

ABSTRACT
Bacillus thuringiensis (Bt) is armed to complete a full cycle in its insect host. During infection, virulence factors are expressed under the control of the quorum sensor PlcR to kill the host. After the host’s death, the quorum sensor NprR controls a necrotrophic lifestyle, allowing the vegetative cells to use the insect cadaver as a bioincubator and to survive. Only a part of the Bt population sporulates in the insect cadaver, and the precise composition of the whole population and its evolution over time are unknown. Using fluorescent reporters to record gene expression at the single-cell level, we have determined the differentiation course of a Bt population and explored the lineage existing among virulent, necrotrophic, and sporulating cells. The dynamics of cell differentiation were monitored during growth in homogenized medium, biofilm formation, and colonization of insect larvae. We demonstrated that in the insect host and in planktonic culture in rich medium, the virulence, necrotrophism, and sporulation regulators are successively activated in the same cell. In contrast, in biofilms, activation of PlcR is dispensable for NprR activation and we observed a greater heterogeneity than under the other two growth conditions. We also showed that sporulating cells arise almost exclusively from necrotrophic cells. In biofilm and in the insect cadaver, we identified an as-yet-uncharacterized category of cells that do not express any of the reporters used. Overall, we showed that PlcR, NprR, and Spo0A act as interconnected integrators to allow finely tuned adaptation of the pathogen to its environment.
IMPORTANCE
Bt is an entomopathogen found ubiquitously in the environment and is a widely used biopesticide. Studies performed at the population level suggest that the infection process of Bt includes three successive steps (virulence, necrotrophism, and sporulation) controlled by different regulators. This study aimed to determine how these phenotypes are activated at the cellular level and if they are switched on in all cells. We used an insect model of infection and biofilms to decipher the cellular differentiation of this bacterium under naturalistic conditions. Our study reveals the connection and lineage existing among virulent, necrotrophic, and sporulating cells. It also shows that the complex conditions encountered in biofilms and during infection generate great heterogeneity inside the population, which might reflect a bet-hedging strategy to ameliorate survival. These data generate new insights into the role of regulatory networks in the adaptation of a pathogen to its host.
INTRODUCTION
Depending on its environment, a clonal bacterial population can be composed of subpopulations of differentiated cells. Several studies have addressed the phenotypic heterogeneity of genetically identical cells within biofilms of the sporulating Gram-positive model organism Bacillus subtilis in which specialized cell types were identified (1–3). B. subtilis cells can differentiate into sporulating, competent, motile, or cannibal cells, as well as produce the lipopeptide surfactin, the biofilm matrix, or exoproteases (1, 2). These differentiations depend on the activity of key transcriptional regulators. The use of fluorescent tools and time-lapse video microscopy helped establish the lineage existing among these cell types in microcolonies or under biofilm conditions (1–3).
The Bacillus cereus group includes the three spore-forming pathogens Bacillus anthracis, B. cereus sensu stricto, and Bacillus thuringiensis (Bt), which are responsible for infections in mammals, humans, and insects, respectively. In addition, B. cereus biofilms are a recurrent problem in the food industry (4). Bacteria in this group and B. subtilis have a large number of transcriptional regulators in common, such as the master regulator of sporulation Spo0A and the transition state regulators SinR and AbrB (5). However, several important regulators, such as the master activators of virulence (PlcR) or necrotrophism (NprR), are specific to the B. cereus group (6, 7) and may reflect an adaptation of B. cereus to animal niches as opposed to plant and soil niches for B. subtilis (5).
In Bt, at least three quorum-sensing systems are involved in the infectious process in a susceptible host (8). The PlcR-PapR system controls the transcription of most of the virulence genes involved in the pathogenicity of Bt in insects infected via the oral route (9). The NprR-NprX system controls the expression of a set of genes encoding degradative enzymes and the machinery for the synthesis of the lipopeptide kurstakin required for survival of the bacterium in the insect cadaver (7, 10, 11). Finally, as demonstrated in B. anthracis, the Rap-Phr system controls the phosphorylation cascade leading to activation of the sporulation regulator Spo0A (12) and to the commitment of part of the bacterial population to sporulation. Interestingly, these three quorum-sensing systems are interconnected (Fig. 1A). The transcription of the nprR-nprX operon is stimulated by the PlcR-PapR complex (10), necrotrophism is linked to sporulation via the activation of nprX expression by Sigma H and Sigma E (10), NprR prevents sporulation by interacting with the phosphorelay (7, 13), and Spo0A-P stops the virulence process by repressing the transcription of plcR (14). Intriguingly, survival does not depend entirely on the sporulation process (11). In fact, analysis of a Bt population colonizing an insect cadaver over a period of 4 days indicated that only 30% of the cells were thermoresistant spores (11).
FIG 1.
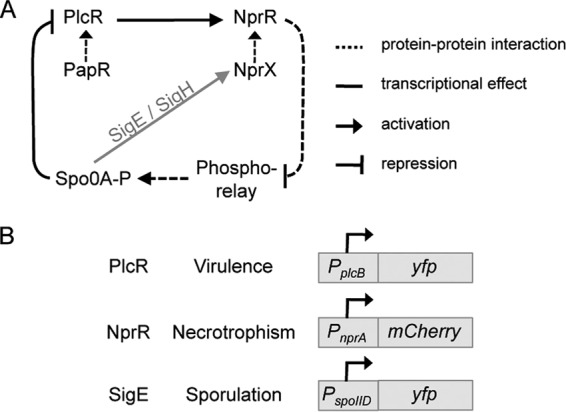
Regulation and monitoring of developmental pathways in Bt. (A) Schematic representation of the regulatory links among PlcR, NprR, and the sporulation regulators. Dashed lines indicate protein-protein or peptide-protein interactions, whereas solid lines indicate a transcriptional effect. Arrows and broken lines represent activation and repression, respectively. (B) Fluorescent reporter constructs used to monitor the activity of the regulators controlling differentiation in a virulent, a necrotrophic, or a sporulating cell type.
Although virulence, necrotrophism, and sporulation are described as being interconnected, the population colonizing an insect cadaver appeared heterogeneous, suggesting that these developmental programs are activated in different subpopulations. We hypothesized that several populations coexist throughout the process of host colonization. Our goal was to decipher the commitment to virulence, necrotrophism, or sporulation at the cell level to determine the composition of a Bt population during infection. We also sought to identify the subpopulation(s) that survived in the host without resorting to sporulation. To reach these objectives, we developed fluorescent reporters specific to the three states mentioned above. We dissected the activation kinetics of PlcR, NprR, and Spo0A in Bt during infection of Galleria mellonella larvae. The composition of the Bt population in the host was compared to that of cells grown in biofilms and in planktonic cultures. Using multifluorescent reporters, we established the lineage existing among cells in these three environments. We showed that necrotrophic cells can arise from either virulent or avirulent cells but, surprisingly, commitment to sporulation occurred almost exclusively in cells that went through a necrotrophic stage. Phenotypic variability in B. cereus populations is poorly documented. Few studies have addressed gene expression by using a single-cell approach in B. cereus populations grown under planktonic or biofilm conditions (15–17). Although numerous publications on genes induced in bacterial pathogens in vivo are available (18–20), only a few reports have described the course of gene expression at the cell level in a complex and dynamic environment such as an animal model of infection. In Vibrio cholerae, the expression pattern of virulence genes was investigated in the rabbit ileal loop model of infection (21), and in Yersinia pseudotuberculosis, the expression of virulence genes was monitored in a murine model of infection (22). To our knowledge, ours is the first study to address the question of the evolution of a bacterial population’s composition during host infection at the cellular level by using multiple developmental markers.
