

ABSTRACT
Bacteria coordinate a variety of social behaviors, important for both environmental and pathogenic bacteria, through a process of intercellular chemical signaling known as quorum sensing (QS). As microbial resistance to antibiotics grows more common, a critical need has emerged to develop novel anti-infective therapies, such as an ability to attenuate bacterial pathogens by means of QS interference. Rgg quorum-sensing pathways, widespread in the phylum Firmicutes, employ cytoplasmic pheromone receptors (Rgg transcription factors) that directly bind and elicit gene expression responses to imported peptide signals. In the human-restricted pathogen Streptococcus pyogenes, the Rgg2/Rgg3 regulatory circuit controls biofilm development in response to the short hydrophobic peptides SHP2 and SHP3. Using Rgg-SHP as a model receptor-ligand target, we sought to identify chemical compounds that could specifically inhibit Rgg quorum-sensing circuits. Individual compounds from a diverse library of known drugs and drug-like molecules were screened for their ability to disrupt complexes of Rgg and FITC (fluorescein isothiocyanate)-conjugated SHP using a fluorescence polarization (FP) assay. The best hits were found to bind Rgg3 in vitro with submicromolar affinities, to specifically abolish transcription of Rgg2/3-controlled genes, and to prevent biofilm development in S. pyogenes without affecting bacterial growth. Furthermore, the top hit, cyclosporine A, as well as its nonimmunosuppressive analog, valspodar, inhibited Rgg-SHP pathways in multiple species of Streptococcus. The Rgg-FITC-peptide-based screen provides a platform to identify inhibitors specific for each Rgg type. Discovery of Rgg inhibitors constitutes a step toward the goal of manipulating bacterial behavior for purposes of improving health.
IMPORTANCE
The global emergence of antibiotic-resistant bacterial infections necessitates discovery not only of new antimicrobials but also of novel drug targets. Since antibiotics restrict microbial growth, strong selective pressures to develop resistance emerge quickly in bacteria. A new strategy to fight microbial infections has been proposed, namely, development of therapies that decrease pathogenicity of invading organisms while not directly inhibiting their growth, thus decreasing selective pressure to establish resistance. One possible means to this goal is to interfere with chemical communication networks used by bacteria to coordinate group behaviors, which can include the synchronized expression of genes that lead to disease. In this study, we identified chemical compounds that disrupt communication pathways regulated by Rgg proteins in species of Streptococcus. Treatment of cultures of S. pyogenes with the inhibitors diminished the development of biofilms, demonstrating an ability to control bacterial behavior with chemicals that do not inhibit growth.
INTRODUCTION
Intercellular chemical signaling among bacteria (quorum sensing [QS]) provides communities of microbes the opportunity to coordinate gene expression to facilitate group behavior. Bacteria establish communication networks by emitting signaling molecules (here referred to as pheromones) to be detected by other members of the community, eliciting a response. Quorum sensing influences a variety of behaviors, and in some species, it may facilitate the development or dispersal of biofilms, may promote an aggressive attack on neighbors or coordinate a community defense system, or may foster symbiotic relationships with a host or engender pathological consequences (1–4). In cases where QS contributes to behaviors that are detrimental to the health of humans or animals, it may be beneficial to identify methods that disrupt active QS circuits (5). Furthermore, as antibiotic-resistant bacteria continue to threaten health, new sustainable strategies to combat microbial infections are needed, thus presenting an opportunity to target virulence through methods like QS interference that do not rely on impeding bacterial growth.
Streptococcus pyogenes (group A Streptococcus [GAS]) is a human-restricted pathogen responsible for a variety of diseases that range in severity from localized, superficial infections like impetigo and pharyngitis to highly aggressive, invasive infections like necrotizing fasciitis and toxic shock (6–8). Immune responses to GAS infections in some instances generate autoantibodies and immune complexes that direct immune responses toward tissues of the heart (acute rheumatic fever) and kidney (glomerulonephritis) (6, 9–12). GAS infections cause more than 500,000 deaths annually, ranking this pathogen among the most common infectious agents worldwide with significant morbidity and mortality (13). β-Lactam antibiotics are generally the first choice in treatment for GAS-related disease (14), and resistance to this class of drug has, remarkably, not yet emerged; however, treatment failure is common, perhaps due to a variety of reasons (15–17). For preferred alternative drugs, like macrolides, used for penicillin-allergic patients, treatment failures, and cases of severe nonpurulent infection, resistance has emerged (18, 19). Thus, development of new alternative methods to treat GAS infections is needed and may reveal new strategies to combat other pathogens.
