

Abstract
Purpose
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is characterized by cutaneous leiomyomas, uterine fibroids, and aggressive papillary renal cell carcinoma (RCC). A number of our HLRCC patients were found to have atypical adrenal nodules and which were further evaluated to determine if these adrenal nodules were associated with HLRCC.
Methods
HLRCC patients underwent a comprehensive clinical and genetic evaluation. Clinical presentation, anatomic and functional imaging, endocrine evaluation, pathologic examination and the results from germline mutation testing were reviewed.
Results
Twenty of 255 HLRCC patients (7.8%) were found to have primary adrenal lesions. Among these, three were found to have bilateral adrenal lesions and four were found to have multiple nodules. Two patients had ACTH-independent hypercortisolism. A total of 27 adrenal lesions were evaluated. The imaging characteristics of five (18.5%) of these lesions were not consistent with adenoma by non-contrast CT criteria. PET imaging was positive in 7 of 10 cases (70%). Twelve nodules were surgically resected from ten adrenal glands. Pathologic examination revealed macronodular adrenal hyperplasia in all specimens.
Conclusions
Unilateral and bilateral adrenal nodular hyperplasia was detected in a subset of patients affected with HLRCC. A functional endocrine evaluation is recommended when an adrenal lesion is discovered. Imaging frequently demonstrates lesions that are not typical of adenomas and PET imaging may be positive. To date, no patient has been found to have adrenal malignancy and active surveillance of HLRCC adrenal nodules appears justified.
Keywords: HLRCC, kidney cancer, adrenal nodule, fumarate hydratase
Introduction
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a recently characterized familial cancer syndrome associated with an aggressive form of papillary kidney cancer and the development of cutaneous and uterine leiomyomas.[1, 2] HLRCC is an autosomal dominant hereditary cancer syndrome characterized by a germline mutation of the fumarate hydratase (FH) gene, located at chromosome 1p42.3–43.[3] Fumarate hydratase is a Krebs cycle enzyme that converts fumarate to malate.[4] Loss of the remaining (somatic) FH allele impairs oxidative phosphorylation and alters glucose metabolism.[5, 6] To meet metabolic demand for energy production, the tumor undergoes a metabolic switch to aerobic glycolysis, a phenotype similar to that first demonstrated by Otto Warburg in the 1920’s.[7] The increase in glycolysis leads to a decrease in activation of the cellular energy sensor, AMPK, which results in an increase in mTOR and fatty acid synthesis, resulting in an aggressive kidney cancer phenotype.[5, 6, 8]
HLRCC renal tumors behave in an aggressive fashion and patients may present with locally advanced or disseminated disease.[9] Our experience with the management of HLRCC patients has led to an approach involving periodic monitoring of at-risk individuals and early surgical intervention for patients found to have a renal mass lesion.[2, 9]
Although adrenal involvement in HLRCC has not traditionally been recognized as a part of the HLRCC phenotype, Smit et al reported an adrenal adenoma in an HLRCC patient and Lehtonen et al. identified four patients with asymptomatic, adrenal adenomas and suggested HLRCC patients may have increased risk of these tumors compared to the general population.[10, 11] In a previous report we described an HLRCC patient that had bilateral adrenal hyperplasia with Cushings’ syndrome and demonstrated loss of heterozygocity (LOH) of FH in the adrenal tissue by florescence in situ hybridization (FISH).[12]
While a limited number of HLRCC patients with adrenal lesions have been reported, previous findings suggested that this may represent part of the phenotype. We therefore set out to further characterize adrenal involvement in patients with HLRCC by evaluating demographic information, mutation status, anatomic and functional imaging, adrenal functional studies, surgical management, and pathologic findings.
