

Abstract
Glioblastoma (GBM) is the most aggressive, deadliest, and most common brain malignancy in adults. Despite the advances made in surgical techniques, radiotherapy and chemotherapy, the median survival for GBM patients has remained at a mere 14 months. GBM poses several unique challenges to currently available treatments for the disease. For example, GBM cells have the propensity to aggressively infiltrate/invade into the normal brain tissues and along the vascular tracks, which prevents complete resection of all malignant cells and limits the effect of localized radiotherapy while sparing normal tissue. Although anti-angiogenic treatment exerts anti-edematic effect in GBM, unfortunately, tumors progress with acquired increased invasiveness. Therefore, it is an important task to gain a deeper understanding of the intrinsic and post-treatment invasive phenotypes of GBM in hopes that the gained knowledge would lead to novel GBM treatments that are more effective and less toxic. This review will give an overview of some of the signaling pathways that have been shown to positively and negatively regulate GBM invasion, including, the PI3K/Akt, Wnt, sonic hedgehog-GLI1, and microRNAs. The review will also discuss several approaches to cancer therapies potentially altering GBM invasiveness.
Keywords: glioblastoma, invasion, PI3K, Wnt, hedgehog
1. Introduction
Gliomas are primary brain cancers that arise from non-neural cells called glial cells [1]. In the central nervous system (CNS), there are three types of glial cells: astrocytes, oligodendrocytes, and microglial cells. Oligodendrocytes are responsible for myelination while microglial cells are derived from hematopoietic stem cells and phagocytize microbes in the CNS. Astrocytes, the most abundant type of glial cells in the CNS, are star-shaped cells which establish metabolic homeostasis and can shift to a reactive phenotype in response to pathogens or injury in the CNS. This shift is normally a highly regulated process and its dysregulation has been shown to promote malignancy [2, 3].
Gliomas can be categorized based on the type of glial cells they are most histologically similar to, the location of the tumor, and the aggressiveness of the cancer cells. Tumors most similar to astrocytes are specifically called astrocytomas and can be further classified into grades I–IV based on the criteria set by World Health Organization, with a higher grade corresponding to more aggressive tumors. Grades I and II astrocytomas correspond to low-grade tumors that are mostly non-malignant. Grades III and IV astrocytomas are high-grade, malignant tumors. Grade III astrocytomas are also known as anaplastic astrocytomas (AAs) while Grade IV astrocytomas, commonly referred to as glioblastoma (GBM), are the most aggressive of all gliomas. Unfortunately, GBMs are also the most common type of gliomas with an annual incident rate of 3.19 per 100,000 in the United States [4, 5].
While cancer research has made great strides in the treatment of most cancer types, the median survival of patients with GBM is still only approximately 14 months, despite advances in detection, radiation, chemotherapy, and surgery [6, 7]. The current standard of care for newly diagnosed GBM patients includes surgery to excise as much of the tumor as safely possible and a combination of radiotherapy with temozolomide (TMZ), an oral alkylating agent which can cross the blood-brain barrier. However, treatment of GBM has remained relatively ineffective because of a number of challenges, including tumor hypoxia which contributes to therapeutic resistance and the invasiveness of GBM tumor cells into normal brain tissues which renders tumor removal insufficient. In particular, the subpopulation of GBM with the stem-like self-renewal property has been shown to highly resistant to various therapies [8]. There is no standard of care for recurrent GBM but one option is the use of a targeted drug called bevacizumab (Genentech). This drug, also known as Avastin, is a monoclonal VEGF-A antibody and is currently the only targeted therapy approved by the FDA to treat recurrent GBM [9]. Development of new drugs has been slow in part because of the ineffective delivery of the drug dosages across the blood-brain barrier and blood-brain tumor barrier. For instance, erlotinib, an epidermal growth factor (EGFR) inhibitor, had shown therapeutic promise in vitro but failed to show survival benefits in phase II studies because it could not sufficiently cross the blood-brain barrier [10, 11].
Since the high degree of infiltration is one of the hallmarks of GBM, this review will summarize the complex, multi-step process of GBM invasion, molecular pathways that have been reported to facilitate GBM invasion, microRNAs that have been associated with the process, and current therapies with the propensity to inhibit GBM infiltration.
2. Glioma Invasion
Even with technological advances in surgical techniques and radiation, malignant gliomas often recur within 1–2 cm of the original tumor site because some of the tumor cells invade into the surrounding normal brain tissue where they can hide from surgical removal and radiation therapy [12]. While other aggressive cancers metastasize by traveling through the circulatory or lymphatic systems to organs, high-grade glioma cells rarely metastasize outside of the brain and instead actively migrate through two types of extracellular space in the brain: 1) the perivascular space that is found around all blood vessels, and 2) the spaces in between the neurons and glial cells which makes up the brain parenchyma and white matter fiber tracts. In order to invade through these spaces, glioma cells typically undergo several biological changes, including gaining the mobility, the ability to degrade extracellular matrix (ECM), and the stem cell phenotype.
First, invasive tumor cells become morphologically polarized and develop membrane protrusions allowing the cells to reach forward and attach to the ECM. During this process, invasive glioma cells alter the cell shape and volume in order to move through differently sized spaces, including the extremely small spaces in normal brain [13]. In addition to gaining mobility, invasive glioma cells must be able to interact with multiple components of the ECM. Though the ECM is a physical barrier that glioma cells must get through, it also provides ligands that the tumor cells can anchor to so that they can pull themselves forward. Beyond these physical interactions, the ECM also interacts chemically with glioma cells. Several studies have shown that tumors influence the nearby stromal cells, causing reorganization of the structure and composition of the ECM. These changes in the ECM then further enhance tumor growth and invasion [14]. Cells are inherently motile, but this is tightly regulated in various stages, such as embryological development, and in physiological responses, such as wound healing and immune-response. In glioma cells, motility becomes dysregulated allowing them to be highly migratory [15].