RESULTS
Monitoring of virulence, necrotrophism, and sporulation in Bt cells grown in homogenized medium.
Previous studies have reported that Bt successively displayed virulent and necrotrophic behavior at the population level and was eventually able to sporulate in an insect host (8, 9, 11). PlcR-PapR, NprR-NprX, and the sporulation phosphorelay control these developmental pathways and are linked by transcriptional and posttranscriptional activation or repression (Fig. 1A) (10, 14). To investigate the developmental history of these three cell types, we monitored the expression of genes reflecting the activity of their regulators and commitment to one of these pathways. We fused the promoters of genes controlled by PlcR, NprR, or the sigma factor SigE, active only when cells are irreversibly engaged in sporulation (23), with reporter genes coding for spectrally distinct fluorescent proteins (Fig. 1B). Yellow fluorescent protein (YFP) fluorescence driven by the promoter of the phospholipase C gene (plcB) allowed us to detect the virulent cell type controlled by the PlcR-PapR system (24). mCherry fluorescence controlled by the nprA promoter identified necrotrophic behavior switched on by the transcriptional activity of NprR upon NprX binding (7). Fusion of the spoIID promoter and the yfp gene allowed us to monitor SigE activity and commitment to sporulation (23, 25). Cells in which the PlcR-, NprR-, or SigE-driven promoter is active are referred to here as Vir+, Nec+, or Spo+ cells, respectively. Low-copy-number plasmid pHT304-18 was used to carry transcriptional reporter constructs. This plasmid is stable under the conditions used in this study. The rationale for the use of a plasmid as opposed to chromosomal fusions and for the selection of the best promoter-reporter gene fusions is explained in more detail in the supplemental material (see Fig. S1A and B and Table S1). We also monitored the decay of YFP- and mCherry-dependent fluorescence and showed that its half-life was about 20 and >50 h, respectively, under the conditions tested (see the supplemental material). In view of these technical parameters, we decided to consider throughout this study an on-versus-off state without taking into account the fluorescence intensity and we kept in mind that fluorescent cells were either expressing the fusion or had expressed it.
We first evaluated the timing of the activation of each of the three regulation systems at the cellular level and the size of the population committed to virulence, necrotrophism, or sporulation under standard laboratory conditions such as homogenized liquid medium. plcB-yfp-, nprA-mCherry-, and spoIID-yfp-expressing Bt strains (see Table S3 in the supplemental material) were grown in a rich medium (lysogeny broth [LB] containing Bacto peptone [LBP]), and samples were harvested during growth. Flow cytometry was used to quantify the plcB-, nprA- and spoIID-expressing cells (Fig. 2). Data indicated that plcB expression was induced 1 h before the onset of the stationary phase (T−1), as the first cells expressing the reporter appeared then (Fig. 2A). Our very stringent method of quantifying the number of cells expressing the fluorescent reporter (see the supplemental material) indicated that more than 85% of the cells would express plcB over the duration of the experiment (Fig. 2A). However, analysis of the histograms over time identified only one population of Vir+ cells and the overall behavior of the Bt population regarding the virulence phenotype seemed unimodal. The necrotrophic reporter gene nprA is expressed from T2 in the majority of the cells (to reach 89.5%, on average, at T5), but in contrast to the virulence marker, a few cells remained clearly Nec− at T2 and T5 and presented the same negative signal as the reporterless wild-type strain (Fig. 2B). Flow cytometric analysis of the spoIID-yfp-expressing Bt strain indicated bimodal behavior of the population regarding sporulation, as a subpopulation of Spo+ cells was identified from T4 (shoulder in Fig. 2C). The size of this subpopulation grew over time, as indicated by the presence of two peaks at T5 and later, to reach about 50% at T7 (Fig. 2C).
FIG 2.

plcB, nprA, and spoIID promoter activities in cells growing under homogeneous growth conditions. Flow cytometric analyses of plcB-yfp (A)-, nprA-mCherry (B)-, and spoIID-yfp (C)-expressing Bt cells are shown. Samples were harvested at the time points indicated. Each time point is associated with a unique color and can be identified by using the time map. The gray area is the fluorescence obtained with control cells with no reporter at T0. On the x axis of each histogram is fluorescence intensity in arbitrary units (A.U.) on a logarithmic scale. On the y axis is the cell count. Each histogram represents at least three independent experiments. Analyses of cells harboring the necrotrophic reporter and the virulence (D) or sporulation marker (E) are also shown. Samples of plcB-yfp nprA-mCherry- and spoIID-yfp nprA-mCherry-expressing Bt strains were harvested at the time points indicated. The percentage of the total cell count represented by each population discriminated in cytograms is presented as a function of time. Each population phenotype is associated with a color. Vir, cells expressing the virulence marker; Nec, cells expressing the necrotrophic marker; Vir/Nec, cells expressing the virulence and necrotrophic markers; Spo/Nec, cells expressing the sporulation and necrotrophic markers; Other, cells that do not express any of the markers used. Cells were grown in LBP under agitation at 30°C. Time zero was defined as the onset of stationary phase. The results are mean values from three independent experiments (error bars show the standard deviation from the mean).
The relationship between the regulators was also evaluated by examining the necrotrophic marker and the virulence or sporulation reporters at the same time in Bt strains carrying two fluorescent transcriptional fusions. The plcB-yfp nprA-mCherry- and spoIID-yfp nprA-mCherry-expressing Bt strains (Table S3) were grown under conditions identical to those described above, and the expression of YFP and mCherry in the samples was determined by flow cytometry. The different populations present in the samples were identified in cytograms obtained with the double-labeled strains by using the reporterless strain signal as a negative control. As expected, activation of the virulence regulator (from T−1 in Fig. 2D) preceded activation of the necrotrophic regulator (from T1 in Fig. 2D). Analysis of the cytograms using the 99% division line (as described in the supplemental material) indicates that about 85% of the total population was Nec+ Vir+ at T2 and about 8% was Nec+ Vir− (Fig. 2D; see quadrant 2 [Q2] in Fig. S2A in the supplemental material). However, the analysis of the cytograms (see Q1 and Q2 in Fig. S2A) shows that these two populations belong to the same cloud of cells, suggesting that all of the Nec+ cells emerged from Vir+ cells. Indeed, low fluorescent signal levels such as those obtained with the plcB-yfp reporter fusion might result in the biased detection of Vir− cells by cytometry (Fig. 2D; see Fig. S2A). Therefore, these data suggest that all of the cells committing to a necrotrophic life style differentiated from the virulent population. Analysis of the coexpression of the necrotrophic and sporulation reporters shows that the expression of nprA preceded the expression of spoIID in the same cell (Fig. 2E; see Fig. S2B). These data also show that commitment to sporulation occurred only in cells expressing the necrotrophic reporter gene first (Fig. 2E; see Fig. S2B; note the movement of Nec+ cells [Q1] to Nec+ Spo+ individuals [Q2] in the cytograms). No Spo+-only cells were identified (see Q4 in Fig. S2B).
