

ABSTRACT
West Nile virus (WNV) is an emerging neuroinvasive flavivirus that now causes significant morbidity and mortality worldwide. The innate and adaptive immune responses to WNV infection have been well studied in C57BL/6J inbred mice, but this model lacks the variations in susceptibility, immunity, and outcome to WNV infection that are observed in humans, thus limiting its usefulness to understand the mechanisms of WNV infection and immunity dynamics. To build a model of WNV infection that captures human infection outcomes, we have used the Collaborative Cross (CC) mouse model. We show that this model, which recapitulates the genetic diversity of the human population, demonstrates diversity in susceptibility and outcomes of WNV infection observed in humans. Using multiple F1 crosses of CC mice, we identified a wide range of susceptibilities to infection, as demonstrated through differences in survival, clinical disease score, viral titer, and innate and adaptive immune responses in both peripheral tissues and the central nervous system. Additionally, we examined the Oas1b alleles in the CC mice and confirmed the previous finding that Oas1b plays a role in susceptibility to WNV; however, even within a given Oas1b allele status, we identified a wide range of strain-specific WNV-associated phenotypes. These results confirmed that the CC model is effective for identifying a repertoire of host genes involved in WNV resistance and susceptibility. The CC effectively models a wide range of WNV clinical, virologic, and immune phenotypes, thus overcoming the limitations of the traditional C57BL/6J model, allowing genetic and mechanistic studies of WNV infection and immunity in differently susceptible populations.
IMPORTANCE
Mouse models of West Nile virus infection have revealed important details regarding the innate and adaptive immune responses to this emerging viral infection. However, traditional mouse models lack the genetic diversity present in human populations and therefore limit our ability to study various disease outcomes and immunologic mechanisms subsequent to West Nile virus infection. In this study, we used the Collaborative Cross mouse model to more effectively model the wide range of clinical, virologic, and immune phenotypes present upon West Nile virus infection in humans.
INTRODUCTION
West Nile virus (WNV) is an emerging flavivirus. It is an enveloped virus that encodes a single-stranded RNA (ssRNA) positive-sense genome (1). Since its introduction to North America in 1999, the virus has spread throughout the United States and into Canada, Mexico, and Latin America (1), to now cause significant morbidity and mortality in the Western hemisphere and represent a global public health problem. WNV is neuroinvasive and can cause disease ranging from self-limiting febrile illness to disease of the central nervous system (CNS), including meningitis and encephalitis (1–3). Neuroinvasive infection and CNS disease can be particularly deadly and leave survivors with long-term physical and cognitive disabilities (4). Approximately 20% of infected individuals experience a limited febrile illness, with 1% developing a more severe neuroinvasive disease characterized by encephalitis, meningitis, and acute flaccid paralysis (1–3). Additionally, a more chronic poliomyelitis-like syndrome can occur, in which patients experience neurologic weakness and/or tremor 1 year after their acute illness (4, 5). Several host genetic factors and immune correlates that influence susceptibility or severity of infection have been identified through blood donor screening programs or retrospective studies (Table 1). Specifically, a loss-of-function mutation in CCR5 corresponded to an increased severity in WNV infection, though it was not associated with increased susceptibility (6, 7). A genomics study with more than 1,500 symptomatic patients showed that severe neuroinvasive disease was associated with single nucleotide polymorphisms (SNPs) in genes encoding a sodium channel, SCN1a, and a cell replication factor, RFC1 (8). SNPs in key regulators of immune function may be associated with increased risk of infection and progression to severe neurological disease. Beyond these studies, limited knowledge is available regarding the immune response to WNV in humans, due to the high prevalence of subclinical infection that precludes identification of WNV-infected individuals and subsequent clinical and immune response evaluations. Thus, most knowledge of anti-WNV immunity comes from the study of WNV infection using inbred mouse models of infection, generally using wild-type (WT) and transgenic gene knockout C57BL/6J mice (B6 mice). In Fig. 1, we highlight the immunologic and clinical parameters that can be readily evaluated in human patients with WNV infection compared to WNV-infected B6 mice. Specifically, the B6 model has been invaluable, as it allows for measurement of viral load in tissues, including both the peripheral nervous system and CNS, as well as inflammatory responses. While these properties cannot be measured in human patients, symptoms such as long-term neurological weakness (4, 9–13) and gastrointestinal (GI) involvement (14) are found in human patients but are not observed to occur in the B6 infection model. Currently, immune responses in the B6 model that may also be measured in human studies are limited to peripheral blood T cell responses (15, 16), serum antibody responses (17), and overt signs of clinical disease (16). Table 1 further describes this diversity of outcomes in human infection.
TABLE 1 .
Diversity in WNV outcomes in humansa
| Method | Finding(s) | Study |
|---|---|---|
| Follow-up interviews after blood donor screenings; 3 of 8 indicator symptoms = symptomatic | 26% of WNV-infected individuals become symptomatic | Zou et al. 2010 (44) |
| PBMCs from WNV-infected blood donors screened for WNV-specific IFN-γ response by ELISPOT | The NS4b epitope is widely recognized across responders; the highest-magnitude T cell responses are mediated by CD8+ T cells | Lanteri et al. 2008 (15) |
| Longitudinal neurologic studies in patients with WNV paralysis | 41% of patients experience neurologic weakness and/or tremor 1 yr after acute illness | Hayes et al. 2005 (45), Sejvar 2007 (4) |
| WNV+ and WNV− blood donors compared for CCR5Δ32 distribution | CCR5 deficiency is a risk factor for early clinical manifestations of WNV infection but not viral transmission | Lim et al. 2010 (7) |
| Examination of Treg frequency following acute WNV infection to 1 yr postinfection | Treg frequency increases after WNV infection; symptomatic subjects exhibit lower Treg levels than asymptomatic subjects | Lanteri et al. 2009 (16) |
| Retrospective chart review; 57 patients examined for clinical features of disease | 56% with WNV neuroinvasive disease, 44% with West Nile fever | Petersen et al. 2012 (17) |
Abbreviations: PBMCs, peripheral blood mononuclear cells; ELISPOT, enzyme-linked immunosorbent spot assay.
FIG 1 .

