

Abstract
Although the huntingtin gene is expressed in brain throughout life, phenotypically Huntington's disease (HD) begins only in midlife to affect specific brain regions. Here, to investigate regional vulnerability in the disease, we used functional magnetic resonance imaging (fMRI) to translationally link studies in patients with a mouse model of disease. Using fMRI, we mapped cerebral blood volume (CBV) in three groups: HD patients, symptom-free carriers of the huntingtin genetic mutation, and age-matched controls. In contrast to a region in the anterior caudate, in which dysfunction was linked to genotype independent of phenotype, a region in the posterior body of the caudate was differentially associated with disease phenotype. Guided by these observations, we harvested regions from the anterior and posterior body of the caudate in postmortem control and HD human brain tissue. Gene-expression profiling identified two molecules whose expression levels were most strongly correlated with regional vulnerability — protein phosphatase 1 regulatory subunit 7 (PPP1R7) and Wnt inhibitory factor-1 (WIF1). To verify and potentially extend these findings, we turned to the YAC128 (C57BL/6J) HD transgenic mice. By fMRI we longitudinally mapped CBV in transgenic and wildtype (WT) mice, and over time, abnormally low fMRI signal emerged selectively in the dorsal striatum. A relatively unaffected brain region, primary somatosensory cortex (S1), was used as a control. Both dorsal striatum and S1 were harvested from transgenic and WT mice and molecular analysis confirmed that PPP1R7 deficiency was strongly correlated with the phenotype. Together, converging findings in human HD patients and this HD mouse model suggest an anatomo/functional pattern of caudate vulnerability and that variation in expression levels of herein identified molecules correlate with this pattern of vulnerability.
INTRODUCTION
Huntington's Disease (HD) is a progressive neurodegenerative process that typically presents with abnormal choreic movements indicative of caudate dysfunction, as well as cognitive and psychiatric symptoms (Reiner et al., 1988). Linkage analysis identified disease-causing defects in a gene subsequently called huntingtin, characterized by an expanded number of CAG repeats (MacDonald, 1993). Since then many studies have clarified the function of huntingtin, the clinical consequences of CAG repeat expansions, and in general have informed hypotheses about disease pathogenesis (Kumar et al., 2010). Nevertheless, many questions remain and there is still no effective intervention for HD.
One outstanding question emerges from the observation that huntingtin is expressed early and diffusely throughout the brain, yet typically the disease is expressed in midlife and characterized by differential regional vulnerability. These observations suggest that genotype is necessary but insufficient to account for the disease's anatomical and temporal pattern of dysfunction.
To begin understanding regional vulnerability in the disease we used a variant of functional magnetic resonance imaging (fMRI) that maps basal CBV (cerebral blood volume), employing the steady-state gadolinium enhancement method (Lin et al., 1999). Like other hemodynamic variables, CBV is tightly coupled to brain metabolism (Gonzalez et al., 1995) and was the first variable to be used in fMRI (Belliveau et al., 1991). CBV is notable for its ability to generate functional maps with submillimeter resolution (Lin et al., 1999). High-spatial resolution is useful for identifying dysfunction in small regions of the brain (Moreno et al., 2007; Pereira et al., 2007), thus CBV mapping is particularly well-suited for translational research across species. Indeed, by imaging patients and animal models, CBV mapping employing this approach has been used to characterize hippocampal dysfunction in Alzheimer's disease (Moreno et al., 2007), schizophrenia (Gaisler-Salomon et al., 2009; Schobel et al., 2009), cognitive aging (Small et al., 2004), diabetes (Wu et al., 2008), and brainstem dysfunction in Parkinson's disease (Lewandowski et al., 2010).
In the current study, we relied on CBV mapping in human patients, asymptomatic gene carriers and in the YAC128 (C57BL/6J) mouse model of HD (Bayram-Weston et al., 2012; Brooks et al., 2010; Brooks et al., 2011a; Brooks et al., 2011b) to map a pattern of caudate/striatal dysfunction differentially linked to phenotype versus genotype. We then exploited this differential pattern to isolate, by microarray, molecules whose expression level changes coincide with this pattern of caudate vulnerability in human postmortem brains of symptomatic patients. The molecular hits were then tested and confirmed at the protein level and in the mouse model of disease.
MATERIALS AND METHODS
Human fMRI
Subjects
Study subjects were recruited from the Columbia Health Sciences Huntington's Disease Society of America (HDSA) Center of Excellence and provided informed consent prior to participation. Subjects were included if they had clinically defined HD and a total functional capacity score (TFC) of >7, indicating mild to moderate disease. Subjects were considered asymptomatic HD gene carriers if they had a unified Huntington's disease rating scale (UHDRS) total motor score <10 and received a score of <4 on the diagnosis confidence level question of the UHDRS. Age-matched, healthy control subjects who had no history of HD were also enrolled and underwent MR imaging. Patient breakdown: 20 HD subjects (mean age= 49, 60% men, mean CAG repeats= 44, mean TFC= 9.5); 12 asymptomatic subjects (mean age= 42, 60% men, mean CAG repeats= 41, mean TFC= 12.8); and 20 healthy controls (mean age= 45, 50% men) (Table 1, fMRI subjects demographics).
Table 1.
fMRI subject and postmorten sample demographics
| fMRI subject demographics | |||||
|---|---|---|---|---|---|
| N | Age (S.D.) | Gender (M:F) | CAG repeats (S.D.) | TFC (S.D.) | |
| Controls | 20 | 45 (11.7) | 10:10 | ||
| Asymptomatics | 12 | 42 (10.0) | 7:4 | 42 (1.9) | 12.8 (0.4) |
| Symptomatic | 20 | 49 (10.6) | 12:8 | 44 (3.1) | 9.5 (2.6) |
| Postmortem sample demographics | |||||
| Microarray Analysis | |||||
| Controls | 10 | 70 (8.1) | 6:4 | ||
| HD | 10 | 73 (7.6) | 6:4 | 41 (2.1) | |
| Western Blot Analysis | |||||
| Controls | 4 | 58 (6.0) | 2:2 | ||
| HD | 4 | 56 (7.4) | 3:1 | 46 (3.2) | |
S.D. = Standard Deviation.
TFC = Total functional capacity score.
Imaging
CBV maps were generated as previously described (Moreno et al., 2007). Briefly, imaging was performed with a 1.5 tesla Philips Intera scanner, generating T1-weighted images (time to repeat, 20ms; time to echo, 6ms; flip angle, 25 degrees; in plane resolution, 0.86mm × 0.86mm; slice thickness, 4mm) acquired perpendicular to the long-axis of the caudate before and 4 minutes after intravenous (i.v.) administration of the contrast agent (CA), gadolinium (0.1mmol/kg).