Previously we described a quorum-sensing network conserved in all sequenced genomes of GAS that utilize two Rgg protein family members (Rgg2 and Rgg3) as cytoplasmic receptors of short hydrophobic peptide (SHP) pheromones (20, 21). Rgg family members are widespread among Firmicutes and are ubiquitous among all species of Streptococcus and Lactobacillales, and multiple paralogs are often found within a genome, presumably serving as independently functioning transcriptional regulators. Rgg proteins contain an N-terminal helix-turn-helix (HTH) DNA-binding domain and a C-terminal alpha-helical domain predicted to fold in a fashion similar to that seen with tetratricopeptide repeat (TPR)-containing proteins of the prototypical Rap/NprR/PlcR/PrgX (RNPP) protein family (22–26). Signaling peptides corresponding to the Rgg and RNPP families are produced by the ribosome from coding transcripts as unmodified, linear prepeptides that are secreted and processed through various pathways that release mature pheromones from the cell that can be recovered in culture supernatants. In signal-receiving cells, pheromones must be imported to the cytoplasm, typically by oligopeptide permease (Opp) transporters, where they directly engage and control activity of cytoplasmic receptors (23, 27–29). Consequently, small molecules that disrupt pheromone-receptor interactions must also traverse the cytoplasmic envelope.
In GAS, Rgg2 and Rgg3 regulate expression of polycistronic transcripts encoding SHP pheromones and downstream genes. The effect of SHP pheromones on the transcriptional state is borne out in two ways, by suppressing the repressive activity of Rgg3 and simultaneously enhancing the activating properties of Rgg2, culminating in robust induction of transcription. In the absence of pheromones, transcriptional intensity is maintained at very low levels. The phenotypic effect of SHP pheromone induction can be seen in some GAS strains in the development of biofilms, where addition of as little as 5 nM synthetic SHP leads to surface-associated films (21). GAS biofilms have been suggested to be a contributing cause of antibiotic treatment failures of pharyngeal infections (30–32).
By a method developed to study interactions between Rgg proteins and their cognate peptide ligands (21), an in vitro, small-volume, high-throughput, compound library screen was utilized to identify compounds that specifically interfere with Rgg3-SHP interactions. These compounds were subsequently tested on S. pyogenes bioluminescent reporter cultures and shown to block Rgg-mediated transcription and prevent biofilm formation. Rgg2/3-SHP circuits are well conserved across multiple species of Streptococcus (33, 34), and we found that inhibitors worked to disrupt Rgg-dependent transcription in Streptococcus agalactiae (group B Streptococcus), S. dysgalactiae (group G Streptococcus), and S. porcinus.
RESULTS
A fluorescence polarization high-throughput screen identified Rgg3 antagonists.