Besides being able to migrate, glioma cells must be able to get through the physical barrier, ECM, by degrading extracellular matrix proteins in order to create a path for invasion. Many studies have reported the involvement of matrix-metalloproteinases (MMPs) in this degradation and the overexpression of several MMPs in cancer cells compared to their normal cell counterparts, including glioma cells [16]. Therefore, it is not surprising that many of the pathways that promote GBM invasion also up-regulate the expression of several MMPs [17–19]. Proteolytic enzymes are tightly associated with invasion. For example, heparanase is an endoglycosidase which degrades and remodels ECM by cleaving heparin sulfate and its overexpression promotes invasiveness of tumor cells in vivo [20]. Other proteases implicated in invasiveness include plasmin, cathepsin B, and cathepsin D [21, 22].
Any tumor is a heterogeneous population of cells where cancer cells are at different stages of differentiation. Recently substantial attention has been given to a subpopulation of tumor cells called cancer stem cells (CSCs) which like true stem cells are undifferentiated and self-renewing. For gliomas, these CSCs are called glioma stem cells (GSCs) or glioma initiating cells (GICs). GSCs express nestin and CD133, factors associated with neural stem cells, although there are some GSCs that are CD133-negative [23, 24]. GSCs also share with normal neural stem cells the ability to form neurospheres in serum-free culture condition, self-renew, and differentiate into different neural cells [25]. GSCs derived from primary human tumors have been shown not only to share many characteristics with neural stem cells, but also to retain the genotype, gene expression pattern, and phenotype of the primary tumor [25]. Because GSCs display more traits of GBM such as excessive invasiveness, this unique cell population is of special interests to GBM research and treatment.
GSCs are considered the primary cause of GBM invasion and recurrence [26]. Cancer stem cells (CSCs) are highly resistant to treatment and if there are CSCs that survive treatment, they are capable of initiating and sustaining new tumor growths, causing tumor recurrences. Therefore these cells are important targets for treatment. Several embryonic signaling pathways, such as Notch, Hedgehog, and Wnt/β-catenin have been reported to help maintain these GSCs and thus provide potential targets for treating these especially malignant cancer cells [5].
3. Wnt signaling pathway in glioma invasion
Wingless/Int1 (Wnt) signaling regulates many cellular processes in adulthood and plays an important role during embryogenesis [27, 28]. Several different intracellular signaling pathways have been identified that can be activated by Wnt ligands and Frizzled (Fz), their seven-transmembrane cell surface receptors. These are divided into those that are dependent on β-catenin and those that are independent (Figure 1).
Figure 1.

Three major signaling pathways that have been reported to be involved in the invasiveness of GBM, namely the PI3K-Akt, Wnt and Shh-tGLI1 pathways.
The β-catenin-dependent pathway is also known as the canonical Wnt pathway. When this pathway is not activated, β-catenin is bound to its destruction complex which consists of glycogen synthase kinase-3β (GSK-3β), Axin, and adenomatous polyposis coli (APC). GSK-3β phosphorylates β-catenin, marking it for proteasomal degradation. When one of the Wnt factors binds to Fz, it induces Fz to interact with the co-receptor low-density-lipoprotein-related protein 5/6 (LRP5/6), forming a complex that recruits the cytoplasmic scaffolding protein Dishevelled (Ds). This activation eventually prevents GSK-3β from marking β-catenin for degradation. Since β-catenin is stabilized, it translocates to the nucleus and interacts with T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors to regulate the expression of target genes such as c-Myc, cyclin D1, and MMPs [29, 30]. Wnt factors that are known to activate this β-catenin-dependent pathway include Wnt1, Wnt3a, and Wnt7a [31].
The β-catenin-independent pathways primarily regulate cell motility and polarity and include the planar cell polarity (PCP) pathway and the calcium pathway, although more β-catenin-independent pathways are continuously being reported [32]. In the PCP pathway, Fz activates Jun-N-terminal kinase, a MAP kinase. In the calcium pathway, various Wnt and Fz homologs activate calcium/calmodulin-dependent kinase II and protein kinase C [33]. These pathways have been shown to be upregulated in GBMs and are known to be activated by Wnt2, Wnt4, Wnt5a, Wnt5b, Wnt6, and Wnt11 [31, 34].
The aberrant activation of the Wnt pathway promotes cancer progression in many cancers types [30]. In GBM, several players of the β-catenin-dependent pathways have been shown to be important for invasion. β-catenin is overexpressed in gliomas and its knockdown in vitro reduced the invasiveness of GBM cells [35]. EGFR activation disrupts the association of α-catenin with β-catenin, allowing transactivation of β-catenin [36]. Additionally, c-MET has also been shown to activate the Wnt/β-catenin pathway in GBM [37]. The Wnt ligands Wnt1 and Wnt3a were found to be significantly overexpressed in tumors derived from Grade III gliomas and GBMs. The knockdown of Wnt1 caused formation of smaller intra-cranial tumors in mice that were noninvasive while the knockdown of Wnt3a completely prevented tumor formation [28]. Knockdown of the transcription factor Lef1 has also been shown to inhibit invasion of GBM in vitro [38].
Another protein that has been linked to promoting GBM invasion is FRAT1 (frequently rearranged in advanced T-cell lymphomas-1), a positive regulator of the β-catenin-dependent Wnt pathway that inhibits GSK3β from marking β-catenin for degradation. FRAT1 is significantly increased in glioma and has been shown to promote invasion in GBM cells lines, as well as tumor growth in vivo [39].
Players involved in β-catenin-independent pathways have also been reported to influence invasiveness in GBM. Wnt5a has been shown to be highly overexpressed glioma cells [31, 35] and its knockdown in vitro suppressed the expression of matrix metalloproteinase-2 and invasion in GBM cells. Inhibition of MMP-2 also abrogated Wnt5a-dependent invasion, suggesting that Wnt5a acts through MMP-2 to promote GBM invasion [31]. Wnt2 is also overexpressed in gliomas and its knockdown with siRNA in vitro reduced the invasiveness of GBM cells [35] (Figure 1).