Voxel-based whole brain analysis
Voxel-wise processing and analysis was implemented using a three-step procedure comprising a) calculation of CBV maps in native anatomical space, b) transformation of individual CBV maps to standardized stereotactic space, and c) statistical analysis.
a) Calculation of CBV maps in native space
Each subject's pre-contrast and post-contrast images were coregistered to each other by six degrees of freedom (df) using Affine Inter-Modal Image Registration (FSL's FLIRT) (Smith et al., 2004). Coregistrations were visually inspected for accuracy. The pre-contrast image was subsequently subtracted from the post-contrast image. We used values within the superior sagittal sinus as an estimate of 100% blood for subtraction, following recommendations of Lin and colleagues (Lin et al., 1999). The superior sagittal sinus for each subject was isolated through intensity thresholding on the subtracted image using a custom script written for Matlab software (The Mathworks, Natick, MA). Image processing was performed in three steps. First, the bottom half of the image in coronal orientation was masked out to remove interference of bone and neck tissue and blood. Second, the intensity threshold for the entire image was set at half of the maximum intensity value of the image. Third, a nearest neighbor clustering routine was run to cluster, or group, each voxel with each contiguous voxel that is above the half-maximum threshold. Determination was made of the cluster with the greatest mean intensity value from among those larger than 100 voxels. This cluster has been within the superior sagittal sinus in all images tested with this approach thus far, which has also been confirmed visually. The mean value of the 50 voxels with the highest intensity values within this cluster was taken as the estimate of 100% blood volume. Each voxel value in the subtracted image was then divided by the estimate of 100% blood volume from the sagittal sinus and multiplied by 100, yielding relative CBV maps (rCBV). We established reliability with this automated approach by comparing manually-derived absolute blood volume. rCBV values derived from a region of interest (ROI) placed in the caudate were highly correlated (r=0.978) with rCBV values in the same ROI employing this automated approach.
b) Transformation of individual CBV maps to standardized stereotactic space
Each subject's pre-contrast image was spatially normalized into standardized, high resolution (1×1×1mm) stereotactic space defined by the International Consortium for Brain Mapping (ICBM)-152 anatomical atlas (Mazziotta et al., 2001) using a 12 df FLIRT registration (Shattuck et al., 2008). The transformation matrix derived from this step was applied to the CBV map (i.e., the subtracted image normalized by values in the superior sagittal sinus). A 6 mm Gaussian smoothing kernel was applied to the normalized rCBV map.
c) Statistical analysis
Statistical analyses were conducted with the image analysis software package SPM5 (Wellcome Department of Imaging Neuroscience, London, UK). We used a voxelwise approach similar to voxel-based morphometry used to examine density or volumetric differences between groups (Ashburner and Friston, 2000; Good et al., 2001). Group t-statistic contrasts were used to compare rCBV maps between asymptomatic (n=5) and symptomatic (n=7) huntingtin gene carriers to controls (n=12) using a t-test threshold of 2.52 (α=0.01) and 3.53 (α=0.001), respectively. This analysis identified which individual voxels differed in rCBV as a function of symptomatology or gene carrier status, and generated statistical parametric maps showing the regional distribution of significant voxel differences. No corrections for multiple comparisons were made.
ROI analysis
Image analysis was performed by an investigator who was blinded to subject diagnosis and genetic status. To generate human CBV maps, the precontrast image was subtracted from the postcontrast image and the difference in the sagittal sinus (which serves as an estimate of the image intensity change of 100% blood), was recorded. The subtracted image was then divided by the difference in the top 4 pixels measured from the sagittal sinus and multiplied by 100, yielding relative CBV maps (rCBV) (Moreno et al., 2007).
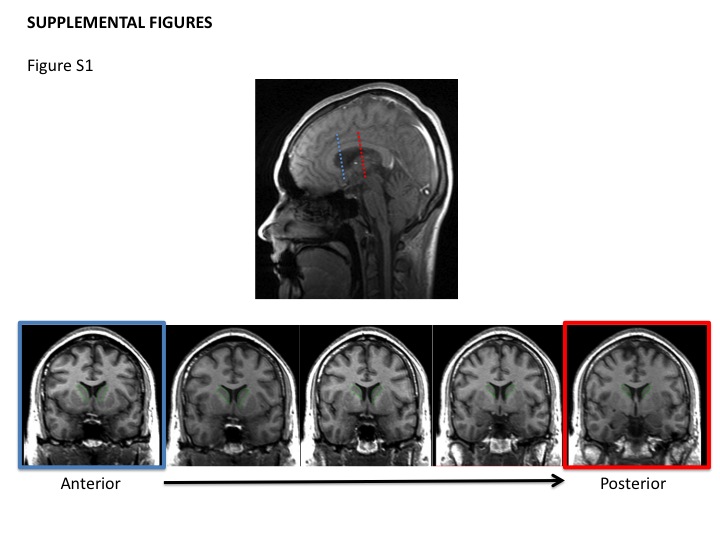
ROIs were manually defined on each coronal pre-contrast T1 image. The caudate nucleus was outlined bilaterally and those ROIs were then transferred onto the rCBV map to obtain normalized CBV values for each region. Analysis was performed on two coronal sections/subject: one at the rostral-most extent of the head of the caudate nucleus (typically 8 mm anterior to the level of the anterior commissure); and the second at the most caudal section of the body of the caudate nucleus that could be visualized. The lateral ventricle was used as the medial border of the structure and the surrounding white matter tracts defined the remainder of the structure (Fig. S1). Mean rCBV in the left and right anterior and posterior caudate was measured for each individual case. Additionally, in the same slices anterior and posterior caudate areas (right and left) were measured as an indirect indicator of tissue changes.
Mouse fMRI
Subjects
The yeast artificial chromosome mouse model of HD (YAC128–C57BL/6J) (Van Raamsdonk et al., 2007) and its WT littermates were obtained from Psychogenics Inc (Tarrytown, NY). Mutant and WT mice were generated crossing mutant male YAC128 congenic C57BL/6J to C57BL/6J females. Mice were group-housed in an enriched environment as described in (Menalled et al., 2009). Food and water were available ad libitum. Genotype was determined at 15 days of age by PCR of tail snips (Morton et al., 2000). CAG repeat lengths were measured by Laragen (USA). All CAG repeat numbers reported here are those determined directly by Genemapper software by Laragen Inc. (Los Angeles, CA). All YAC128 mice selected for this study carried around 119 CAG repeats.