We previously described a method quantifying a direct, reversible interaction between Rgg proteins and SHP peptides using a competitive fluorescence polarization assay (21). From these assays, we found that purified Rgg3 formed complexes with a synthetic fluorescein isothiocyanate (FITC)-labeled SHP2-C8 (comprising the C-terminal eight amino acids encoded by the shp2 gene, DILIIVGG), with an apparent Kd of 0.2 µM (21). We hypothesized that due to the reversibility of this interaction it might be possible to find compounds that displace bound SHP from Rgg–FITC-SHP complexes, and such compounds might therefore interfere with Rgg2/3-regulated pathways, including biofilm development. We employed the competition-fluorescence polarization (FP) assay in a high-throughput fashion (see Materials and Methods) to screen the Prestwick Chemical Library, containing 1,280 agency-approved drugs, to identify compounds that decreased FP values attained by Rgg3–FITC-SHP2-C8 complexes (Fig. 1A). To assist our ability to identify compounds that worked specifically on Rgg3, we developed a second FP assay utilizing the S. mutans ComR protein. ComR is another Rgg-type protein, present among streptococcal species of the pyogenes, mutans, and bovis groups, and binds directly to an XIP (S. mutans GLDWWSL) peptide pheromone (35–37). Direct binding between ComR and FITC-XIP was observed in this assay (Fig. 1C), and the interaction was found also to be reversible, since unlabeled XIP, but not a different peptide with similar properties (ADLAYQSA), displaced FITC-XIP associated with ComR in a competition assay (Fig. 1D). Preformed ComR–FITC-XIP complexes were thus also used to screen the Prestwick Chemical Library. The results of screening, performed in duplicate for each targeted receptor-ligand complex, are presented in Fig. 1B. Five hits were identified that exhibited ≥75% inhibition of FITC-SHP binding to Rgg3 while displaying ≤20% inhibition of FITC-XIP binding to ComR (Fig. 1B; Table 1). Since compounds that specifically bind Rgg3 are the primary focus of this study, ComR antagonists will be reported in detail elsewhere. Compounds that displayed nonspecific antagonism in both assay types were set aside and assumed to interfere with the fluorescent readout of the assay.
FIG 1 .

Identification of Rgg3-specific antagonists from high-throughput screening of the drug compound library. (A) Rgg3–FITC-SHP2-C8 complexes were combined with individual compounds of an arrayed drug library and screened in duplicate by fluorescence polarization (FP) to identify those that disrupted the receptor-ligand interaction. Data from two replicate experiments A and B are plotted to illustrate reproducibility. (B) The library was also screened for disruption of ComR–FITC-XIP complexes as was done for Rgg3. Data points denote the percent disruption (i.e., percent inhibition) for each compound using the average of two FP measurements. The top five compounds that selectively inhibited Rgg3 by ≥75% and ComR by ≤20% are highlighted (red dots). (C) FITC-XIP (10 nM) was titrated with purified His-ComR to generate a binding curve by the direct-FP assay. (D) Synthetic unlabeled XIP or HTS compounds were assessed for their ability to compete with FITC-XIP for binding to ComR..
TABLE 1 .
High-throughput screening hits
| HTS hit | Rgg3 |
ComR |
||
|---|---|---|---|---|
| Avg % inhibition | Z′ | Avg % inhibition | Z′ | |
| Cyclosporine A | 115 | 0.74 | 0 | 0.41 |
| Telmisartan | 99 | 0.74 | 3 | 0.35 |
| Simvastatin | 85 | 0.75 | 10 | 0.39 |
| Saquinavir mesylate | 84 | 0.74 | 13 | 0.35 |
| Olmesartan | 73 | 0.74 | 1 | 0.35 |
Cyclosporine A and a chemical analog bind Rgg3 with an affinity similar to that of SHP.
To validate and characterize the Rgg3-inhibitory activity of each of the top five hits, dose-response experiments were performed using the FP-based binding assay in which a 0.1 to 100 µM concentration of each compound was incubated with 500 nM Rgg3 premixed with 10 nM FITC–SHP2-C8 (Fig. 2A). Cyclosporine A (CsA) bound to Rgg3 with an affinity similar (0.45 µM) to that of SHP2-C8 for Rgg3. The remaining four best compounds displayed at least a 10-fold-lower Kd than CsA (Fig. 2A; Table 2). CsA is a fungus-derived drug with immunosuppressive properties (38) due to its ability to target cyclophilin and inhibit calcineurin and cytokine responses in T cells (39). Nonimmunosuppressive analogs of CsA are well characterized (40, 41), and we hypothesized that one, valspodar (PSC-833) (42), whose structure is highly similar to that of CsA, would possess Rgg3-SHP-disrupting properties. Valspodar also bound to Rgg3 with an affinity (0.55 µM) similar to that of CsA and SHP (Fig. 2A).
FIG 2 .

