

Abstract
The inhibitors of cyclin-dependent kinase (CDK) 4 (INK4) bind CDK4/6 to prevent their association with D-cyclins and G1 cell cycle initiation and progression. We report here that among the seven CDK inhibitors, p18INK4c played an important role in modulating TCR-mediated T cell proliferation. Loss of p18INK4c in T cells led to hyperproliferation in response to CD3 stimulation. p18INK4c-null mice developed lymphoproliferative disorder and T cell lymphomas. Expression of IL-2, IL-2R-a, and the major G1 cell cycle regulatory proteins was not altered in p18-null T cells. Both FK506 and rapamycin efficiently inhibited proliferation of p18-null T cells. In activated T cells, p18INK4c remained constant, and preferentially associated with and inhibited CDK6 but not CDK4. We propose that p18INK4c sets an inhibitory threshold in T cells and one function of CD28 costimulation is to counteract the p18INK4c inhibitory activity on CDK6-cyclin D complexes. The p18INK4c protein may provide a novel target to modulate T cell immunity.
Quiescent (G0) T cells respond to Ag stimulation by entering into cell cycle, producing cytokine, and undergoing clonal expansion. Signal transduction pathways leading to cytokine (IL-2) induction have been extensively studied (1, 2). Efficient activation of quiescent T cells requires two signals (3, 4). The primary signal involves the TCR to interact with specific Ags in the MHC complex on APCs. The major costimulatory molecule is CD28 on T cells, which interacts with the B7.1 or B7.2 molecules on APC. TCR activation leads to activation of nonreceptor tyrosine kinases such as lck, fyn, and ZAP70 that activate the NFATc via phospholipase Cγ1 and calcineurin, and induction of the Fos/Jun (AP1) transcription factor via the Ras and mitogen-activated protein kinase pathway (1, 5). Activated NFATc and AP1 bind to the IL-2 promoter to induce IL-2 expression. The CD28-specific signaling pathway is not well defined, probably involving activation of the phosphatidylinositol 3 kinase and Akt kinase pathway (6, 7), the Rho family GTPase Rac, and NF-KB (8). CD28 costimulation enhances the TCR-mediated signals in IL-2 expression by enhancing the stability of the IL-2 mRNA as well as IL-2 transcription. Blocking of CD28 costimulation suppresses T cell proliferation and induces unresponsiveness and cell death (9).
Relatively less is known about how the G0/G1 cell cycle regulatory molecules function during T cell activation. IL-2-mediated mitogenic signaling has been suggested to lead to clonal expansion of activated T cells (10, 11) and Stat5a/b are required for IL-2-induced cell cycle progression of peripheral T cells (12). However, IL-2-independent cell proliferation induced by TCR and CD28 costimulation has also been reported (13). Entry of quiescent (G0) cells into G1 phases of the cell cycle involves induction of a number of cell cycle activators, including cyclin-dependent kinases (CDKs)4 4/6 and D-type cyclins. Activation of cyclin D-CDK4/6, in conjunction with subsequent activation of cyclin E-CDK2, results in phosphorylation of members of retinoblastoma product family proteins and progression through the G1 phase of the cell cycle (14–16). Mammalian CDKs are also negatively regulated by CDK inhibitors (17). Currently, seven CDK inhibitors have been identified in mammalian cells. They consist of two distinct multigene families that differ in both structure and mechanism of action. Members of the CIP/KIP family (p21CIP1, p27KIP1, and p57KIP2) inhibit activity of all CDKs by forming a ternary complex with cyclin-CDK, whereas members of the INK4 family (p16INK4a, p15INK4b, p18INK4c, and p19INK4d) specifically inactivate CDK4 and CDK6 activity by forming a binary INK4-CDK4/6 complex (17). Thus, CDK4/6 are regulated by both families of CDK inhibitors and by mitogen-induced D-cyclins, and may serve as integrators for various signaling pathways of G1 cell cycle control.
In T cells, CDK4/6 and D-type cyclins are induced by T cell activation via TCR and CD28 costimulation (13, 18, 19), or by chemical or growth factor stimulations (20–22). Cyclin D3 has been reported to be the major inducible D-type cyclin in T cells (19, 21, 23, 24). Among the CIP/KIP family of CDK inhibitors, p21 (25) and p57 (26) are not significantly expressed in lymphoid organs, and mutations of the genes show no effect on T cell proliferation. Induction of p21 during T cell activation has recently been reported, and autoimmune diseases develop in old female mice (27). p27 is preferentially expressed in lymphoid organs and cells (28, 29). T cell activation with mitogens and IL-2 leads to a decrease of steady-state p27 level in T cells (30, 31) and to a higher rate of proliferation of p27-deficient mouse T cells and thymocytes (32). Thus, a potential role of p27 in the negative modulation of T cell proliferation has been proposed. However, other studies report that p27-deficient mouse peripheral T cells from the spleen or lymph node (LN) show normal proliferation in response to CD3/CD28 stimulation (33) or to CD3/IL-2 stimulation (34). These discrepant reports indicate that the role of p27 in modulating TCR-mediated T cell proliferation is unclear at present. A recent report has implicated p27 in the induction of T cell unresponsiveness to stimulation (T cell anergy, Ref. 35). Therefore, although not directly involved in regulating initial T cell clonal expansion, p21 and p27 may be involved in modulating some aspects of T cell immunity.
Of the four INK4 CDK inhibitors, p16INK4a expression is not detected and p15INK4b is expressed at low levels in lymphoid organs (36). The p18INK4c and p19INK4d genes are both highly expressed in lymphoid organs/cells (37–42). Inactivation of p15INK4b was recently shown to have little or no significant effect on T cell proliferation (43). In B lymphocytes, p19 is induced after B cell activation (44), and T cells with a mutant p19 allele show normal T lymphocyte proliferation (45). In addition to expression in lymphoid organs, a role of p18 in regulating T cell proliferation is also suggested by the observations that mice lacking p18 develop enlarged lymphoid organs and that p18-deficient lymphocytes show increased proliferation to lectin-mediated stimulation (43, 46).