Imaging
The following procedures were performed in accordance with approval from the Institutional Animal Care and Use Committee (IACUC) at Columbia University Medical Center. YAC128 mice and WT littermates (sex matched between groups) were imaged at 20 weeks of age and then repeatedly at 12 week intervals (n=10 per group); 32 weeks (n=10 per group), 44 weeks (n=9 per group), and 56 weeks (n=9 WT; 7 YAC128). As previously described (Moreno et al., 2007), mice were imaged with a Bruker AVANCE 400WB spectrometer (Bruker NMR, Inc., Billerica, MA) outfitted with an 89 mm bore 9.4 Tesla vertical Bruker magnet (Oxford Instruments Ltd., UK), a 30 mm-i.d birdcage RF probe, and a shielded gradient system (100 G/cm). Like other fMRI studies in rodents, we used isoflurane for anesthesia (induction phase 3 vol % and maintenance 1.1- 1.5 vol % at 1 l/min air flow, via a nose cone).
An investigator blinded to subject grouping performed all imaging processing. Although anesthesia and soft head fixation minimized head motion, the AIR program was used to co-register the images. A Gnu plot was generated to assess the quality of the co-registration, and an individual study was rejected if a shift greater than 1 voxel dimension was detected. CBV was mapped as changes of transverse relaxation rate (ΔR2) induced by the CA. Note that R2 = 1/T2. When the contrast agent reaches uniform distribution, then CBV maps can be measured from steady state T2-weighted images as: CBV α ΔR2= ln (Spre/Spost)/TE (Belliveau et al., 1991). Where TE is the effective echo time, Spre is the T2-weighted signal before CA administration, and Spost is the T2-weighted signal after CA reaches steady state. We have previously studied the kinetics of intraperitoneal gadolinium generated CBV maps in C57BL/6J mice and identified the 37.5 minute time-point as the optimum post-contrast time interval (Moreno et al., 2006). The derived ΔR2 maps were then divided by 4 voxels with the highest ΔR2 measured from the posterior cerebral vein, yielding rCBV maps. We have published a detailed discussion on the necessity for normalization of CBV maps (Moreno et al., 2006; Moreno et al., 2007).
Dorsal Striatum (DS)
The lateral boundary follows the ventromedially facing concavity of the external capsule, which is inserted between its dorsal, lateral and anterior margins and the neocortex. The medial boundary is the lateral ventricle, with the globus pallidus, overlapping ventrally the ventral striatum (VS) and, the amygdale extending caudally. Given these anatomical references an axial slice corresponding to the stereotaxic coordinates [Interaural = 4.36 mm, bregma = -5.64mm], was selected to identify a DS-ROI.
Ventral Striatum
VS comprises the ventromedial caudate-putamen, the nucleus accumbens and part of the olfactory tubercule (Kelley et al., 1982). VS is considered by some to be the ventral continuation of the dorsal striatum because no clear boundary can be drawn between the regions. We selected an axial slice located ventrally form DS, where the nucleus accumbens and olfactory tubercule were identified, corresponding to the stereotactic coordinates [interarural=3.76 mm, bregma=-6.24]. Note that in the sagittal slice we also selected the nucleus accumbens, which is beneath the rostral caudate-putamen and the external capsule forms the boundaries.
Primary Somatosensory Cortex
The three subdivisions of S1 (forelimb, hind-limb and head representation areas) were included. S1 is bordered caudally by Par 2 and rostrally by the agranular insular cortex. Border zones were avoided in an axial slice corresponding to the stereotactic coordinates [interaural=-4.44 mm, bregma=5.56 mm] (Fig. S2A). Note that for the analysis we selected and analyzed right and left structures, but only report mean value of both, because no laterality differences were found.
Stereotactic coordinates for all ROIs were based on the mouse brain library (MBL) atlases for C57BL/6J (Rosen et al., 2000; Williams, 2000).
Structural Imaging
The MRIs generated for functional analysis were used to obtain anatomical volumes as well. 2D-fast spin echo (FSE) were used to generate brain volumes utilizing the ITK-SNAP program (Yushkevich et al., 2006), which allows for active contour generation. For the present work manual boundary drawing was used, which involved slice-by-slice boundary drawing on available orthogonal views. This method has a coupling cursor between 2D slices and the 3D display, which significantly helps to reduce slice-by-slice jitter that occurs in this type of segmentation. Manual segmentation was performed by three highly trained raters using a standard anatomical atlas. We had previously compared 2D-FSE and 3D- gradient echo (GE) generated structural data, although there was a high correlation between 2D and 3D MRI measurements (r=0.87) reported in (Moreno et al., 2011), 2D images consistently provided slightly higher volumes. The volumes measured in 2D images were obtained by multiplying the pixel size by the interslice distance (SI=0.7mm). A slight overestimation may have resulted because the volume from where the signal is obtained reflects a smaller slice thickness (ST=0.6mm). Therefore we calculated the average height in the slice dimension for a given ROI (Hs) and multiplied the 2D volumes by the following correction factor: (1-(1-ST/SI)/Hs)). For instance Hs=1.2: (1-(0.6/0.7)/1.2=0.88; Hs = 2.34: (1-(1-0.6/0.7)/2.34)=0.94. We therefore used this correction factor in all data presented. Both rCBV-ROIs and anatomical ROIs- segmentations were compared between the three highly trained raters using common intraclass correlation statistics. The analysis showed high agreement between the raters for all the ROIs analyzed with values between 0.891 and 0.976. Striatum ROI selection included VS and DS using the criteria described above.
Gene-expression studies
All samples from the New York Brain Bank (NYBB), and were flash frozen in liquid nitrogen and stored at −80°C. Guided by our imaging findings, we harvested the posterior and anterior caudate, as well as the S1 region, using strict anatomical criteria following NYBB procedures from each of the 20 brains: 10 control (mean age: 70; 60% male) and 10 HD brains (mean age= 73, 60% male, mean CAG repeats = 41, rated at pathological stages 1 or 2, as previously described (Vonsattel et al., 1985)). Using an Affymetrix chip platform (GeneChip; Affymetrix, Santa Clara, CA) we generated gene-expression profiles in each of these 60 brain samples. Total RNA was extracted from each of the 60 tissue samples with TRIzol reagent (Invitrogen, Carlsbad, CA) and purified with RNeasy mini columns (Qiagen, Valencia, CA). All subsequent steps followed, Affymetrix's Eukaryotic target preparation protocol found in the GeneChip Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA). RNA and microarray workup was preformed as previously described (Lewandowski et al., 2010). The microarray data has been submitted in a MIAME-compliant format to Gene Expression Omnibus (GEO) via NCBI, series accession # GSE11897.
Western blot analysis
A new set of 4 HD (mean age= 56, 75% male, mean CAG repeats= 46, rated pathological grade 2 or 3 (Vonsattel et al., 1985)) and 4 control (mean age=58, 50% male) brain samples were obtained from the NYBB, following the criteria described above. Within each brain the posterior caudate and the S1 region were identified and sectioned using strict anatomical criteria (total number of samples, n=14). Methods were preformed as previously described (Lewandowski et al., 2010), with primary antibodies for PPP1R7 (Zymed-Invitrogen custom made). Full-length human PPP1R7 was used to generate polyclonal antibodies, which were affinity isolated. WIF-1 antibodies obtained from (Novus Biologicals) were also used. All samples loading were normalized to tubulin expression. This protocol was repeated on laterally pooled striatum and S1 from 48 week old (7 YAC128 and 7 wildtype, total of 28 samples) and 28 week old (4 YAC128 and 4 wildtype, total of 8 samples) mouse brains (See supplementary information for details).