Immunologic parameters in mice versus humans. Parameters that can be measured in the traditional B6 mouse model, in human subjects, and in both populations are shown. Symptoms such as chronic infection, gastrointestinal (GI) symptoms, and long-term neurological weakness are not observed in the B6 model but are present among human patients with WNV infection. CSF, cerebrospinal fluid;
Compared to this diverse breadth of clinical outcomes and immune responses observed in the human population, the B6 mouse model of WNV infection is much less variable. Specifically, when infected subcutaneously in the footpad with the contemporary emerging and neuroinvasive strain of WNV, approximately 30% of infected mice succumb to infection within 10 to 12 days postinfection, depending on the inoculum dose (18–22). While this outcome captures the processes of neuroinvasive disease of the CNS observed in human West Nile neuroinvasive disease (WNND), B6 mice generally display extremely similar immunophenotypes with a repeatable fraction of animals transitioning to symptomatic infection. Upon subcutaneous infection, WNV replicates locally in myeloid cells, including macrophages and dendritic cells (DCs), which initiate cell-intrinsic innate immune responses that can restrict infection. These cells also migrate to the draining lymph nodes (dLNs) and subsequently initiate adaptive immune responses. Infected cells first sense the presence of WNV through the actions of RIG-I-like receptors (RLRs), RIG-I, and MDA5 expressed within the cell cytoplasm and subsequently through endosomal Toll-like receptor 3 (TLR3) (19, 23). Virus sensing drives RLR signaling to downstream activation of interferon (IFN) regulatory factor 3 (IRF3) and NF-κB for induction of direct gene targets. These genes encode antiviral proteins that confer antiviral actions to restrict WNV replication and cytokines and chemokines that serve to modulate the innate and adaptive immune response to infection. Type I IFN is a major component of this response which facilitates an antiviral state through autocrine and paracrine signaling actions that induce hundreds of interferon-stimulated genes (ISGs) that have antiviral and immunomodulatory activities to drive systemic antiviral actions. Studies with the B6 model have shown that innate and adaptive immune responses orchestrate protection and control of WNV infection and disease (24). RLR-mediated innate immunity and IFN actions are essential for restricting WNV replication and neuroinvasion to the CNS (18, 20, 21, 25–27), while humoral immunity is involved in peripheral viral clearance. Moreover, T cells are critical for viral clearance in the CNS. Specifically, the induction of virus-specific IgM early postinfection with WNV limits viremia and spread to the CNS, thereby helping to protect against lethal infection (28, 29). A role for cytotoxic T lymphocytes (CTLs) in immunity to WNV infection was demonstrated via adoptive transfer of WNV-specific CD8+ T cells, which led to prolonged survival after WNV infection in recipient mice. Importantly, CD8+ T cells were found to infiltrate the infected brains, suggesting that these T cells could be involved in recovery from encephalitis (30). However, the in vivo dynamics of these innate and adaptive immune responses that are associated with differential outcomes of WNV infection in the human population have not been effectively modeled and therefore remain poorly understood.
Given the breadth in symptoms and disease severity seen in human WNV infection that are not effectively captured in the B6 model, we turned to the newly developed Collaborative Cross (CC) mouse to better model WNV infection and understand the role of host genetics in the immune response to and control of WNV. The CC model includes eight founder mouse strains: five classical inbred strains (C57BL/6J, A/J, 129S1/SvImJ, NOD/ShiLtJ, and NZO/H1LuJ) and three wild-type-derived strains (CAST/EiJ, PWK/PhJ, and WSB/EiJ) (31–33). The founder strains represent the three major Mus musculus subspecies, and genotyping has revealed that the founder strains capture nearly 90% of common genetic variation in Mus musculus strains (>40 million SNPs, >4 million small insertion/deletions), with this variation randomly distributed across the genome (34). Recombinant inbred (RI) strains were created by three generations of funnel breeding to incorporate all founder lines, followed by at least 20 generations of inbred mating to develop each CC RI line (33) (Fig. 2). For this study, F1 progeny from crosses between CC RI strains (called discovery recombinant intercross, or dRIX, lines) were used. These dRIX lines were heterozygous for the H-2b major histocompatibility complex (MHC) haplotype and were created by crossing two independent RI lines, one of which had the H-2b allele, in order to allow for the detection of WNV-specific T cells by using MHC class I tetramer. Following MHC WNV infection and a complete phenotyping analysis of the innate and adaptive immune responses, various immune responses associated with a range of clinical outcomes were observed in the dRIX lines.
FIG 2 .
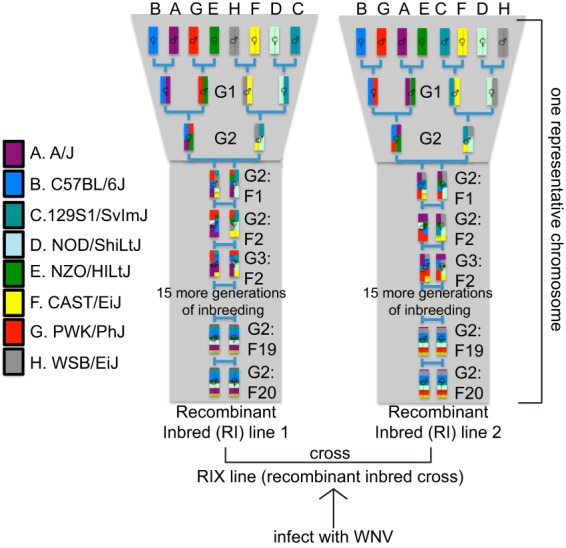
Derivation of the Collaborative Cross mouse model. In a funnel breeding scheme, genetic contributions of all eight founder strains are incorporated after the G2 generation. After 20 generations of inbreeding, a recombinant inbred line is created. The funnel code is shifted (A to H), and each founder strain occupies the same position an equal number of times. Two RI lines crossed together produced the dRIX lines used in this study.
Here, we demonstrate that the CC model can be used as a resource to model WNV infection in humans, such that infection and comparison of CC mouse lines improves upon information from inbred mouse models to better capture the diversity of WNV infection outcomes observed across the genetically diverse human population. We studied mice over a time course designed to encompass the range of WNV-induced disease outcomes as well as innate and adaptive immune response induction, peak response, and immune memory cell differentiation. Using these approaches, we identified three broad categories of WNV susceptibility (asymptomatic, symptomatic, and asymptomatic with CNS involvement) within CC mice that mirrored the range of disease states in human infection. We show that the CC offers an effective model of WNV infection that captures the outcome diversity observed in the human population and that can be further used to identify unique correlates of protection from WNV infection and disease.