Rgg antagonist activities. (A) The top five compounds identified from screening, as well as the CsA analog valspodar, were assessed for their ability to compete with FITC-SHP2-C8 for binding to Rgg3. Complexes of His6-SUMO-Rgg3–FITC-SHP2-C8 (premixed at 160 nM Rgg3, 10 nM FITC-SHP2-C8) were titrated with the HTS hits. (B to D) Relative luminescence activity of the Pshp2-luxAB reporter in response to a single concentration (10 µM) of test compounds (B), a range of concentrations in the wild-type genetic background (BNL148) (C), or the activity in a Δrgg3 strain that is incapable of producing endogenous SHP pheromones (BNL178) (D). (E) Optical densities of S. pyogenes cultures grown at 37°C in the presence of sSHP2-C8 (10 nM) with Rgg antagonists provided at 10 µM. (F) Relative luminescence of PsigX-luxAB in S. pyogenes and S. mutans cultures in response to CsA and valspodar.
TABLE 2 .
HTS hits and Kd values as obtained from fluorescence polarization and IC50s of HTS hits obtained from luciferase assay dose-response curves
| HTS hit | Kd (µM) for Rgg3 | IC50 (nM)a |
|---|---|---|
| SHP2-C8 | 0.22 | |
| Cyclosporine A | 0.45 | 203.3 |
| Valspodar | 0.55 | 251.5 |
| Telmisartan | 8.3 | NT |
| Simvastatin | 9.7 | >10 µM |
| Saquinavir mesylate | 8.5 | >10 µM |
| Olmesartan | 19.8 | NT |
NT, not tested.
Validation of high-throughput screeing (HTS) hits using a cell-based luciferase reporter.
Since Rgg proteins interact with pheromones in the bacterial cytoplasm, the cell surface and envelope are likely significant barriers to compounds that disrupt these receptor-ligand interactions. To test the ability of Rgg antagonists to surmount these barriers, a bioluminescent reporter strain (BNL148) containing an Rgg-regulated promoter, Pshp2-luxAB, was employed that enables the quantification of Rgg activity in culture. Addition of synthetic pheromone to BNL148 cultures leads to robust induction profiles that exceed the basal level of transcription more than 100-fold. We asked if Rgg antagonists could block SHP-induced light activity by providing BNL148 with 10 nM SHP2-C8 together with a 1,000-fold excess (10 µM) of each antagonist compound (Fig. 2B). At these concentrations, CsA and valspodar completely abolished light production, and a third hit, saquinavir, inhibited transcription by approximately 90%. The remaining compounds telmisartan, olmesartan, and simvastatin were less effective, inhibiting light production by less than 50% (Fig. 2B).
To further evaluate their specific activities, antagonist compounds were titrated into cultures of the synthetic SHP2-C8 (sSHP)-induced bioluminescent reporter. The relative luminescence activity that followed was used to generate dose-response curves and to calculate the inhibitory concentrations resulting in 50% reduction of the log maximum luminescence activity that was generated when sSHP2-C8 was provided alone (IC50) (Fig. 2C; Table 2). CsA and valspodar were found to be the most active antagonists and had similar submicromolar IC50s, whereas saquinavir was 10 times less active. A direct correlation was observed between apparent binding constants of antagonists toward Rgg3, as determined by FP assays, and the compound bioactivities as seen in transcriptional reporter measurements, with CsA and valspodar displaying the lowest Kd and IC50s. Therefore, only these compounds were pursued in further analyses. Because Rgg2 and Rgg3 both contribute to shp promoter regulation, we also tested CsA and valspodar on a reporter strain in which only Rgg2 was present (BNL178). Both compounds were effective at inhibiting transcription in this genetic background, indicating that the inhibitors identified using Rgg3 are also capable of inhibiting Rgg2 (Fig. 2D). Importantly, even at these high concentrations, none of the compounds, with the exception of simvastatin, inhibited culture growth (Fig. 2E).
