoding cSHMT in relation to NTD risk. However, the scope and size of the studies thus far has been inadequate to fully determine a potential association with NTD risk (Heil et al., 2001; Relton et al., 2004a,b). Interestingly, the biochemical effect of the L474F cSHMT variant has recently been determined. The L474F variant is impaired in its ability to undergo SUMO modification and SUMO-dependent nuclear localization (Woeller et al., 2007). These data suggest that the SNP may interfere with the ability of cSHMT to preferentially partition one-carbon units to thymidylate biosynthesis, ultimately modifying the utilization of folate cofactors in the cytoplasm. Further investigation of the SNP in relation to NTDs in humans should shed light on the importance of cSHMT-mediated regulation of folate utilization in human NTD risk.
Although the incidence of NTDs in response to genetic deletion of folate-related genes mediating de novo thymidylate biosynthesis in mice has not yet been explored, there is some evidence from other folate-responsive mouse models to suggest that impairments in de novo thymidylate biosynthesis might underlie NTD pathogenesis. Fleming and Copp (1998) investigated metabolic alterations and folate responsiveness in the splotch mouse model of NTDs. Homozygous splotch embryos, which display a completely penetrant NTD phenotype, exhibited impairments in de novo thymidylate biosynthesis as evidenced by a reduction in deoxyuridine suppression values. Supplementation of culture media with either folic acid or thymidine prevented NTDs in homozygous splotch embryos, directly implicating impairments in thymidylate biosynthesis in NTD pathogenesis in this folate-responsive mouse model. Surprisingly, supplementation of cultured splotch embryos with methionine exacerbated impairments in deoxyuridine suppression and caused NTDs in heterozygous splotch embryos, which do not otherwise develop NTDs. Rescue of NTDs in homozygous splotch embryos by folic acid supplementation and exacerbation by methionine supplementation was also observed in another study in vivo (Wlodarczyk et al., 2005). More recently, impaired de novo thymidylate biosynthesis was also observed in human embryos with NTDs (Dunlevy et al., 2007). Collectively, these data provide preliminary evidence that de novo thymidylate biosynthesis is crucial for proper neural tube closure. In addition, maintaining a balance of utilization between various folate-dependent anabolic pathways in the cytoplasm (e.g. methionine biosynthesis and thymidylate biosynthesis) may influence neural tube closure via direct effects on the thymidylate biosynthesis pathway.
Homocysteine Remethylation/Methylation Cycle
Much of the focus on identifying a metabolic basis for folate-responsive NTDs has centered on the role of homocysteine and the methylation cycle, which is consistent with the results from human epidemiologic studies that have linked moderately elevated maternal homocysteine with NTD risk. Supplementation with folic acid alleviates hyperhomocysteinemia, leading to the hypothesis that impairments in the homocysteine remethylation cycle were causal in NTD pathogenesis. In addition to being a biomarker for impaired folate status and reduced methylation potential (the AdoMet/AdoHcy ratio), homocysteine at elevated levels is cytotoxic (Frandsen et al., 1993; Huang et al., 2001), and also negatively affects cellular methylation through the accumulation of AdoHcy, which is an inhibitor of cellular methylation reactions (Hoffman et al., 1980; Finkelstein, 1998; Clarke, 2001). Altered gene expression resulting from chromatin hypomethylation has been proposed as a mechanism underlying many folate-related pathologies, including NTDs (Beaudin and Stover, 2007). Likewise, the direct modification of proteins by homocysteine has also been implicated in these developmental anomalies (Taparia et al., 2007). The regulation of homocysteine remethylation and AdoMet bio-synthesis is complex and includes feedback mechanisms that prevent both the accumulation of homocysteine and maintenance of both AdoMet and AdoHcy concentrations for cellular methylation reactions (Finkelstein, 1998). For example, AdoMet is an allosteric, feedback inhibitor of MTHFR (Kutzbach and Stokstad, 1971; Jencks and Mathews, 1987), and its accumulation inhibits MTHFR activity, thereby preventing the synthesis of 5-methyl-THF from the methylene-THF cofactor when AdoMet levels are adequate for cellular methylation reactions.
In addition to the folate-dependent remethylation of homocysteine to methionine, two other folate-independent pathways exist that function to prevent the accumulation of homocysteine in certain tissues and cell types: homocysteine can be remethylated to methionine by the transfer of a one-carbon moiety from betaine, catalyzed by the enzyme betaine homocysteinemethyltransferase (BHMT), and homocysteine can be eliminated via the transsulfuration pathway, in which cystathionine β-synthase (CBS) catalyzes the condensation of serine and homocysteine to form cystathionine. Transsulfuration simultaneously degrades homocysteine while removing a source for methionine biosynthesis in the cell. It is important to note that BHMT and CBS are not expressed in all tissues (Sunden et al., 1997; Finkelstein, 1998; Jhee and Kruger, 2005), whereas folate-dependent methionine synthase expression is more ubiquitous (Jhee and Kruger, 2005). The activities of the BHMT-mediated methylation pathway and the transsulfuration pathway are also regulated via feedback mechanisms associated with the cellular methylation cycle; accumulation of AdoMet inhibits BHMT-mediated methionine biosynthesis while stimulating CBS-mediated transsulfuration (Okada et al., 1981).