RESULTS
Disease phenotypes in CC dRIX mice.
To evaluate WNV disease in the CC mice as well as their associated immune response phenotype, we challenged cohorts of CC dRIX lines with WNV-TX, and we identified three broad categories of WNV disease outcomes based on weight loss and clinical disease score, viral loads in WNV target organs, including the spleen, brain, and kidney, and brain pathology based on histologic assessment (Table 2; Fig. 3). Strains classified as asymptomatic showed virtually no outward signs of disease, as seen by an absence of weight loss (Fig. 3A) and consistent clinical scores of zero. In these RIX animals, virus was quickly cleared from the spleen by day 7 postinfection, with minimal viral escape to the CNS at early time points and only one mouse showing low levels of virus reaching the brain at day 12. Despite a robust innate immune response, some mice might have experienced viral escape to the CNS, marked by IFN-β and IFIT1 (interferon-induced tetracopeptide 1 gene) induction [CC(017 × 004)F1], but brain histopathology showed few to no signs of inflammation or parenchymal damage (Fig. 4). In contrast, symptomatic RIX mice began losing significant weight and showed increased clinical scores through the early infection time points, beginning at days 7 to 12 postinfection. In symptomatic RIX lines, viral loads in the CNS increased from day 4 to day 7 postinfection, and there was neuropathology characterized by perivascular mononuclear cuffing and infiltration with parenchymal lesions (combinations of degeneration, malacia, and apoptosis/necrosis) (Fig. 3C). The third category of WNV disease revealed by the RIX infection model was a response defined as “asymptomatic with CNS involvement.” Outwardly, these mice appeared to be asymptomatic, as there was little to no weight loss or signs of clinical disease, and limited viral escape resulted in IFN-β and IFIT1 induction in the CNS and viral clearance, as in asymptomatic mice. However, brain histology examining immunopathology in the CNS, along with brain flow cytometry assays, revealed the presence of antigen-specific CD8+ immune cells in the CNS, as seen in neuroinvasive WNV infection (Fig. 3C and 5). However, despite neuroinvasive infection and CNS immunopathology, the mice showed no outward signs of disease. Thus, the outcome termed asymptomatic with CNS involvement represents a previously unmodeled but critical disease outcome that may represent a correlate to WNV-infected humans with limited disease symptoms or subclinical infection.
TABLE 2 .
Phenotypes measured in the CC lines
| Phenotype | Result measured | Organ(s) analyzed |
|---|---|---|
| Clinical disease | Weight loss, clinical score | Whole mouse |
| Viral replication | Viral load | Spleen, kidney, brain |
| Innate immune response | IFN-β, IFIT1 | Spleen, kidney, brain |
| Histology | Inflammatory infiltrates | Spleen, brain |
| T-cell response | Antigen-specific and bystander activation, short-lived effector cells and memory precursors, Treg activation, T-cell activation and trafficking, cytokine response, CD4 and CD8 T-cell memory response | Spleen, brain |
| Humoral response | WNV-specific IgG and IgM ELISA | Serum |
| Immune response | Gene expression profiling | Spleen, brain |
FIG 3 .

Overview of the three phenotypes observed in the CC dRIX mice. CC dRIX mice were infected with 100 PFU WNV in the footpad as previously described and monitored throughout the time course of infection. Outward signs of disease were measured by weight loss (A) and clinical score. (B) Viral load in the kidney, brain, and spleen. (C) Representative hematoxylin and eosin-stained sections of formalin-fixed paraffin-embedded brains from d12 CC dRIX mice, as indicated. Histology of asymptomatic lines CC(041 × 012)F1 and (017 × 004)F1 was within normal limits. The meninges, midbrain, and cerebellum of these lines were indistinguishable from mock control tissues. Lesions were noted in the CC(024 × 023)F1 (minimal to mild), CC(005 × 001)F1 (mild), and CC(004 × 011)F1 (moderate to severe) groups. Virus-induced changes included infiltration of mononuclear cells in the meninges (arrowheads), perivascular spaces (arrows), and parenchyma with glial nodules (circles). Higher magnifications of boxed regions [CC(004 × 011)F1] illustrate changes, including cortical parenchymal rarefication (meninges), neuronophagia and apoptosis (midbrain), and severe mononuclear perivascular cuffing (cerebellum). Original magnification, ×200 (all panels); ×400 (insets).
FIG 4 .

Differential innate immune response among CC lines in association with clinical outcome. Total RNA was isolated from spleens, brains, and kidneys from CC dRIX mice infected with WNV for the indicated times. Plots show qPCR results for IFN-β (A) and IFIT1 (B) expression in asymptomatic lines CC(041 × 012)F1 and CC(017 × 004)F1, symptomatic lines CC(005 × 001)F1 and CC(024 × 023)F1, and an asymptomatic line with CNS involvement, CC(004 × 011)F1.
Differential innate immune responses among CC dRIX lines are associated with clinical outcome.
The extent of innate immune activation in the different RIX lines was measured by assessing relative expression levels of the IFN-β and IFIT1 genes. While both IFN-β and IFIT1 are IRF3 target genes (35) induced by WNV infection, IFIT1 is further induced by IFN signaling, thus allowing us to mark induction of IRF-3 activation and the IFN response. We monitored the expression of each gene in various mouse tissues to mark sites of IRF-3 activity and IFN response. In the asymptomatic lines CC(041 × 012)F1 and CC(017 × 004)F1, early IFN-β expression was observed in the kidney along with increased IFIT1 expression in the spleen (Fig. 4A and B). At later time points, days 7 and 12 postinfection, IFN-β and IFIT1 expression levels were generally lower except for in the brains of the asymptomatic line CC(017 × 004)F1 (Fig. 4A, bottom). Higher (>50× relative expression) IFN-β or IFIT1 expression levels in the brains at later time points were associated with WNV susceptibility, as was seen in the symptomatic lines CC(005 × 001)F1 and CC(024 × 023)F1 (Fig. 4). In the asymptomatic with CNS involvement line [CC(004 × 011)F1], high IFIT1 expression was observed early (day 2) and expression was lower by day 7 and day 12, with IFN-β expression elevated in the spleen and kidney by day 12. In summary, early innate responses that resolved by day 7 were observed in asymptomatic lines, whereas the presence of a prolonged innate response (day 7 through day 12) correlated with a lack of peripheral virus control and viral spread to the brain.