Though Wnt signaling has an important role in cancers and other diseases, targeting the aberrant signaling in cancers has been difficult because Wnt signaling also partakes in many crucial physiological processes of normal adults. For example, Wnt is involved is hair and skin cell regeneration, maintenance of homeostasis, and hematopoiesis. Therefore inhibitors of Wnt signaling can have multiple side effects such as muscle spasms and cramps, alopecia (type of hair loss), fatigue, weight loss, and bone loss or breakage [40]. In addition to the processes that Wnt regulates, several players of this pathway also cross-talk with different essential pathways including the growth factor signaling pathways, making it even more difficult to avoid unintended side effects. There are currently no therapeutic agents against Wnt signaling that have been approved by the FDA for any type of cancer.
While there are many inhibitors of the Wnt pathway, few have been tested in GBM and their effects on invasion were not measured. For instance, SEN461 (Siena), a small-molecule inhibitor of the Wnt pathway, has been shown to inhibit canonical Wnt signaling by stabilizing Axin and increasing β-catenin degradation in vitro and in vivo. This inhibitor’s effect on GBM invasion has not been tested yet but it has been reported to reduce growth of GBM xenograft models [41]. Therefore, SEN461 may be useful for potential therapy of GBM, although no clinical trials have begun to test it [42].
4. PI3K/Akt signaling pathway in glioma invasion
Another pathway that influences the balance between degradation and stabilization of β-catenin is the PI3K/Akt pathway (Figure 1). This pathway is activated by growth factors and other extracellular stimuli, and regulates many biological processes such as cell metabolism, growth, and survival, including several receptor tyrosine kinases such as EGFR. Akt, also known as protein kinase B (PKB), is a cytoplasmic Ser/Thr kinase which is phosphorylated by phosphoinositide-dependent protein kinase 1 (PDK1) when they are both recruited to the cell membrane by phosphosphatidyl-3,4,5-triphosphates (PIP3). PIP3 is converted from phosphotidylinositol-3,4-bisphosphate (PIP2) by phosphoinositide-3-kinase (PI3K) and back by phosphatase and tensin homologue (PTEN) [43]. Therefore, PI3K activates Akt signaling while PTEN suppresses it. Once Akt is activated by phosphorylation, it can phosphorylate many other molecules including GSK3β which leads to the stabilization of β-catenin.
In many cancers, the PI3K/Akt signaling pathway is overactivated, often through the deletion or mutation of PTEN, or the hyperactivation of PI3K. In GBM, Akt signaling increases MMP-2 and MMP-9 activity, especially in the cells at the border between tumor and normal brain tissue, giving these tumor cells the proteolytic capability to invade normal brain [18]. PTEN has been reported to suppress GBM invasion by inhibiting MMP-2 through its transcription [17] (Figure 1).
Bcl-w, a prosurvival Bcl-2 protein, has been reported to promote invasion of GBM by activating Akt signaling, leading to the phosphorylation of GSK3β and nuclear translocation of β-catenin. Once in the nucleus, β-catenin can upregulate its target genes, which includes MMP-2, and promote invasion as well as other mesenchymal traits such as the increased levels of Twist1 and Snail in vitro [19].
The effects of several Akt-targeted drugs on GBM invasion have been investigated pre-clinically and clinically. Sulindac (Merck), also known as Clinoril, is a non-steroidal anti-inflammatory drug (NSAID) and its metabolites have been shown to inhibit GBM invasion in vitro by dephosphorylation of Akt at Ser473, which caused a decrease in MMP-2 gene expression and activity. When sulindac was combined with LY294002, a PI3K inhibitor, they synergistically inhibited GBM invasion [44]. Another NSAID that is able to inhibit GBM invasion is celecoxib and its effect also involves diminishing activity of the Akt signaling pathway. Other NSAIDs, including aspirin, ketoprofen, ketorolac, and naproxen, were not able to inhibit GBM invasion [44]. Furthermore, arsenic-derived compounds have shown success in clinical trials in treating various cancers including gliomas. Arsenic trioxide (ATO), which has been approved by the FDA for treating promyelocytic leukemia, has been shown to accumulate more in brain tumors than in normal human brain tissues. In phase I studies, ATO was well-tolerated with TMZ and radiotherapy against malignant gliomas [45, 46]. Tetra-arsenic oxide (TAO), another arsenic compound, is less toxic than ATO and has anti-angiogenic, anti-proliferative, and anti-invasive effects at lower concentrations than ATO. TAO has been reported to decreases GBM invasion in vitro by inhibiting Akt phosphorylation at Ser473 and by down-regulating MMP-2 expression and activity [47].
BKM120 (Novartis), also known as Buparlisib, is a pan-class I PI3K inhibitor in glioma cells that can cross the blood-brain barrier. It has shown effective inhibition of the PI3K/Akt signaling pathway in vitro and in vivo. Treatment of various GBM cell lines showed a dose-dependent inhibition of growth while treatment of mice with intracranial U87MG xenografts increased the median survival without any obvious adverse effects, such as weight loss or decreased activeness [48]. Currently, BKM120 is being studied in a phase II trial in patients with recurrent GBMs (NCT01339052). There are also studies investigating the efficacy of this drug in combination with current first-line GBM treatments. There is an ongoing phase I dose-escalation study for newly diagnosed GBMs that combines BKM120 with radiotherapy and TMZ (NCT01473901). BKM120 is also combined with bevacizumab in a phase I/II study with relapsed/refractory GBMs (NCT01329660). In addition, there are studies verifying the efficacy of BKM120 in combination with other inhibitors, including a phase Ib/II study of INC280, a c-Met inhibitor, and BKM120 in recurrent GBM (NCT01870726) and another phase Ib/II study of BKM120 and one of the alkylating agents, carboplatin or lomustine, also in recurrent GBM (NCT0193461).