The activation of SigE in the mother cell compartment of nprA-expressing cells was further visualized by time-lapse microscopy. The spoIID-yfp nprA-mCherry-expressing Bt strain was allowed to develop in a microcolony under the microscope. Protocols previously published for B. subtilis (26, 27) were not adapted to Bt, as also observed by Eijlander and Kuipers (16). For this reason, we set up a protocol in which the cells were grown on a solid medium composed of polyacrylamide mixed with LBP. Polyacrylamide offered the advantage of favoring growth in a monolayer. We used the same medium as in agitated liquid culture, but to promote sporulation while limiting the size of the microcolony, we diluted the LBP 100-fold. Our observations indicated that activation of NprR preceded activation of the sporulation regulator (Fig. 3; see Movie S1 in the supplemental material). We also observed that all of the cells in the microcolony expressed nprA during stationary phase and that expression of spoIID occurred in 83% of these cells (Fig. 3; see Movie S1). The level of expression of the two reporters was heterogeneous inside the cells in the microcolony, and this might be attributable to the use of a plasmid reporter construct.
FIG 3.

nprA and spoIID promoter activities in cells developing in a microcolony. Cells of the spoIID-yfp nprA-mCherry-expressing Bt strain were allowed to grow at 30°C on a solid medium containing LBP diluted 1:100, and mCherry and YFP expression was monitored by time-lapse microscopy. The fluorescent mCherry and YFP signals were false colored red and green, respectively. Snapshots are taken from Movie S1 in the supplemental material. Time zero is the first frame of the movie. The numbers in the left panels are frame numbers. Merge, merged image of the signals in the fluorescent channels and the phase-contrast image; nprA, mCherry fluorescence associated with the activity of the nprA promoter; spoIID, YFP fluorescence associated with the activity of the spoIID promoter; overlay, overlay of the signals in both fluorescent channels. Bars, 10 µm.
Altogether, our observations indicate that bacteria growing in rich medium sequentially activate PlcR, NprR, and SigE in the same cell. This correlates with the timing of expression obtained with the lacZ reporter gene in whole populations (10, 25, 28). We also observed that only a part of the population differentiated into spore formers that originated exclusively from Nec+ cells. Time-lapse observations of cells growing in a microcolony further confirmed these observations even when the growth environment was not homogeneous.
Differentiation of Bt cells in a biofilm.
Biofilms are complex communities where cells can undergo various developmental pathways (3, 29) and in which cell-cell signaling has been shown to promote cellular differentiation (30, 31). The relative immobility of the cells embedded in the exopolymer matrix and the diffusion properties of molecules may trigger preferential and localized differentiation (32). We assessed the composition of a Bt biofilm developing in glass tubes at the air-liquid interface of LBP, the same rich medium as above, which promotes biofilm formation in static cultures (33). This biofilm development is illustrated in Fig. S3 in the supplemental material. The expression of the virulence reporter gene was highly variable in 24-h-old biofilms (error bar in Fig. 4D). However, the kinetics of plcB expression was the same in all experiments. The highest number of Vir+ cells according to the 99% division line method was found in 36-h-old biofilms (Fig. 4D). About 35% of the cells expressed the virulence reporter in 24- and 36-h-old biofilms (Fig. 4A and D; see Fig. S4A). This Vir+ population remained detectable in older biofilms (on average, 25 and 16% of the total population at 48 and 72 h postinoculation, respectively) (Fig. 4A and D; see microscopic images in Fig. S4A). The overall profile of the histograms shows a unimodal albeit broadly heterogeneous expression pattern, except in 24-h-old biofilms, where it is clearly bimodal (Fig. 4A). Cytograms also showed this bimodality, notably at 24 and 36 h (see Fig. S4A in the supplemental material). Bimodality of necrotrophic behavior and sporulation was observed throughout the experiment. A subpopulation of Nec+ cells is present at all of the time points examined (Fig. 4B, D, and E; see Fig. S4). The size of this subpopulation grew over time and reached 74%, on average, in 72-h-old biofilms. A subpopulation of Spo+ cells was also identified at all times (Fig. 4C and E; see Fig. S4B) but represented only 15% of the cells.
FIG 4.

plcB, nprA, and spoIID promoter activities in cells isolated from a biofilm grown in LBP. Shown are flow cytometric analyses of plcB-yfp (A)-, nprA-mCherry (B)-, and spoIID-yfp (C)-expressing Bt cells isolated from a biofilm grown in LBP at the air-liquid interface in glass tubes. Biofilms were allowed to develop at 30°C for 72 h, and samples were harvested at the time points indicated after LBP inoculation. Histograms are representative of at least three independent experiments. A.U., arbitrary units. Also shown are flow cytometric analyses of plcB-yfp nprA-mCherry (D)- and spoIID-yfp nprA-mCherry (E)-expressing Bt cells harvested at the time points indicated. For full descriptions of the histograms, graphs, and color code used, see the legend to Fig. 2. The results are mean values from three independent experiments (error bars show the standard deviation from the mean).
These results showed that, in biofilms, a high level of heterogeneity is reached, as all four cell types (virulent, necrotrophic, virulent/necrotrophic, and necrotrophic/sporulating) were identified (Fig. 4; see Fig. S4). However, it is worth noting that a significant part of the bacterial population, about 25 to 50%, depending on the time of sampling, did not express any reporter genes, suggesting the presence of cells at another physiological stage (Fig. 4; see Fig. S4). Our results also indicate that under biofilm-promoting growth conditions, the three regulators are activated in a smaller part of the population than under homogenized growth conditions. In addition, results obtained with the multifluorescent strains indicate that in young biofilms necrotrophic cells arose mainly from the virulent population. However, a small Nec+ Vir− subpopulation seems to emerge in 24- and 36-h-old plcB-yfp nprA-mCherry-expressing Bt strain biofilms (cells circled in Q1 and identified by an arrow in the microscopic images in Fig. S4A). This is different from the results obtained with homogenized medium, where all of the Nec+ cells emerged from the same cloud of Vir+ cells (see Fig. S2A). Later, the necrotrophic population will differentiate from the nonidentified cell type (see the increase in Nec+ cells in Q1 and the decrease in cells in Q3 between 48- and 72-h-old biofilms in Fig. S4A, as well as the constant decrease in the Vir+ population after 36 h in Fig. 4D). It has been previously reported that the activity of PlcR is not required for NprR activation in homogenized growth cultures in a sporulation-promoting medium (10). In contrast, commitment to sporulation was observed almost exclusively in cells previously differentiated as necrotrophic (Fig. 4E; see Fig. S4B).
Differentiation of Bt cells in their insect host.