We report here that the p18-deficient T cells were hyperproliferative in response to TCR stimulation in the absence or presence of CD28 costimulation. These findings establish a role of p18 in modulating TCR-mediated T cell proliferation, and we suggest that one important function of CD28 costimulation (or strong stimulation signals) is to antagonize the p18 function. T cells lacking p27 or p19 exhibited normal levels of T cell proliferation after activation with CD3 and CD28, indicating a specificity of p18 in the modulation of T cell proliferation. Although anti-CD3 mAb treatment induced efficient expression of CDK4/6, full induction of cyclin D3 required CD28 costimulation. During activation of T cells, the level of p18 proteins remained constant. Induction of CDK4/6 correlated with an increase of p18-CDK6 complex but not p18-CDK4 complex. In p18-null T cells activated with anti-CD3, CDK6- but not CDK4-associated kinase activity was elevated over wild-type (WT) controls. Our results suggest a model in which p18 functions as an inhibitory threshold in quiescent T cells and modulates proliferation of T cells.
Materials and Methods
Mutant mice and reconstituted SCID mice
WT and p18-null mice were maintained as described (46). Littermates or age-matched mice (2–3 mo of age) were used in each experiment. For T cell lymphoma development, age-matched WT or p18-null mice with similar genetic breeding history were kept in microisolator cages for 12–18 mo and analyzed for different types of tumors. T cell lymphomas were defined as T cell tumors in multiple organs including the LNs, spleen, and liver, and by histopathology. The tumor cells were analyzed by FACS for T and B cell markers.
To reconstitute T cells in SCID mice, WT or p19-null fetal liver cells from 14-day-old embryos (or WT and p18-null bone marrow cells) were injected into irradiated scid/scid CB17 mice (0.5 × 106 fetal liver cells or 2 × 106 bone marrow cells per mouse) as described (47). LN T cells in the reconstituted mice were harvested at 8 wk post reconstitution, and standard T cell proliferation assays were performed. Normal T cell reconstitution was observed with p19-null fetal liver cells or with p18-null bone marrow cells (data not shown). No significant number of T cells is recoverable from nonreconstituted SCID mice.
Abs and FACS assays
Monoclonal Abs used for immunofluorescence staining include hamster anti-mouse CD3-FITC (500-A2), rat anti-mouse CD4-FITC (CT-CD4), and rat anti-mouse CD25 IL-2R-PE (PC61 5.3) (Caltag Laboratories, Burlingame, CA). LN cells were stained and analyzed on a FACScan flow cytometer (BD Biosciences, Mountain View, CA). Nonviable cells were excluded by propidium iodide staining and light scatter profiles. The FACS data were analyzed with Cyclops 2000 version 4 data analysis software (Cytomation, Fort Collins, CO).
LN T cell preparation, activation, and proliferation assays
Complete medium (RPMI 1640 supplemented with 10% FCS, 2 mM L-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin) was used for cell preparation and culture. Cervical, umbilical, and axial LNs from each mouse were harvested and pooled. CD3+ T cells were purified by negative enrichment (B220 depletion) with Immuno-columns (Biotex Laboratories, Edmonton, Canada) according to manufacturer’s protocol. For stimulation, T cells (2 × 106 total LN cells or 8 × 105 purified CD3+ cells) were stained with 100 μl of anti-CD3 (0.2 μg/ml) alone or in combination with anti-CD28 (1 μg/ml) for 30 min on ice. Total LN cells (5 × 105) (or 2 × 105 purified CD3+ T cells) were added to plates coated with goat anti-hamster Ab (50 μg/ml; Caltag Laboratories). Cells were incubated at 37°C for 36 h and pulsed for 12 h with 1 μCi [3H]thymidine (NEN, Boston, MA) per well. All assays were performed in triplicate. Student’s t test was used for the statistical analysis.
Various concentrations of rapamycin and FK506 were added to activated T cell cultures. For the anti-CD3 titration assay, LN cells were plated in wells previously coated with different concentrations of anti-mouse CD3 mAb as described (48).
CD25 expression and IL-2 production assay
LN T cells were unstimulated, stimulated with either anti-CD3 alone, or stimulated with both anti-CD3 and anti-CD28 in triplicate wells. The IL-2 level in the culture supernatant were measured by an ELISA kit at 12 and 24 h poststimulation (BioSource International, Camarillo, CA). The LN T cells were also analyzed for CD25 expression (by FACS) and for proliferation by [3H]thymidine incorporation.
Western, immunoprecipitation (IP)-Western blot, and IP-kinase assays
Antisera for p18, p27, CDK2, CDK4, CDK6, and tubulin have been previously described (46, 49). IP, immunoblotting, and IP-kinase assay procedures were performed as described previously (46, 50, 51). LN cells from age-matched WT or p18−/− (null) mice were pooled and stimulated with anti-CD3, anti-CD28, or both. Cells treated with either no Ab or a low concentration of anti-CD3 (~1 ng/106 cells) that did not induce activation in either WT or p18-null cells (data not shown) were used as negative controls. Cells were activated as described above and harvested at 36 h post activation for biochemistry assays. Protein concentrations were determined by the Bradford assay and equal amounts of proteins were used for each experiment. T cell activation was confirmed by measuring [3H]thymidine incorporation and CD25 expression. The kinase activity was quantified by a PhosphorImager with ImageQuant 5.0 software (Molecular Dynamics, Sunnyvale, CA). Densitometry was used for quantifying Western blots with NIH Image 1.61 software.
Results
Hyperproliferative responsiveness of p18-null T cells to TCR stimulation
Mice lacking p18 develop enlarged lymphoid organs, and p18-deficient lymphocytes exhibit an increased proliferation rate in response to lectin stimulation (46). We further investigated the role of p18 in modulating T cell proliferation in response to TCR-mediated activation. We demonstrated that activation of TCR with the anti-CD3 mAb led to hyperproliferation in the absence of CD28 costimulation (Fig. 1). LN T cells from WT mice showed low or no detectable levels of proliferation when activated with only anti-CD3 mAb. However, LN T cells from p18-null mice efficiently incorporated [3H]thymidine when stimulated with the same concentrations of anti-CD3 mAb (3- to 5-fold over WT, Fig. 1). Costimulation with anti-CD3/CD28 mAbs led to proliferation of both WT and p18-null T cells, and p18-null T cells showed further enhanced proliferation than WT T cells (Fig. 1). Analysis of data from 19 independent experiments with 43 mice of each genotype showed that p18-null T cells consistently exhibited an average of 4-fold higher rate of proliferation than the WT T cells in response to anti-CD3 stimulation (Fig. 1B, p < 0.0005). Under the condition of costimulation with anti-CD3 and anti-CD28 mAbs, p18-null T cells still proliferated at an average of 2-fold higher rate than the WT T cells ( p < 0.05). Therefore, p18-deficient LN T cells responded to TCR (anti-CD3) stimulation more efficiently than WT T cells. This suggests that one of the costimulation functions of CD28 in T cells may be to counteract the inhibitory activity of p18.