Statistics
Standard statistical methods were used for the data. These included generalized estimated equations (GEE) (as detailed in supplementary material), t-tests, analysis of variance (ANOVA) and repeated measures ANOVA. The specific selection of variables and methods is specified for each experiment in the Results section. Outcome variables were normally distributed.
RESULTS
Mapping a pattern of regional vulnerability within the caudate in HD
Steady-state gadolinium enhanced CBV (Moreno et al., 2007) maps were generated in three groups: 20 symptomatic patients with HD (mean age = 49, 60% male, mean CAG repeats = 44, mean Total functional capacity score (TFC) = 9.5); 12 asymptomatic subjects with expanded CAG repeats (mean age = 42, 60% male, mean CAG repeats = 41, mean TFC = 12.8), and 20 healthy controls (mean age = 45, 50% male).
Coregistration of brains into a unified space facilitated a whole brain voxel-based analysis comparing HD patients to controls. Consistent with previous functional imaging studies (Kuhl et al., 1985; Kuhl et al., 1984; Paulsen, 2009) and those using fluoro-deoxyglucose positron emission tomography (Kuhl et al., 1982), significantly lower CBV values were observed in the caudate of HD patients, indicating a functional abnormality (Fig. 1A). Previous studies have suggested a posterior-anterior gradient of caudate pathology in HD (Kassubek et al., 2004; Vonsattel and DiFiglia, 1998; Vonsattel et al., 1985), and because coregistration reduces spatial resolution, a “region of analysis” approach was used to investigate the long axis of the caudate. ROIs were identified in the anterior and posterior body of the caudate (Fig. S1), the most posterior aspect of the caudate that could reliably be visualized in HD patients with atrophy. ROIs were applied to each subject, and data were analyzed with repeated measures ANOVA, including region (anterior and posterior) and laterality (left and right) as within-subject factors, group (symptomatic, asymptomatic, controls) as a between-group factor, and age and sex as covariates. A significant group-by-region interaction (F=3.8, p=0.024) was observed (Fig. 1B and C), which was confirmed by post-hoc comparisons (Bonferroni test). To address whether caudate atrophy may be contributing to the effect observed, we replicated the analysis but included measures of caudate area (see Methods) as covariates. Results still showed a significant group-by-region interaction (F=4.7, p=0.013). Nota Bene: No gender effects were noted within these comparisons.
Figure 1. Mapping an anatomical phenotype of caudate dysfunction in HD.

(A) Voxel-based analysis of rCBV in whole brain maps comparing HD patients to controls reveals greatest rCBV decreases in the caudate.
(B) ROIs in the caudate reveals decreased rCBV in the anterior caudate in asymptomatic gene positive subjects (light gray) and HD patients (dark gray) compared to controls (black), while decreased rCBV in the posterior body of the caudate was found only in HD patients. Shown are normalized mean rCBV values and standard deviation (SD).
(C) Individual representative examples of rCBV maps in the posterior body of the caudate of a control subject (upper panel), an asymptomatic gene positive subject (middle panel), and a HD patient (lower panel).
* denotes p≤0.05. rCBV maps in A and C were color coded, such that warmer colors represent higher rCBV values.
The interaction was driven by rCBV differences measured in the posterior caudate between the symptomatic and asymptomatic groups (both with expanded repeats) (F=4.3, p=0.031), and by rCBV differences measured in the anterior caudate between the asymptomatic and control groups (F=5.7, p=0.024). These findings suggest that abnormally low rCBV values in the anterior caudate are linked to genotype independent of phenotype (both groups asymptomatic), while dysfunction in the posterior caudate is selectively linked to the clinical phenotype (symptomatic vs. asymptomatic). In anticipation of our molecular studies, we were interested in identifying a region that was unaffected by genotype or phenotype. In a secondary analysis, therefore, ROIs were identified in the primary somatosensory cortex (S1), a region reported to be relatively less affected than the caudate nucleus in HD (de la Monte et al., 1988). An ANOVA revealed no significant difference between the three subject groups in this region (F=0.15, p=0.87) (Fig. S2A).
Molecules linked to regional vulnerability of the caudate
Ten healthy brains (mean age=70; 60% male) and ten HD brains (mean age=73, 60% male, mean CAG repeats= 41, rated at pathological stages 1 or 2) with minimal gliosis were identified. Guided by the imaging findings, 3 regions were harvested from each brain—the anterior caudate, posterior body of the caudate, and the S1 region of cortex. Affymetrix microarray chips were used to generate gene expression profiles in each of the 60 tissue samples.
As with imaging, two analyses were performed. First, applying the same statistical model used with fMRI, caudate expression levels were analyzed using a repeated-measures ANOVA, including region (anterior vs. posterior caudate) as a within subject factor and group (healthy vs. HD) as a between group factor. A group-by-region interaction was identified for 12 transcripts (p<0.05), (Table 2). Second, an ANOVA was used to compare expression levels of the 12 transcripts in S1 between HD and healthy brains. Nine transcripts were found to have abnormal expression levels in S1 (Table 2), suggesting that they are not selectively linked to HD-related caudate dysfunction. Based on both analyses, three transcripts were identified whose expression levels best tracked with regional vulnerability in the disease: Wnt inhibitory factor-1 (WIF1), protein phosphatase 1 regulatory subunit 7 (PPP1R7) (Fig. 2), and an uncharacterized hypothetical protein FLJ10955. We concentrated in the study of PPP1R7 and WIF-1. When comparing controls vs. HD patients expression level of these two transcripts—ANOVA analysis showed that they were significantly lower in HD cases only in posterior caudate for both PPP1R7 (F= 4.67; p=0.027) and WIF-1 (F=5.36 p=0.021), while no significant differences were observed when comparing expression levels in anterior and posterior caudate of HD patients for either PPP1R7 (F=0.16; p=0.86) or WIF-1 (F=2.6; p=0.061).
Table 2.