Because of the strong association between elevated homocysteine and NTD risk, genes involved in homocysteine metabolism and the methylation cycle have been at the center of the search for candidate genes involved in NTD pathogenesis. The most prominent of these is MTHFR, for which two human SNPs have been identified that are associated with NTD risk, MTHFR 677C>T and MTHFR 1298A>C. The MTHFR 677C>T SNP has been well-characterized and codes for a thermolabile enzyme with a 50 to 60% reduction in enzymatic activity in individuals homozygous for the T allele (Kang et al., 1988; Frosst et al., 1995; van der Put et al., 1996). To date, the 677C>T SNP has been widely considered the most attractive genetic candidate underlying folate-responsive NTDs based on three findings: (1) it has consistently demonstrated a strong association with NTD occurrence in the human epidemiologic literature, with both maternal and fetal alleles contributing to risk (van der Put et al., 1997a; Botto and Yang, 2000; Blom et al., 2006); (2) it is associated with elevated homocysteine, a predictor of NTD risk (Engbersen et al., 1995; van der Put et al., 1995; Harmon et al., 1996); and (3) it interacts with folate status in determining both impairments in metabolism and NTD risk (Jacques et al., 1996; Malinow et al., 1997; Christensen et al., 1999; Friso et al., 2002; Volcik et al., 2003a; Kim et al., 2004). Furthermore, the strength of the association between the MTHFR 677C>T SNP and NTD risk has reciprocally bolstered the hypothesis that metabolic disruptions in homocysteine homeostasis underlie NTD pathogenesis.
Despite MTHFR having the strongest association in the literature with NTD risk, the prevalence of the SNP cannot account for NTD incidence in the population. The frequency of the T allele in the general population does not account for the level of reduction of NTD risk observed in response to folic acid supplementation (Molloy et al., 1998). Furthermore, there is no direct evidence that the association between the MTHFR 677C>T SNP and NTD risk is related to metabolic perturbations of the homocysteine remethylation cycle. Deletion of the MTHFR gene in the mouse germ line is associated with both elevated homocysteine as well as alterations in cellular methylation potential and global DNA hypomethylation (Chen et al., 2001), yet does not result in NTDs in vivo, even in response to maternal folate deficiency (Li et al., 2005). Therefore, there is no causal evidence that reduced MTHFR activity induces NTD risk by impairing the homocysteine remethylation cycle. However, in addition to its inhibitory effects on homocysteine remethylation, the MTHFR SNP also impairs folate status. Data from several studies have revealed that the T allele is associated with reductions in serum and red blood cell folate levels (Nelen et al., 1998; Ashfield-Watt et al., 2002; de Bree et al., 2003; Narayanan et al., 2004). The observed reduction of total folates in individuals with reduced MTHFR activity may result from the accumulation of the less stable formylated derivatives of folate at the expense of 5-methyl-THF, resulting in increased folate turnover (Bagley and Selhub, 1998; Smulders et al., 2007). Therefore, the 677C>T SNP may influence NTD risk by impairing folate status, as opposed to impairing folate metabolism via the methylation cycle. This suggestion has been corroborated by the finding that MTHFR knockout mice also display reduced plasma folate levels, in addition to an alteration in percentage of methylated folate derivatives (Ghandour et al., 2004). Because folate deficiency alone does not induce NTDs in mice (Heid et al., 1992; Burgoon et al., 2002;), data from studies of the MTHFR knockout mouse model support the notion that alterations in MTHFR activity in humans may influence neurulation indirectly via alterations in folate status and/or the distribution of folate cofactors that ultimately impinge upon other folate-dependent anabolic pathways beyond the methylation cycle.
Other studies support the conclusion that the accumulation of homocysteine is not an underlying cause of NTDs. Examination of polymorphisms in other genes regulating cellular homocysteine accumulation, including MS, CBS, and BHMT, has not provided convincing evidence that these genes are associated with NTD risk in humans (Morrison et al., 1997; van der Put et al., 1997b; Morrison et al., 1998; Speer et al., 1999; Richter et al., 2001; Morin et al., 2003b; Zhu et al., 2005). Similarly, mouse models with deletions of genes that regulate homocysteine accumulation fail to support the hypothesis that homocysteine accumulation underlies NTD pathogenesis. Genetic deletion of CBS in mice produces highly elevated homocysteine levels, but these mice do not exhibit NTDs (Watanabe et al., 1995). Another mouse model with reduced expression of the gene MTRR, which regulates methionine synthase activity, also produces a robust metabolic phenotype, including elevated plasma homocysteine, reduced plasma methionine, and tissue-specific alterations in methylation potential, but does not exhibit a developmental phenotype (Elmore et al., 2007). Lastly, hyperhomocysteinemia induced in culture or in vivo by nutritional manipulation does not affect neural tube closure in mice (Greene et al., 2003; Hansen et al., 2001; Bennett et al., 2006). Together, these data do not provide support for a direct role of homocysteine in NTD pathogenesis and instead indicate that elevated homocysteine is merely a biomarker for impairments in folate status, which disrupt the function of the entire folate-dependent OCM network.
Notwithstanding the lack of evidence for a role of homocysteine in NTD pathogenesis, vitamin B12 status has been found to be moderately associated with NTD risk (Ray and Blom, 2003; Gaber et al., 2007; Ray et al., 2007). Many of these studies have been limited by the failure or inability to statistically account for maternal folate status, which is often closely linked to maternal B12 status. Recently, the association between vitamin B12 status and NTD risk was examined in a folic acid–fortified population. Vitamin B12 levels in the lowest quartile were associated with a tripling of NTD risk (Ray et al., 2007). Severe vitamin B12 deficiency impairs the activity of MS and leads to impairment of the homocysteine remethylation cycle and impaired nucleotide biosynthesis. The impairment in nucleotide biosynthesis is indirect; lack of MS activity can result in the accumulation of cellular folate as 5-methyl-THF at the expense of other folate cofactors, a condition referred to as a methyl trap. The methyl trap occurs because the reduction of methylene-THF to 5-methyl-THF catalyzed by MTHFR is irreversible, and MS is the only folate-dependent enzyme to use 5-methyl-THF as a cofactor (Fig. 2). Vitamin B12 deficiency also diminishes AdoMet levels, resulting in the activation of MTHFR activity thereby enhancing the methyl trap. Therefore, vitamin B12 deficiency may influence NTD risk by altering the distribution of folate cofactors, thereby affecting the entire OCM network. Future investigation of the association between vitamin B12 deficiency, cellular folate utilization, and NTD risk is required to determine the precise metabolic perturbation underlying NTD pathogenesis.