FIGURE 1.

Hyperproliferative responses of p18−/− T cells to TCR stimulation. A, Enhanced proliferation of p18−/− LN cells was observed in response to stimulation with anti-CD3 or to costimulation with anti-CD3 and anti-CD28 mAbs. LN T cells from each mouse were assayed in triplicate. Data are from one representative experiment with two mice of each genotype, and error bars indicate SD of the average between the two mice. LN T cells cultured without stimulation were used as background controls (Bkg). B, Data from 19 independent experiments (43 mice of each genotype) are summarized. The average value obtained from WT T cells in each experiment is equalized to 1. The fold of increased proliferation of p18-null T cells is calculated. Anti-CD3 stimulation of p18−/− T cells led to a 4-fold hyperproliferation (p < 0.0005). Costimulation with anti-CD3/CD28 led to a 2.7-fold enhanced proliferation of p18-null T cells (p < 0.05). Error bars indicate SE values (n = 19). C, Enriched CD3+ T cells from WT and p18-null mice were used for the proliferation assays as described in A. The experiment was performed two times with similar results. D, Loss of p27 in T cells did not lead to hyperproliferation in response to TCR stimulation. LN T cells from p27 mutant or WT littermate mice were isolated and analyzed as above. Three independent experiments were performed with similar results. E and F, Loss of p19INK4d did not lead to TCR-mediated T cell hyperproliferation. T cells derived from SCID mice reconstituted with p19-null or WT fetal liver cells showed similar proliferation in response to anti-CD3 stimulation and anti-CD3/CD28 costimulation (E). Data with LN T cells from two reconstituted SCID mice are shown. SD values are shown as error bars. T cells derived from SCID mice reconstituted with p18-null bone marrow cells were hyperproliferative in response to TCR (anti-CD3) stimulation or CD28 costimulation (F). Shown are data from triplicate samples with LN T cells from reconstituted SCID mice. SD values are shown as error bars.
Although the majority of total LN cells are T lymphocytes, lymphocytes from LNs also contain a small percentage of B cells. To show that the hyperproliferation is intrinsic to p18-null T lymphocytes, we purified the CD3+ T cells from WT or p18-null LNs by negatively depleting B cells. Their proliferation rate in response to anti-CD3 stimulation and anti-CD3/anti-CD28 costimulation was determined. Consistent with the results obtained from total LN lymphocytes, a higher rate of T cell proliferation was seen in p18-null CD3+ T cells in response to anti-CD3 stimulation or to costimulation with anti-CD3 and anti-CD28 (Fig. 1C). Therefore, the p18-null T cells are intrinsically hyperproliferative in response to stimulation via their TCRs.
p18 plays an important role in the modulation of T cell proliferation
T cell activation with mitogens and IL-2 leads to a decrease of steady-state p27 level in T cells (30, 31) and to a higher rate of proliferation of p27-deficient mouse T cells and thymocytes (32). Thus, a potential role of p27 in the negative modulation of T cell proliferation has been implicated. However, other reports show that p27-deficient mouse peripheral T cells from the spleen or LN proliferate normally in response to CD3/CD28 stimulation (33) or to CD3/IL-2 stimulation (34). We tested the proliferation of p27-null LN T cells and showed that WT and p27-null T cells proliferated at the same levels in response to CD3 stimulation or to CD3/CD28 costimulation (Fig. 1D).
The p19 gene, like p18, is highly expressed in lymphoid cells/tissues (38–41). We generated two lines of p19-null mice with a p19 mutation that deleted a larger portion of the p19 coding region than the one reported with an insertion mutation (45) and resulted in lethality in homozygous embryos at 12–14 days postgestation (M. Nicholas and Y. Xiong, unpublished data). To further confirm the specificity of p18 in modulating T cell proliferation, we derived T cells from scid/scid mice reconstituted with the p19-null and WT littermate fetal liver cells and tested for their proliferation responses to anti-CD3 or anti-CD3/CD28 stimulation. In contrast to the p18-null T cells, there was no detectable increase of proliferation of reconstituted p19 mutant T cells in response to either CD3 alone or CD3/CD28 costimulation (Fig. 1E). To control for the effect of SCID reconstitution on T cells, bone marrow cells from WT or p18-null mice were also used to reconstitute scid/scid mice to derive T cells that were similarly analyzed. Consistent with previous studies with T cells directly isolated from p18-null mice, reconstituted p18-null T cells showed hyperproliferative responses to either CD3 single stimulation or CD3/CD28 costimulation (Fig. 1F). These results, together with the lack of any detectable effect on TCR-mediated T cell proliferation by the loss of p21 (52, 53), p57 (54), and p27 (Refs. 33 and 34 and Fig. 1D), indicate the specificity of p18 as a unique CDK inhibitor in regulating peripheral T cell proliferation in response to TCR stimulation.
Lymphoproliferative disorder and T cell lymphomas in old p18-null mice
Consistent with hyperproliferation of p18-null T cells, p18-null mice developed lymphoproliferative disorders illustrated by enlarged secondary lymphoid organs (Fig. 2A, spleen) (46). In the spleen, expansion of the white pulp was largely confined to the periarteriolar lymphatic sheaths (indicated by arrows in Fig. 2C). A small number of p18-null mice (3 of 24, 12%) at 12–18 mo of age developed T cell lymphomas in multiple organs including the LNs, spleen, and liver. None of the age-matched WT control mice observed in the same period developed lymphomas (n = 20). Histopathology analysis of the lymphomas revealed massive lymphocytic infiltration of the liver, LNs, and spleen with effacement of normal architecture. The lymphomas were composed of monomorphic lymphocytes with irregularly oval nuclei, a single, often central nucleolus, finely clumped chromatin, and scant cytoplasm (Fig. 2D, and data not shown). Most cells from the p18-null lymphoma tissues expressed the CD3 marker (>90% T cells) indicating their T cell origin (Fig. 2E). Development of T cell lymphomas is consistent with the increased cellularity of T lymphocytes in p18-null mice and with their hyperresponsiveness to TCR stimulation (Fig. 1), and suggests a role of p18 in suppressing T cell lymphoma development. Compared with development of pituitary tumors in the intermediate lobe observed in the majority of p18 mutant mice (43, 46), the development of T cell lymphomas occurred at a lower frequency and in older animals. The low penetrance suggests that additional genetic mutation(s) is required to collaborate with the p18 loss in the development of T cell lymphomas.