Materials and Methods
Patients suspected of being affected with Hereditary Leiomyomatosis Renal Carcinoma (HLRCC) were evaluated on an NCI IRB approved protocol (NCT00055627). Family history, clinical phenotype, tumor characteristics, and mutational assessment were evaluated. From 1990 to 2010, a total of 255 patients affected with HLRCC were evaluated. All HLRCC patients were either confirmed by FH germline testing or were found to have clinical manifestations characteristic of HLRCC. Germline fumarate hydratase (FH) mutation testing was performed using Clinical Laboratory Improvement Amendments (CLIA) certified laboratories. All patients underwent clinical, including dermatologic, evaluation as well as abdominal imaging with a CT, or MRI. In recent years, FDG-PET imaging has been increasingly performed in patients with HLRCC. While this imaging modality has variable sensitivity in detecting metastatic clear cell RCC, we have found FDG-PET quite useful in HLRCC as its sensitivity for the detection of distant disease is over 90% (Shuch, B et al. Manuscript in Preparation). To evaluate the presence of metastatic HLRCC renal cancer or to characterize suspicious sites (including the adrenal gland), FDG-PET imaging was performed.
Abdominal CT scans were evaluated to determine lesion laterality, size, and degree of enhancement by Hounsfield units (HU). Those with FDG-PET imaging were further characterized by their uptake of FDG using standardized uptake values (SUV). Adrenal lesions were interpreted as FDG positive or negative by considering the maximal adrenal SUV value versus that of liver parenchyma.
Patients with adrenal lesions were recommended to undergo a biochemical evaluation consisting of either plasma-free or 24-hour urinary metanephrines. Plasma or urinary cortisol, aldosterone, and renin levels were also obtained. Additional testing including evaluation of sex steroids and ACTH was performed at the discretion of the treating physician when clinically indicated. Those with available functional studies were reviewed to determine the incidence of a functional adrenal lesion.
Surgical management in patients with HLRCC was dictated by the manifestations detected on imaging. In patients with a renal mass lesion as well as an associated adrenal lesion, both were addressed at the time of surgery. For those with an isolated, suspicious adrenal lesion, surgical management was recommended if the adrenal lesion was positive on FDG-PET imaging. Partial adrenalectomy was performed by a laproscopic or robotic approach when feasible.[13, 14] A single genitourinary pathologist (MM) reviewed all surgical cases. Cortical adrenal hyperplasia was characterized by the presence of increased numbers of adrenal cortical cells and designated as diffuse or nodular. Macronodular was defined if a distinct nodule (>0.5 cm) was present on the H&E slide. Cases with micronodular adrenal hyperplasia were those in which the adrenal tissue demonstrated nodular hyperplasia in areas distinct from a gross nodule. Germline FH mutation status was analyzed to determine if there was a specific genotype-phenotype association.
Results
A total of 22 patients were identified as having adrenal lesions on imaging. Of those, two patients were excluded from analysis as they had widely metastatic RCC with extension of bulky disease to the ipsilateral adrenal gland. Of the remaining 20 patients (7.8% of HLRCC patients) there were seven men and 13 women, with a median age of 51.5 (20–77) years (Table 1).
Table 1.
Patient Demographics and adrenal lesion laterality/multifocality
| # | % | ||
|---|---|---|---|
| Sex | Male | 7 | 30% |
| Female | 13 | 70% | |
| Age | Range | 20–77 | - |
| Median | 51.1 | - | |
| Mean | 51.5 | SD 15.2 | |
| Side | Right | 10 | 37.00% |
| Left | 17 | 63.00% | |
| Laterality | Unilateral | 17 | 85.00% |
| Bilateral | 3 | 15.00% | |
| Lesions | 1 | 15 | 78.90% |
| >1 | 4 | 21.10% |
Sixteen of the 20 patients had one or more biochemical studies available for review (Table 2). Two of the 20 patients (10.0%) had clinical manifestations of hypercorticolism. One patient, as we previously reported in Matyakhina, et al.[12], presented with Cushing’s syndrome, hypertension and obesity, and was found to have bilateral, massive, macronodular adrenal hyperplasia, An additional patient with a unilateral adrenal lesion had evidence of subclinical Cushing’s syndrome. Neither case of hypercorticolism was associated with an elevated ACTH. No patient was found to have abnormal catecholamines (n=16), mineralocorticoids (n=14), or sex steroid levels (n=7).
Table 2.