CONCLUSIONS
Although maternal folic acid supplementation or fortification is effective for the prevention of NTDs, the mechanisms underlying folate-responsive NTDs remain unknown. Continued investigation of gene-gene interactions and gene-nutrient interactions in humans will provide greater insight into the influence of genetic perturbations on both folate status and folate utilization in NTD pathogenesis.
The examination of potential genetic risk factors is currently limited by gaps in knowledge of the enzymes and associated genes involved in OCM, especially in mitochondria, and the metabolic sequelae associated with specific gene-nutrient interactions. Furthermore, there is still much knowledge to be gained concerning the regulation of cellular folate status, metabolism, utilization, and the effects of alterations in the flux of folates through the metabolic network on all metabolic, genomic, and cellular outcomes. As an example, it is still unclear whether the 677C>T polymorphism in MTHFR increases human NTD risk by influencing homocysteine levels, the distribution of folate cofactors in the cell, and/or depressing cellular folate status. In addition, the generation and detailed metabolic characterization of genetically altered mice that model impairments in specific folate-dependent anabolic pathways and their regulation is necessary to provide greater insight into the effects of altered folate metabolism on genomic, cellular, and developmental outcomes and ultimately establish the underlying mechanisms of developmental anomalies. Determining the precise metabolic impairments that result in NTDs will enable the design of improved nutritional interventions that target both susceptible subgroups of the population and the metabolic pathway that causes neural tube closure defects.
Acknowledgments
This study was funded by PHS grant HD059120.
REFERENCES
- Afman LA, Trijbels FJ, Blom HJ. The H475Y polymorphism in the glutamate carboxypeptidase II gene increases plasma folate without affecting the risk for neural tube defects in humans. J Nutr. 2003;133:75–77. doi: 10.1093/jn/133.1.75. [DOI] [PubMed] [Google Scholar]
- Andersen S, Heine T, Sneve R, et al. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26(3):547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- Anderson DD, Woeller CF, Stover PJ. Small ubiquitin-like modifier-1 (SUMO-1) modification of thymidylate synthase and dihydrofolate reductase. Clin Chem Lab Med. 2007;45:1760–1763. doi: 10.1515/CCLM.2007.355. [DOI] [PubMed] [Google Scholar]
- Anguera MC, Suh JR, Ghandour H, et al. Methenyltetrahydrofolate synthetase regulates folate turnover and accumulation. J Biol Chem. 2003;278:29856–29862. doi: 10.1074/jbc.M302883200. [DOI] [PubMed] [Google Scholar]
- Appling DR. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991;5:2645–2651. doi: 10.1096/fasebj.5.12.1916088. [DOI] [PubMed] [Google Scholar]
- Ashfield-Watt PA, Pullin CH, Whiting JM, et al. Methylenetetrahydrofolate reductase 677C−>T genotype modulates homocysteine responses to a folate-rich diet or a low-dose folic acid supplement: a randomized controlled trial. Am J Clin Nutr. 2002;76:180–186. doi: 10.1093/ajcn/76.1.180. [DOI] [PubMed] [Google Scholar]
- Bagley PJ, Selhub J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc Natl Acad Sci U S A. 1998;95:13217–13220. doi: 10.1073/pnas.95.22.13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Ghoshal K, Datta J, et al. DNA methyltransferase 3b regulates nerve growth factor-induced differentiation of PC12 cells by recruiting histone deacetylase 2. Molecular and cellular biology. 2005;25:751–766. doi: 10.1128/MCB.25.2.751-766.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bailey LB. Folate requirements and dietary recommendations. In: Bailey LB, editor. Folate in health and disease. Marcel Dekker; New York: 1995. [Google Scholar]
- Barber R, Shalat S, Hendricks K, et al. Investigation of folate pathway gene polymorphisms and the incidence of neural tube defects in a Texas hispanic population. Mol Genet Metab. 2000;70:45–52. doi: 10.1006/mgme.2000.2991. [DOI] [PubMed] [Google Scholar]
- Barber RC, Shaw GM, Lammer EJ, et al. Lack of association between mutations in the folate receptor-alpha gene and spina bifida. Am J Med Genet. 1998;76:310–317. [PubMed] [Google Scholar]
- Beaudin AE, Stover PJ. Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res C Embryo Today. 2007;81:183–203. doi: 10.1002/bdrc.20100. [DOI] [PubMed] [Google Scholar]
- Bennett GD, Vanwaes J, Moser K, et al. Failure of homocysteine to induce neural tube defects in a mouse model. Birth Defects Res B Dev Reprod Toxicol. 2006;77:89–94. doi: 10.1002/bdrb.20071. [DOI] [PubMed] [Google Scholar]
- Bertrand R, Jolivet J. Methenyltetrahydrofolate synthetase prevents the inhibition of phosphoribosyl 5-aminoimidazole 4-carboxamide ribonucleotide formyltransferase by 5-formyltetrahydrofolate polyglutamates. J Biol Chem. 1989;264:8843–8846. [PubMed] [Google Scholar]
- Blom HJ, Shaw GM, den Heijer M, Finnell RH. Neural tube defects and folate: case far from closed. Nat Rev Neurosci. 2006;7:724–731. doi: 10.1038/nrn1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount BC, Mack MM, Wehr CM, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151:862–877. doi: 10.1093/oxfordjournals.aje.a010290. [DOI] [PubMed] [Google Scholar]