Some PI3K inhibitors also inhibit the mTOR pathway since the catalytic domains of mTOR and of the PI3K subunit p110 are structurally similar [9]. XL147 (Sanofi), also referred to as SAR245408, is one of these dual PI3K/mTOR inhibitors and has been shown to have cytotoxic effects on GBM cells and anti-cancer effects in nude mice with intracranial xenografts. Furthermore, XL147 and TMZ had additive effects in vivo with no obvious adverse effects on the mice, suggesting the potential of combining these two drugs in treating GBM [49]. There has been a phase I study of combining XL765 with TMZ with malignant tumors (NCT00704080). However, the results of the trial have not been published yet.
5. Hedgehog-GLI1 signaling pathway in gliomal invasion
In vertebrates, Hedgehog (Hh) signaling can be initiated by three ligands: Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh). These ligands will bind to the 12-transmembrane receptor Patched (PTCH), abrogating its repression of Smoothened (SMO), a 7-transmembrane receptor-like protein. No longer repressed, SMO prevents the cleavage of GLI1 which allows it to localize to the nucleus and act as a transcription factor, upregulating the transcription of genes including GLI1, PTCH1, cyclin D, Bcl-2, and VEGF [50–54]. GLI1 has also been shown to be activated by multiple non-canonical pathways including the PI3K/Akt pathway, which can be activated by multiple receptor tyrosine kinases. Additionally, GLI1 can be activated by MEK, which is activated by multiple receptor tyrosine kinases such as EGFR and PDGFR [55].
During embryogenesis, the Hedgehog signaling pathway is a key regulator of important developmental events of the central nervous system, and when it does not function properly, major defects such as microencephaly or cyclopia can occur [56]. In adults, this pathway is involved in normal tissue and stem cell maintenance. This pathway is also often up-regulated in many cancers, including gliomas, and plays a role in both tumorigenesis and tumor progression [51].
The GLI family of Zinc-finger transcription factors, GLI1, GLI12 and GLI3, are the nuclear effectors of the Shh pathway. Although GLI1 was identified as an amplified gene in GBM[57], this gene amplification is rare in GBM. Somatic mutations have never been reported in any cell or cancer types. Our laboratory recently identified a novel isoform of GLI, namely, truncated GLI1 (tGLI1) has been linked to increasing motility and invasion in GBM and invasive breast cancer [58–61]. tGLI1 is a product of alternative splicing of the full-length GLI1 that lacks exon 3 and part of exon 4 corresponding to 123 nucleotides and 41 amino acids [60]. The tGLI1 variant is expressed in most GBM specimens and patient-derived xenografts, but undetectable in normal brain cells or tissues [60, 61]. In addition to promoting motility and invasion, tGLI1 was found to render GBM xenografts more vascularized than GLI1, in part, through upregulating expression of VEGF-A and heparanase [61] (Figure 1).
tGLI1 has also been reported to upregulate the transcription of heparanase, another gene associated with invasion, in GBM and in breast cancer [58, 59, 61]. In addition to remodeling the ECM, it also releases angiogenic factors from the ECM, eliciting an angiogenic response from the tGLI1-expressing GBM cells. Additionally, tGLI1 has also been shown in GBM and in breast cancer to upregulate the transcription of vascular endothelial growth factor-A (VEGF-A) in vitro and in vivo [58, 59, 61]. Because tGLI1 is specifically expressed in aggressive cancer cells and not in normal cells, it can be a specific therapeutic target against GBM, although nothing has been developed yet to specifically target tGLI1.
Like therapeutic agents of the Wnt pathway, inhibitors of the Hedgehog pathway produce many unintentional side effects because this pathway is involved in many normal cell processes. Currently the inhibitors of the Hh pathway that have made it to clinical trials are Smo inhibitors. A naturally-occurring inhibitor is cyclopamine which had anti-tumor effects in vitro and in vivo [62]. However, due to poor bioavailability, attention has been shifted to semi-synthetic and synthetic derivatives of cyclopamine which are more potent and more readily available. One derivative is GDC0449 (Genentech), also referred to as Erivedge or vismodegib, which is able to cross the blood-brain barrier. GDC0449 is also being evaluated in other types of cancer and was the first drug approved by the FDA to treat basal cell carcinoma [40].
LDE225 (Novartis), also known as sonidegib or erismodegib, is another Smo inhibitor that can cross the blood-brain barrier and is being studied in GBM. Treatment of glioblastoma stem cells with this inhibitor is able to suppress invasion in vitro by upregulating the miR-200 family, as well as suppressing migration, neurosphere formation, and cell viability [63]. This inhibitor has also been shown to effectively work with the PI3K inhibitor BKM120 to reduce the growth of PTEN-deficient GBMS in vitro and in vivo [64]. LDE225 is currently being studied in combination with BKM120 in a phase Ib dose-escalating study with advanced solid tumors, including recurrent GBM (NCT01576666).
6. microRNAs in glioma invasion
MicroRNAs (miRNAs) are short, endogenous, noncoding RNAs that are about 19–25 nucleotides in length [65]. They regulate expression of their target genes by binding to the 3′ untranslated regions (3′-UTRs) of the genes’ mRNA. This binding will suppress translation or induce degradation of the mRNA. One miRNA can regulate the expression of many genes, and depending on its target genes, a miRNA can act as a tumor suppressor or tumor promoter. Many cellular processes have been linked to miRNA regulation including cell differentiation, proliferation, apoptosis, metabolism, and stem cell maintenance [63]. Some of the invasion-associated miRNAs are mentioned below and are depicted in Figure 2.
Figure 2.
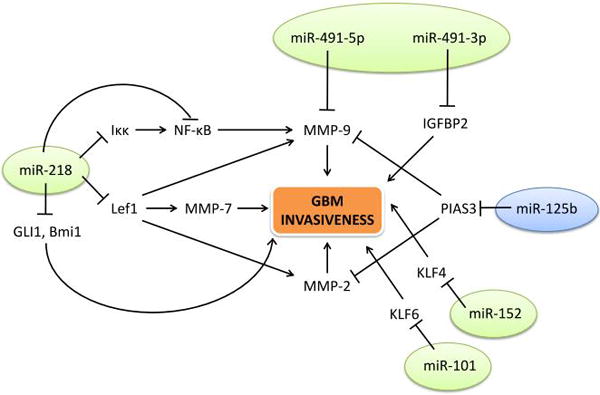
Several miRNAs that have been shown to influence invasiveness in GBM and their targets. Green circles mark the microRNAs that act as tumor suppressors while the blue circle indicates the miRNA that acts as an onco-miR.