Previous studies carried out at the population scale found that genes belonging to the PlcR and NprR regulons are sequentially activated during the infectious cycle and that nonsporulating cells coexist with thermoresistant spores in the insect cadaver (8, 10, 11). Here we used the G. mellonella insect model to determine, for the first time at the single-cell level, the activation process of PlcR, NprR, and SigE during infection. G. mellonella larvae were infected by intrahemocoelic injection with a plcB-yfp nprA-mCherry- or spoIID-yfp nprA-mCherry-expressing Bt strain, as well as with strains harboring the corresponding single transcriptional fusions. Bacteria were isolated from larvae sacrificed over a period of 72 h. Analysis of plcB expression identified one population of Vir+ cells at 18 and 24 h postinjection (Fig. 5A), with the highest fluorescence intensity of the virulence reporter at the 18-h time point. The low plcB-driven YFP signal overlapped the control, and while the expression of the virulence reporter seemed unimodal in the population, we cannot exclude the possibility that a part of the population remained Vir− (histogram overlaps in Fig. 5A; see the cells localized in Q3 of Fig. S5A in the supplemental material). Indeed, a part of the population of the Bt plcB-yfp strain (12%, on average, in samples harvested at 18 h postinjection) gave the same fluorescent signal as the unmarked strain used as a control. We noted that the background signal of the reporterless strain decreased in cells isolated from cadavers starting at the 40-h time point (dark gray peak in Fig. 5A). The shorter length of the bacteria in these samples (see the microscopic images in Fig. S5A) may explain this shift. Indeed, at the same fluorescence signal level per volume unit, a bigger cell will give a stronger fluorescence signal than a smaller cell. At these time points, the fluorescence signals of the plcB-yfp-expressing Bt strain correspond to the background fluorescence intensity (Fig. 5A). A minor subpopulation of necrotrophic cells appeared at 18 h postinjection (stretch on the right side of the curve in Fig. 5B). The two populations were clearly identified starting at 24 h postinjection on the histograms (Fig. 5B). From 16 to 37% of the cells, depending on the time point, remained Nec−, indicating that NprR was not active in all of the bacteria present in the insect cadaver. Cells expressing the sporulation marker, as well as spores, were identified from 40 h postinjection (Fig. 5C; see microscopic images in Fig. S5B). This subpopulation peaked at 40 and 48 h postinjection and constituted 55% of the population (Fig. 5C). Coexpression analysis of the different markers indicated that Nec+ cells of the plcB-yfp nprA-mCherry-expressing Bt strain identified within the first 24 h postinjection were also Vir+ (Fig. 5D; see Fig. S5A). Data obtained with the spoIID-yfp nprA-mCherry-expressing Bt samples indicate that sporulating cells differentiated almost exclusively from cells expressing the necrotrophic marker (Fig. 5E; see Fig. S5B). At 48 h postinjection, the population of Nec+ Spo+ cells represented about 50% of the total population, whereas about 30% of the cells were Nec+ only. Taken together, analyses of the samples isolated from an insect cadaver suggest that sequential activation of PlcR, NprR, and SigE also occurs in the insect in the same cell. However, not all Vir+ cells become Nec+ and not all Nec+ cells become Spo+. Cells differentiating into a necrotrophic lifestyle coexist with cells committed to sporulation, as well as cells having already completed the sporulation pathway (see the presence in the same field of necrotrophic Nec+ cells, Nec+ Spo+ cells, and spores from 40 h postinjection on in Fig. S5B in the supplemental material). As in a biofilm, at least another physiological cell type was present and was represented by the population of cells that did not express any reporter gene.
FIG 5.

plcB, nprA, and spoIID promoter activities in cells isolated from an insect cadaver. Shown are flow cytometric analyses of plcB-yfp (A)-, nprA-mCherry (B)-, and spoIID-yfp (C)-expressing Bt cells. Bacteria were isolated from cadavers of G. mellonella larvae infected by intrahemocoelic injection and incubated at 30°C. Samples were harvested 18, 24, 40, 48, 66, and 72 h after injection. Peaks in light and dark gray are the fluorescence levels obtained with control cells with no reporter harvested at 18 and 48 h postinjection, respectively. Histograms are representative of at least three independent experiments. A.U., arbitrary units. Also shown are analyses of plcB-yfp nprA-mCherry (D)- and spoIID-yfp nprA-mCherry (E)-expressing Bt cells harvested from infected G. mellonella larvae at the time points indicated. For full descriptions of the histograms, graphs, and color code used, see the legend to Fig. 2. The results are mean values from three independent experiments (error bars show the standard deviation from the mean).
DISCUSSION
Our work presents the activation patterns of the PlcR, NprR, and SigE regulators characterizing the virulent, necrotrophic, and sporulation states, respectively, in three different environments at the cell level. We notably established the course of differentiation of a Bt population and determined the lineage existing among the subpopulations emerging throughout the infectious process. We compared these results to two standard laboratory conditions, a homogenized liquid culture and biofilm growth.
These results allowed us to refine the model of regulator activation during infection proposed in previous studies (10, 11). We also propose a model of regulator activation at the cell level in an air-liquid interface biofilm and in homogenized medium (Fig. 6). Monitoring of the activities of PlcR, NprR, and SigE in cells grown under the latter conditions revealed that the three regulators were successively activated in the cells (Fig. 1 and 6A). The linear and sequential activation of the three regulators reflects the homogeneity of the medium in which the cells grow when cultivated in shaken flasks. However, while the activation of PlcR was unimodal, we observed two populations regarding the commitment to sporulation and, to a lesser extent, the activation of NprR. Bimodality in sporulation might reflect the complex activation mode of Spo0A-P, which depends on quorum sensing, on the phosphorelay, on autoregulation, and on the activity of several regulators, as described in B. subtilis (34–36). Interestingly, we observed that activation of the SigE-dependent promoter was heterogeneous in a microcolony and did not arise in foci, which could have been expected since quorum sensing is involved in the regulation of sporulation.
FIG 6.
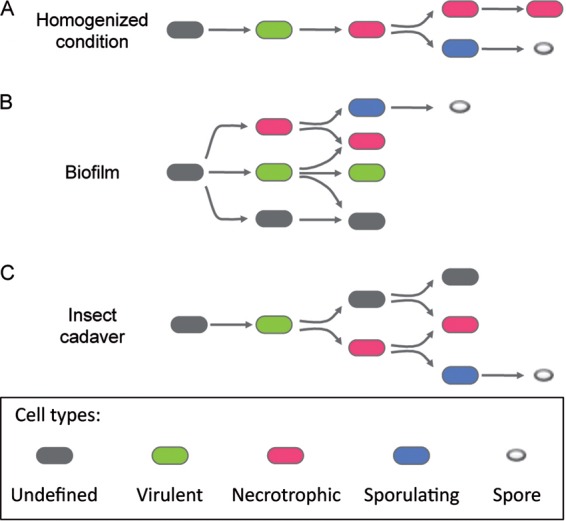
Model of Bt cellular differentiation. The differentiation pathways of Bt growing as planktonic cells (A), in a biofilm in LBP (B), or in an insect cadaver (C) are presented in this schematic. Green ovals represent cells in a virulent state. Red ovals represent cells displaying a necrotrophic life style. Blue ovals represent cells committed to sporulation. Gray ovals represent cells that do not fit into the three differentiated stages mentioned above.
NprR activity is not affected by entry into sporulation (7). However, commitment to sporulation depends on the activity of NprR as a transcriptional activator. This suggests that the level of Spo0A-P required for a cell to sporulate is reached only in a necrotrophic bacterium. In an nprR-nprX mutant strain, the level of sporulation is lower than in the wild-type strain in an insect cadaver but also in LB (11, 37). This is in agreement with the presumed role of NprR-NprX in the modulation of the Spo0A phosphorylation rate (13, 37).
Growth in a biofilm offered the highest level of heterogeneity, as at least five subpopulations (virulent, virulent/necrotrophic, necrotrophic, necrotrophic/sporulating, and an undefined physiological stage) were identified at the same time (Fig. 6B). In sharp contrast, PlcR, NprR, and SigE were successively activated in the same cell when bacteria were grown in shaken cultures. Our results suggest that in biofilm, the activity of these regulators is concomitant and happens in different subpopulations.