Diversity in adaptive immune responses to WNV infection.
Following WNV infection, CC dRIX mice were also evaluated for WNV-specific adaptive immune responses though day 28 postinfection, a time at which immunological memory forms. WNV-specific IgM was variable among groups; while one symptomatic strain showed no increase in IgM concentration over the time course of infection [CC(024 × 023)F1], a second symptomatic line [CC(005 × 001)F1] had higher baseline titers that increased through the early immune response (days 7 to 12) (Fig. 5A) and then returned to baseline by days 21 to 28. In the two asymptomatic strains, as well as in the asymptomatic with CNS involvement line, IgM levels increased from baseline levels early and then quickly dropped again, presumably as the infection was cleared from the periphery.
FIG 5 .

Diversity in the adaptive immune response to WNV infection in mice. Mice were infected as previously described. (A) WNV-specific IgM levels in serum at the indicated time points after infection. (B and C) Antigen-specific CD8+ cells in the spleen (B) and brain (C) following infection. (D and E) Numbers of Foxp3+ regulatory T cells present in the spleen (D) and brain (E) after infection.
Antigen-specific T cells were quantified in the spleen (Fig. 5B) and brain (Fig. 5C) by MHC class I tetramer staining. In the two asymptomatic lines, CD8+ tetramer+ cells specific to the immunodominant NS4B epitope expanded in the spleen at days 7 to 12 and then this cell population contracted by day 28. The dRIX line presenting with asymptomatic WNV infection with CNS involvement, CC(004 × 011)F1, showed high numbers of bulk CD8 T cells as well as antigen-specific T cells in the brain throughout the adaptive immune response to infection, as the cells remained in the brain at elevated levels through the day 28 time point. Again, we observed differences among phenotypes with the two symptomatic lines: while line CC(005 × 001)F1 showed higher levels of antigen-specific cells in both the spleen and brain at later time points (days 21 to 28), tetramer-positive cells failed to reach the brain in most mice of line CC(024 × 023)F1 by day 12 postinfection. In fact, only one mouse survived beyond the day 12 time point in this line, and high levels of antigen-specific CD8 T cells persisted in the brain at day 28.
Regulatory T cells (Tregs) were also quantified in the spleen (Fig. 5D) and brain (Fig. 5E). Interestingly, we observed a difference in baseline levels of Tregs in the brains of strains tested, as the asymptomatic strain CC(017 × 004)F1 had nearly a 1.5-log-fold increase in Tregs in naive mice. In symptomatic strain CC(005 × 001)F1, Tregs expanded in the brain at later time points, while Tregs failed to expand in either the periphery or CNS for the other symptomatic strain, with the exception of one mouse that recovered by the day 28 time point.
Genetic control of WNV susceptibility.
Oas1b has previously been shown to play a major role in susceptibility to WNV and other flaviviruses in mice (36, 37). We therefore investigated the potential impact of Oas1b alleles on WNV susceptibility (Fig. 6) through investigation of the founder strain sequences at the Oas1b allele (38). While the 5 classic inbred strains (A/J, C57BL/6J, 129s1/SvImJ, NOD/HILtJ, and NZO/ShILtJ) have identical sequences at Oas1b, the 3 wild-type-derived strains (CAST/EiJ, PWK/PhJ, and WSB/EiJ) have a number of amino acid-changing mutations; importantly these three wild-type-derived strains all share a mutation that causes an extended coding sequence (Fig. 6A). When we assessed the relationship between these two major allele classes and the above-described symptomatic and asymptomatic disease outcomes, we found a highly significant (P < 1e−08; analysis of variance [ANOVA], F2,43 = 30.06) relationship between allele class and disease outcome such that those CC-RIX mice homozygous for the classic inbred allele showed an enhanced propensity for symptomatic disease outcomes. In contrast, those CC-RIX mice homozygous for a wild-type-derived allele showed asymptomatic disease outcomes. These observations serve to validate the important previous studies that first defined the genetic relationship of Oas1b with enhanced susceptibility to WNV (5, 35, 39), and thus underscore the promising application of the CC mouse model for future genetic studies to identify host genes of WNV susceptibility and resistance.
FIG 6 .