FIGURE 2.

Lymphoproliferative disorder and T cell lymphomas in p18−/− mice. A, Enlargement of spleens from p18-null mice. Photographs of spleens of a WT or a p18−/− mouse (2-mo old) indicate their relative sizes. B and C, Hyperplastic white pulp in the p18-null spleen. Histology (stained with H&E) of the WT (B) and p18−/− (C) spleens shows enlarged T cells zone (periarteriolar lymphatic sheaths) (light blue area of follicles surrounding the center arterioles shown by arrows) in the p18-null spleen. D, Lymphomas in the p18 mouse. Histopathology of a spleen lymphoma from a 15-mo-old p18-null mouse shows effacement of red and white pulp with densely packed, monomorphic lymphocytes. E, Flow cytometric analysis of lymphocytes obtained from the lymphoma tissues. The cells were stained with anti-CD3 and anti-B220. The majority of the cells are T cells (94% CD3+ cells).
p18-null T cells show normal expression of IL-2 and IL-2Rα
IL-2-mediated mitogenesis has been suggested to play a major role in T cell proliferation after activation (10, 12). To test the possibility that elevated expression of IL-2 and/or IL-2 receptors may contribute to the hyperproliferation of p18-null T cells in response to CD3-mediated signaling, we analyzed expression of the high-affinity IL-2R-a (CD25) and IL-2 production in LN T cells. Freshly isolated LN cells or LN cells from WT and p18 mutant mice cultured in the absence of Ab stimulation showed similarly low levels of CD25 expression (Fig. 3A, top panels, and data not shown). When activated with either CD3 alone or with both CD3 and CD28 Abs, no significant differences were observed between WT and p18-null T cells for CD25 expression at 12 or 24 h post stimulation (Fig. 3A). Similarly, IL-2 production with either CD3 or with both CD3 and CD28 mAbs was the same for WT and p18-null T cells (Fig. 3B). The same WT or p18-null cells were also tested for proliferation, and p18-null T cells proliferated efficiently in response to CD3 stimulation despite low levels of IL-2 production (data not shown).
FIGURE 3.

Normal IL-2 production and IL-2 dependence of p18−/− T cells. A, Normal IL-2-Ra (CD25) expression in p18−/− T lymphocytes. LN cells from WT (left) and p18−/− (right) mice were unstimulated (background, upper panels), or activated with anti-CD3 (middle panels) or anti-CD3/CD28 (lower panels). CD25 expression was monitored at 12 and 24 h post activation. The experiments were repeated three times. B, Normal IL-2 induction in p18−/− T cells. Cells similarly stimulated as above were used for measuring IL-2 production (in picograms per milliliter) in the culture supernatant by ELISA. Data represent results from triplicate samples, and SD is shown as error bars. Three independent experiments were performed with similar results. C, Normal responses of p18-null T cells to inhibitors of T cell activation. LN T cells were costimulated with anti-CD3/anti-CD28 mAbs and cultured with various concentrations of FK 506 (left panel) and rapamycin (right panel). Two mice were used for each genotype. The SD is shown as error bars. The experiment was repeated three times with similar results.
Both FK506 and rapamycin efficiently inhibit p18-null T cell proliferation
CD28 costimulation appears to be required to counteract the p18 activity for efficient T cell proliferation. TCR-mediated signaling pathway via the Ca2+-dependent (calcineurin) NFAT activation and IL-2 expression can be inhibited by FK506 (1, 55), and IL-2-induced proliferation is sensitive to rapamycin (31, 56, 57). When tested, either FK506 or rapamycin efficiently inhibited the proliferation of both WT and p18-null T cells (Fig. 3C). Therefore, TCR-mediated signaling via calcineurin is essential for the proliferation of both WT and p18-null T cells. The rapamycin-sensitive IL-2 signaling pathways are also required for the proliferation of p18-null T cells.
Expression of G1 regulatory proteins in p18-null T cells is unchanged
To define the biochemical mechanism of p18-null T cell hyperproliferation, we examined the induction of three major G1 CDKs (CDK2, CDK4, and CDK6), the major D-type cyclin induced in T cells (cyclin D3), and CDK inhibitor p27KIP1, during activation of WT and p18-deficient LN T cells (Fig. 4). Anti-CD3 alone, as well as anti-CD3/CD28 costimulation, efficiently induced expression of CDK2, CDK4, and CDK6, and a significant degradation of p27. However, cyclin D3 was only induced weakly by anti-CD3 stimulation. Costimulation with anti-CD28 resulted in a 4-fold increase of cyclin D3 than anti-CD3 alone (Fig. 4 and data not shown). These results indicate that whereas the expression of G1 CDK proteins is primarily controlled by anti-CD3 (TCR) pathway, the expression of cyclin D3 is regulated by both anti-CD3 and anti-CD28 signaling pathways and may be required to counteract the p18 inhibitory activity in T cells. The expression patterns of these five G1 regulatory proteins were consistent with published results and indistinguishable between the WT and p18-deficient T cells. These results suggest that loss of p18 did not affect the expression and steady levels of CDK2, CDK4/6, cyclin D3, and p27.
FIGURE 4.
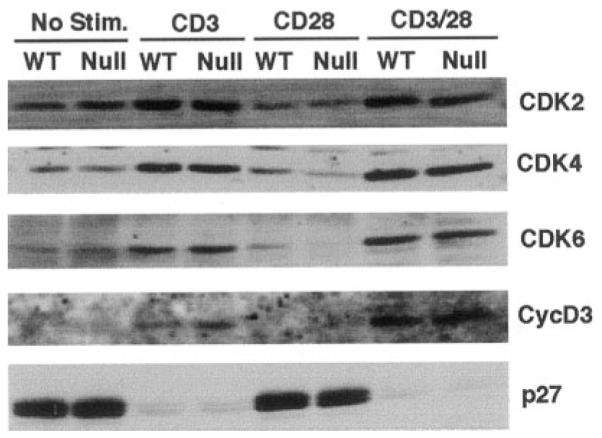
Normal induction of G1 cell cycle regulators in p18−/− T cells. LN T cells from WT or p18−/− (Null) mice were incubated without Ab activation (No Stim.), stimulated with anti-CD3, anti-CD28, or both (CD3/CD28). Cells were harvested at 36 h post culture for Western blot assays with various Abs (labeled on the right). T cell activation was confirmed by measuring [3H]thymidine incorporation (data not shown). Equal amounts of protein from each sample were analyzed.