The activation of PlcR, NprR, and SigE (through the modulation of phosphorylation of Spo0A via the Rap phosphatases) depends on the interaction of the regulators with cognate peptides that are secreted by the bacteria and reimported into the cytoplasm of the cell. Poor dispersal of the signaling molecules that are exported into the surrounding medium because of the presence of a matrix might create local conditions favorable for the fast but confined activation of a regulator, thereby promoting heterogeneity within the population as a whole. This contrasts with the shaken condition, in which secreted peptides are homogeneously distributed in the medium and are able to elicit a more synchronous response. However, the spatial repartition of active regulators was not investigated here. This additional information would bring valuable clues to the effect of biofilm architecture on cell fate regarding the three differentiation states studied here.
Monitoring of the physiological stages of Bt during the infection process in an insect larva showed that activation of the virulence regulator PlcR occurred in the majority of the cells constituting the population isolated shortly after the death of the larva (Fig. 6C). In contrast, commitment to a necrotrophic lifestyle was observed in only a part of the virulent population (Fig. 6C). Activation of NprR was not required in all cells for the whole population to survive. This suggests that the Nec+ population that produces the survival factors shares them with the community, allowing the non-NprR-activated population to benefit from them. A ΔnprR-nprX mutant strain is unable to survive in the insect cadaver, but coculture of a ΔnprR-nprX mutant with the wild-type strain allowed the mutant strain to survive (11). It should be noted that the NprR regulon contains many secreted molecules such as degradative enzymes and the lipopeptide kurstakin, which might be shared inside the community (11). Differentiation into a spore takes place almost exclusively in necrotrophic cells, similarly to what we observed when cells grew in a biofilm and homogenized medium (Fig. 6C). Finally, we showed that in the late stages of infection, necrotrophic cells, cells undergoing sporulation, and an undefined cell type coexisted with thermoresistant spores (see the microscopic images in Fig. S5 in the supplemental material).
Very few studies have addressed the in vivo expression of multiple genes by pathogenic bacteria by using fluorescent reporters, mainly because of technical problems such as the fluorescent background coming from host cells or tissue debris. To circumvent this problem, the bacteria were isolated from insect cell debris. However, this procedure resulted in loss of the spatial dimension of regulator activation inside the insect body. Addressing the localization of the different subpopulations will help us understand if physicochemical constraints such as limited access to oxygen for cells localizing far from the insect cuticle has an influence on their differentiation process.
This study presents the first data on the cell fate of a pathogen obtained by using multiple developmental markers under different conditions, including infection. The results indicate that differentiation during the stationary phase is dynamic throughout development in the insect and during biofilm formation. We still need to understand in which physiological state cells that do not express any of the reporters used are. Understanding the regulation network controlling virulence but also the mechanisms allowing persistence of bacteria and its forms of dissemination is an important issue for the basic knowledge of Bt. These data may also be a source of information for the improvement of Bt as a biopesticide and also for a better understanding of the persistence of B. cereus group food-borne pathogens.
MATERIALS AND METHODS
Bacterial strains and general growth conditions.
Cry− Bt strain 407 is an acrystalliferous strain cured of its cry plasmid (38). Escherichia coli K-12 strain DH5α was used as the host for plasmid construction and cloning experiments. Plasmid DNA for Bt electroporation was prepared from Dam− Dcm− E. coli strain ET12567 (Stratagene, La Jolla, CA). E. coli cells were chemically transformed as previously described (39). Bt cells were transformed by electroporation as previously described (38, 39). For routine growth, E. coli and Bt strains were propagated in LB at 37 and 30°C, respectively. Growth kinetics in liquid culture were monitored in LBP (33).
The antibiotics were used for bacterial selection were ampicillin at 100 µg/ml for E. coli and spectinomycin at 300 µg/ml and erythromycin at 10 µg/ml for Bt.
DNA manipulations.
Chromosomal DNA was extracted from Bt cells with Puregene Yeast/Bact kit B (Qiagen, France). Plasmid DNA was extracted from E. coli by a standard alkaline lysis procedure with QIAprep spin columns (Qiagen, France). PCR-amplified fragments and digested fragments separated on 0.8% agarose gels were purified with kits from Qiagen (France). Restriction enzymes, T4 DNA ligase, and Taq or Phusion high-fidelity polymerase (New England Biolabs, France) were used in accordance with the manufacturer’s recommendations. Oligonucleotides (see Table S2 in the supplemental material) were synthesized by Sigma-Proligo (Paris, France) or Eurofins-MWG (Paris, France). PCRs were performed in a Applied Biosystems 2720 Thermak thermocycler (ABI). Nucleotide sequences of all constructs were determined by Beckman Coulter Genomics (Takeley, United Kingdom).
Design of the mCherryLGC protein.
The mCherry fluorescent protein (DNA coding sequence present in pAH9 [40] and derived from pRSET-mCherry [41]) did not yield enough fluorescence for our purpose. We designed a protein optimized for Bt. The DNA sequence coding for the protein was synthesized by Eurofins (France) and delivered cloned into a pEX-A backbone. The plasmid was named pEX-mCherryLGC. For optimization details, see the supplemental material.
Plasmid constructions.
All of the plasmids constructed for this study are listed in Table S3. For details of plasmid construction, see the supplemental material.
In vivo experiments.
Intrahemocoelic injection experiments with G. mellonella were carried out as described previously (9). For each strain, each larva was injected with 2 × 104 bacteria and kept at 30°C for 72 h. At 18 h after injection, surviving insects were eliminated. At each time point, Bt cells were harvested from dead insects as follows. A larva was cut open and transferred to a 1.5-ml Eppendorf tube containing 1 ml of phosphate-buffered saline (PBS). After vigorous shaking, cells were fixed for 7 min in PBS–4% formaldehyde and then washed in PBS. The suspension was then loaded onto a cotton filter set in a 1-ml syringe in order to isolate bacterial cells from cadaver debris. Cells were then concentrated by centrifugation, resuspended in GTE buffer (3), and kept at 4°C until flow cytometric analysis or microscopy.
Biofilm assays.
Biofilm formation at the air-liquid interface in a glass tube was carried out in LBP as described in reference 17, as were cell harvesting and sample fixation. Prior to flow cytometric analysis and for further disruption of aggregates and chains into single cells, the cell suspension was sonicated with a Branson Sonifier 250. The parameters applied were two rounds of 12 1-s pulses (each pulse at a 10% duty cycle) at 20% power (protocol adapted in our lab from reference 42).
Flow cytometric analysis.
For flow cytometric analysis, cells were diluted in filter-sterilized PBS to remove particles. Fluorescence was measured on a CyFlow Space cytometer (Partec, France). For details of the parameters used to collect fluorescence and the software used to analyze data, see the supplemental material. The subpopulations were identified by using histograms or cytograms. YFP- or mCherry-expressing cells were identified as cells giving a higher signal intensity than the reporterless cells used as a control. Data were analyzed by using statistical tables that indicate the percentages of fluorescent cells determined by each detector.
Microscopy.
Cells were observed with a Zeiss AxioObserver.Z1 inverted fluorescence microscope equipped with a Zeiss AxioCam MRm digital camera and Zeiss fluorescence filters. For the filters used to image fluorescence, see the supplemental material. Images were processed with the Zeiss ZEN software package. Wild-type cells containing no fluorescent fusions were analyzed under each culture condition (liquid culture, biofilm, or insect cadaver) to determine the background fluorescence of YFP or mCherry and the optimal exposure time.