- Boyles AL, Billups AV, Deak KL, et al. Neural tube defects and folate pathway genes: family-based association tests of gene-gene and gene-environment interactions. Environ Health Perspect. 2006;114:1547–1552. doi: 10.1289/ehp.9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody LC, Conley M, Cox C, et al. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet. 2002;71:1207–1215. doi: 10.1086/344213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoon JM, Selhub J, Nadeau M, Sadler TW. Investigation of the effects of folate deficiency on embryonic development through the establishment of a folate deficient mouse model. Teratology. 2002;65:219–227. doi: 10.1002/tera.10040. [DOI] [PubMed] [Google Scholar]
- Castilla EE, Orioli IM, Lopez-Camelo JS, et al. Preliminary data on changes in neural tube defect prevalence rates after folic acid fortification in South America. Am J Med Genet A. 2003;123A:123–128. doi: 10.1002/ajmg.a.20230. [DOI] [PubMed] [Google Scholar]
- Chen Z, Karaplis AC, Ackerman SL, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- Christensen B, Arbour L, Tran P, et al. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;84:151–157. doi: 10.1002/(sici)1096-8628(19990521)84:2<151::aid-ajmg12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Christensen KE, MacKenzie RE. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays. 2006;28:595–605. doi: 10.1002/bies.20420. [DOI] [PubMed] [Google Scholar]
- Christensen KE, Patel H, Kuzmanov U, et al. Disruption of the mthfd1 gene reveals a monofunctional 10-formyltetrahydrofolate synthetase in mammalian mitochondria. J Biol Chem. 2005;280:7597–7602. doi: 10.1074/jbc.M409380200. [DOI] [PubMed] [Google Scholar]
- Christensen KE, Rohlicek CV, Andelfinger GU, Michaud J, Bigras JL, Richter A, Mackenzie RE, Rozen R. The MTHFD1 p. Arg653Gln variant alters enzyme function and increases risk for congenital heart defects. Hum Mutat. 2008 doi: 10.1002/humu.20830. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Clarke S, Banfield K. S-adenosylmethionine-dependent methyltransferases. In: Carmel RJD, editor. Homocysteine in health and disease. Cambridge Press; Cambridge: 2001. [Google Scholar]
- Czeizel AE, Dudas I. Prevention of the first occurrence of neuraltube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832–1835. doi: 10.1056/NEJM199212243272602. [DOI] [PubMed] [Google Scholar]
- de Bree A, Verschuren WM, Bjorke-Monsen AL, et al. Effect of the methylenetetrahydrofolate reductase 677C−>T mutation on the relations among folate intake and plasma folate and homocysteine concentrations in a general population sample. Am J Clin Nutr. 2003;77:687–693. doi: 10.1093/ajcn/77.3.687. [DOI] [PubMed] [Google Scholar]
- De Marco P, Merello E, Calevo MG, et al. Evaluation of a methylenetetrahydrofolate-dehydrogenase 1958G>A polymorphism for neural tube defect risk. J Hum Genet. 2006;51:98–103. doi: 10.1007/s10038-005-0329-6. [DOI] [PubMed] [Google Scholar]
- Dunlevy LP, Chitty LS, Burren KA, et al. Abnormal folate metabolism in foetuses affected by neural tube defects. Brain. 2007;130:1043–1049. doi: 10.1093/brain/awm028. Pt 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore CL, Wu X, Leclerc D, et al. Metabolic derangement of methionine and folate metabolism in mice deficient in methionine synthase reductase. Mol Genet Metab. 2007;91:85–97. doi: 10.1016/j.ymgme.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwood JM, Little J, Elwood JH. Epidemiology and control of neural tube defects: monographs in epidemiology and biostatistics. Oxford University Press; New York: 1992. [Google Scholar]
- Engbersen AM, Franken DG, Boers GH, et al. Thermolabile 5,10-methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet. 1995;56:142–150. [PMC free article] [PubMed] [Google Scholar]
- Field MS, Szebenyi DM, Stover PJ. Regulation of de novo purine biosynthesis by methenyltetrahydrofolate synthetase in neuroblastoma. J Biol Chem. 2006;281:4215–4221. doi: 10.1074/jbc.M510624200. [DOI] [PubMed] [Google Scholar]
- Finkelstein JD. The metabolism of homocysteine: pathways and regulation. Eur J Pediatr. 1998;157(Suppl 2):S40–44. doi: 10.1007/pl00014300. [DOI] [PubMed] [Google Scholar]
- Finnell RH, Spiegelstein O, Wlodarczyk B, et al. DNA methylation in Folbp1 knockout mice supplemented with folic acid during gestation. J Nutr. 2002;132(8 Suppl):2457S–2461S. doi: 10.1093/jn/132.8.2457S. [DOI] [PubMed] [Google Scholar]
- Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280:2107–2109. doi: 10.1126/science.280.5372.2107. [DOI] [PubMed] [Google Scholar]
- Frandsen A, Schousboe A, Griffiths R. Cytotoxic actions and effects on intracellular Ca2+ and cGMP concentrations of sulphur-containing excitatory amino acids in cultured cerebral cortical neurons. J Neurosci Res. 1993;34:331–339. doi: 10.1002/jnr.490340310. [DOI] [PubMed] [Google Scholar]
- Friso S, Choi SW, Girelli D, et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99:5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- Gaber KR, Farag MK, Soliman SE, et al. Maternal vitamin B12 and the risk of fetal neural tube defects in Egyptian patients. Clin Lab. 2007;53:69–75. [PubMed] [Google Scholar]