To rule out the possibility that CsA and valspodar inhibited light production in BNL148 by a mechanism independent of Rgg-SHP disruption, perhaps either by blocking SHP-peptide uptake through the oligopeptide permease transporter (Opp) or by disrupting the luciferase enzymatic reaction, we tested the antagonists in the ComR-XIP QS system, since it also relies on Opp for transport of its cognate peptide XIP and, upon activation, triggers expression from the sigX gene promoter, which we have previously studied using luciferase as an assay readout (29). The PsigX-luxAB reporter strains S. pyogenes [MGAS315(pWAR200)] and S. mutans [UA159(pWAR304)] were each incubated with 10 µM CsA or valspodar in the presence of 100 nM XIP, but light production was not affected (Fig. 2F). Additionally, to test the possibility that the studied antagonists could alter signaling by the remaining Rgg-family protein in GAS, RopB, we tested their effects using a luciferase reporter of the speB promoter, whose transcription requires RopB. The PspeB-luxAB reporter strain [NZ131(pWAR193)] was grown on C medium, which promotes RopB-dependent activation of PspeB (Fig. 2G). Addition of 10 µM CsA or valspodar was unable to block activation of PspeB, while addition of 300 mM NaCl, a known culturing condition that suppresses speB expression (43), abolished luciferase activity. Therefore, we conclude that CsA and valspodar are inhibitors specific for the Rgg2/3 quorum-sensing pathway and its Rgg-SHP interaction.
Antagonists block SHP-dependent biofilm development.
We previously reported that addition of SHP pheromones to cultures of S. pyogenes strain NZ131 stimulates development of biofilms (20, 21), and therefore we asked if the Rgg antagonists were capable of blocking this phenotype. As previously documented, addition of 10 nM sSHP2-C8 peptide to cultures was sufficient to induce formation of biofilm mats of S. pyogenes along the bottom surface of culture wells. Concentrations as low as 0.625 µM CsA added to cultures together with SHP peptides were unable to develop biofilms (Fig. 3). Similar results are shown for valspodar; a concentration of 0.312 µM was sufficient to inhibit the biomass increase (Fig. 3). Additionally, we tested strains that exhibit high levels of basal biofilm formation, including an NZ131 Δrgg3 mutant in which SHP pheromone production is rendered constitutive due the lack of the Rgg3 repressor (20), and two other GAS serotypes, GA19681(M6) and MGAS5005(M1). As shown in Fig. 4A, CsA is able to abolish activation of the Pshp2-luxAB reporter in all tested strains. However, the degree to which SHPs induce biofilm development, and hence the ability of CsA to inhibit biofilms, was more variable (Fig. 4B and C). Crystal violet staining was sufficient to observe modest inhibition by CsA on SHP-dependent biofilm development; however, MGAS5005, which produces high levels of biofilm irrespective of SHP pheromone, was unaffected by CsA. These results indicate that, whereas QS signaling, as well as QS inhibition by CsA is functional in all three GAS serotypes tested, the physiological consequence of SHP signaling resulting in biofilm development remains a strain-dependent phenomenon.
FIG 3 .