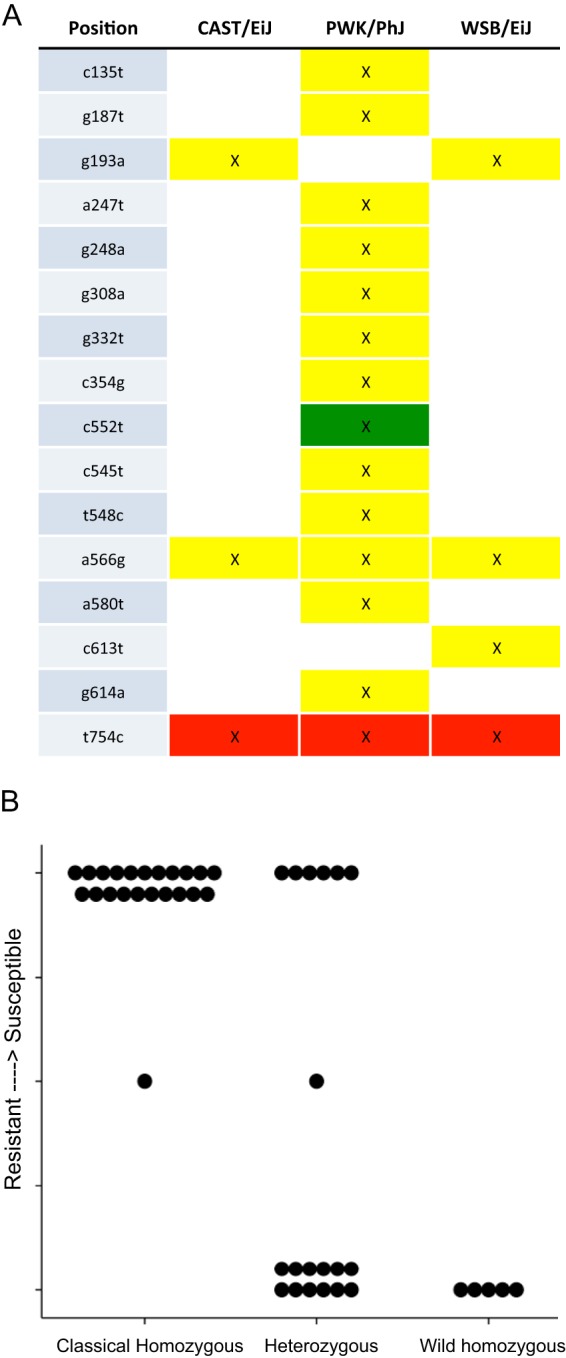
Genetic variation at Oas1b strongly correlates with disease outcome. (A) Genetic variation within the antiflavivirus Oas1b gene (5). C57BL/6J, 129s1/SvImJ, NOD/ShILtJ, and NZO/HILtJ strains (classical strains) showed no differences in sequence from each other. In contrast, CAST/EiJ, PWK/PhJ, and WSB/EiJ (wild-type-derived strains) had a number of differences relative to the classical strains (yellow, missense mutation; green, splice variant; red, stop codon lost), most importantly, a stop codon was lost in all three strains, suggesting a larger, mature Oas1b protein. (B) When we grouped CC-RIX mice by their disease outcomes (y axis, symptomatic versus asymptomatic responses), as well as whether each CC-RIX line showed a classical (short), wild-type-derived (larger; mature Oas1b), or heterozygous haplotype state at the Oas1b locus (x axis) (see also Table S1 in the supplemental material), we identified a highly significant relationship between WNV susceptibility and the founder strain allele class possessed by each RIX.
DISCUSSION
We have identified three broadly categorized WNV disease outcomes, including asymptomatic, symptomatic, and asymptomatic with CNS involvement, modeled by multiple dRIX lines that well represent the wide range of WNV disease states and immune variability observed in the human population compared to the C57BL/6J mouse model. Importantly, we show that multiple CC lines model each of the disease categories, and there are differences in WNV immune parameters both between these disease states that track with outcome as well as differences within each state that reveal a continuum of innate and adaptive immune responses that are associated with differential WNV susceptibility and disease. More generally, these responses suggest that complex genetic interactions drive these differential initial/innate antiviral responses, as well as the overall control of in vivo viral disease responses. Thus, our studies highlight the need for WNV disease models wherein a wider heterogeneity of responses is recapitulated, such as the CC model used in this study.
We measured IFIT1 and IFN-β induction as markers of innate immune response activation in WNV target organs, including the spleen and brain, as well as in the kidney as a representative organ that becomes involved during unrestrained viral replication. Early induction of IFN-β and/or IFIT1 occurred in the kidney and spleen of lines that were either asymptomatic or asymptomatic with CNS involvement (Fig. 4A and B). These observations might simply reflect the host response to continued low-level viral replication suppressed by the innate immune response in these organs, or they could imply broader innate immune regulation wherein the dRIX mice differentially regulate their respective innate immune response via altered expression of negative regulatory factors of innate immune signaling and/or differential sensitivity to viral products that confer RLR or TLR signaling. The CC model allows for identification of either possibility, based on our ongoing studies to define expression of quantitative trait loci and individual genes of innate immune regulation that impart differential outcomes to WNV infection.
We hypothesized that due to the genetic heterogeneity among CC dRIX animals, we would observe differences in immune responses both between, and more importantly within, our disease state groups. For example, while our asymptomatic lines were comparable when assessed by weight loss, viral titers, and IFN-β expression in spleen, the IFN-β expression in the brain in line CC(017 ×004)F1 showed continued expression through day 12 (Fig. 4A, bottom). While the significance of this nuance in IFN-β gene induction is unclear at this point, we speculate that it could impart differentially broad ISG expression set points among mouse lines with altered WNV susceptibility. These results demonstrate that there are intragroup differences in our broad “asymptomatic,” “symptomatic,” and “asymptomatic with CNS involvement” classifications that would more accurately capture the breadth of phenotypes observed in human populations than by current mouse models.
Our analysis of the role of Oas1b status further highlights the importance of other genetic factors in driving a range of WNV responses. As mentioned above, polymorphisms at Oas1b have been shown to drive resistance to flaviviral infections in mice (37, 40). Furthermore, genetic variation in OAS family members, such as Oas1, the putative human homologue to Oas1b, have been shown to influence WNV disease (41), as well as dengue (42) and hepatitis C virus (39) in humans. However, despite the dominant role of Oas1/Oas1b in these populations in driving overall symptoms/susceptibility to WNV, there remains a range of pathogenic differences in both the human population as well as in our CC RIX lines, even among CC RIX group with identical Oas1b allele classifications. This can be seen most clearly in the heterozygous CC RIX class Oas1b. These RIXes all have one wild-type, or functional, Oas1b allele yet show a range of susceptibility responses, including the highly novel CC RIX line that showed CNS involvement without clinical symptoms. Such divergent outcomes despite sharing Oas1b alleles reinforces the notion that the ISG expression set point or altered innate immune regulation among hosts can likely contribute to differential outcome of infection and immunity among a population outbreak and that Oas1b is often not entirely sufficient to completely protect against WNV infection. These various responses mirror the variation seen within the human population and highlight the critical nature of population-based approaches, such as CC studies, in better understanding the underlying genetic architecture driving wide ranges of disease responses, even within individuals sharing major resistance loci (43).
Our adaptive immune response studies also revealed intra- and interdisease state heterogeneity in immune responses. For example, in some asymptomatic strains there are large numbers of WNV-specific CD8+ T cells present within the central nervous system, while in some there are not, and the same is true for symptomatic strains. Additionally, the kinetics and functionality of the T-cell response vary by strain and within disease state categories. The humoral immune response to WNV is also variable, depending on the strain examined. Though in general symptomatic strains have a lower WNV-specific IgM response in the blood at 7 days postinfection, this correlation is not always present, suggesting that WNV-specific IgM can be a correlate of protection but is not the only critical player in a successful immune response to WNV (Fig. 5). Previous studies have shown that higher levels of Tregs after infection protect against severe WNV disease in both mice and humans (16). Interestingly, one of our asymptomatic strains presented with high levels of Tregs at baseline in the CNS, which is unique for this immune-privileged environment. After infection, the Tregs expanded even further, and there was no sign of CNS immunopathology by histology or via an expansion of antigen-specific CD8 T cells, potentially suggesting a mechanism by which Tregs limit the immune response to WNV infection in the CNS when infection is controlled in the periphery by innate and humoral responses.