- Ghandour H, Chen Z, Selhub J, Rozen R. Mice deficient in methylenetetrahydrofolate reductase exhibit tissue-specific distribution of folates. J Nutr. 2004;134:2975–2978. doi: 10.1093/jn/134.11.2975. [DOI] [PubMed] [Google Scholar]
- Green JM, MacKenzie RE, Matthews RG. Substrate flux through methylenetetrahydrofolate dehydrogenase: predicted effects of the concentration of methylenetetrahydrofolate on its partitioning into pathways leading to nucleotide biosynthesis or methionine regeneration. Biochemistry. 1988;27:8014–8022. doi: 10.1021/bi00421a007. [DOI] [PubMed] [Google Scholar]
- Greene ND, Dunlevy LE, Copp AJ. Homocysteine is embryotoxic but does not cause neural tube defects in mouse embryos. Anat Embryol (Berl) 2003;206:185–191. doi: 10.1007/s00429-002-0284-3. [DOI] [PubMed] [Google Scholar]
- Gregory JF., 3rd Case study: folate bioavailability. J Nutr. 2001;131(4 Suppl):1376S–1382S. doi: 10.1093/jn/131.4.1376S. [DOI] [PubMed] [Google Scholar]
- Hansen DK, Grafton TF, Melnyk S, James SJ. Lack of embryotoxicity of homocysteine thiolactone in mouse embryos in vitro. Reprod Toxicol. 2001;15:239–244. doi: 10.1016/s0890-6238(01)00133-2. [DOI] [PubMed] [Google Scholar]
- Harmon DL, Woodside JV, Yarnell JW, et al. The common ‘thermolabile’ variant of methylene tetrahydrofolate reductase is a major determinant of mild hyperhomocysteinaemia. QJM. 1996;89:571–577. doi: 10.1093/qjmed/89.8.571. [DOI] [PubMed] [Google Scholar]
- Heid MK, Bills ND, Hinrichs SH, Clifford AJ. Folate deficiency alone does not produce neural tube defects in mice. J Nutr. 1992;122:888–894. doi: 10.1093/jn/122.4.888. [DOI] [PubMed] [Google Scholar]
- Heil SG, Van der Put NM, Waas ET, et al. Is mutated serine hydroxymethyltransferase (SHMT) involved in the etiology of neural tube defects? Mol Genet Metab. 2001;73:164–172. doi: 10.1006/mgme.2001.3175. [DOI] [PubMed] [Google Scholar]
- Herbig K, Chiang EP, Lee LR, et al. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem. 2002;277:38381–38389. doi: 10.1074/jbc.M205000200. [DOI] [PubMed] [Google Scholar]
- Herrera E, Samper E, Blasco MA. Telomere shortening in mTR−/− embryos is associated with failure to close the neural tube. Embo J. 1999;18:1172–1181. doi: 10.1093/emboj/18.5.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbard BM. The role of folic acid in pregnancy; with particular reference to anaemia, abruption and abortion. J Obstet Gynaecol Br Commonw. 1964;71:529–542. doi: 10.1111/j.1471-0528.1964.tb04317.x. [DOI] [PubMed] [Google Scholar]
- Hibbard BM. Defective folate metabolism in pathological conditions of pregnancy. Acta Obstet Gynecol Scand. 1967;46(Suppl 7):47–59. doi: 10.3109/00016346709157073. [DOI] [PubMed] [Google Scholar]
- Hoffman DR, Marion DW, Cornatzer WE, Duerre JA. S-Adenosylmethionine and S-adenosylhomocystein metabolism in isolated rat liver. Effects of L-methionine, L-homocystein, and adenosine. J Biol Chem. 1980;255:10822–10827. [PubMed] [Google Scholar]
- Hol FA, van der Put NM, Geurds MP, et al. Molecular genetic analysis of the gene encoding the trifunctional enzyme MTHFD (methylenetetrahydrofolate-dehydrogenase, methenyltetrahydrofolate-cyclohydrolase, formyltetrahydrofolate synthetase) in patients with neural tube defects. Clin Genet. 1998;53:119–125. doi: 10.1111/j.1399-0004.1998.tb02658.x. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Sheikh MS, Bulavin DV, et al. Genomic instability in Gadd45a-deficient mice. Nat Genet. 1999;23:176–184. doi: 10.1038/13802. [DOI] [PubMed] [Google Scholar]
- Huang RF, Huang SM, Lin BS, et al. Homocysteine thiolactone induces apoptotic DNA damage mediated by increased intracellular hydrogen peroxide and caspase 3 activation in HL-60 cells. Life Sci. 2001;68:2799–2811. doi: 10.1016/s0024-3205(01)01066-9. [DOI] [PubMed] [Google Scholar]
- Jacques PF, Bostom AG, Williams RR, et al. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation. 1996;93:7–9. doi: 10.1161/01.cir.93.1.7. [DOI] [PubMed] [Google Scholar]
- Jencks DA, Mathews RG. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J Biol Chem. 1987;262:2485–2493. [PubMed] [Google Scholar]
- Jhee KH, Kruger WD. The role of cystathionine beta-synthase in homocysteine metabolism. Antioxid Redox Signal. 2005;7:813–822. doi: 10.1089/ars.2005.7.813. [DOI] [PubMed] [Google Scholar]
- Johnson WG, Stenroos ES, Spychala JR, et al. New 19 bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR): a risk factor for spina bifida acting in mothers during pregnancy? Am J Med Genet A. 2004;124:339–345. doi: 10.1002/ajmg.a.20505. [DOI] [PubMed] [Google Scholar]
- Kamen BA, Wang MT, Streckfuss AJ, et al. Delivery of folates to the cytoplasm of MA104 cells is mediated by a surface membrane receptor that recycles. J Biol Chem. 1988;263:13602–13609. [PubMed] [Google Scholar]
- Kang SS, Wong PW, Zhou JM, et al. Thermolabile methylenetetrahydrofolate reductase in patients with coronary artery disease. Metabolism. 1988;37:611–613. doi: 10.1016/0026-0495(88)90076-5. [DOI] [PubMed] [Google Scholar]
- Kim KN, Kim YJ, Chang N. Effects of the interaction between the C677T 5,10-methylenetetrahydrofolate reductase polymorphism and serum B vitamins on homocysteine levels in pregnant women. Eur J Clin Nutr. 2004;58:10–16. doi: 10.1038/sj.ejcn.1601729. [DOI] [PubMed] [Google Scholar]