Twelve molecular transcripts that emerged from the microarray analysis
| Gene name | GenBank # | Region X group | S1 cortex |
|---|---|---|---|
| WIF1 | NM_007191.1 | p = 0.003 | Ns* |
| PP1R7 | BF718769 | p = 0.02 | Ns* |
| FLJ10955 | NM_018282.1 | p = 0.03 | Ns* |
| PRO0518 | AF090934.1 | p = 0.006 | p = 0.009 |
| DDX21 | NM_004728.1 | p = 0.009 | p = 0.02 |
| SNW1 | NM_012245.1 | p = 0.01 | p = 0.004 |
| NFYB | AI804118 | p = 0.02 | p = 0.02 |
| HSPC069 | AI761110 | p = 0.02 | p = 0.01 |
| BIRC2 | NM_001166.2 | p = 0.03 | p = 0.01 |
| FBRNP | AA528233 | p = 0.03 | p = 0.001 |
| PTPRK | NM_002844.1 | p = 0.04 | p = 0.01 |
| PLOD2 | NM_000935.1 | p = 0.04 | p = 0.003 |
mRNA levels of 12 molecules conformed to the fMRI spatial profile of Huntington's disease (columns 1-3). A secondary analysis identified three molecules whose expression deficit was restricted to the posterior caudate (column 4, Ns). These hits are considered to best-tracked fMRI-phenotype.
Group represents healthy vs. control. Region x group is the interaction term, including region (anterior vs. posterior caudate) and group.
Ns= Not Significant
Figure 2. Molecules that track the anatomical phenotype of HD-related caudate dysfunction in humans.

The expression profiles of PPP1R7 and WIF1 measured in the anterior and posterior body of the caudate in HD (gray) and control (black) brains.
* denotes p≤0.05, ** denotes p≤0.01
Evaluating caudate function and structure with MRI in a HD mouse model
To confirm and extend the human fMRI findings, we imaged a mouse model of HD. Transgenic mice YAC128 in C57BL/6J background strain were selected for this study. This mouse line expresses a full-length genomic construct under the control of the human HD gene promoter and has been well characterized in prior studies (Brooks et al., 2010; Brooks et al., 2011a; Brooks et al., 2011b; Lerch et al., 2008b; Van Raamsdonk et al., 2007).
Steady-state gadolinium enhanced rCBV maps were generated using a 9.4 tesla dedicated rodent scanner, as previously described (Moreno et al., 2007). Ten YAC128 mice and 10 WT littermates (gender-matched between groups) were imaged beginning at 20 weeks, prior to the observed onset of significant neurodegeneration (Slow et al., 2003) and then repeatedly at 12 weeks intervals; 32 weeks (n=10 per group), 44 weeks (n=9 per group), and 56 weeks (n=9 WT; n=7 YAC128).
Unlike human imaging, mice were imaged using a 2D acquisition. The acquisition protocol contained a 0.1mm inter-slice interval (ISI) in an attempt to minimize the effects of partial volume sampling in our dataset, as well as, interslice cross talk. Considering the relatively large ISI and its implications on the inference of anatomical and CBV changes, whole brain voxel-level analysis was not performed. Thus a ROI-based analysis was performed instead. Guided by the human imaging study, ROIs were identified in the ventral and dorsal striatum (VS and DS), equivalent to the anterior and posterior body of the caudate in primates, and in S1.
To map functional changes over time we used a GEE model, a statistical approach used for longitudinal data analysis (Zeger et al., 1988). This statistical method takes into account the multiple observations per subject which are likely to be correlated, and treats them as clusters. rCBV values from the 3 ROIs were included as the dependent variables, genotype was included as the independent variable, and genotype-by-time was included as the interaction variable. Gender was included as a covariate.
In the DS a significant effect was found for time (Beta=-0.002, Wald chi square=31.71, p=0.0001), and for the genotype-by-time interaction (Beta=-0.001, Wald chi square=9.64, p=0.002). The negative beta values indicate an age-related decline in rCBV for both groups, but more importantly the interaction variable indicates that transgenics had an accelerated decline in the dorsal striatum compared to WT littermates (Fig. 3A and 3B) without gender differences. For the VS, a significant time effect was observed (Beta=-0.002, Wald chi square=3.98, p=0.047) but no significant genotype-by-time interaction was observed (Fig. 3C and 3D). For S1, no significant effects were found for genotype, time, or genotype-by-time (Fig S2B).
Figure 3. Age-dependent dorsal striatum dysfunction in the YAC128 mouse model.

(A) rCBV imaged longitudinally over time reveals an accelerated decline in rCBV in the dorsal striatum of YAC128 mice (broken line) compared to WT littermates (solid line). Shown are mean normalized ΔR2 values (see Methods) and SD vs. age in weeks.
(B) Representative rCBV pseudocolor maps of the dorsal striatum shown for a 44 week old YAC128 mouse and a WT littermate. Warmer colors represent higher rCBV values.
(C) rCBV imaged longitudinally over time reveals no difference in the ventral striatum of YAC128 mice (broken line) compared to WT littermates (solid line). Shown are mean normalized ΔR2 values and SD vs. age.
(D) Representative rCBV pseudocolor maps of the ventral striatum shown for a 44 week old YAC128 mouse and a WT littermate. Warmer colors represent higher rCBV values
Although, structural volumes of striatum in this mouse model have been reported before (Lerch et al., 2008a; Lerch et al., 2008b), we have measured MR striatum volumes longitudinally. This may provide more detail information on possible age-dependent structural changes and— more importantly help identify the influence, if any, of the structural changes on the rCBV measurements. We used the same horizontal 2D-T2 images used for the fMRI-analysis, and the same YAC128 and WT groups used for the fMRI analysis were used here [see Methods for details]. Striatum volume analysis employed the same GEE model described above. Volumes in mm3 were included as the dependent variable, genotype was included as the independent variable, and genotype-by-time was included as the interaction variable. Gender was included as a covariate.
In striatum volume, only a significant effect was found for time (Beta=-2.648, Wald chi square=53.81, p<0.001), the negative beta value indicates an age-related volume loss in striatum for both groups. Nevertheless no significant effect was found for the genotype-by-time interaction (Beta=-0.391, Wald chi square=0.254, p=0.614), indicating that if there is striatum volume loss, aged-transgenics have no worsening of caudate atrophy. To clarify these findings we evaluated each age group independently. Although volumes tended to be lower in YAC128 mice after 32 weeks (Fig. 4). ANOVA analysis revealed that only at age 44 weeks was there a significantly lower volume in YAC128 mice in striatum (F= 4.98, p=0.04).
Figure 4. Striatum Atrophy in YAC128 mice.

Striatum volumes in YAC128 mice (gray) and WT littermates (black) were determined, demonstrating a significant atrophy only at the 44 weeks time point in YAC128 mice. Shown are mean values in mm3 and SD.
* denotes p≤0.05
Finally, we replicated the rCBV GEE analysis in VS and DS, including as covariates the striatum volumes. The results were comparable. In DS time had values of (Beta=-0.012, Wald chi square=43.18, p<0.0001), and for the genotype-by-time interaction (Beta=-0.0231, Wald chi square= 5.64, p=0.036). For the VS the time effect was (Beta=-0.0017, Wald chi square=2.97, p=0.049) with no significant genotype-by-time interaction.