Rgg3 antagonists prevent biofilm formation. S. pyogenes wild-type strain NZ131 cultures, grown in 24-well plates, were treated with 10 nM SHP2-C8 and various concentrations of CsA and valspodar. At 20 h, biofilms were assessed by a standard crystal violet staining method. Error bars indicate standard errors from a minimum of three independent experiments.
FIG 4 .
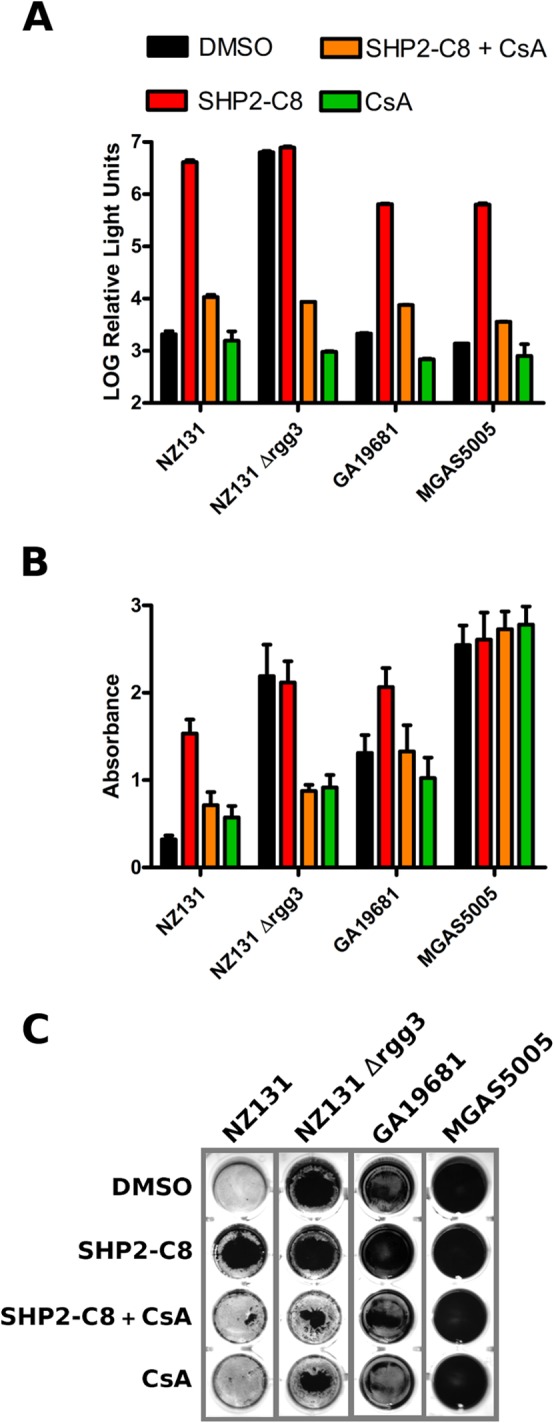
Effects of CsA on SHP signaling and biofilm formation in different S. pyogenes strains. (A) Relative luminescence activity of the Pshp2-luxAB reporter in response to the addition of 10 nM SHP2-C8 pheromone and 5 µM CsA. Bars show standard deviations. (B) S. pyogenes strains grown in 24-well plates were treated with 10 nM SHP2-C8 and 5 µM CsA. At 20 h, biofilms were assessed by a standard crystal violet staining method. Error bars indicate standard errors from a minimum of three independent experiments. (C) Picture of one representative biofilm experiment.
Rgg antagonists block quorum sensing in several Streptococcus species.
Rgg-SHP quorum-sensing circuits are conserved in multiple species of Streptococcus (20, 29, 34, 35), where sequence alignments show more than 50% identity and more than 70% similarity (33). We therefore predicted that Rgg antagonists might block Rgg quorum-sensing pathways in other species of streptococci. To test this, the inhibitory activities of CsA and valspodar were assessed using Pshp-luxAB bioluminescent reporters constructed in S. agalactiae (group B Streptococcus [GBS]), S. dysgalactiae (group G Streptococcus [GGS]), and S. porcinus (group E Streptococcus [GES]), all important pathogens in their own right. Reporters in GBS and GGS strains, unlike in S. pyogenes, were found to be induced even in the absence of exogenously provided SHP peptides, indicating that these strains produce endogenous levels of SHP that are sufficient to induce Rgg-dependent promoters under the growth conditions tested. S. porcinus remained uninduced in the absence of exogenous SHP, and therefore sSHP was needed to stimulate light production. Under conditions found to produce luciferase in each of these species, CsA and valspodar were able to inhibit light production at least 5-fold in each (Fig. 5); however, valspodar had only a moderate effect on the S. porcinus strain.
FIG 5 .

Rgg3 antagonists are effective in multiple streptococcal species. Relative luminescence activity of Pshp-luxAB transcriptional reporters in multiple streptococcal species in response to Rgg antagonists (5 µM).
DISCUSSION
Antimicrobials, whether bactericidal or growth inhibitory, place strong selective pressures on bacteria to develop resistance, and their widespread use has accelerated the emergence of resistant pathogens. Though there is a desperate need for the discovery and production of novel antimicrobials, an investment in developing new strategies that target mechanisms of virulence rather than bacterial growth may offer therapeutic potential that is more sustainable. These so-called anti-infective therapies (44–46) would ideally target bacterial pathways that lead to disease but not interfere with bacterial growth. Impeding quorum-sensing pathways is a strategy that conforms to antivirulence approaches and therefore could lead to an effective therapeutic tactic.