Adrenal functional assessments documented for each patient
| Hormonal Test | Abnormal | Normal | % Abnormal |
|---|---|---|---|
| Cortisol | 2 | 15 | 13.30% |
| Aldosterone | 0 | 14 | 0% |
| Catecholamines | 0 | 16 | 0% |
A total of 27 adrenal nodules were identified in these 20 patients. Cross sectional and PET imaging characteristics are presented in Table 3. Adrenal lesions were found on the left and right side in 17 (63.0%) and 10 (37.0%), respectively. Four patients (20%) had multifocal adrenal lesions, of which three (15.0%) were bilateral. Median nodule size in maximal dimension was 1.6 cm (range 0.7–5.5) and 1.2 cm (0.6–3.2) for the secondary dimension. Non-contrast CT HU’s ranged from −17 to +23, with a mean and median of 0.9 and 0, respectively. No nodules were found with calcifications or irregular borders suggestive of invasion into adjacent structures. Based on non-contrast enhancement characteristics reported previously (<10 HU), a total of 22 (81.5%) of the adrenal lesions would be characterized as adenomas.[15] All 27 lesions were found to have brisk contrast enhancement; most were homogeneous, and none showed evidence of necrosis. In the 10 cases where PET imaging was performed, the adrenal lesions were PET positive in 7 (70%). PET positive adrenal lesions demonstrated SUV max values ranging from 3.9 to 6.6 and were more intense than the liver in all cases.
Table 3.
Cross sectional CT scan and nuclear imaging findings for each adrenal lesion
|
Size X & Y |
Range | X= 0.7–5.5 | Y= 0.6–3.2 |
| Median | 1.6 | 1.2 | |
| Mean | 2.0 (1.25 SD) | 1.4 (0.57 SD) | |
| Non-Contrast HU | Range | −17 to +23 | |
| Median | 0 | ||
| Mean | 0.9 | SD 9.9 | |
| Adenoma by CT* | No | 5 | 18.50% |
| Yes | 22 | 81.5% | |
|
Adrenal PET Activity (n=10) |
Positive | 7 | 63.0% |
| Range | 3.9–6.6 SUVs | ||
| Negative | 3 | 42.9% | |
| Range | 1.5–2.8 SUVs |
Per 2002 NIH Consensus Statement14
A total of 9 patients with 12 adrenal lesions were treated surgically with adrenalectomy, including 3 patients with that underwent a partial adrenalectomy. As reported by Matyakhina, et al., the single patient with symptomatic Cushing’s syndrome improved dramatically after surgery.[12] Macronodular hyperplasia was found in all ten adrenal glands (12 nodules) resected; in five glands, additional areas of micronodular hyperplasia were identified. In no cases were pigmented spots of blue nevi, as seen in primary pigmented nodular adrenocortical disease (PPNAD), found. The adrenal cortical cells appeared uniform in size and morphology with normal appearing nuclei and no areas of necrosis were identified. One tumor had a small, sub-centimeter myelolipoma adjacent to the region of nodular hyperplasia, however, this was considered to be an incidental finding. As demonstrated in Table 4, the histology was similar regardless of level of CT attenuation, size of nodule, or FDG uptake.
Table 4.