PPP1R7 protein deficiency correlates with striatum vulnerability in YAC128 mice
To confirm and extend the human microarray findings we performed molecular studies in brain tissue harvested from the mouse model. As in humans, the striatum was the brain region of interest, but because of the lack of clear boundaries between VS and DS in rodents (Heimer et al., 1995) the whole structure was harvested. Guided by the fMRI results, the striatum was harvested at 48 weeks of age, a time point at which lower striatal rCBV values had already emerged (Fig. 3A). The S1 cortex was also harvested as a within-brain control. Western blot analysis was used to measure expression levels of the two genes identified in the human microarray study—WIF1 and PPP1R7. Results showed that PPP1R7 levels in the striatum were significantly reduced in the YAC128 mice compared to WT littermates (F=19.3, p=0.0046) (Fig. 5A), while no significant difference was found for WIF1 (F=1.49, p=0.25). Additionally, no between-group differences were found for PPP1R7 levels measured from S1 (F=0.97, p=0.34). Finally, PPP1R7 levels measured in the striatum harvested from 28 week old mice, prior to the time at which striatal rCBV decline is observed (Fig. 3A), revealed no difference between YAC128 mice and their WT littermates (F=0.163, p=0.87) (Fig. 5A). In order to further confirm the PPP1R7 finding in light of reported mismatches in similar studies between mRNA and protein expression (Lewandowski and Small, 2005), we completed a second molecular study in human brains. Tissue was harvested from the posterior body of the caudate and S1 from a new group of human brains with [n=4 (mean age=56, 75% male, mean CAG repeats=46, rated pathological grade 2 or 3 (Vonsattel et al., 1985)] and without HD [n=4 (mean age=58, 50% male)]. Analysis by Western blot revealed that PPP1R7 protein levels were significantly reduced in HD compared to controls (F=18.62, p=0.007), with no difference found in the S1 (F=0.088, p=0.71) (Fig. 5B).
Figure 5. PPP1R7 deficiency correlates with anatomical phenotype.

(A) As shown in individual blots and by group data, PPP1R7 deficiency is observed in the striatum, but not the S1, in 48 weeks old YAC128 mice compared to WT littermates (48 weeks- upper panel). In contrast, 28 weeks old YAC128 mice compared to WT littermates showed no difference in PPP1R7 expression within the striatum (28 weeks- lower panel).
(B) As shown in individual blots and by group data, PPP1R7 deficiency is observed in the posterior body of the caudate, but not the S1, in HD human brains compared to controls. Lower panels in A and B show PPP1R7/Tubulin mean ratios and SE.
* denotes p≤0.05, ** denotes p≤0.01
DISCUSSION
Relying on the high resolution capabilities of CBV mapping with fMRI, our first study in human HD patients suggests that dysfunction in the posterior body of the caudate is differentially associated with phenotype, not only genotype. Nevertheless, there are obvious limitations to human studies. In particular, it is difficult to account for all the potential confounds, such as medication use or lifestyle differences, which might affect brain function independent of disease. Relying on the translational capabilities of CBV mapping (Moreno et al., 2007), the fMRI findings in the YAC128 (C57BL/6J) mouse model, in which there is better experimental control, addresses these concerns. By finding differential abnormalities in the dorsal striatum, a region homologous to the posterior caudate in humans, the mouse fMRI study provides confirmation of the human data.
Despite convergent findings, fMRI discrepancies were noted between patients and mice. In particular, despite the link between reductions in anterior caudate CBV and genotype in humans, an equivalent group-specific reduction was not found in VS of YAC128 mice. The difference could have been masked by an aging effect, since significant age-dependent rCBV decline was observed in both transgenic and WT mice in the VS. Similarly, differences between the axes of striatum atrophy have been found between human HD and YAC128 mice in FVB/n background strain (Lerch et al., 2008b).
On the other hand cerebral blood flow (CBF) signals have been shown to correlate strongly with local field potentials [LFPs] (reflecting neurovascular coupling), which perhaps indicate the level of neuronal synchronization in a circuit (Chaigneau et al., 2007; Lauritzen, 2005). Given the aforementioned consideration and the relationship between CBV and CBF (Rostrup et al., 2005), we expected that decreased regional rCBV might reveal abnormal baseline LFPs activity, if neurovascular coupling to neuronal energy demand was indeed preserved in mice and humans with or without HD. Since brain rCBV is a hemodynamic measure that indirectly informs on neuronal metabolism and function (Cepeda-Prado et al., 2012), minor differences in microcirculation (in particular venular), circuit activity or glucose utilization may produces differences in the final readout between species.
MRI has facilitated longitudinal analysis of neuropathology in vivo, because the same group of mice can be imaged on several occasions (Bock et al., 2006). In fact, longitudinal structural MRI has been used in a HD mouse model, YAC128 FVB/n, (Lerch et al., 2008a; Lerch et al., 2008b), but to our knowledge this is the first longitudinal brain fMRI study in any HD mouse model. The fact that functional changes (rCBV) emerged over time further supports the interpretation that dysfunction in the dorsal striatum/posterior caudate is associated with disease phenotype, not merely genotype. Longitudinal behavioral studies in C57BL/6J YAC128 mouse line identified that mutant mice had significant deficits detectable in the rotarod test from 4 months of age, in the balance beam from 10 months of age (Brooks et al., 2010; Brooks et al., 2011a), and cognitive impairment from around 6 months of age. Consistently with the present MRI structural findings, non-progressive striatal atrophy and cell loss has recently been reported in this mouse line (Bayram-Weston et al., 2012).
Whether observed reductions in an indirect indicator of metabolic function (rCBV) arise from tissue loss, as has been suggested by PET-FDG metabolic studies (Karow et al., 2010), was addressed both in the human and mice groups by covarying the data analysis with caudate area (humans) or with striatum volumes (mice, see Methods) of the identified abnormal ROIs. covariation of rCBV results with structural data did not affect rCBV findings, which suggests that rCBV abnormalities are not the result of the structural changes observed in HD.
The longitudinal mice fMRI study also demonstrated that S1 region did not differ significantly between mutant and WT mice, findings consistent with the human study. Mapping a unified fMRI- based anatomical pattern of caudate dysfunction in both patients and a mouse model formed the basis of the subsequent translational studies. Microarray analysis was combined with in vivo brain imaging in an attempt to: 1) maximize signal amplitude in the microarray experiment; 2) reduce sources of noise; and 3) address the high rate of false positive that occurs with multiple comparisons (Lewandowski and Small, 2005), HD is a regionally selective brain disorder, and it is likely that microarray analysis performed on brain tissue most affected by the disease should result in increased signal magnitude. Microarray analysis performed on large sections of brain tissue containing a mixture of affected, less affected, and unaffected tissue is likely to degrade the strength of the signal of an already low but meaningful abnormality in expression.