The p18 protein remains constant during T cell activation, and is associated with and inhibitory to CDK6 but not CDK4 in activated T cells
We next examined the steady-state level of p18 during T cell activation. Freshly isolated LN T cells or LN T cells cultured in the absence of stimulation showed very low levels of CDK4 and CDK6, and high levels of p27 (Figs. 4 and 5A, lanes 1 and 2). In LN cells activated with anti-CD3 and anti-CD28, both CDK4 and CDK6 were induced and p27 was degraded (Fig. 5A, lane 3). However, the level of p18 was not significantly altered after T cell activation (Fig. 5A).
FIGURE 5.

p18 is stable and associated preferentially with CDK6 in T cells. A, Stable levels of p18 during T cell activation. Total levels of CDK6, CDK4, p27, and p18 in freshly isolated LN T cells (lane 1) or cultured for 36h (5 × 106/ml) without (lane 2) and with (lane 3) CD3/CD28 activation were determined by IP-Western blot analysis with specific Ab. B, Preferential association of p18 with CDK6 in activated T cells. Coimmunoprecipitation of p18 with CDK4 (lane 1) and CDK6 (lane 2) in CD3/CD28-activated T cells (from Fig. 5A) was performed. The IP pellets were immunoblotted with anti-CDK4 (top left), anti-CDK6 (top right), or anti-p18 Ab (bottom panels). C, Preferential association of CDK6 and p18 in primary T cells stimulated with anti-CD3 and/or anti-CD28 mAbs. LN T cells were cultured in the absence (lane 1) or presence of anti-CD3 (lane 2), anti-CD28 (lane 3), and both anti-CD3 and anti-CD28 (lane 4). The cells were harvested at 36 h post activation and analyzed by IP (with Abs labeled on top of the panels) and Western blot (labeled on the left and right) assays. Densitometry results (mean density) are presented. ■, Level of p18 associated with CDK6 (bottom right panel); □, p18 associated with CDK4 (bottom left panel). The band below the CDK6 band in the CDK6 IP was not related to CDK4, because immunoblotting of the anti-CDK6 IP with the anti-CDK4 Ab did not detect this band.
When its association with CDK4 or CDK6 was analyzed by coimmunoprecipitation, we found that p18 was preferentially associated with CDK6 in activated T cells (Fig. 5, B and C). Only a minor amount of p18 was detected in association with CDK4 in resting or activated T cells (Fig. 5B, left panel and C, lowest left panel) despite readily detectable levels of CDK4 and p18 (Fig. 5, B and C). Preferential association of p18 with CDK6 in vivo is consistent with previous findings that p18 has higher affinity for CDK6 than CDK4 in vitro (37). The band below the CDK6 band in the anti-CDK6 IP (Fig. 5C, upper right panel), although running close to the same size as CDK4, is not related to CDK4 because it was not detected with the anti-CDK4 Ab (data not shown).
When the kinase activity of CDK4 and CDK6 was analyzed, we found that CDK4 activity in WT T cells was efficiently induced by CD3 stimulation alone, and full activation by CD3 and CD28 costimulation showed no further enhancement (Fig. 6A). The slight reduction of CDK4 activity in p18-null T cells after CD3 stimulation was not consistently observed. Consistent with lack of significant p18 association with CDK4 (Fig. 5), p18-null T cells showed no increased CDK4 activity either stimulated with CD3 or costimulated with CD3 and CD28 (Fig. 6A). In contrast, the kinase activity of CDK6 in WT T cells was only weakly induced by CD3 stimulation, and full induction of CDK6 activity required CD3 and CD28 costimulation (Fig. 6B). Consistent with its preferential association with p18, p18-null T cells activated with anti-CD3, at a concentration that induced proliferation of p18-null but not WT T cells, displayed a 3-fold increase of elevated CDK6 kinase activity over WT T cells (Fig. 6C). Like unstimulated T cells, WT or p18-null T cells stimulated with low concentrations of anti-CD3 (that did not induce either WT or p18 mutant T cell proliferation) showed no significant induction of CDK6 activity and no proliferation (Fig. 6C and data not shown). A slight but reproducible reduction of CDK6 activity in p18-null T cells after CD3/CD28 costimulation was observed, but its significance on T cell proliferation was not clear.
FIGURE 6.

p18-null T cells activated with anti-CD3 showed enhanced CDK6 activity. A, Normal CDK4 activity in p18-null T cells. WT or p18-null LN T cells were cultured without stimulation (unstimulated), stimulated with anti-CD3 mAb, or costimulated with anti-CD3 and anti-CD28 mAbs (anti-CD3/CD28) for 36 h. The competing peptide (+) for the anti-CDK4 Ab was used as specificity control. The IP pellets were assayed for CDK4 kinase activity with recombinant GST-retinoblastoma product as substrates. PhosphorImager quantitation represents the relative activity of CDK4 in WT (□) and p18−/− cells (■). B, Full induction of CDK6 activity in T cells required CD3/CD28 costimulation. WT LN T cells were treated as in Fig. 6A, except an anti-CDK6 Ab was used for the IP-kinase assay. The competing peptide (+) for the anti-CDK6 Ab was used as specificity control. ■, Relative CDK6 activity as a result of PhosphorImager quantitation. C, Enhanced CDK6 kinase activity in CD3-activated p18-null T cells. WT or p18-null LN T cells were cultured with low (anti-CD3 low = 1 ng/106 cells, no proliferation induced for either WT or p18-null T cells) or high (anti-CD3 high = 2.5 ng/106 cells, induced proliferation of p18-null but not WT T cells) concentrations of anti-CD3, or stimulated with anti-CD3 and anti-CD28 mAbs for 36 h. CDK6 kinase activity assay was performed by IP of CDK6 as above. PhosphorImager quantitation is shown for WT (□) and p18−/− (■) T cells.