- Kirke PN, Molloy AM, Daly LE, et al. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q J M. 1993;86:703–708. [PubMed] [Google Scholar]
- Konrad C, Muller GA, Langer C, et al. Plasma homocysteine, MTHFR C677T, CBS 844ins68bp, and MTHFD1 G1958A polymorphisms in spontaneous cervical artery dissections. J Neurol. 2004;251:1242–1248. doi: 10.1007/s00415-004-0523-z. [DOI] [PubMed] [Google Scholar]
- Kutzbach C, Stokstad EL. Mammalian methylenetetrahydrofolate reductase. Partial purification, properties, and inhibition by S-adenosylmethionine. Biochim Biophys Acta. 1971;250:459–477. doi: 10.1016/0005-2744(71)90247-6. [DOI] [PubMed] [Google Scholar]
- Li D, Pickell L, Liu Y, et al. Maternal methylenetetrahydrofolate reductase deficiency and low dietary folate lead to adverse reproductive outcomes and congenital heart defects in mice. Am J Clin Nutr. 2005;82:188–195. doi: 10.1093/ajcn.82.1.188. [DOI] [PubMed] [Google Scholar]
- Lin BF, Shane B. Expression of Escherichia coli folylpolyglutamate synthetase in the Chinese hamster ovary cell mitochondrion. J Biol Chem. 1994;269:9705–9713. [PubMed] [Google Scholar]
- Macfarlane AJ, Liu X, Perry CA, et al. Cytoplasmic serine hydroxymethyltransferase regulates themetabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008;283(38):25846–25853. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow MR, Nieto FJ, Kruger WD, et al. The effects of folic acid supplementation on plasma total homocysteine are modulated by multivitamin use and methylenetetrahydrofolate reductase genotypes. Arterioscler Thromb Vasc Biol. 1997;17:1157–1162. doi: 10.1161/01.atv.17.6.1157. [DOI] [PubMed] [Google Scholar]
- Mills JL, McPartlin JM, Kirke PN, et al. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet. 1995;345:149–151. doi: 10.1016/s0140-6736(95)90165-5. [DOI] [PubMed] [Google Scholar]
- Mills JL, Signore C. Neural tube defect rates before and after food fortification with folic acid. Birth Defects Res A Clin Mol Teratol. 2004;70:844–845. doi: 10.1002/bdra.20075. [DOI] [PubMed] [Google Scholar]
- Mills JL, Tuomilehto J, Yu KF, et al. Maternal vitamin levels during pregnancies producing infants with neural tube defects. J Pediatr. 1992;120:863–871. doi: 10.1016/s0022-3476(05)81951-1. [DOI] [PubMed] [Google Scholar]
- Mitchell LE, Adzick NS, Melchionne J, et al. Spina bifida. Lancet. 2004;364:1885–1895. doi: 10.1016/S0140-6736(04)17445-X. [DOI] [PubMed] [Google Scholar]
- Molloy AM, Kirke P, Hillary I, et al. Maternal serum folate and vitamin B12 concentrations in pregnancies associated with neural tube defects. Arch Dis Child. 1985;60:660–665. doi: 10.1136/adc.60.7.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy AM, Mills JL, Kirke PN, et al. Low blood folates in NTD pregnancies are only partly explained by thermolabile 5,10-methylenetetrahydrofolate reductase: low folate status alone may be the critical factor. Am J Med Genet. 1998;78:155–159. [PubMed] [Google Scholar]
- Morin I, Devlin AM, Leclerc D, et al. Evaluation of genetic variants in the reduced folate carrier and in glutamate carboxypeptidase II for spina bifida risk. Mol Genet Metab. 2003a;79:197–200. doi: 10.1016/s1096-7192(03)00086-6. [DOI] [PubMed] [Google Scholar]
- Morin I, Platt R, Weisberg I, et al. Common variant in betaine-homocysteine methyltransferase (BHMT) and risk for spina bifida. Am J Med Genet A. 2003b;119:172–176. doi: 10.1002/ajmg.a.20115. [DOI] [PubMed] [Google Scholar]
- Morrison K, Edwards YH, Lynch SA, et al. Methionine synthase and neural tube defects. J Med Genet. 1997;34:958. doi: 10.1136/jmg.34.11.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison K, Papapetrou C, Hol FA, et al. Susceptibility to spina bifida: an association study of five candidate genes. Ann Hum Genet. 1998;62:379–396. doi: 10.1046/j.1469-1809.1998.6250379.x. Pt 5. [DOI] [PubMed] [Google Scholar]
- Motokawa Y, Kikuchi G. Glycine metabolism in rat liver mitochondria. V. Intramitochondrial localization of the reversible glycine cleavage system and serine hydroxymethyltransferase. Arch Biochem Biophys. 1971;146:461–464. doi: 10.1016/0003-9861(71)90149-4. [DOI] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131–137. [PubMed] [Google Scholar]
- Narayanan S, McConnell J, Little J, et al. Associations between two common variants C677T and A1298C in the methylenetetrahydrofolate reductase gene and measures of folate metabolism and DNA stability (strand breaks, misincorporated uracil, and DNA methylation status) in human lymphocytes in vivo. Cancer Epidemiol Biomarkers Prev. 2004;13:1436–1443. [PubMed] [Google Scholar]
- Nelen WL, Blom HJ, Thomas CM, et al. Methylenetetrahydrofolate reductase polymorphism affects the change in homocysteine and folate concentrations resulting from low dose folic acid supplementation in women with unexplained recurrent miscarriages. J Nutr. 1998;128:1336–1341. doi: 10.1093/jn/128.8.1336. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Mills JL, Kirke PN, et al. Analysis of the human folate receptor beta gene for an association with neural tube defects. Mol Genet Metab. 2003;79:129–133. doi: 10.1016/s1096-7192(03)00075-1. [DOI] [PubMed] [Google Scholar]