6.1. miR-218
miR-218 is highly expressed in developing and normal cells of the central nervous system [65]. Its expression has been found to be significantly down-regulated in GBM cell lines and human GBM tissues samples [29, 66]. miR-218 expression also correlates negatively with glioma grade [67]. Several studies have shown that miR-218 is a negative regulator for invasion in GBM through various pathways [29, 66, 67]. It has been reported to target the mRNA of Lef1, the transcription factor that is upregulated by β-catenin [29]. Suppression of Lef1 leads to the reduction of MMP-2, MMP-7, and MMP-9 activity and inhibition of invasion in vitro [29]. Another mechanism through which miR-218 negatively regulates GBM invasion is targeting IKKβ mRNA along with reducing the transcription of NF-κB. Both of these actions will decrease the activity of NF-κB, a transcription factor that is important in many cellular processes and has been strongly linked to migration and invasion of difference cancer cells [66]. NF-κB’s target genes include MMP-9, so its inhibition by miR-218 causes a decrease in MMP-9 levels and in invasion. Furthermore, the mRNA of Bmi1 (a regulator of stemness in glioma cells) and GLI1 have been shown to be targets of miR-218. These studies had not confirmed that Bmi1 and GLI1are necessary for miR-218 to reduce invasion in GBM cells, but other studies have reported that Bmi1 and GLI1 regulate invasiveness [60, 67–69]. It should be noted that most studies do not distinguish between GLI1 and the splice variant tGLI1.
6.2. miR-101 and miR-152
miR-101 is a tumor suppressor that is down-regulated in many cancers, including GBM [70–72]. It has been shown to down-regulate invasion of GSCs, as well as proliferation and migration, by targeting the transcription factor Kruppel-like factor 6 (KLF6). This suppression of KLF6 reduced the expression of Chitinase-3-like protein 1 (CHI3L1) and inactivation of MEK1/2 and PI3K signaling [72]. However, whether KLF6-regulated invasiveness depends on CHI3L1 or MEK1/2 and PI3K signaling was not investigated. miR-101 down-regulation has been shown to result in EZH2-induced proliferation, migration, and angiogenesis in GBM [70].
Similarly, miR-152 is also a tumor suppressor that is down-regulated often in cancers, including in GBM. It was shown to suppress invasion of GSCs, as well as cell proliferation, migration, invasion, and apoptosis. It has been reported that miR-152 exerts its tumor suppressing effects by targeting the transcription factor Kruppel-like factor 4 (KLF4). This suppression of KLF4 causes the transcriptional downregulation of galectin-3 (LGALS3) and the inactivation of MEK1/2 and PI3K signaling [73]. However, it was not shown whether KLF4 acts through LGALS3 and/or MEK1/2 and PI3K signaling to promote invasiveness in GSCs.
6.3. miR-491
miR-491 is commonly deleted in many cancers, including GBM, producing two mature miRNAs: miR-491-5p and miR-491-3p. Both of these miRNAs are down-regulated in GBMs compared to normal brain tissue. miR-491-5p expression has been reported to reduce cell proliferation and invasion by targeting MMP9 [74]. miR-491-3p also reduces the invasiveness of GBM cells by targeting the mRNA of insulin-like growth factor binding protein 2 (IGFBP2). Therefore, the two products of the miR-491 gene act as tumor suppressors that inhibits GBM invasion.
6.4. miR-125b
Unlike the previously mentioned miRNAs, miR-125b acts as an oncogene instead of a tumor suppressor. It is commonly overexpressed in GSCs and its inhibition leads to suppression of proliferation and invasion of primary GSCs [75]. In GSCs, miR-125b confers resistance to the first-line chemotherapy drug TMZ [76]. Inhibition of miR-125b with shRNA has been shown to sensitize GSCs to TMZ by down-regulating MMP-2 and MMP-9 through PIAS3, an inhibitor of STAT3 signaling, in vitro and in vivo [77]. Other miRNAs have been reported to confer drug resistance. Therefore it is worth considering miRNAs, such as miR-125b, in the treatment of GBM.
6. Conclusion
For patients diagnosed with GBM, it is not a question of if the cancer will progress, but when it will. The current first-line treatment for GBM follows the Stupp protocol which consists of surgery to remove as much of the tumor as safely possible, followed by a combination of radiotherapy and TMZ treatment [5, 7]. TMZ is a drug that easily crosses the blood-brain barrier and had anti-tumor effects. The administration of TMZ in addition to radiotherapy has been reported in a phase 3 trial to improve the median survival from 12 months to <15 months. However, tumors still recur in all cases [26].
In addition to TMZ, other agents that have been approved for the treatment of cancer by the FDA include BCNU (Emcure), also known as carmustine, and bevacizumab. BCNU is another alkylating agent showing limited improvement in survival [78, 79]. Bevacizumab received accelerated approval in 2009 for GBM [80]; however, bevacizumab monotherapy has shown only modest benefits. One of the two Phase III studies that examined adding benvacizumab to the current Stupp protocol showed increased progression-free survival but no significant effect on overall survival [5, 26]. It is also worth mentioning that in addition to being used for first-line treatment, bevacizumab has shown promise in preventing GBM recurrence by improving the outcome of re-irradiation, likely because it reduces VEGF signaling and thus increases tumor sensitivity to radiotherapy [81, 82].
While the current standard first-line regiment has moderately prolonged survival, the problem of tumor recurrence has not been solved. Because GBM is so invasive, these cancer cells can move into normal brain tissue where they escape surgery and/or radiation therapy. Therefore, there is an urgent need for developing new treatment options that can suppress the invasiveness of GBM cells, and the first step is to gain a better understanding of the molecular pathways involved in mediating intrinsic and post-treatment invasion of GBM.