Discussion
We report here that among the seven CDK inhibitor genes, p18INK4c was uniquely involved in negatively modulating T cell-proliferative responses to TCR and CD28 costimulation. Loss of p18 in T cells reduced the requirement of CD28 costimulation for efficient T cell proliferation (Fig. 1). Lymphoproliferative disorder and lymphomas developed in old p18-null mice (Fig. 2). Furthermore, our biochemical analyses of the G1 regulatory proteins indicated that p18 was stable and preferentially inhibitory to CDK6 but not CDK4 activity in activated T cells (Figs. 5 and 6). The data suggest that p18INK4c may set an inhibitory threshold in resting T cells. One of the functions of CD28 costimulation is to overcome the p18 inhibitory threshold, probably by fully inducing cyclin D, which competes with p18 to form active CDK6-cyclin D complexes.
p18 is a CDK inhibitor involved in modulating T cell proliferation
The presence of seven distinct CDK inhibitor genes with apparently overlapping activity suggests that individual CDK inhibitor genes may function in specific cell types. Indeed, individual CDK inhibitors are expressed in tissue-specific fashions (58, 59). However, multiple CDK inhibitors are detected in the same cell types. For example, p18, p19, and p27 are all highly expressed in lymphoid organs and lymphocytes (28–30, 38–40). Three lines of evidence support a specific and unique role of the p18 protein in modulating T cell proliferation. First, p18-null T cells exhibit hyperproliferative responses to anti-CD3 single stimulation and to anti-CD3/anti-CD28 costimulation (Fig. 1) or to nonspecific mitogen stimulation (46). Loss of p15 (or of both p15 and p18) in T cells has no significant (or further) effect on T cell proliferation to mitogenic activation (43). Although also highly expressed in lymphoid cells and tissues, loss of either p27KIP1 (Fig. 1D and Refs. 33, 34) or p19INK4d (Fig. 1E and Ref. 45) showed no detectable effects on T cell proliferation in response to TCR stimulation. The hyperproliferation of total p27-null thymocytes in response to CD3 and IL-2 stimulation (32) may indicate a different role of p27 in thymocytes from its role in mature peripheral T lymphocytes. Second, p18-null mice developed enlarged lymphoid organs and increased lymphocyte cellularity (Fig. 2 and Ref. 46). Consistent with the hyperproliferative response of p18-deficient T cells, old p18-null mice developed lymphoproliferative disorders and T cell lymphomas (Fig. 2 and Ref. 43). Third, p18 was associated with CDK6 in T cells and loss of p18 resulted in elevated CDK6 activity and T cell hyperproliferation (Figs. 5 and 6). Therefore, p18 is a unique CDK inhibitor involved in modulating proliferation of T cells in response to TCR stimulation.
Induction of CDK6 activity is correlated with T cell proliferation and is the major target of p18 in peripheral T cells
Quiescent T cells respond to efficient Ag stimulation by cell cycle entry (proliferation-competence), cytokine production (Refs. 1, 2, and see introduction), and cell cycle progression (clonal expansion). Relatively little is known about how G0/G1 cell cycle regulatory molecules function during T cell activation. CDK4/6 and D-type cyclins are induced by T cell activation signals such as TCR and CD28 costimulation (13, 18, 19) or by chemical activation (21, 22, 60, 61). Stimulation with anti-CD3 alone leads to efficient induction of a number of G1 regulatory proteins, including CDK2, CDK4, and CDK6 (Fig. 4 and Ref. 12). However, full induction of D-cyclins depended on TCR and CD28 costimulation (Refs. 13, 18; and Fig. 4). This G0-G1 “cell cycle entry” step can occur independently of the IL-2 activity (10, 60), although CD3-induced CDK6 and D-cyclins appear to depend on Stat5a/b (12). It is conceivable that other cytokines that signal through Stat5a/b (62, 63) may also be involved. IL-2-mediated signaling is clearly required for the proliferation of p18-null T cells, because p18-null T cells produced WT levels of IL-2 and IL-2R (CD25), and showed normal sensitivity to FK506 or rapamycin (Fig. 3).
To elucidate the biochemical mechanism of G0/G1 regulation during T cell activation, we conducted a comprehensive analysis of three families of G1 cell cycle regulatory proteins during T cell activation with anti-CD3 and/or anti-CD28 stimulation (Figs. 4 – 6). In LN T cells, all three G1 CDKs (CDK2, CDK4, and CDK6) and cyclin D3 were induced, although full induction of cyclin D3 required CD28 costimulation. Correlated with T cell proliferation, efficient induction of CDK6 (but not CDK4) kinase activity required CD28 costimulation. p27 was significantly degraded and p18 remained unchanged in response to anti-CD3 alone and to anti-CD3/CD28 costimulation. Interestingly, p18 was preferentially associated with CDK6 in activated T cells. Only a minor amount of p18 was detected in association with CDK4 in T cells stimulated with CD3 or CD28 alone, or with both CD3 and CD28, despite readily detectable levels of CDK4 and p18 (Fig. 5 and data not shown). Consistent with the p18-CDK6 complex, CDK6 kinase activity, but not CDK4 activity, was hyperelevated in p18-null T cells (Fig. 6). Therefore, the major target of p18 inhibition in primary T cells is CDK6. Interestingly, induction of CDK4 and CDK6 in T cells requires distinct signaling pathways because Stat5a/b are required for the induction of CDK6, but not CDK4, by anti-CD3 stimulation (12).
In cell lines, p18 has been detected in association with both CDK4 and CDK6 (24, 39, 49, 64, 65), although p18 has higher affinity for CDK6 than CDK4 in vitro (37). In an Ag-dependent T cell line, we showed that p18 was associated with both CDK4 and CDK6. After activation of the T cell line with anti-CD3 and anti-CD28 mAbs, there was an increase of p18-CDK6 complex and decrease of p18-CDK4 association coincident with CDK6 induction (G. Kovalev and L. Su, unpublished results). The constitutively high levels of CDK4 in T cell lines cultured in vitro may have contributed to the detectable p18-CDK4 complexes in cell lines.
Modulation of CDK6 activity and T cell proliferation by p18
Based on the T cell proliferation study and biochemistry of G1 regulatory proteins, we propose that p18 sets an inhibitory threshold in resting T cells to maintain a stable quiescence state and to ensure that only significant stimulation of T cells leads to cell cycle activation and clonal expansion. In addition, the presence of p18 in activated T cells can down-modulate their proliferation to prevent overreactive immune responses. TCR (anti-CD3) signaling alone can induce expression of CDK4 and CDK6 proteins, but only low levels of cyclin D3 (Fig. 4 and data not shown), which are below the threshold to counteract the p18 activity for a proliferation response. Only in T cells coactivated with both TCR and CD28 can the p18 inhibition be overcome (by the full induction of D-cyclins, and CDK6). Loss of p18 removes the inhibitory threshold, allowing the assembly of stable, active CDK6-cyclin D complexes after TCR stimulation. Therefore, modulation of the p18 activity in T cells may provide a way to modulate T cell immunity.