Many additional immune phenotypes of WNV infection can be been measured, in both the periphery and CNS, to give a more complete understanding of the adaptive immune response to WNV. Through the use of the CC mice, we have observed a much wider range in phenotypes than previously seen in the B6 model, and importantly, these phenotypes mirror human symptoms and immune responses. While a limited discussion of the broad classifications of immune phenotypes is presented here, many additional aspects of the innate and adaptive immune response are being measured and can be combined with genomics analysis in future downstream studies. Our findings demonstrate that with the genetic diversity provided by the CC model, we can tease out several specific phenotypes that are present in humans but previously unable to be studied in a mouse model, such as asymptomatic mice with CNS involvement. The CC model allows us to link these outcomes with host genotype to identify genes of susceptibility and protection from WNV infection and disease. Extensive analyses of the CC and other mouse strains with rare phenotypes may elucidate how genetic differences affect both susceptibility and the kinetics of the innate and adaptive response to viral infection. Such approaches will serve to assist in identifying unique correlates of protection from neuroinvasive viral infections to inform therapeutic and vaccine strategies for controlling WNV infection in humans.
MATERIALS AND METHODS
Virus.
West Nile virus TX-2002-HC (WN-TX) was propagated as previously described (26). Working stocks were generated from supernatants collected from infected Vero cell lines and stored at −80°C. One hundred PFU WN-TX was used as the infectious dose in all studies.
CC dRIX lines and disease definitions.
The clinical scoring system to evaluate WNV-infected mice was as follows: 0, healthy mouse (baseline); 1, ruffled fur, lethargy, hunched posture, no paresis, normal gait; 2, altered gait, limited movement in one hind limb; 3, lack of movement, paresis in one or both hind limbs; 4, moribund. Based on weight loss, clinical scoring, and brain histology, CC dRIX lines segregated into three broad categories: asymptomatic (asympt), symptomatic (sympt), and asymptomatic with CNS involvement (asympt-CNS). Symptomatic was defined as a weight loss ≥10% and/or any death, whereas asymptomatic was defined as a weight loss of >10% and no death. CNS histopathologic involvement was evaluated by a veterinary pathologist. The CC dRIX lines featured in this study were CC(041 × 012)F1 and CC(017 × 004)F1 (asympt), CC(005 × 001)F1 and CC(024 × 023)F1 (sympt), and CC(004 × 011)F1 (asympt-CNS).
Mice and infection.
CC dRIX lines were bred at the University of North Carolina at Chapel Hill under specific-pathogen-free (SPF) conditions. Male mice were transferred to the University of Washington at 6 to 8 weeks old and housed directly in a biosafety level 2+ (BSL-2+) laboratory within an SPF barrier facility. Age- and sex-matched 8- to 10-week-old mice were subcutaneously inoculated in the rear footpad with 100 PFU WN-TX. Mice were monitored daily for morbidity (percentage of initial weight loss) and clinical disease scores. Mice were housed under BSL-3 conditions throughout the experiments, and tissues were processed under BSL-3 conditions. All animal experiments were approved by the University of Washington Institutional Animal Care and Use Committee. The Office of Laboratory Animal Welfare of the National Institutes of Health (NIH) has approved the University of Washington (a3464-01), and this study was carried out in strict compliance with the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals.
Histology.
Tissues were perfused with 20 ml phosphate-buffered saline (PBS), whole brain was isolated and one-half of the brain tissue was suspended in PBS–4% paraformaldehyde, pH 7.3. Brains were embedded in paraffin, and 4- to 6-µm sections were prepared and stained with hematoxylin and eosin (H&E) by the UW Histology and Imaging Core. Sections were analyzed using a Nikon Eclipse E600 microscope. All micrograph panels have an original magnification of ×200; for the insets, the original magnification was ×400.
RNA extraction and analysis.
Spleens, kidneys, and brains were removed from mock-infected or WNV-infected mice after perfusion as described above. Organs were suspended in RNA-later and stored, following which they were suspended in PBS, and homogenized using a Precellys 24 machine (Bertin Technologies, France) at 1,500 rpm for 20 s. Total RNA was extracted using the RNeasy kit (Qiagen), DNase treated (Ambion), and evaluated for WNV by using a probe specific for the E gene, IFIT1, and IFN-β1 RNA expression and SYBR green for RT-qPCR. Specific primer sets for WNV, IFIT1, and IFN-β were the following: WNV, 5′-TCAGCGATCTCTCCACCAAAG and 3′-GGGTCAGCCGTTTGTCATTG; mIFIT1, 5′-CTGAGATGTCACTTCACATGGAA and 3′-GTGCATCCCCAATGGGTTCT; mIFNβ1, 5′-CAGCTCCAAGAAAGGACGAAC and 3′-GGCAGTGTAACTCTTCTGCAT.
Cell preparation for flow cytometry assays.
Following euthanasia, mice were perfused with 10 ml PBS to remove any residual intravascular leukocytes. Spleens were homogenized, treated with ammonium-chloride-potassium (ACK) lysis buffer to remove red blood cells, washed, and resuspended in fluorescence-activated cell sorting (FACS) buffer (1× PBS, 0.5% fetal bovine serum [FBS]). To obtain lymphocytes from the CNS, brains were harvested into RPMI and a suspension was created through mechanical disruption. The suspension was added to hypertonic Percoll to create a 30% Percoll solution, vortexed, and centrifuged at 1,250 rpm for 30 min at 4°C. After aspirating the supernatant, any remaining red blood cells in the cell pellet were lysed with ACK, washed, passed through a 70-µm nylon mesh, and resuspended in FACS buffer. Cells were counted by using a hemacytometer and trypan blue exclusion.
Flow cytometry analysis.