On the other hand pinpointing regions within the same brain structure that are differentially targeted by and relatively resistant to a disease can be used (identified by fMRI). Specifically, a 2 × 2 factorial ANOVA was designed which included both within and between-group factors, and this was effective in improving signal-to-noise in the microarray experiment as described in detail below.
As mentioned above the human study was guided by the fMRI results, in which we performed 60 microarray analyses in three regions of the brain harvested from brains with and without HD. In the primary analysis we used expression levels from the anterior caudate as a within-brain control in a repeated measures ANOVA, identifying a group of molecules whose expression levels were differentially affected in the posterior caudate. As previously described (Lewandowski and Small, 2005; Small et al., 2005), using a within-brain control to perform a “double subtraction” analysis is an effective statistical approach for constraining sources of noise when microarray is applied to the postmortem brain (Lewandowski and Small, 2005). Previous microarray studies have successfully used this analytic approach to isolate pathogenic defects in retromer sorting and trafficking in Alzheimer's disease (Small et al., 2005) and the polyamine pathway in Parkinson's disease (Lewandowski et al., 2010).
Expression levels from tissue harvested from S1 were used in a secondary analysis to further filter the list of candidate molecules. These two analyses identified a small list of molecules whose expression levels best tracked with the anatomical phenotype of the disease. In microarrays, generated from postmortem human brains, it is difficult to account for the inter- individual variance that may affect expression levels independent of disease; therefore, we exploited the anatomical phenocopy between human patients and the mouse model to address these concerns. Indeed, molecular analysis in the striatum of YAC128, at a time point in midlife when a decline in rCBV value in the striatum was detected by fMRI, strongly suggests that PPP1R7 deficiency is linked to the anatomical phenotype of the disease. An important related finding is that PPP1R7 do not vary prior to the observed decline in function (at 28 weeks). This finding was further evaluated in a second human study, showing that PPP1R7 protein is selectively deficient in the posterior caudate of HD patients.
Taken together, our human and mouse molecular analyses show that PPP1R7 deficiency is one molecular defect that coincides with the fMRI-detected anatomical phenotype in HD. PPP1R7 is one of the many proteins that regulate the function of protein phosphatase 1 (PP1) by inhibiting its activity (Ceulemans et al., 1999). PP1 regulates the phosphorylation state of many receptors and channels and its activity has been implicated in many neuronal processes, including long-term potentiation and long-term depression (Greengard et al., 1999). Playing a role in dopaminergic signal transduction, PP1 is a key enzyme in medium spiny neurons of the caudate, a dominant site of dopaminergic projections (Greengard et al., 1999). Interestingly, previous studies have identified deficiencies in other proteins that inhibit PP1 activity in the striatum of HD mouse models, including a decrease in protein phosphatase 1 inhibitor in the R6/2 mouse model (Luthi-Carter et al., 2000), and a decrease in DARPP32 in the R6/1 mouse model (van Dellen et al., 2000) also called ‘protein phosphatase 1 regulatory subunit 1b’. Specifically, in YAC128 mice FVB/n, downregulation of DARPP-32 in the lateral striatum is correlated with nuclear mutant htt expression (Van Raamsdonk et al., 2005). Together with our results, a convergence of studies suggests that the huntingtin mutation causes deficiencies in inhibitory proteins, leading to abnormal upregulation of PP1 activity. To our knowledge, the present findings are the first to link a deficiency in a PP1 regulatory protein to the human disease.
In conclusion, our results demonstrate that in vivo fMRI-CBV is capable of detecting regional specific brain abnormalities both in HD patients and a mouse model of the disease, supporting the view that YAC128 mice can model adult HD appropriately. From the molecular point of view, although our findings suggest that the observed PPP1R7 deficiency is related to huntingtin mutations, specific cellular and molecular mechanisms mediating this potential link remain unknown. Nevertheless, our findings open the possibility of exploring in more detail the role of PP1 activity in HD pathogenesis, and its potential as a target for drug discovery.
Supplementary Material
{kind=link}
Acknowledgments
We thank K. Hess and F. Hua for animal MRI prep and acquisition, A. Muhammad and A. Bender for WB prep, and our profound gratitude to all the subjects who willingly participated in the studies presented herein. Funding: This work was supported in part by awards from the CHDI Foundation, Inc. (formerly, High Q Foundation, Inc.), Huntington's Disease Society of America, Inc. (HDSA), and the Family Fund.
Footnotes
Author contributions: N.M.L designed, preformed research, analyzed data, and wrote the paper. Y.B. preformed research and analyzed data with human fMRI. A.M.B. analyzed human fMRI data. S.A. acquired mouse fMRI data, J.M. technical and analysis in human fMRI. P.W. support guidance. L.M. analytical tools. J.P.V. postmortem analysis and analytical tools. K.M. designed research and analytical tools. H.M. designed, analyzed data for mouse fMRI, and wrote the paper. S.A.S designed research, analyzed data, and wrote the paper.
Competing interests: The authors declare no conflicts of interest. Accession numbers: The microarray data has been submitted in a MIAME- compliant format to Gene Expression Omnibus (GEO) via NCBI (www.ncbi.nih.gov/geo), series accession # GSE21897.