In this study, we identified inhibitors of the Rgg-SHP quorum-sensing signaling pathway found in streptococcal species. An in vitro fluorescence polarization assay was used to screen compounds from the Prestwick Chemical Library that specifically target Rgg3 and compete with SHP binding. Our previous studies found that SHP peptides bind to both Rgg2 and Rgg3 with equivalent affinities (21), and though we would predict that using either Rgg2 or Rgg3 would suffice to identify compounds that disrupt SHP binding, it is possible that competitors may be selective for one receptor or the other. Rgg3 was used in this study for practical reasons, as the His6-SUMO tag provided a convenient method for purification with robust tag removal. The Prestwick Chemical Library contains compounds approved for human use by various agencies, including the FDA. The library’s drug diversity provides a foundation on which drug optimization can proceed by medicinal chemistry approaches. Some of the compounds identified by our screen exhibited submicromolar affinity to Rgg3 and blocked transcription of Rgg-controlled promoters without affecting bacterial growth. The most potent inhibitor identified from the screen, based on the measured binding affinity to Rgg3 and the IC50 of luciferase transcriptional reporters, was cyclosporine A. Cyclosporine was first isolated from the fungus Tolypocladium inflatum and shown to contain immunosuppressive activities in the 1970s (47, 48). CsA targets cyclophilin and modulates its interaction with calcineurin, inhibiting signal transduction of T-cell receptor signaling to induce cytokine expression (49–51). A variety of CsA analogs exist, many without immunosuppressive activities, such as valspodar (PSC 833, [3′-keto-Bmt1-Val2]-cyclosporine) (52, 53). Since valspodar is structurally similar to CsA, it is perhaps not surprising that its Rgg-inhibitory activity nearly matched that of CsA (IC50 = 0.5 µM). Both inhibitors were found to act specifically on Rgg2 and Rgg3 receptors and did not bind to ComR (an Rgg-family homolog whose cognate peptide ligand, XIP, is unlike SHP2/3) or interfere with ComR-controlled transcription. CsA and valspodar also prevented SHP-dependent biofilm formation in S. pyogenes and blocked Rgg-SHP-mediated QS pathways in several other species of Streptococcus, indicating the effectiveness of this screening platform, even on a relatively small library (1,280 compounds), to identify inhibitors of multiple organisms containing Rgg quorum-sensing pathways. Further screening of larger chemical libraries might yield novel scaffolds with lower IC50s. The screen also identified inhibitors capable of specifically blocking ComR and other compounds that inhibited both ComR and Rgg3 proteins and may act as generalist compounds capable of binding either type of Rgg protein. It will be of particular interest to elucidate the mechanism underlying nonspecific Rgg inhibitors, considering that the mechanism of action leading to their identification relies on competition with peptide pheromone binding to the receptors. Since SHP and XIP ligands are highly dissimilar, it is not clear at this stage of understanding how a competitive inhibitor is able to substitute for either ligand.
Rgg proteins likely switch between two functional states whose conformation is determined by SHP binding. Our studies indicate that Rgg3 binds to DNA and represses transcription of target promoters in the absence of pheromone. It is predicted that upon binding SHP, Rgg3 undergoes a conformational change that makes DNA binding unfavorable, preventing Rgg3 from acting as a transcriptional repressor. Rgg2 behaves differently from Rgg3, since the Rgg2-SHP complex binds DNA and induces transcriptional activation. Our studies indicate that affinity of Rgg2 for DNA is not affected by SHP; instead, SHP likely drives a conformational change in Rgg2 that favors positive interaction with RNA polymerase (54). Thus, for both Rgg2 and Rgg3 proteins, SHP pheromones promote a functional state, resulting in increased transcription of target promoters. Consequently, preventing Rgg-SHP complexes has the combined effect of blocking Rgg2-dependent transcriptional activation while simultaneously favoring Rgg3-induced repression. CsA and valspodar each appear to compete directly with SHP binding to both Rgg2 and Rgg3. Because Rgg inhibitors were selected by their ability to displace FITC-SHP, it is likely that CsA and valspodar share the same or overlapping binding sites on Rgg. Elucidation of the exact SHP and CsA binding sites on Rgg proteins would greatly facilitate the design of enhanced anti-Rgg compounds but will likely require cocomplex structure elucidation and targeted mutagenesis to fully understand the nature of interactions.