Slides for time-lapse microscopy were prepared according to the protocols described in references 16 and 27, with some adjustments. An LBP-polyacrylamide solution (1% LBP, 10% polyacrylamide solution [acrylamide/bisacrylamide ratio, 37.5:1], 0.1% ammonium persulfate, 2 µl of N,N,N′,N′-tetramethylethylenediamine [TEMED]) was used to prepare the solid medium. According to the previously mentioned protocol, a 125-µl GeneFrame (Thermo Scientific ABgene) was filled with the LBP-polyacrylamide solution. After polymerization, 1.5-mm-wide strips were cut and washed four times for 30 min in LBP diluted 1:100. Two strips were then symmetrically placed in a gene frame in the first and third quarters of the width of the frame. A 2-µl volume of an exponentially growing cell suspension (optical density at 600 nm of 1) diluted 20× were pipetted onto the strips before the frame was sealed with a coverslip. Bt cell development at 30°C in a temperature-controlled chamber was monitored with the microscope described above. Phase-contrast and fluorescence images were taken every 15 min for 20 h.
Nucleotide sequence accession number.
The DNA sequence of the codon-optimized mCherryLGC gene has been submitted to GenBank and assigned accession number KJ547675.
SUPPLEMENTAL MATERIAL
Expression kinetics of nprA with mCherry or lacZ as a reporter. Comparison of the plasmid and chromosomal PnprA-mCherry transcriptional fusions by flow cytometry (A). Samples of cells carrying transcriptional fusions on a plasmid (green) or at the α-amylase locus (pink) were harvested at T0 (dotted lines), T2 (dashed lines), and T4 (solid lines). The gray area is the fluorescence level obtained with control cells with no reporter at T0. On the x axis of each histogram is the fluorescence intensity in arbitrary units (A.U.) on a logarithmic scale. On the y axis is the cell count. At least 40,000 cells were counted. Microscopy analysis of the plasmid or chromosomal PnprA-mCherry transcriptional fusions (B). Fluorescent signals (false colored red) obtained with cells grown in LBP at 30°C carrying the PnprA-mCherry reporter plasmid (top) were compared with signals obtained with cells harboring the chromosomal construct (bottom). Membranes were stained with FM4-64 (light blue). Time zero was defined as the onset of stationary phase. Bars, 5 µm. (C) nprA promoter activity in LBP with lacZ as a reporter gene. pHT304-18ΩPnprA-lacZ-carrying Bt cells grown in LBP at 30°C under shaking were harvested, and their β-galactosidase activities were measured as previously described (2). The activity of the nprA promoter was detected approximately 1.5 h after entry into stationary phase, similarly to the expression kinetics obtained with the mCherry reporter. These results validated codon-optimized mCherry as an efficient reporter of gene expression in Bt. Growth is indicated by gray squares, and promoter activity is indicated by black diamonds (error bars are standard deviations of the mean values calculated from two independent experiments). Time zero was defined as the onset of stationary phase (i.e., the time at which the slope of the growth curve decreases). OD600nm, optical density at 600 nm. Download
Cytograms and microscopic images of the plcB-yfp nprA-mCherry (A)- and spoIID-yfp nprA-mCherry (B)-expressing Bt strains grown in LBP at 30°C. Samples were harvested and analyzed at the time points indicated. Time zero (T0) was defined as entry into stationary phase. On cytograms, YFP fluorescence intensity reflecting the expression of plcB (A) or spoIID (B) is on the x axis and mCherry fluorescence intensity, reflecting the expression of nprA, is on the y axis. The cytograms presented represent at least three independent experiments. The microscopic images obtained with the mCherry and YFP filters are in black and white. On the overlay images, mCherry fluorescence is false colored red and YFP fluorescence is false colored green. Cells expressing both mCherry and YFP are various shades of yellow, depending on the intensity of mCherry and YFP fluorescence. Download
Evolution of the biomass (ring and pellicle) of Bt strain 407 growing at the air-liquid interface of LBP in glass tubes at 30°C. Time zero is the time of inoculation. Pictures of the corresponding biofilms are presented. Download
Cytograms and microscopic images of dual-reporter plcB-yfp nprA-mCherry (A) and spoIID-yfp nprA-mCherry (B) Bt strains grown in a biofilm at the air-liquid interface of LBP in glass tubes. Cells were harvested at the time points indicated. Time zero is the time of medium inoculation. A description of the graphs is presented in the legend to Fig. S2. Arrows point to cells expressing mCherry only. Circled cells are Nec+ cells that did not emerge from a Vir+ population. Download
Cytograms and fluorescence microscopic images of dual-reporter plcB-yfp nprA-mCherry (A)- and spoIID-yfp nprA-mCherry (B)-expressing Bt strains isolated from G. mellonella larva cadavers. Samples were harvested and analyzed at the time points indicated. Time zero is the time of injection. A description of the graphs is presented in the legend to Fig. S2. Download
Time-lapse microscopy of a developing spoIID-yfp nprA-mCherry-expressing Bt cell microcolony on a solid medium containing LBP diluted 1:100 at 30°C. Merged images from phase-contrast and fluorescent channels. mCherry and YFP fluorescence was false colored red and green, respectively. Cells expressing both mCherry and YFP present are yellow. Images were acquired every 15 min. Download
Plasmid stability in Bt cells. Maintenance of the pHT304-18ΩPspoIID-yfp-PnprA-mCherry plasmid in Bt cells harvested from LBP liquid cultures, biofilms, and insect cadavers was investigated. LB, number of CFU on LB plates. LB Erm, number of CFU on LB plates supplemented with erythromycin; %, normalized ratio of the number of ErmR CFU to the CFU count on LB. The time of sampling is in hours after inoculation of medium or infection of larvae.
Primers used in this study. Bold letters indicate restriction sites, and italic letters indicate the sequences of the restriction sites present in the multiple cloning sites of pAMY-spec. The underlined sequences are complementary sequences used for splicing by overlap extension as described in Materials and Methods.
Plasmids and strains used in this study.
Supplemental results and experimental procedures. Download
ACKNOWLEDGMENTS
We thank Christina Nielsen-Leroux and Stéphane Perchat for discussion and Christelle Lemy and Christophe Buisson for technical assistance.
This study was funded by the French Agence Nationale de la Recherche (Cell.com; no. ANR-09-Blan-0253).
We gratefully acknowledge funding from the DIM Astrea (French regional program: Ast11 0137) for the CyFlow Space flow cytometer used in this work.
D.L., L.S., M.G., and E.V. are responsible for the concept and design of the study. E.V., L.S., and D.L. designed the experiments. E.V. performed the experiments. E.V. and L.S. analyzed the data. E.V., L.S., M.G., and D.L. wrote the paper.
Footnotes
Citation Verplaetse E, Slamti L, Gohar M, Lereclus D. 2015. Cell differentiation in a Bacillus thuringiensis population during planktonic growth, biofilm formation, and host infection. mBio 6(3):e00138-15. doi:10.1128/mBio.00138-15.