- Okada G, Teraoka H, Tsukada K. Multiple species of mammalian S-adenosylmethionine synthetase. Partial purification and characterization. Biochemistry. 1981;20:934–940. doi: 10.1021/bi00507a045. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Kirke PN, Mills JL, et al. Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur J Hum Genet. 2006;14:768–772. doi: 10.1038/sj.ejhg.5201603. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Pangilinan F, Mills JL, et al. The 19-bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR) may decrease rather than increase risk for spina bifida in the Irish population. Am J Med Genet A. 2007;143:1174–1180. doi: 10.1002/ajmg.a.31725. [DOI] [PubMed] [Google Scholar]
- Pei L, Zhu H, Ren A, et al. Reduced folate carrier gene is a risk factor for neural tube defects in a Chinese population. Birth Defects Res A Clin Mol Teratol. 2005;73:430–433. doi: 10.1002/bdra.20130. [DOI] [PubMed] [Google Scholar]
- Piedrahita JA, Oetama B, Bennett GD, et al. Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nat Genet. 1999;23:228–232. doi: 10.1038/13861. [DOI] [PubMed] [Google Scholar]
- Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Rampersaud E, Melvin EC, Siegel D, et al. Updated investigations of the role of methylenetetrahydrofolate reductase in human neural tube defects. Clin Genet. 2003;63:210–214. doi: 10.1034/j.1399-0004.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- Ray JG, Blom HJ. Vitamin B12 insufficiency and the risk of fetal neural tube defects. QJM. 2003;96:289–295. doi: 10.1093/qjmed/hcg043. [DOI] [PubMed] [Google Scholar]
- Ray JG, Wyatt PR, Thompson MD, et al. Vitamin B12 and the risk of neural tube defects in a folic-acid-fortified population. Epidemiology. 2007;18:362–366. doi: 10.1097/01.ede.0000257063.77411.e9. [DOI] [PubMed] [Google Scholar]
- Reidy JA. Folate- and deoxyuridine-sensitive chromatid breakage may result from DNA repair during G2. Mutat Res. 1987;192:217–219. doi: 10.1016/0165-7992(87)90059-5. [DOI] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Laffling AJ, et al. Low erythrocyte folate status and polymorphic variation in folate-related genes are associated with risk of neural tube defect pregnancy. Mol Genet Metab. 2004a;81:273–281. doi: 10.1016/j.ymgme.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Pearce MS, et al. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet. 2004b;41:256–260. doi: 10.1136/jmg.2003.010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Stegmann K, Roper B, et al. Interaction of folate and homocysteine pathway genotypes evaluated in susceptibility to neural tube defects (NTD) in a German population. J Hum Genet. 2001;46:105–109. doi: 10.1007/s100380170096. [DOI] [PubMed] [Google Scholar]
- Sayed AR, Bourne D, Pattinson R, et al. Decline in the prevalence of neural tube defects following folic acid fortification and its cost-benefit in South Africa. Birth Defects Res A Clin Mol Teratol. 2008;82:211–216. doi: 10.1002/bdra.20442. [DOI] [PubMed] [Google Scholar]
- Schirch V, Strong WB. Interaction of folylpolyglutamates with enzymes in one-carbon metabolism. Arch Biochem Biophys. 1989;269:371–380. doi: 10.1016/0003-9861(89)90120-3. [DOI] [PubMed] [Google Scholar]
- Scott JM, Dinn JJ, Wilson P, Weir DG. Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet. 1981;2:334–337. doi: 10.1016/s0140-6736(81)90649-8. [DOI] [PubMed] [Google Scholar]
- Selhub J. Homocysteine metabolism. Annu Rev Nutr. 1999;19:217–246. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- Shane B. Folylpolyglutamate synthesis and role in the regulation of one-carbon metabolism. Vitam Horm. 1989;45:263–335. doi: 10.1016/s0083-6729(08)60397-0. [DOI] [PubMed] [Google Scholar]
- Shane B. Folate chemistry and metabolism. In: Bailey LP, editor. Folate in health and disease. Marcel Dekker; New York: 1995. pp. 1–22. [Google Scholar]
- Shaw GM, Lammer EJ, Zhu H, et al. Maternal periconceptional vitamin use, genetic variation of infant reduced folate carrier (A80G), and risk of spina bifida. Am J Med Genet. 2002;108:1–6. doi: 10.1002/ajmg.10195. [DOI] [PubMed] [Google Scholar]
- Smithells R, Sheppard S, Schorah J. Vitamin deficiency and neural tube defects. Arch Dis Child. 1976;51:944–950. doi: 10.1136/adc.51.12.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulders YM, Smith DE, Kok RM, et al. Red blood cell folate vitamer distribution in healthy subjects is determined by the methylenetetrahydrofolate reductase C677T polymorphism and by the total folate status. J Nutr Biochem. 2007;18:693–699. doi: 10.1016/j.jnutbio.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Speer MC, Nye J, McLone D, et al. NTD Collaborative Group Possible interaction of genotypes at cystathionine beta-synthase and methylenetetrahydrofolate reductase (MTHFR) in neural tube defects. Clin Genet. 1999;56:142–144. doi: 10.1034/j.1399-0004.1999.560208.x. [DOI] [PubMed] [Google Scholar]
- Steegers-Theunissen RP, Boers GH, Trijbels FJ, Eskes TK. Neuraltube defects and derangement of homocysteine metabolism. N Engl J Med. 1991;324:199–200. doi: 10.1056/NEJM199101173240315. [DOI] [PubMed] [Google Scholar]