Acknowledgments
We thank Dr. Roland Tisch for critically reading the manuscript, Dr. Jenny Ting for discussions, Dr. Virginia Godfrey for help with the analysis of lymphomas, Mike Nichols for providing WT and p19INK4d embryos, Tamara Simon for the reconstitution experiments, and Jonathan Smith, Liping Wang, and Maria Rodriguez for technical assistance. We also thank members of the Su laboratory for discussions and the University of North Carolina Division of Laboratory Animal Medicine and FACS core facilities for their help with animal cares and FACS analysis.
Footnotes
This study was supported by National Institutes of Health Grants AI41356 (to L.S.) and CA68377 (to Y.X.). Y.X. was a Pew Scholar in Biomedical Science and recipient of an American Cancer Society Junior Faculty award.
- CDK
- cyclin-dependent kinase
- INK4
- inhibitor of CDK4
- LN
- lymph node
- WT
- wild type
References
- 1.Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 2.Germain RN, Stefanova I. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu. Rev, Immunol. 1999;17:467. doi: 10.1146/annurev.immunol.17.1.467. [DOI] [PubMed] [Google Scholar]
- 3.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996;14:233. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 4.Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr. Opin. Cell Biol. 1999;11:203. doi: 10.1016/s0955-0674(99)80027-1. [DOI] [PubMed] [Google Scholar]
- 5.Masuda ES, Imamura R, Amasaki Y, Arai K, Arai N. Signalling into the T-cell nucleus: NFAT regulation. Cell. Signal. 1998;10:599. doi: 10.1016/s0898-6568(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 6.Parry RV, Reif K, Smith G, Sansom DM, Hemmings BA, Ward SG. Ligation of the T cell co-stimulatory receptor CD28 activates the serine-threonine protein kinase protein kinase B. Eur. J. Immunol. 1997;27:2495. doi: 10.1002/eji.1830271006. [DOI] [PubMed] [Google Scholar]
- 7.Kane L, Andres P, Howland K, Abbas A, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-'Y but not Th2 cytokines. Nat. Immunol. 2001;2:37. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- 8.Rudd CE. Upstream-downstream: CD28 cosignaling pathways and T cell function. Immunity. 1996;4:527. doi: 10.1016/s1074-7613(00)80479-3. [DOI] [PubMed] [Google Scholar]
- 9.Powell JD, Ragheb JA, Kitagawa-Sakakida S, Schwartz RH. Molecular regulation of interleukin-2 expression by CD28 co-stimulation and anergy. Immunol. Rev. 1998;165:287. doi: 10.1111/j.1600-065x.1998.tb01246.x. [DOI] [PubMed] [Google Scholar]
- 10.Modiano JF, Domenico J, Szepesi A, Terada N, Lucas JJ, Gelfand EW. Symmetry of the activation of cyclin-dependent kinases in mitogen and growth factor-stimulated T lymphocytes. Ann. NY Acad. Sci. 1995;766:134. doi: 10.1111/j.1749-6632.1995.tb26657.x. [DOI] [PubMed] [Google Scholar]
- 11.Mills GB, Girard P, Grinstein S, Gelfand EW. Interleukin-2 induces proliferation of T lymphocyte mutants lacking protein kinase C. Cell. 1988;55:91. doi: 10.1016/0092-8674(88)90012-8. [DOI] [PubMed] [Google Scholar]
- 12.Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, Hoffmeyer A, van Deursen J, Sangster MY, Bunting KD, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- 13.Boonen GJ, van Dijk AM, Verdonck LF, van Lier RA, Rijksen G, Medema RH. CD28 induces cell cycle progression by IL-2-independent down-regulation of p27kip1 expression in human peripheral T lymphocytes. Eur. J. Immunol. 1999;29:789. doi: 10.1002/(SICI)1521-4141(199903)29:03<789::AID-IMMU789>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 15.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 16.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 18.Kwon TK, Buchholz MA, Ponsalle P, Chrest FJ, Nordin AA. The regulation of p27Kip1 expression following the polyclonal activation of murine G0 T cells. J. Immunol. 1997;158:5642. [PubMed] [Google Scholar]
- 19.Nagasawa M, Melamed I, Kupfer A, Gelfand EW, Lucas JJ. Rapid nuclear translocation and increased activity of cyclin-dependent kinase 6 after T cell activation. J. Immunol. 1997;158:5146. [PubMed] [Google Scholar]
- 20.Lucas JJ, Szepesi A, Domenico J, Tordai A, Terada N, Gelfand EW. Differential regulation of the synthesis and activity of the major cyclin-dependent kinases, p34cdc2, p33cdk2, and p34cdk4, during cell cycle entry and progression in normal human T lymphocytes. J. Cell. Physiol. 1995;165:406. doi: 10.1002/jcp.1041650222. [DOI] [PubMed] [Google Scholar]
- 21.Laliberte J, Yee A, Xiong Y, Mitchell BS. Effects of guanine nucleotide depletion on cell cycle progression in human T lymphocytes. Blood. 1998;91:2896. [PubMed] [Google Scholar]
- 22.Ajchenbaum F, Ando K, DeCaprio JA, Griffin JD. Independent regulation of human D-type cyclin gene expression during G1 phase in primary human T lymphocytes. J. Biol. Chem. 1993;268:4113. [PubMed] [Google Scholar]
- 23.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21CIP1 and p27KIP1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parry D, Mahony D, Wills K, Lees E. Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol. Cell. Biol. 1999;19:1775. doi: 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- 26.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57Kip2, a structurally distinct member of the p21CIP1 CDK inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 27.Balomenos D, Martin-Caballero J, Garcia MI, Prieto I, Flores JM, Serrano M, Martinez AC. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat. Med. 2000;6:171. doi: 10.1038/72272. [DOI] [PubMed] [Google Scholar]
- 28.Polyak K, Lee M-H, Erdjument-Bromage H, Koff A, Roberts J, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 29.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk proteins kinase activity, is related to p21. Cell. 1994;78:67. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 30.Firpo EJ, Koff A, Solomon MJ, Roberts JM. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol. Cell. Biol. 1994;14:4889. doi: 10.1128/mcb.14.7.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nourse J, Firpo E, Flanagan MW, Coats S, Polyak K, Lee M-H, Massague J, Crabtree GR, Roberts JM. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 32.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai L-H, Broudy V, Perlmutter RM, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell. 1996;85:733. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 34.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996;85:721. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 35.Boussiotis VA, Freeman GJ, Taylor PA, Berezovskaya A, Grass I, Blazar BR, Nadler LM. functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat. Med. 2000;6:290. doi: 10.1038/73144. [DOI] [PubMed] [Google Scholar]