Highlights.
Glioblastoma (GBM) is the deadliest and most common brain malignancy in adults with a dismal median survival of about 14 months.
Because of its tendency to aggressively invade throughout the brain, surgical resection and localized radiotherapy cannot completely target all malignant GBM cells.
GBM invasion can be regulated by the PI3K/Akt, Wnt, sonic hedgehog-GLI1, and microRNAs.
Acknowledgments
This study was supported by the NIH grants 7R01NS087169-02 (to HWL), P30CA012197 (to Wake Forest University Comprehensive Cancer Center), and 5T32CA079448 (to HWL and RLC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
None
References
- 1.Ferluga S, Debinski W. Ephs and Ephrins in malignant gliomas. Growth factors (Chur, Switzerland) 2014;32:190–201. doi: 10.3109/08977194.2014.985787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biasoli D, Sobrinho MF, da Fonseca AC, de Matos DG, Romao L, de Moraes Maciel R, Rehen SK, Moura-Neto V, Borges HL, Lima FR. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis. 2014;3:e123. doi: 10.1038/oncsis.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang C, Rahimpour S, Yu AC, Lonser RR, Zhuang Z. Regulation and dysregulation of astrocyte activation and implications in tumor formation. Cellular and molecular life sciences : CMLS. 2013;70:4201–4211. doi: 10.1007/s00018-013-1274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-oncology. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA neurology. 2014;71:1437–1444. doi: 10.1001/jamaneurol.2014.1701. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. Journal of neuro-oncology. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 7.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 9.Wen PY, Lee EQ, Reardon DA, Ligon KL, Alfred Yung WK. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-oncology. 2012;14:819–829. doi: 10.1093/neuonc/nos117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agarwal S, Manchanda P, Vogelbaum MA, Ohlfest JR, Elmquist WF. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: findings in an orthotopic rat xenograft model of glioma. Drug metabolism and disposition: the biological fate of chemicals. 2013;41:33–39. doi: 10.1124/dmd.112.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou LC, Veeravagu A, Hsu AR, Tse VC. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurgical focus. 2006;20:E5. doi: 10.3171/foc.2006.20.4.2. [DOI] [PubMed] [Google Scholar]
- 13.Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nature reviews Neuroscience. 2014;15:455–465. doi: 10.1038/nrn3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 15.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. Journal of neuro-oncology. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 16.Tonn JC, Goldbrunner R. Mechanisms of glioma cell invasion. Acta neurochirurgica. Supplement. 2003;88:163–167. doi: 10.1007/978-3-7091-6090-9_22. [DOI] [PubMed] [Google Scholar]
- 17.Koul D, Parthasarathy R, Shen R, Davies MA, Jasser SA, Chintala SK, Rao JS, Sun Y, Benvenisite EN, Liu TJ, Yung WK. Suppression of matrix metalloproteinase-2 gene expression and invasion in human glioma cells by MMAC/PTEN. Oncogene. 2001;20:6669–6678. doi: 10.1038/sj.onc.1204799. [DOI] [PubMed] [Google Scholar]
- 18.Kubiatowski T, Jang T, Lachyankar MB, Salmonsen R, Nabi RR, Quesenberry PJ, Litofsky NS, Ross AH, Recht LD. Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. Journal of neurosurgery. 2001;95:480–488. doi: 10.3171/jns.2001.95.3.0480. [DOI] [PubMed] [Google Scholar]
- 19.Lee WS, Woo EY, Kwon J, Park MJ, Lee JS, Han YH, Bae IH. Bcl-w Enhances Mesenchymal Changes and Invasiveness of Glioblastoma Cells by Inducing Nuclear Accumulation of beta-Catenin. PloS one. 2013;8:e68030. doi: 10.1371/journal.pone.0068030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. The Journal of clinical investigation. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazar AP, Ahn RW, O’Halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Current pharmaceutical design. 2011;17:1970–1978. doi: 10.2174/138161211796718152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins mediate tumor metastasis. World journal of biological chemistry. 2013;4:91–101. doi: 10.4331/wjbc.v4.i4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer research. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 24.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS, Modrusan Z, Nacu S, Guerrero S, Edgar KA, Wallin JJ, Lamszus K, Westphal M, Heim S, James CD, VandenBerg SR, Costello JF, Moorefield S, Cowdrey CJ, Prados M, Phillips HS. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Xie Q, Mittal S, Berens ME. Targeting adaptive glioblastoma: an overview of proliferation and invasion. Neuro-oncology. 2014;16:1575–1584. doi: 10.1093/neuonc/nou147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dijksterhuis JP, Petersen J, Schulte G. WNT/Frizzled signalling: receptor-ligand selectivity with focus on FZD-G protein signalling and its physiological relevance: IUPHAR Review 3. British journal of pharmacology. 2014;171:1195–1209. doi: 10.1111/bph.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur N, Chettiar S, Rathod S, Rath P, Muzumdar D, Shaikh ML, Shiras A. Wnt3a mediated activation of Wnt/beta-catenin signaling promotes tumor progression in glioblastoma. Molecular and cellular neurosciences. 2013;54:44–57. doi: 10.1016/j.mcn.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Yan W, Zhang W, Chen L, You G, Bao Z, Wang Y, Wang H, Kang C, Jiang T. MiR-218 reverses high invasiveness of glioblastoma cells by targeting the oncogenic transcription factor LEF1. Oncology reports. 2012;28:1013–1021. doi: 10.3892/or.2012.1902. [DOI] [PubMed] [Google Scholar]
- 30.Nager M, Bhardwaj D, Canti C, Medina L, Nogues P, Herreros J. beta-Catenin Signalling in Glioblastoma Multiforme and Glioma-Initiating Cells. Chemotherapy research and practice. 2012;2012:192362. doi: 10.1155/2012/192362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamino M, Kishida M, Kibe T, Ikoma K, Iijima M, Hirano H, Tokudome M, Chen L, Koriyama C, Yamada K, Arita K, Kishida S. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer science. 2011;102:540–548. doi: 10.1111/j.1349-7006.2010.01815.x. [DOI] [PubMed] [Google Scholar]