REFERENCES AND NOTES
- Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11:805–21. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- Bayram-Weston Z, et al. Light and electron microscopic characterization of the evolution of cellular pathology in YAC128 Huntington's disease transgenic mice. Brain Res Bull. 2012;88:137–47. doi: 10.1016/j.brainresbull.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Belliveau JW, et al. Functional mapping of the human visual cortex by magnetic resonance imaging. Science. 1991;254:716–9. doi: 10.1126/science.1948051. [DOI] [PubMed] [Google Scholar]
- Bock NA, et al. In vivo magnetic resonance imaging and semiautomated image analysis extend the brain phenotype for cdf/cdf mice. J Neurosci. 2006;26:4455–9. doi: 10.1523/JNEUROSCI.5438-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks S, et al. Longitudinal analysis of the behavioural phenotype in YAC128 (C57BL/6J) Huntington's disease transgenic mice. Brain Res Bull. 2010 doi: 10.1016/j.brainresbull.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Brooks SP, et al. Selective cognitive impairment in the YAC128 Huntington's disease mouse. Brain Res Bull. 2011a doi: 10.1016/j.brainresbull.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Brooks SP, et al. Longitudinal analyses of operant performance on the serial implicit learning task (SILT) in the YAC128 Huntington's disease mouse line. Brain Res Bull. 2011b. doi: 10.1016/j.brainresbull.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Cepeda-Prado E, et al. R6/2 Huntington's disease mice develop early and progressive abnormal brain metabolism and seizures. J Neurosci. 2012;32:6456–67. doi: 10.1523/JNEUROSCI.0388-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans H, et al. Structure and splice products of the human gene encoding sds22, a putative mitotic regulator of protein phosphatase-1. Eur J Biochem. 1999;262:36–42. doi: 10.1046/j.1432-1327.1999.00344.x. [DOI] [PubMed] [Google Scholar]
- Chaigneau E, et al. The relationship between blood flow and neuronal activity in the rodent olfactory bulb. J Neurosci. 2007;27:6452–60. doi: 10.1523/JNEUROSCI.3141-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, et al. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol. 1988;47:516–25. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- Gaisler-Salomon I, et al. Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacology. 2009;34:2305–22. doi: 10.1038/npp.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RG, et al. Functional MR in the evaluation of dementia: correlation of abnormal dynamic cerebral blood volume measurements with changes in cerebral metabolism on positron emission tomography with fludeoxyglucose F 18. AJNR Am J Neuroradiol. 1995;16:1763–70. [PMC free article] [PubMed] [Google Scholar]
- Good CD, et al. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage. 2001;14:21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- Greengard P, et al. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–47. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Heimer L, et al. Basal Ganglia. In: Paxinos G, editor. The Rat Nervous System. Academic Press; San Diego: 1995. pp. 579–628. [Google Scholar]
- Karow DS, et al. Relative capability of MR imaging and FDG PET to depict changes associated with prodromal and early Alzheimer disease. Radiology. 2010;256:932–42. doi: 10.1148/radiol.10091402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassubek J, et al. Topography of cerebral atrophy in early Huntington's disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry. 2004;75:213–20. [PMC free article] [PubMed] [Google Scholar]
- Kelley AE, et al. The amygdalostriatal projection in the rat--an anatomical study by anterograde and retrograde tracing methods. Neuroscience. 1982;7:615–30. doi: 10.1016/0306-4522(82)90067-7. [DOI] [PubMed] [Google Scholar]
- Kuhl DE, et al. Local cerebral glucose utilization in symptomatic and presymptomatic Huntington's disease. Res Publ Assoc Res Nerv Ment Dis. 1985;63:199–209. [PubMed] [Google Scholar]
- Kuhl DE, et al. Patterns of cerebral glucose utilization in Parkinson's disease and Huntington's disease. Ann Neurol. 1984;15(Suppl):S119–25. doi: 10.1002/ana.410150723. [DOI] [PubMed] [Google Scholar]
- Kuhl DE, et al. Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann Neurol. 1982;12:425–34. doi: 10.1002/ana.410120504. [DOI] [PubMed] [Google Scholar]
- Kumar P, et al. Huntington's disease: pathogenesis to animal models. Pharmacol Rep. 2010;62:1–14. doi: 10.1016/s1734-1140(10)70238-3. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Reading vascular changes in brain imaging: is dendritic calcium the key? Nat Rev Neurosci. 2005;6:77–85. doi: 10.1038/nrn1589. [DOI] [PubMed] [Google Scholar]
- Lerch JP, et al. Cortical thickness measured from MRI in the YAC128 mouse model of Huntington's disease. Neuroimage. 2008a;41:243–51. doi: 10.1016/j.neuroimage.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Lerch JP, et al. Automated deformation analysis in the YAC128 Huntington disease mouse model. Neuroimage. 2008b;39:32–9. doi: 10.1016/j.neuroimage.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Lewandowski NM, et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A. 2010;107:16970–5. doi: 10.1073/pnas.1011751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski NM, Small SA. Brain microarray: finding needles in molecular haystacks. J Neurosci. 2005;25:10341–6. doi: 10.1523/JNEUROSCI.4006-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, et al. Regional cerebral blood volume: a comparison of the dynamic imaging and the steady state methods. J Magn Reson Imaging. 1999;9:44–52. doi: 10.1002/(sici)1522-2586(199901)9:1<44::aid-jmri6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–71. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- MacDonald ME. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Mazziotta J, et al. A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos Trans R Soc Lond B Biol Sci. 2001;356:1293–322. doi: 10.1098/rstb.2001.0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled L, et al. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–36. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno H, et al. The absence of the calcium-buffering protein calbindin is associated with faster age-related decline in hippocampal metabolism. Hippocampus. 2011 doi: 10.1002/hipo.20957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno H, et al. Longitudinal mapping of mouse cerebral blood volume with MRI. NMR Biomed. 2006;19:535–43. doi: 10.1002/nbm.1022. [DOI] [PubMed] [Google Scholar]
- Moreno H, et al. Imaging the Abeta-related neurotoxicity of Alzheimer disease. Arch Neurol. 2007;64:1467–77. doi: 10.1001/archneur.64.10.1467. [DOI] [PubMed] [Google Scholar]
- Morton AJ, et al. Progressive formation of inclusions in the striatum and hippocampus of mice transgenic for the human Huntington's disease mutation. J Neurocytol. 2000;29:679–702. doi: 10.1023/a:1010887421592. [DOI] [PubMed] [Google Scholar]
- Paulsen JS. Functional imaging in Huntington's disease. Exp Neurol. 2009;216:272–7. doi: 10.1016/j.expneurol.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AC, et al. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2007;104:5638–43. doi: 10.1073/pnas.0611721104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–7. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GD, et al. The Mouse Brain Library ©. Int Mouse Genome Conference; 2000. p. 166. www.mbl.org. [Google Scholar]
- Rostrup E, et al. The relationship between cerebral blood flow and volume in humans. Neuroimage. 2005;24:1–11. doi: 10.1016/j.neuroimage.2004.09.043. [DOI] [PubMed] [Google Scholar]
- Schobel SA, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–46. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattuck DW, et al. Construction of a 3D probabilistic atlas of human cortical structures. Neuroimage. 2008;39:1064–80. doi: 10.1016/j.neuroimage.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–67. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Small SA, et al. Imaging correlates of brain function in monkeys and rats isolates a hippocampal subregion differentially vulnerable to aging. Proc Natl Acad Sci U S A. 2004;101:7181–6. doi: 10.1073/pnas.0400285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, et al. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–19. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Smith SM, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- van Dellen A, et al. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. Neuroreport. 2000;11:3751–7. doi: 10.1097/00001756-200011270-00032. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25:4169–80. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–84. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, et al. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Williams RW. Mapping genes that modulate mouse brain development: a quantitative genetic approach. In: Rakic P, Goffinet A, editors. Mouse Brain Development. Springer; New York: 2000. pp. 21–49. [DOI] [PubMed] [Google Scholar]
- Wu W, et al. The brain in the age of old: the hippocampal formation is targeted differentially by diseases of late life. Ann Neurol. 2008;64:698–706. doi: 10.1002/ana.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–28. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zeger SL, et al. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44:1049–60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.