- 36.Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- 37.Guan K-L, Jenkins CW, Li Y, Nichols MA, Wu X, O’Keefe CL, Matera AG, Xiong Y. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 38.Chan FKM, Zhang L, Chen L, Shapiro DN, Winoto A. Identification of human/mouse p19, a novel cdk4/cdk6 inhibitor with homology to p16ink4. Mol. Cell. Biol. 1995;15:2682. doi: 10.1128/mcb.15.5.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirai H, Roussel MF, Kato J, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell. Biol. 1995;15:2672. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan KL, Jenkins CW, Li Y, O’Keefe CL, Noh S, Wu X, Zariwala M, Matera AG, Xiong Y. Isolation and characterization of p19INK4d, a p16-related inhibitor specific to CDK6 and CDK4. Mol. Biol. Cell. 1996;7:57. doi: 10.1091/mbc.7.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia. 1998;12:845. doi: 10.1038/sj.leu.2401043. [DOI] [PubMed] [Google Scholar]
- 42.Zariwala M, Liu E, Xiong Y. Mutational analysis of the p16 family cyclin-dependent kinase inhibitors p15INK4b and p18INK4c in tumor-derived cell lines and primary tumors. Oncogene. 1996;12:451. [PubMed] [Google Scholar]
- 43.Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of P15INK4b and P18INK4c cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solvason N, Wu WW, Kabra N, Wu X, Lees E, Howard MC. Induction of cell cycle regulatory proteins in anti-immunoglobulin-stimulated mature B lymphocytes. J. Exp. Med. 1996;184:407. doi: 10.1084/jem.184.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zindy F, van Deursen J, Grosveld G, Sherr C, Roussel M. INK4d-deficient mice are fertile despite testicular atrophy. Mol. Cell. Biol. 2000;20:372. doi: 10.1128/mcb.20.1.372-378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franklin DS, Godfrey VL, Lee H, I. Kovalev G, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18INK4c and p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su L, Lee R, Bonyhadi M, Matsuzaki H, Forestell S, Escaich S, Bohnlein E, Kaneshima H. Hematopoietic stem cell-based gene therapy for acquired immunodeficiency syndrome: efficient transduction and expression of RevM10 in myeloid cells in vivo and in vitro. Blood. 1997;89:2283. [PubMed] [Google Scholar]
- 48.Liao XC, Fournier S, Killeen N, Weiss A, Allison JP, Littman DR. Itk negatively regulates induction of T cell proliferation by CD28 costimulation. J. Exp. Med. 1997;186:221. doi: 10.1084/jem.186.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franklin DS, Xiong Y. Induction of p18INK4c and its predominant association with CDK4 and CDK6 during myogenic differentiation. Mol. Biol. Cell. 1996;7:1587. doi: 10.1091/mbc.7.10.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol. Cell. Biol. 2000;20:6147. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phelps DE, Xiong Y. Assay for activity of mammalian cyclin D-dependent kinases CDK4 and CDK6. Methods Enzymol. 1997;283:194. doi: 10.1016/s0076-6879(97)83016-9. [DOI] [PubMed] [Google Scholar]
- 52.Ravitz MJ, Wenner CE. Cyclin-dependent kinase regulation during G1 phase and cell cycle regulation by TGF-13. Adv. Cancer Res. 1997;71:165. doi: 10.1016/s0065-230x(08)60099-8. [DOI] [PubMed] [Google Scholar]
- 53.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 54.Yan Y, Frisen J, Lee M-H, Massague J, Barbacid M. Ablation of the CDK inhibitor p57Kip1 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11:973. doi: 10.1101/gad.11.8.973. [DOI] [PubMed] [Google Scholar]
- 55.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol. Today. 1992;13:136. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 56.Flanagan WM, Crabtree GR. Rapamycin inhibits p34cdc2 expression and arrests T lymphocyte proliferation at the G1/S transition. Ann. NY Acad. Sci. 1993;696:31. doi: 10.1111/j.1749-6632.1993.tb17139.x. [DOI] [PubMed] [Google Scholar]
- 57.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. USA. 1990;87:9231. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 59.Xiong Y. Why are there so many CDK inhibitors? Biochim. Biophys. Acta. 1996;1288:01. doi: 10.1016/0304-419x(96)00012-1. [DOI] [PubMed] [Google Scholar]
- 60.Lucas JJ, Szepesi A, Modiano JF, Domenico J, Gelfand EW. Regulation of synthesis and activity of the PLSTIRE protein (cyclin-dependent kinase 6 (cdk6)), a major cyclin D-associated cdk4 homologue in normal human T lymphocytes. J. Immunol. 1995;154:6275. [PubMed] [Google Scholar]
- 61.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994;14:2077. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lischke A, Moriggl R, Brandlein S, Berchtold S, Kammer W, Sebald W, Groner B, Liu X, Hennighausen L, Friedrich K. The interleukin-4 receptor activates STAT5 by a mechanism that relies upon common γ-chain. J. Biol. Chem. 1998;273:31222. doi: 10.1074/jbc.273.47.31222. [DOI] [PubMed] [Google Scholar]
- 63.Moriggl R, Sexl V, Piekorz R, Topham D, Ihle JN. Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity. 1999;11:225. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- 64.Ragione FD, Russo GL, Oliva A, Mercurio C, Mastropietro S, Pietra VD, Zappia V. Biochemical characterization of p16INK4- and p18-containing complexes in human cell lines. J. Biol. Chem. 1996;271:15942. doi: 10.1074/jbc.271.27.15942. [DOI] [PubMed] [Google Scholar]
- 65.Thullberg M, Bartkova J, Khan S, Hansen K, Ronnstrand L, Lukas J, Strauss M, Bartek J. Distinct versus redundant properties among members of the INK4 family of cyclin-dependent kinase inhibitors. FEBS Lett. 2000;470:161. doi: 10.1016/s0014-5793(00)01307-7. [DOI] [PubMed] [Google Scholar]