Radiologic/pathologic correlation for each patient
| Adrenal | Patient | Functional | PET Positive | Non-Contrast (HU) | Size (cm) | Pathology |
|---|---|---|---|---|---|---|
| 1 | 1 | Yes | - | 9 | 4.7×2.7 | Macronodular Adrenal Hyperplasia |
| 2 | - | 11 | 4.6×2.0 | Macronodular Adrenal Hyperplasia | ||
| 3 | - | 5 | 1.2×0.9 | Macronodular Adrenal Hyperplasia | ||
| 4 | - | 1 | 1.1×1.0 | Macronodular Adrenal Hyperplasia | ||
| 5 | 2 | No | No | 0 | 2.3×1.6 | Micro and Macronodular Hyperplasia |
| 6 | 3 | No | Yes | −3 | 1.7×1.4 | Micro and Macronodular Hyperplasia |
| 7 | 4 | Yes | Yes | 15 | 2.7×2.2 | Macronodular Adrenal Hyperplasia |
| 8 | 5 | No | Yes | −3 | 1.6×1.1 | Macronodular Adrenal Hyperplasia |
| 9 | 6 | No | Yes | 14 | 3.0×1.6 | Macronodular Adrenal Hyperplasia |
| 10 | 7 | No | - | −12 | 1.5×1.1 | Micro and Macronodular Hyperplasia |
| 11 | 8 | No | - | −12 | 1.5×1.1 | Micro and Macronodular Hyperplasia |
| 12 | 9 | No | - | 0 | 5.5×3.2 | Macronodular Adrenal Hyperplasia |
Figure 1 demonstrates a representative patient (Table 4, patient 6) who was found to have a 3 cm adrenal nodule that was suspicious for a non-adenoma on CT scan (1a) and was positive on PET scan (1b) who underwent a right-sided, robotic, partial adrenalectomy. Intraoperative ultrasound helped identify the adrenal mass within Gerota’s fascia that was distinct from normal adrenal limbs (1c). The adrenal mass was resected, sparing the normal adrenal gland. Gross examination revealed an encapsulated, yellow-tan lesion measuring greater than 3 cm in size (1d). Microscopically the adrenal tissue was found to contain both micro and macronodular adrenal hyperplasia (1e).
Figure 1.

a: CT Scan of 3 × 1.6 cm right Adrenal lesion with non-contrast enhancement of 14 HU
b: PET/CT Scan demonstrating an active foci with maximal SUV of 5.9 corresponding to the adrenal lesion.
c: Intra-operative ultrasound performed during right sided robotic partial adrenalectomy demonstrating normal adrenal limbs (red arrows) and a large central adrenal mass (yellow arrow).
d: Gross pathological examination of the bi-valved adrenal mass after right sided robotic partial adrenalectomy
e: Microscopic examination demonstrating adrenal pathology to be both micro and macronodular adrenal hyperplasia
Assessment of germline FH mutation in the affected patients from 10 different families was performed. Figure 2 shows the mutational map of the FH gene with site of mutation. Germline alterations were observed in seven of the ten exons. Only exons 1, 2, and 4 were not associated with this phenotype. Four microdeletions were seen in addition to 1 nonsense and 5 missense mutations. No region or regulatory site alterations appeared to predispose to this phenotype.
Figure 2.
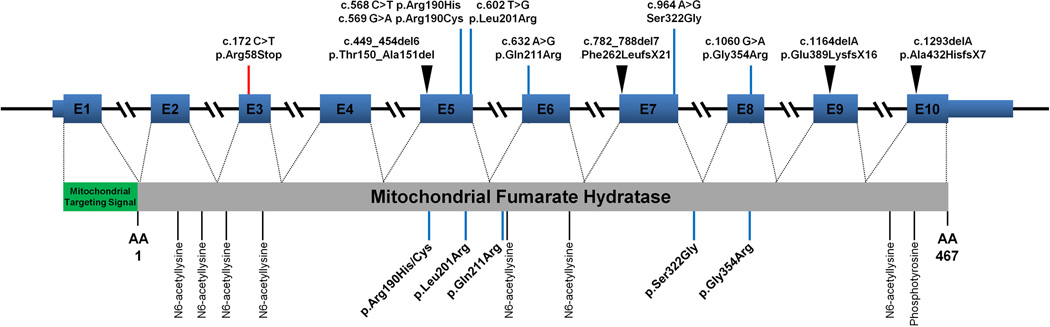
Fumarate Hydratase Germline Mutational Map
Discussion
In the general population adrenal lesions can represent a wide range of pathology ranging from benign conditions such as a non-functional adrenal adenoma to aggressive pathologies including adrenal cortical carcinoma, malignant pheochromocytoma, or adrenal gland metastases. With increased utilization of cross sectional imaging, many adrenal lesions are now detected incidentally (0.35–4.36%).[16] Most of these adrenal nodules are found to be benign and are managed conservatively. In our HLRCC patient population, the incidence of benign adrenal nodules was nearly 8%, with the surgically resected cases demonstrating nodular hyperplasia.