Following preparation of single-cell suspensions, cells were plated at 1 × 106 cells/well and stained for surface markers for 15 min on ice. For tetramer staining, cells were stained with the WNV NS4b-H2Db tetramer (generated by the Immune Monitoring Lab, Fred Hutchinson Cancer Research Center). Cells were subsequently fixed, permeabilized (Foxp3 fixation/permeabilization concentrate and diluent; eBioscience), and stained intracellularly with antibodies for 30 min on ice. Flow cytometry was performed on a BD LSRII machine and with BD FACSDiva software. Analysis was performed using FlowJo software.
The following directly conjugated antibodies were used: CD3-ECD (143-2C11), CD4-BV605 (RM4-5), CD8-BV650 (53-6.7), Foxp3-Alexa700 (FJK-16S), and NS4b class I tetramer-allophycocyanin. The Foxp3 intracellular staining kit (eBioscience) was used for fixation/permeabilization and all intracellular staining. AmCyan Live/Dead stain (Invitrogen) was used in all panels for identification of live cells.
Intracellular cytokine staining.
Splenocytes or CNS lymphocytes were stimulated with either medium, the WNV NS4b peptide (SSVWNATTAI), heat-inactivated WNV (multiplicity of infection, 5) or polyclonal stimulation using anti-CD3/CD28, and incubated for 5 h at 37°C. Cells were washed and stained with fixable viability stain (AmCyan Live/Dead stain; Invitrogen) for 30 min on ice, followed by surface staining for 15 min on ice. The cells were then fixed, permeabilized, and stained intracellularly with antibodies according to the manufacturer’s protocol (eBioscience). After staining, the cells were analyzed as described above.
WNV IgM ELISA.
Serum from each animal was collected at the designated endpoint and stored at −80°C until an enzyme-linked immunosorbent assay (ELISA) was performed. Briefly, sample wells were coated with 106 PFU WNV/well and incubated at 4°C overnight. The next day, the plates were washed 4 times, and standards and samples (in duplicate) were incubated for 2 h at room temperature, followed by incubation with goat anti-mouse IgM–horseradish peroxidase (HRP; eBioscience). 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate was used, and the plate was read via a spectrophotometer at 450 nm.
IFN-β ELISA.
Blood was collected from a cohort of WNV-infected CC founder lines at 24, 48, or 72 h postinfection, and serum was assayed for IFN-β in an ELISA as described elsewhere (26).
Statistical analysis.
When comparing groups, two-tailed unpaired Student’s t tests were conducted, with P values of <0.05 considered significant. Error bars show standard errors of the means. For WNV plaque assays and IFN-β ELISA, one-way ANOVA with the Newman-Keuls multiple comparison test was performed.
SUPPLEMENTAL MATERIAL
Oas1b haplotypes and susceptibility scores
ACKNOWLEDGMENTS
Funding for this study was provided by NIH grants U19AI100625 (Projects 3 and 4), R01AI04002, and U19AI083019 (Project 1). We also thank the James B. Pendleton Charitable Trust for their generous equipment donation.
We thank our collaborators in the Systems Immunogenetics Group for helpful discussions and provision of mice.
Footnotes
Citation Graham JB, Thomas S, Swarts J, McMillan AA, Ferris MT, Suthar MS, Treuting PM, Ireton R, Gale M, Jr, Lund JM. 2015. Genetic diversity in the Collaborative Cross model recapitulates human West Nile virus disease outcomes. mBio 6(3):e00493-15. doi:10.1128/mBio.00493-15.
REFERENCES
- 1.Colpitts TM, Conway MJ, Montgomery RR, Fikrig E. 2012. West Nile virus: biology, transmission, and human infection. Clin Microbiol Rev 25:635–648. doi: 10.1128/CMR.00045-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond MS, Mehlhop E, Oliphant T, Samuel MA. 2009. The host immunologic response to West Nile encephalitis virus. Front Biosci 14:3024–3034. doi: 10.2741/3432. [DOI] [PubMed] [Google Scholar]
- 3.Samuel MA, Diamond MS. 2006. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol 80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sejvar JJ. 2007. The long-term outcomes of human West Nile virus infection. Clin Infect Dis 44:1617–1624. doi: 10.1086/518281. [DOI] [PubMed] [Google Scholar]
- 5.Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, Fleischauer A, Leis AA, Stokic DS, Petersen LR. 2003. Neurologic manifestations and outcome of West Nile virus infection. JAMA 290:511–515. doi: 10.1001/jama.290.4.511. [DOI] [PubMed] [Google Scholar]
- 6.Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, McDermott DH, Murphy PM. 2008. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis 197:262–265. doi: 10.1086/524691. [DOI] [PubMed] [Google Scholar]
- 7.Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, Stramer SL, Murphy PM. 2010. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis 201:178–185. doi: 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeb M, Eskandarian S, Rupp M, Fishman N, Gasink L, Patterson J, Bramson J, Hudson TJ, Lemire M. 2011. Genetic variants and susceptibility to neurological complications following West Nile virus infection. J Infect Dis 204:1031–1037. doi: 10.1093/infdis/jir493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carson PJ, Konewko P, Wold KS, Mariani P, Goli S, Bergloff P, Crosby RD. 2006. Long-term clinical and neuropsychological outcomes of West Nile virus infection. Clin Infect Dis 43:723–730. doi: 10.1086/506939. [DOI] [PubMed] [Google Scholar]
- 10.Gottfried K, Quinn R, Jones T. 2005. Clinical description and follow-up investigation of human West Nile virus cases. South Med J 98:603–606. doi: 10.1097/01.SMJ.0000155633.43244.AC. [DOI] [PubMed] [Google Scholar]
- 11.Klee AL, Maidin B, Edwin B, Poshni I, Mostashari F, Fine A, Layton M, Nash D. 2004. Long-term prognosis for clinical West Nile virus infection. Emerg Infect Dis 10:1405–1411. doi: 10.3201/eid1008.030879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leis AA, Stokic DS, Webb RM, Slavinski SA, Fratkin J. 2003. Clinical spectrum of muscle weakness in human West Nile virus infection. Muscle Nerve 28:302–308. doi: 10.1002/mus.10440. [DOI] [PubMed] [Google Scholar]
- 13.Marciniak C, Sorosky S, Hynes C. 2004. Acute flaccid paralysis associated with West Nile virus: motor and functional improvement in 4 patients. Arch Phys Med Rehabil 85:1933–1938. doi: 10.1016/j.apmr.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Siddharthan V, Hall JO, Morrey JD. 2011. Autonomic nervous dysfunction in hamsters infected with West Nile virus. PLoS One 6:e19575. doi: 10.1371/journal.pone.0019575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanteri MC, Heitman JW, Owen RE, Busch T, Gefter N, Kiely N, Kamel HT, Tobler LH, Busch MP, Norris PJ. 2008. Comprehensive analysis of West Nile virus-specific T cell responses in humans. J Infect Dis 197:1296–1306. doi: 10.1086/586898. [DOI] [PubMed] [Google Scholar]