- Stover PJ. Physiology of folate and vitamin B12 in health and disease. Nutr Rev. 2004;62(6):S3–S13. doi: 10.1111/j.1753-4887.2004.tb00070.x. Pt 2. [DOI] [PubMed] [Google Scholar]
- Stover PJ, Garza C. Bringing individuality to public health recommendations. J Nutr. 2002;132:2476S–2480S. doi: 10.1093/jn/132.8.2476S. [DOI] [PubMed] [Google Scholar]
- Stover P, Schirch V. Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate. J Biol Chem. 1990;265:14227–14233. [PubMed] [Google Scholar]
- Suh JR, Herbig AK, Stover PJ. New perspectives on folate catabolism. Annu Rev Nutr. 2001;21:255–282. doi: 10.1146/annurev.nutr.21.1.255. [DOI] [PubMed] [Google Scholar]
- Sunden SL, Renduchintala MS, Park EI, et al. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch Biochem Biophys. 1997;345:171–174. doi: 10.1006/abbi.1997.0246. [DOI] [PubMed] [Google Scholar]
- Tamura T, Stokstad EL. The availability of food folate in man. Br J Haematol. 1973;25:513–532. doi: 10.1111/j.1365-2141.1973.tb01763.x. [DOI] [PubMed] [Google Scholar]
- Taparia S, Gelineau-van Waes J, Rosenquist TH, Finnell RH. Importance of folate-homocysteine homeostasis during early embryonic development. Clin Chem Lab Med. 2007;45:1717–1727. doi: 10.1515/CCLM.2007.345. [DOI] [PubMed] [Google Scholar]
- van der Linden IJ, Heil SG, Kouwenberg IC, et al. The methylenetetrahydrofolate dehydrogenase (MTHFD1) 1958G>A variant is not associated with spina bifida risk in the Dutch population. Clin Genet. 2007a;72:599–600. doi: 10.1111/j.1399-0004.2007.00904.x. [DOI] [PubMed] [Google Scholar]
- van der Linden IJ, Nguyen U, Heil SG, et al. Variation and expression of dihydrofolate reductase (DHFR) in relation to spina bifida. Mol Genet Metab. 2007b;91:98–103. doi: 10.1016/j.ymgme.2007.01.009. [DOI] [PubMed] [Google Scholar]
- van der Put NM, Eskes TK, Blom HJ. Is the common 677C−>T mutation in the methylenetetrahydrofolate reductase gene a risk factor for neural tube defects? A meta-analysis. QJM. 1997a;90:111–115. doi: 10.1093/qjmed/90.2.111. [DOI] [PubMed] [Google Scholar]
- van der Put NM, Steegers-Theunissen RP, Frosst P, et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet. 1995;346:1070–1071. doi: 10.1016/s0140-6736(95)91743-8. [DOI] [PubMed] [Google Scholar]
- van der Put NM, van den Heuvel LP, Steegers-Theunissen RP, et al. Decreased methylene tetrahydrofolate reductase activity due to the 677C−>T mutation in families with spina bifida offspring. J Mol Med. 1996;74:691–694. doi: 10.1007/s001090050073. [DOI] [PubMed] [Google Scholar]
- van der Put NM, van der Molen EF, Kluijtmans LA, et al. Sequence analysis of the coding region of human methionine synthase: relevance to hyperhomocysteinaemia in neural-tube defects and vascular disease. QJM. 1997b;90:511–517. doi: 10.1093/qjmed/90.8.511. [DOI] [PubMed] [Google Scholar]
- Volcik KA, Shaw GM, Lammer EJ, et al. Evaluation of infant methylenetetrahydrofolate reductase genotype, maternal vitamin use, and risk of high versus low level spina bifida defects. Birth Defects Res A Clin Mol Teratol. 2003a;67:154–157. doi: 10.1002/bdra.10008. [DOI] [PubMed] [Google Scholar]
- Volcik KA, Shaw GM, Zhu H, et al. Associations between polymorphisms within the thymidylate synthase gene and spina bifida. Birth Defects Res A Clin Mol Teratol. 2003b;67:924–928. doi: 10.1002/bdra.10029. [DOI] [PubMed] [Google Scholar]
- Wagner C. Biochemical role of folate in cellular metabolism. In: Bailey LP, editor. Folate in health and disease. Marcel Dekkar; New York: 1995. pp. 23–42. [Google Scholar]
- Wang X, Wang RH, Li W, et al. Deng CX. Genetic interactions between Brca1 and Gadd45a in centrosome duplication, genetic stability, and neural tube closure. J Biol Chem. 2004;279:29606–29614. doi: 10.1074/jbc.M312279200. Jr. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding CS, Relton CL, Sutton MJ, et al. Thymidylate synthase repeat polymorphisms and risk of neural tube defects in a population from the northern United Kingdom. Birth Defects Res A Clin Mol Teratol. 2004;70:483–485. doi: 10.1002/bdra.20038. [DOI] [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C. Identification of the folate-binding proteins of rat liver mitochondria as dimethylglycine dehydrogenase and sarcosine dehydrogenase. Purification and folate-binding characteristics. J Biol Chem. 1981;256:4102–4108. [PubMed] [Google Scholar]
- Wlodarczyk BJ, Tang LS, Triplett A, et al. Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicol Appl Pharmacol. 2006;213:55–63. doi: 10.1016/j.taap.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Woeller CF, Anderson DD, Szebenyi DM, Stover PJ. Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J Biol Chem. 2007;282:17623–17631. doi: 10.1074/jbc.M702526200. [DOI] [PubMed] [Google Scholar]
- Zhao R, Russell RG, Wang Y, et al. Rescue of embryonic lethality in reduced folate carrier-deficient mice by maternal folic acid supplementation reveals early neonatal failure of hematopoietic organs. J Biol Chem. 2001;276:10224–10228. doi: 10.1074/jbc.c000905200. [DOI] [PubMed] [Google Scholar]
- Zhu H, Curry S, Wen S, et al. Are the betaine-homocysteine methyltransferase (BHMT and BHMT2) genes risk factors for spina bifida and orofacial clefts? Am J Med Genet A. 2005;135:274–277. doi: 10.1002/ajmg.a.30739. [DOI] [PubMed] [Google Scholar]