The incidence of nodular hyperplasia in the general population is unknown and is likely under-reported due to the fact that many patients with nodules do not require surgical resection. In non-HLRCC patients, adrenal nodular hyperplasia is believed to be associated with aging and hypertension.[17] Adrenal hyperplasia has also been associated with other hereditary syndromes. Primary pigmented nodular adrenocortical disease (PPNAD) is a familial syndrome associated with Carney Complex and is associated with germline mutation in PRKAR1A[18, 19]. In Carney complex, histologically black pigment and nevi are found in association with predominantly micronodular disease, but, macronodules can also occur.[20] Another form of hereditary hyperplasia is an ACTH-independent macronodular adrenal hyperplasia (AIMAH) and is associated with several known conditions.[21]
In the radiologic evaluation of adrenal lesions several studies have utilized non-contrast CT attenuation as a predictive tool for adrenal lesion characterization. The 2002 NIH consensus statement on incidental adrenal nodules reported “a homogeneous mass with a smooth border and an attenuation value of less than 10 HU on an unenhanced CT study strongly suggests the diagnosis of a benign adrenal adenoma.”[15] In the HLRCC population this panel recommendation may not be applicable as no patients were found with an adenoma despite over 80% meeting this criterion. More recent studies however, expanded the non-contrast values of 20 HU or less to include both hyperplasias in this category.[22] Of all characterized lesions, 26 of 27 (96.3%) met these criteria.
Despite being benign on pathologic examination, 70% of the adrenal lesions in our HLRCC patients evaluated with FDG PET imaging showed abnormal FDG uptake greater than the liver. Such a finding would normally raise the suspicion for malignancy. The HLRCC associated tumors (kidney and both uterine and cutaneous leiomyomas) demonstrate LOH of the wild type FH allele, thus, impairing the Krebs’ cycle and causing a metabolic shift towards glycolysis.[4, 23, 24] The inefficient energy production leads to a critical dependence on glucose uptake and up-regulation of GLUT1 expression.[25, 26] While the genetic alterations of HLRCC in adrenal nodular hyperplasia need further elucidation, our prior case report also suggests LOH as the mechanism. Thus as FDG PET imaging reflects glucose uptake, it’s not surprising that many HLRCC associated tumors show increased tracer activity.
The majority of ACTH-independent hypercortisolism cases (accounting for 20% of patients with Cushings’ Syndrome) are related to adrenal adenomas and very rarely adrenocortical carcinoma. The glycolytic phenotype associated with HLRCC tumors may be responsible for the infrequent development of ACTH-independent, hypercortisolism in this population. This unusual type of pituitary-independent, hyperplastic, hypercortisolism has been observed with the other hereditary syndromes mentioned above. Hypercortisolism has been reported to occur in 50% of Carney Complex patients with PPNAD.[18] The other conditions related to AIMAH can also present in a similar fashion.[21] It is plausible that the glycolytic shift in the adrenal cortical nodule leads to a cortisol-mediated response.
There are limitations to this study. This report describes a small number of patients (20) and a limited number of surgically resected adrenal nodules (12) evaluated histologically. Our experience with HLRCC has evolved of the past decade and we now incorporate FDG-PET imaging when clinically indicated. Thus, functional imaging was not performed in many of the early patients in our series. While all patients were recommended to undergo biochemical evaluation, not all patients had results available for review. Due to the referral pattern at our center, many patients had testing performed at their home institution. While the remaining patients were reported to have normal results, patients were only included when documented lab values were available for verification. Further studies including FISH and/or FH sequencing will be required to better characterize whether or not biallelic inactivation of fumarate hydratase is associated with adrenal tumor formation. Analysis of a larger cohort to determine genotype-phenotype is planned to further characterize which patients manifest this particular HLRCC phenotype.