REFERENCES
- 1.López D, Vlamakis H, Kolter R. 2010. Biofilms. Cold Spring Harb Perspect Biol 2:a000398. doi: 10.1101/cshperspect.a000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marlow VL, Cianfanelli FR, Porter M, Cairns LS, Dale JK, Stanley-Wall NR. 2014. The prevalence and origin of exoprotease-producing cells in the Bacillus subtilis biofilm. Microbiology 160:56–66. doi: 10.1099/mic.0.072389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vlamakis H, Aguilar C, Losick R, Kolter R. 2008. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev 22:945–953. doi: 10.1101/gad.1645008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zottola EA, Sasahara KC. 1994. Microbial biofilms in the food processing industry—should they be a concern? Int J Food Microbiol 23:125–148. doi: 10.1016/0168-1605(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 5.Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, Bhattacharyya A, Reznik G, Mikhailova N, Lapidus A, Chu L, Mazur M, Goltsman E, Larsen N, D’Souza M, Walunas T, Grechkin Y, Pusch G, Haselkorn R, Fonstein M, Ehrlich SD, Overbeek R, Kyrpides N. 2003. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423:87–91. doi: 10.1038/nature01582. [DOI] [PubMed] [Google Scholar]
- 6.Agaisse H, Gominet M, Okstad OA, Kolstø AB, Lereclus D. 1999. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol Microbiol 32:1043–1053. doi: 10.1046/j.1365-2958.1999.01419.x. [DOI] [PubMed] [Google Scholar]
- 7.Perchat S, Dubois T, Zouhir S, Gominet M, Poncet S, Lemy C, Aumont-Nicaise M, Deutscher J, Gohar M, Nessler S, Lereclus D. 2011. A cell-cell communication system regulates protease production during sporulation in bacteria of the Bacillus cereus group. Mol Microbiol 82:619–633. doi: 10.1111/j.1365-2958.2011.07839.x. [DOI] [PubMed] [Google Scholar]
- 8.Slamti L, Perchat S, Huillet E, Lereclus D. 2014. Quorum sensing in Bacillus thuringiensis is required for completion of a full infectious cycle in the insect. Toxins (Basel) 6:2239-2255. doi: 10.3390/toxins6082239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salamitou S, Ramisse F, Brehélin M, Bourguet D, Gilois N, Gominet M, Hernandez E, Lereclus D. 2000. The PlcR regulon is involved in the opportunistic properties of Bacillus thuringiensis and Bacillus cereus in mice and insects. Microbiology 146:2825–2832. [DOI] [PubMed] [Google Scholar]
- 10.Dubois T, Perchat S, Verplaetse E, Gominet M, Lemy C, Aumont-Nicaise M, Grenha R, Nessler S, Lereclus D. 2013. Activity of the Bacillus thuringiensis NprR-NprX cell-cell communication system is co-ordinated to the physiological stage through a complex transcriptional regulation. Mol Microbiol 88:48–63. doi: 10.1111/mmi.12168. [DOI] [PubMed] [Google Scholar]
- 11.Dubois T, Faegri K, Perchat S, Lemy C, Buisson C, Nielsen-LeRoux C, Gohar M, Jacques P, Ramarao N, Kolstø AB, Lereclus D. 2012. Necrotrophism is a quorum-sensing-regulated lifestyle in Bacillus thuringiensis. PLoS Pathog 8:e1002629. doi: 10.1371/journal.ppat.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bongiorni C, Stoessel R, Shoemaker D, Perego M. 2006. Rap phosphatase of virulence plasmid pXO1 inhibits Bacillus anthracis sporulation. J Bacteriol 188:487–498. doi: 10.1128/JB.188.2.487-498.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabrera R, Rocha J, Flores V, Vázquez-Moreno L, Guarneros G, Olmedo G, Rodríguez-Romero A, de la Torre M. 2014. Regulation of sporulation initiation by NprR and its signaling peptide NprRB: molecular recognition and conformational changes. Appl Microbiol Biotechnol 98:9399–9412. doi: 10.1007/s00253-014-6094-8. [DOI] [PubMed] [Google Scholar]
- 14.Lereclus D, Agaisse H, Grandvalet C, Salamitou S, Gominet M. 2000. Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int J Med Microbiol 290:295–299. doi: 10.1016/S1438-4221(00)80024-7. [DOI] [PubMed] [Google Scholar]
- 15.Ceuppens S, Timmery S, Mahillon J, Uyttendaele M, Boon N. 2013. Small Bacillus cereus ATCC 14579 subpopulations are responsible for cytotoxin K production. J Appl Microbiol 114:899–906. doi: 10.1111/jam.12096. [DOI] [PubMed] [Google Scholar]
- 16.Eijlander RT, Kuipers OP. 2013. Live-cell imaging tool optimization to study gene expression levels and dynamics in single cells of Bacillus cereus. Appl Environ Microbiol 79:5643–5651. doi: 10.1128/AEM.01347-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fagerlund A, Dubois T, Økstad OA, Verplaetse E, Gilois N, Bennaceur I, Perchat S, Gominet M, Aymerich S, Kolstø AB, Lereclus D, Gohar M. 2014. SinR controls enterotoxin expression in Bacillus thuringiensis biofilms. PLoS One 9:e87532. doi: 10.1371/journal.pone.0087532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merrell DS, Camilli A. 2000. Detection and analysis of gene expression during infection by in vivo expression technology. Philos Trans R Soc Lond B Biol Sci 355:587–599. doi: 10.1098/rstb.2000.0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fedhila S, Daou N, Lereclus D, Nielsen-LeRoux C. 2006. Identification of Bacillus cereus internalin and other candidate virulence genes specifically induced during oral infection in insects. Mol Microbiol 62:339–355. doi: 10.1111/j.1365-2958.2006.05362.x. [DOI] [PubMed] [Google Scholar]
- 20.Slauch JM, Mahan MJ, Mekalanos JJ. 1994. In vivo expression technology for selection of bacterial genes specifically induced in host tissues. Methods Enzymol 235:481–492. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. 2010. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog 6:e1001102. doi: 10.1371/journal.ppat.1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uliczka F, Pisano F, Kochut A, Opitz W, Herbst K, Stolz T, Dersch P. 2011. Monitoring of gene expression in bacteria during infections using an adaptable set of bioluminescent, fluorescent and colorigenic fusion vectors. PLoS One 6:e20425. doi: 10.1371/journal.pone.0020425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker GF, Daniel RA, Errington J. 1996. Timing and genetic regulation of commitment to sporulation in Bacillus subtilis. Microbiology 142:3445–3452. doi: 10.1099/13500872-142-12-3445. [DOI] [PubMed] [Google Scholar]
- 24.Gohar M, Faegri K, Perchat S, Ravnum S, Økstad OA, Gominet M, Kolstø AB, Lereclus D. 2008. The PlcR virulence regulon of Bacillus cereus. PLoS One 3:e2793. doi: 10.1371/journal.pone.0002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bravo A, Agaisse H, Salamitou S, Lereclus D. 1996. Analysis of cryIAa expression in sigE and sigK mutants of Bacillus thuringiensis. Mol Gen Genet 250:734–741. [DOI] [PubMed] [Google Scholar]
- 26.Veening JW, Stewart EJ, Berngruber TW, Taddei F, Kuipers OP, Hamoen LW. 2008. Bet-hedging and epigenetic inheritance in bacterial cell development. Proc Natl Acad Sci U S A 105:4393–4398. doi: 10.1073/pnas.0700463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Jong IG, Beilharz K, Kuipers OP, Veening JW. 2011. Live cell imaging of Bacillus subtilis and Streptococcus pneumoniae using automated time-lapse microscopy. J Vis Exp 53:3145. doi: 10.3791/3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lereclus D, Agaisse H, Gominet M, Salamitou S, Sanchis V. 1996. Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. J Bacteriol 178:2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rani SA, Pitts B, Beyenal H, Veluchamy RA, Lewandowski Z, Davison WM, Buckingham-Meyer K, Stewart PS. 2007. Spatial patterns of DNA replication, protein synthesis, and oxygen concentration within bacterial biofilms reveal diverse physiological states. J Bacteriol 189:4223–4233. doi: 10.1128/JB.00107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López D, Kolter R. 2010. Extracellular signals that define distinct and coexisting cell fates in Bacillus subtilis. FEMS Microbiol. Rev. 34:134–149. doi: 10.1111/j.1574-6976.2009.00199.x. [DOI] [PubMed] [Google Scholar]