- 16.Lanteri MC, O’Brien KM, Purtha WE, Cameron MJ, Lund JM, Owen RE, Heitman JW, Custer B, Hirschkorn DF, Tobler LH, Kiely N, Prince HE, Ndhlovu LC, Nixon DF, Kamel HT, Kelvin DJ, Busch MP, Rudensky AY, Diamond MS, Norris PJ. 2009. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J Clin Invest 119:3266–3277. doi: 10.1172/JCI39387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petersen LR, Fischer M. 2012. Unpredictable and difficult to control—the adolescence of West Nile virus. N Engl J Med 367:1281–1284. doi: 10.1056/NEJMp1210537. [DOI] [PubMed] [Google Scholar]
- 18.Daffis S, Samuel MA, Keller BC, Gale M Jr, Diamond MS. 2007. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog 3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS. 2008. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M Jr, Diamond MS. 2008. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal west Nile virus infection. J Virol 82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, Silverman RH, Gale M Jr, Diamond MS. 2006. PKR and RNase L contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol 80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M Jr. 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog 6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. 2007. Antiviral signaling through pattern recognition receptors. J Biochem 141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 24.Suthar MS, Diamond MS, Gale M Jr. 2013. West Nile virus infection and immunity. Nat Rev Microbiol 11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- 25.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M Jr. 2006. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol 80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M Jr. 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog 6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr. 2013. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol 87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. 2003. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol 77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med 198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Lobigs M, Lee E, Koskinen A, Müllbacher A. 2006. CD8(+) T cell-mediated immune responses in West Nile virus (Sarafend strain) encephalitis are independent of gamma interferon. J Gen Virol 87:3599–3609. doi: 10.1099/vir.0.81306-0. [DOI] [PubMed] [Google Scholar]
- 31.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, Bleich A, Bogue M, Broman KW, Buck KJ, Buckler E, Burmeister M, Chesler EJ, Cheverud JM, Clapcote S, Cook MN, Cox RD, Crabbe JC, Crusio WE, Darvasi A, Deschepper CF, Doerge RW, Farber CR, Forejt J, Gaile D, Garlow SJ, Geiger H, Gershenfeld H, Gordon T, Gu J, Gu W, de Haan G, Hayes NL, Heller C, Himmelbauer H, Hitzemann R, Hunter K, Hsu HC. 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 32.Threadgill DW, Churchill GA. 2012. Ten years of the Collaborative Cross. G3 2:153–156. doi: 10.1534/g3.111.001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Threadgill DW, Miller DR, Churchill GA, de Villena FP. 2011. The Collaborative Cross: a recombinant inbred mouse population for the systems genetic era. ILAR J 52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- 34.Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. 2007. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome 18:473–481. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Courtney SC, Di H, Stockman BM, Liu H, Scherbik SV, Brinton MA. 2012. Identification of novel host cell binding partners of Oas1b, the protein conferring resistance to flavivirus-induced disease in mice. J Virol 86:7953–7963. doi: 10.1128/JVI.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon-Chazottes D, Frenkiel MP, Montagutelli X, Guénet JL, Desprès P, Panthier JJ. 2011. Transgenic expression of full-length 2′,5′-oligoadenylate synthetase 1b confers to BALB/c mice resistance against West Nile virus-induced encephalitis. Virology 417:147–153. doi: 10.1016/j.virol.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellåker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assuncao JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imran M, Manzoor S, Khattak NM, Tariq M, Khalid M, Javed F, Bhatti S. 2014. Correlation of OAS1 gene polymorphism at exon 7 splice accepter site with interferon-based therapy of HCV infection in Pakistan. Viral Immunol 27:105–111. doi: 10.1089/vim.2013.0107. [DOI] [PubMed] [Google Scholar]
- 40.Urosevic N, Silvia OJ, Sangster MY, Mansfield JP, Hodgetts SI, Shellam GR. 1999. Development and characterization of new flavivirus-resistant mouse strains bearing Flv(r)-like and Flv(mr) alleles from wild or wild-derived mice. J Gen Virol 80:897–906. [DOI] [PubMed] [Google Scholar]
- 41.Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, Rutherford A, Astakhova NM, Perelygin AA, Busch MP, Murray KO, Sejvar JJ, Green S, Kriesel J, Brinton MA, Bamshad M. 2011. Host genetic risk factors for West Nile virus infection and disease progression. PLoS One 6:e24745. doi: 10.1371/journal.pone.0024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thamizhmani R, Vijayachari P. 2014. Association of dengue virus infection susceptibility with polymorphisms of 2′-5′-oligoadenylate synthetase genes: a case-control study. Braz J Infect Dis 18:548–550. doi: 10.1016/j.bjid.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, Bradel-Tretheway B, Bryan JT, Buus RJ, Gralinski LE, Haagmans BL, McMillan L, Miller DR, Rosenzweig E, Valdar W, Wang J, Churchill GA, Threadgill DW, McWeeney SK, Katze MG, de Villena P-M F, Baric RS, Heise MT. 2013. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLOS Pathog 9:e1003196. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou S, Foster GA, Dodd RY, Petersen LR, Stramer SL. 2010. West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis 202:1354–1361. doi: 10.1086/656602. [DOI] [PubMed] [Google Scholar]
- 45.Hayes EB, Sejvar JJ, Zaki SR, Lanciotti RS, Bode AV, Campbell GL. 2005. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis 11:1174–1179. doi: 10.3201/eid1108.050289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oas1b haplotypes and susceptibility scores