Conclusions
HLRCC is characterized by cutaneous and uterine leiomyomas and aggressive papillary kidney cancer. Adrenal macro and micro-nodular hyperplasia may appear in a small percentage of patients affected with HLRCC. Patients may rarely develop hypercortisolism and, therefore, a functional workup is recommended in affected individuals found with an adrenal nodule. The fact that HLRCC-associated tumors are characterized by aerobic glycolysis may explain while many of these tumors have an abnormal appearance on PET scans. After surgical removal of ten adrenal lesions, in which no patient has been found to have adrenal malignancy, we recommend periodic surveillance for HLRCC adrenal tumors. If an interval change prompts concern and surgical intervention is considered, partial adrenalectomy is recommended as these patients may be at risk for contralateral recurrence.
Acknowledgements
Supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Reference List
- 1.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98:3387-2. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merino MJ, Torres-Cabala C, Pinto PA, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–1585. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 3.Alam NA, Bevan S, Churchman M, et al. Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3-q43. Am J Hum Genet. 2001;68:1264–1269. doi: 10.1086/320124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 5.Tong WH, Sourbier C, Kovtunovych G, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases metabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullen AR, Wheaton WW, Jin ES, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 8.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature Reviews Urology. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grubb RL, III, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177:2074–2080. doi: 10.1016/j.juro.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 10.Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet. 2006;43:523–526. doi: 10.1136/jmg.2005.036400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49–59. doi: 10.1111/j.1399-0004.2010.01486.x. [DOI] [PubMed] [Google Scholar]
- 12.Matyakhina L, Freedman RJ, Bourdeau I, et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical Cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab. 2005;90:3773–3779. doi: 10.1210/jc.2004-2377. [DOI] [PubMed] [Google Scholar]
- 13.Benhammou JN, Boris RS, Pacak K, et al. Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol. 2010;184:1855–1859. doi: 10.1016/j.juro.2010.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boris RS, Gupta G, Linehan WM, et al. Robot-assisted Laparoscopic Partial Adrenalectomy: Initial Experience. Urol. 2010 doi: 10.1016/j.urology.2010.07.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.NIH state-of-the-science statement on management of the clinically inapparent adrenal mass ("incidentaloma") NIH Consens State Sci Statements. 2002;19:1–25. [PubMed] [Google Scholar]
- 16.Kloos RT, Gross MD, Francis IR, et al. Incidentally discovered adrenal masses. Endocr Rev. 1995;16:460–484. doi: 10.1210/edrv-16-4-460. [DOI] [PubMed] [Google Scholar]
- 17.Dobbie JW. Adrenocortical nodular hyperplasia: the ageing adrenal. J Pathol. 1969;99:1–18. doi: 10.1002/path.1710990102. [DOI] [PubMed] [Google Scholar]
- 18.Groussin L, Kirschner LS, Vincent-Dejean C, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet. 2002;71:1433–1442. doi: 10.1086/344579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes WS, Davidson AJ, Grimley PM, et al. Extraadrenal retroperitoneal paraganglioma: clinical, pathologic, and CT findings. AJR Am J Roentgenol. 1990;155:1247–1250. doi: 10.2214/ajr.155.6.2173385. [DOI] [PubMed] [Google Scholar]
- 20.Travis WD, Tsokos M, Doppman JL, et al. Primary pigmented nodular adrenocortical disease. A light and electron microscopic study of eight cases. Am J Surg Pathol. 1989;13:921–930. [PubMed] [Google Scholar]
- 21.Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:245–259. doi: 10.1016/j.beem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Hamrahian AH, Ioachimescu AG, Remer EM, et al. Clinical utility of noncontrast computed tomography attenuation value (hounsfield units) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab. 2005;90:871–877. doi: 10.1210/jc.2004-1627. [DOI] [PubMed] [Google Scholar]
- 23.Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 24.Vanharanta S, Pollard PJ, Lehtonen HJ, et al. Distinct expression profile in fumarate-hydratase-deficient uterine fibroids. Hum Mol Genet. 2006;15:97–103. doi: 10.1093/hmg/ddi431. [DOI] [PubMed] [Google Scholar]
- 25.Pollard PJ, Spencer-Dene B, Shukla D, et al. Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell. 2007;11:311–319. doi: 10.1016/j.ccr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Sudarshan S, Sourbier C, Kong HS, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and HIF-1 alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;15:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]