- 31.Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu Rev Microbiol 56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 32.Horswill AR, Stoodley P, Stewart PS, Parsek MR. 2007. The effect of the chemical, biological, and physical environment on quorum sensing in structured microbial communities. Anal. Bioanal. Chem. 387:371–380. doi: 10.1007/s00216-006-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auger S, Krin E, Aymerich S, Gohar M. 2006. Autoinducer 2 affects biofilm formation by Bacillus cereus. Appl Environ Microbiol 72:937–941. doi: 10.1128/AEM.72.1.937-941.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perego M, Glaser P, Hoch JA. 1996. Aspartyl-phosphate phosphatases deactivate the response regulator components of the sporulation signal transduction system in Bacillus subtilis. Mol Microbiol 19:1151–1157. doi: 10.1111/j.1365-2958.1996.tb02460.x. [DOI] [PubMed] [Google Scholar]
- 35.Chastanet A, Vitkup D, Yuan GC, Norman TM, Liu JS, Losick RM. 2010. Broadly heterogeneous activation of the master regulator for sporulation in Bacillus subtilis. Proc. Natl Acad Sci U S A 107:8486–8491. doi: 10.1073/pnas.1002499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veening JW, Hamoen LW, Kuipers OP. 2005. Phosphatases modulate the bistable sporulation gene expression pattern in Bacillus subtilis. Mol Microbiol 56:1481–1494. doi: 10.1111/j.1365-2958.2005.04659.x. [DOI] [PubMed] [Google Scholar]
- 37.Perchat S, Zouhir S, Poncet S, Gohar M, Nessler S, Lereclus D. 2013. NprR is a bifunctional quorum sensor that couples necrotrophism and cell development in bacteria of the Bacillus cereus group, abstr 76. Abstr. 7th International Conference on Gram-positive Microorganisms—17th International Conference on Bacilli. Montecatini Terme, Tuscany, Italy. [Google Scholar]
- 38.Lereclus D, Arantès O, Chaufaux J, Lecadet M. 1989. Transformation and expression of a cloned delta-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol Lett 51:211–217. doi: 10.1016/0378-1097(89)90511-9. [DOI] [PubMed] [Google Scholar]
- 39.Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. 2000. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol 328:575–592. doi: 10.1016/S0076-6879(00)28419-X. [DOI] [PubMed] [Google Scholar]
- 40.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 42.Branda SS, Chu F, Kearns DB, Losick R, Kolter R. 2006. A major protein component of the Bacillus subtilis biofilm matrix. Mol Microbiol 59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression kinetics of nprA with mCherry or lacZ as a reporter. Comparison of the plasmid and chromosomal PnprA-mCherry transcriptional fusions by flow cytometry (A). Samples of cells carrying transcriptional fusions on a plasmid (green) or at the α-amylase locus (pink) were harvested at T0 (dotted lines), T2 (dashed lines), and T4 (solid lines). The gray area is the fluorescence level obtained with control cells with no reporter at T0. On the x axis of each histogram is the fluorescence intensity in arbitrary units (A.U.) on a logarithmic scale. On the y axis is the cell count. At least 40,000 cells were counted. Microscopy analysis of the plasmid or chromosomal PnprA-mCherry transcriptional fusions (B). Fluorescent signals (false colored red) obtained with cells grown in LBP at 30°C carrying the PnprA-mCherry reporter plasmid (top) were compared with signals obtained with cells harboring the chromosomal construct (bottom). Membranes were stained with FM4-64 (light blue). Time zero was defined as the onset of stationary phase. Bars, 5 µm. (C) nprA promoter activity in LBP with lacZ as a reporter gene. pHT304-18ΩPnprA-lacZ-carrying Bt cells grown in LBP at 30°C under shaking were harvested, and their β-galactosidase activities were measured as previously described (2). The activity of the nprA promoter was detected approximately 1.5 h after entry into stationary phase, similarly to the expression kinetics obtained with the mCherry reporter. These results validated codon-optimized mCherry as an efficient reporter of gene expression in Bt. Growth is indicated by gray squares, and promoter activity is indicated by black diamonds (error bars are standard deviations of the mean values calculated from two independent experiments). Time zero was defined as the onset of stationary phase (i.e., the time at which the slope of the growth curve decreases). OD600nm, optical density at 600 nm. Download
Cytograms and microscopic images of the plcB-yfp nprA-mCherry (A)- and spoIID-yfp nprA-mCherry (B)-expressing Bt strains grown in LBP at 30°C. Samples were harvested and analyzed at the time points indicated. Time zero (T0) was defined as entry into stationary phase. On cytograms, YFP fluorescence intensity reflecting the expression of plcB (A) or spoIID (B) is on the x axis and mCherry fluorescence intensity, reflecting the expression of nprA, is on the y axis. The cytograms presented represent at least three independent experiments. The microscopic images obtained with the mCherry and YFP filters are in black and white. On the overlay images, mCherry fluorescence is false colored red and YFP fluorescence is false colored green. Cells expressing both mCherry and YFP are various shades of yellow, depending on the intensity of mCherry and YFP fluorescence. Download
Evolution of the biomass (ring and pellicle) of Bt strain 407 growing at the air-liquid interface of LBP in glass tubes at 30°C. Time zero is the time of inoculation. Pictures of the corresponding biofilms are presented. Download
Cytograms and microscopic images of dual-reporter plcB-yfp nprA-mCherry (A) and spoIID-yfp nprA-mCherry (B) Bt strains grown in a biofilm at the air-liquid interface of LBP in glass tubes. Cells were harvested at the time points indicated. Time zero is the time of medium inoculation. A description of the graphs is presented in the legend to Fig. S2. Arrows point to cells expressing mCherry only. Circled cells are Nec+ cells that did not emerge from a Vir+ population. Download
Cytograms and fluorescence microscopic images of dual-reporter plcB-yfp nprA-mCherry (A)- and spoIID-yfp nprA-mCherry (B)-expressing Bt strains isolated from G. mellonella larva cadavers. Samples were harvested and analyzed at the time points indicated. Time zero is the time of injection. A description of the graphs is presented in the legend to Fig. S2. Download
Time-lapse microscopy of a developing spoIID-yfp nprA-mCherry-expressing Bt cell microcolony on a solid medium containing LBP diluted 1:100 at 30°C. Merged images from phase-contrast and fluorescent channels. mCherry and YFP fluorescence was false colored red and green, respectively. Cells expressing both mCherry and YFP present are yellow. Images were acquired every 15 min. Download
Plasmid stability in Bt cells. Maintenance of the pHT304-18ΩPspoIID-yfp-PnprA-mCherry plasmid in Bt cells harvested from LBP liquid cultures, biofilms, and insect cadavers was investigated. LB, number of CFU on LB plates. LB Erm, number of CFU on LB plates supplemented with erythromycin; %, normalized ratio of the number of ErmR CFU to the CFU count on LB. The time of sampling is in hours after inoculation of medium or infection of larvae.
Primers used in this study. Bold letters indicate restriction sites, and italic letters indicate the sequences of the restriction sites present in the multiple cloning sites of pAMY-spec. The underlined sequences are complementary sequences used for splicing by overlap extension as described in Materials and Methods.
Plasmids and strains used in this study.
Supplemental results and experimental procedures. Download