- 32.Schulte G. International Union of Basic and Clinical Pharmacology. LXXX. The class Frizzled receptors. Pharmacological reviews. 2010;62:632–667. doi: 10.1124/pr.110.002931. [DOI] [PubMed] [Google Scholar]
- 33.Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 34.Hara K, Kageji T, Mizobuchi Y, Kitazato KT, Okazaki T, Fujihara T, Nakajima K, Mure H, Kuwayama K, Hara T, Nagahiro S. Blocking of the interaction between Wnt proteins and their co-receptors contributes to the anti-tumor effects of adenovirus-mediated DKK3 in glioblastoma. Cancer letters. 2015;356:496–505. doi: 10.1016/j.canlet.2014.09.045. [DOI] [PubMed] [Google Scholar]
- 35.Pu P, Zhang Z, Kang C, Jiang R, Jia Z, Wang G, Jiang H. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer gene therapy. 2009;16:351–361. doi: 10.1038/cgt.2008.78. [DOI] [PubMed] [Google Scholar]
- 36.Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K, Lu Z. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Molecular cell. 2009;36:547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim KH, Seol HJ, Kim EH, Rheey J, Jin HJ, Lee Y, Joo KM, Lee J, Nam DH. Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro-oncology. 2013;15:161–171. doi: 10.1093/neuonc/nos299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao X, Mi Y, Ma Y, Jin W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:11505–11511. doi: 10.1007/s13277-014-2466-z. [DOI] [PubMed] [Google Scholar]
- 39.Guo G, Kuai D, Cai S, Xue N, Liu Y, Hao J, Fan Y, Jin J, Mao X, Liu B, Zhong C, Zhang X, Yue Y, Liu X, Ma N, Guo Y. Knockdown of FRAT1 expression by RNA interference inhibits human glioblastoma cell growth, migration and invasion. PloS one. 2013;8:e61206. doi: 10.1371/journal.pone.0061206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kahn M. Can we safely target the WNT pathway? Nature reviews Drug discovery. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Robertis A, Valensin S, Rossi M, Tunici P, Verani M, De Rosa A, Giordano C, Varrone M, Nencini A, Pratelli C, Benicchi T, Bakker A, Hill J, Sangthongpitag K, Pendharkar V, Liu B, Ng FM, Then SW, Jing Tai S, Cheong SM, He X, Caricasole A, Salerno M. Identification and characterization of a small-molecule inhibitor of Wnt signaling in glioblastoma cells. Molecular cancer therapeutics. 2013;12:1180–1189. doi: 10.1158/1535-7163.MCT-12-1176-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO molecular medicine. 2014;6:1359–1370. doi: 10.15252/emmm.201302627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sami A, Karsy M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013;34:1991–2002. doi: 10.1007/s13277-013-0800-5. [DOI] [PubMed] [Google Scholar]
- 44.Lee HC, Park IC, Park MJ, An S, Woo SH, Jin HO, Chung HY, Lee SJ, Gwak HS, Hong YJ, Yoo DH, Rhee CH, Hong SI. Sulindac and its metabolites inhibit invasion of glioblastoma cells via down-regulation of Akt/PKB and MMP-2. Journal of cellular biochemistry. 2005;94:597–610. doi: 10.1002/jcb.20312. [DOI] [PubMed] [Google Scholar]
- 45.Cohen KJ, Gibbs IC, Fisher PG, Hayashi RJ, Macy ME, Gore L. A phase I trial of arsenic trioxide chemoradiotherapy for infiltrating astrocytomas of childhood. Neuro-oncology. 2013;15:783–787. doi: 10.1093/neuonc/not021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, Jovanovic B, McCarthy K, Raizer JJ. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. Journal of neuro-oncology. 2012;110:237–243. doi: 10.1007/s11060-012-0957-6. [DOI] [PubMed] [Google Scholar]
- 47.Gwak HS, Park MJ, Park IC, Woo SH, Jin HO, Rhee CH, Jung HW. Tetraarsenic oxide-induced inhibition of malignant glioma cell invasion in vitro via a decrease in matrix metalloproteinase secretion and protein kinase B phosphorylation. Journal of neurosurgery. 2014;121:1483–1491. doi: 10.3171/2014.8.JNS131991. [DOI] [PubMed] [Google Scholar]
- 48.Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, Kim YW, Liu JL, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira SM, Yung WK. Antitumor activity of NVP-BKM120–a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT, Prados MD, Haas-Kogan DA. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-oncology. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bigelow RL, Chari NS, Unden AB, Spurgers KB, Lee S, Roop DR, Toftgard R, McDonnell TJ. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. The Journal of biological chemistry. 2004;279:1197–1205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- 51.Carpenter RL, Lo HW. Identification, functional characterization, and pathobiological significance of GLI1 isoforms in human cancers. Vitam Horm. 2012;88:115–140. doi: 10.1016/B978-0-12-394622-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T, Isner JM. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nature medicine. 2001;7:706–711. doi: 10.1038/89083. [DOI] [PubMed] [Google Scholar]
- 53.Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. The Journal of biological chemistry. 2002;277:5548–5555. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 54.Zhu H, Lo HW. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Current genomics. 2010;11:238–245. doi: 10.2174/138920210791233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, Epstein E, McCormick F. A role of PDGFRalpha in basal cell carcinoma proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9255–9259. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choudhry Z, Rikani AA, Choudhry AM, Tariq S, Zakaria F, Asghar MW, Sarfraz MK, Haider K, Shafiq AA, Mobassarah NJ. Sonic hedgehog signalling pathway: a complex network. Annals of neurosciences. 2014;21:28–31. doi: 10.5214/ans.0972.7531.210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science (New York, N.Y.) 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 58.Cao X, Geradts J, Dewhirst MW, Lo HW. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene. 2012;31:104–115. doi: 10.1038/onc.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dikshit B, Irshad K, Madan E, Aggarwal N, Sarkar C, Chandra PS, Gupta DK, Chattopadhyay P, Sinha S, Chosdol K. FAT1 acts as an upstream regulator of oncogenic and inflammatory pathways, via PDCD4, in glioma cells. Oncogene. 2013;32:3798–3808. doi: 10.1038/onc.2012.393. [DOI] [PubMed] [Google Scholar]
- 60.Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69:6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu H, Carpenter RL, Han W, Lo HW. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014;343:51–61. doi: 10.1016/j.canlet.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie J. Hedgehog signaling pathway: development of antagonists for cancer therapy. Current oncology reports. 2008;10:107–113. doi: 10.1007/s11912-008-0018-7. [DOI] [PubMed] [Google Scholar]
- 63.Fu J, Rodova M, Nanta R, Meeker D, Van Veldhuizen PJ, Srivastava RK, Shankar S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro-oncology. 2013;15:691–706. doi: 10.1093/neuonc/not011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gruber Filbin M, Dabral SK, Pazyra-Murphy MF, Ramkissoon S, Kung AL, Pak E, Chung J, Theisen MA, Sun Y, Franchetti Y, Sun Y, Shulman DS, Redjal N, Tabak B, Beroukhim R, Wang Q, Zhao J, Dorsch M, Buonamici S, Ligon KL, Kelleher JF, Segal RA. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nature medicine. 2013;19:1518–1523. doi: 10.1038/nm.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao X, Jin W. The emerging role of tumor-suppressive microRNA-218 in targeting glioblastoma stemness. Cancer letters. 2014;353:25–31. doi: 10.1016/j.canlet.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 66.Song L, Huang Q, Chen K, Liu L, Lin C, Dai T, Yu C, Wu Z, Li J. miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-beta. Biochemical and biophysical research communications. 2010;402:135–140. doi: 10.1016/j.bbrc.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 67.Peng B, Li D, Qin M, Luo D, Zhang X, Zhao H, Hu S. MicroRNA218 inhibits glioma migration and invasion via inhibiting glioma-associated oncogene homolog 1 expression at N terminus. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:3831–3837. doi: 10.1007/s13277-013-1507-3. [DOI] [PubMed] [Google Scholar]
- 68.Jiang L, Wu J, Yang Y, Liu L, Song L, Li J, Li M. Bmi-1 promotes the aggressiveness of glioma via activating the NF-kappaB/MMP-9 signaling pathway. BMC cancer. 2012;12:406. doi: 10.1186/1471-2407-12-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tu Y, Gao X, Li G, Fu H, Cui D, Liu H, Jin W, Zhang Y. MicroRNA-218 inhibits glioma invasion, migration, proliferation, and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer research. 2013;73:6046–6055. doi: 10.1158/0008-5472.CAN-13-0358. [DOI] [PubMed] [Google Scholar]
- 70.Smits M, Nilsson J, Mir SE, van der Stoop PM, Hulleman E, Niers JM, de Witt Hamer PC, Marquez VE, Cloos J, Krichevsky AM, Noske DP, Tannous BA, Wurdinger T. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget. 2010;1:710–720. doi: 10.18632/oncotarget.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Visani M, de Biase D, Marucci G, Cerasoli S, Nigrisoli E, Bacchi Reggiani ML, Albani F, Baruzzi A, Pession A. Expression of 19 microRNAs in glioblastoma and comparison with other brain neoplasia of grades I-III. Molecular oncology. 2014;8:417–430. doi: 10.1016/j.molonc.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao YL, Ma J, Wang P, Xue YX, Li Z, Zhao LN, Li ZQ, Feng TD, Liu YH. miR-101 Acts as a Tumor Suppressor by Targeting Kruppel-like Factor 6 in Glioblastoma Stem Cells. CNS neuroscience & therapeutics. 2015;21:40–51. doi: 10.1111/cns.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mehran R, Nilsson M, Khajavi M, Du Z, Cascone T, Wu HK, Cortes A, Xu L, Zurita A, Schier R, Riedel B, El-Zein R, Heymach JV. Tumor endothelial markers define novel subsets of cancer-specific circulating endothelial cells associated with antitumor efficacy. Cancer Res. 2014;74:2731–2741. doi: 10.1158/0008-5472.CAN-13-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, Liu Y, Granberg KJ, Wang Q, Moore LM, Ji P, Gumin J, Sulman EP, Calin GA, Haapasalo H, Nykter M, Shmulevich I, Fuller GN, Lang FF, Zhang W. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene. 2014 doi: 10.1038/onc.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi L, Zhang J, Pan T, Zhou J, Gong W, Liu N, Fu Z, You Y. MiR-125b is critical for the suppression of human U251 glioma stem cell proliferation. Brain research. 2010;1312:120–126. doi: 10.1016/j.brainres.2009.11.056. [DOI] [PubMed] [Google Scholar]
- 76.Shi L, Zhang S, Feng K, Wu F, Wan Y, Wang Z, Zhang J, Wang Y, Yan W, Fu Z, You Y. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. International journal of oncology. 2012;40:119–129. doi: 10.3892/ijo.2011.1179. [DOI] [PubMed] [Google Scholar]
- 77.Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y. miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2014;28:41–54. doi: 10.1007/s40259-013-0053-2. [DOI] [PubMed] [Google Scholar]
- 78.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. Jama. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 79.Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jaaskelainen J, Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-oncology. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. The oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 81.Shapiro LQ, Beal K, Goenka A, Karimi S, Iwamoto FM, Yamada Y, Zhang Z, Lassman AB, Abrey LE, Gutin PH. Patterns of failure after concurrent bevacizumab and hypofractionated stereotactic radiation therapy for recurrent high-grade glioma. International journal of radiation oncology, biology, physics. 2013;85:636–642. doi: 10.1016/j.ijrobp.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 82.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma–are we there yet? Neuro-oncology. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]