A primary aim in the development of antivirulence therapeutics is to decrease the pathogenic potential of bacteria without directly inhibiting their growth. Cyclosporine A and valspodar do not display toxic effects on GAS growth at concentrations at least 20-fold greater (10 µM) than the effective concentrations needed to inhibit transcription and biofilm development. Even if a genetic variant resistant to CsA or other inhibitors were to emerge at these elevated concentrations in bacterial culture, there is no apparent reason why it would exhibit a growth advantage. However, our observations indicate that SHPs give rise to unknown changes of the bacterial surface that culminate in the development of biofilms (20). Though the physiological significance of biofilm development remains unclear for GAS, it has been proposed that biofilms could account for antibiotic treatment failure and recurrence of GAS infections (55). If Rgg-dependent quorum sensing contributes to biofilm development in vivo, and if Rgg inhibitors block this process, then GAS quorum-sensing inhibitor (QSI)-resistant variants that emerge as ligand-blind bypass mutants would be unable to regulate the processes enabling these bacteria to enter, and possibly exit, the biofilm state. Though such resistant mutants would be nonresponsive to a QS inhibitor, they would also be incapacitated in the ability to respond properly to regulatory systems that presumably provide an evolutionary advantage, given the absolute conservation of these pathways in all sequenced genomes. However, in the face of antibiotic challenge, a QSI-resistant variant that constitutively conforms to a biofilm lifestyle may have enhanced resistance to antibiotic treatment but may also be less pathogenic. A primary concern yet to be addressed in any GAS study is the consequence of misregulation of any quorum-sensing pathway of GAS in pathogenesis. It should be cautioned that disruption of biofilms may not be ideal, as it has been demonstrated that dispersion of GAS biofilms as a consequence of enhanced activity of the SpeB protease (a primary transcriptional target of RopB [Rgg1]) in a skin infection model led to enhanced lesions (56).
MATERIALS AND METHODS
Bacterial strains, plasmids, and culturing conditions.
Bacterial strains and plasmids used in this study are listed in Table 3. S. pyogenes, S. mutans, S. agalactiae (group B Streptococcus [GBS]), S. dysgalactiae subsp. equisimilis (group G Streptococcus [GGS]), and S. porcinus were routinely grown in Todd-Hewitt medium (BD Biosciences) supplemented with 0.2% (wt/vol) yeast extract (AMRESCO) (THY) or in a chemically defined medium (CDM) (20) containing 1% (wt/vol) glucose. Luciferase reporter assays were performed by growing reporter strains in CDM. When necessary, antibiotics were included at the following concentrations for S. pyogenes: chloramphenicol (Cm), 3 µg ml−1; erythromycin (Em), 0.5 µg ml−1; spectinomycin (Spec), 100 µg ml−1. Escherichia coli strains DH10β (Invitrogen) and BH10C (57) were used for cloning purposes and were grown in Luria broth (LB) or on Luria agar with antibiotics at the following concentrations: chloramphenicol, 10 µg ml−1; erythromycin, 500 µg ml−1; spectinomycin, 100 µg ml−1, ampicillin, 100 µg ml−1. The E. coli expression strain C41(DE3) (58) was maintained on LB agar with ampicillin.
TABLE 3 .
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| NZ131 | Wild-type S. pyogenes M49 strain | 64, 65 |
| BNL148 | NZ131 integrated with pBL111 Pshp2-luxAB reporter; Ermr | 20] |
| BNL178 | NZ131 Δrgg3 shp3GGG shp2GGG::Pshp2-luxAB | 21 |
| MGAS315(pWAR200) | S. pyogenes M3 isolate with PsigX-luxAB reporter on plasmid pWAR200 | 37, 66 |
| UA159(pWAR304) | S. mutans isolate with PsigX-luxAB on plasmid pWAR304 | 35, 67 |
| NZ131(pWAR193) | S. pyogenes M49 strain with PspeB-luxAB reporter on plasmid pWAR193; Ermr | This study |
| A909(pSar110) | Wild-type S. agalactiae A909 clinical isolate with Pshp1520-luxAB cloned in pLZ12-spec | 33, 68, 69 |
| GGS-LT1(pLC301) | S. dysgalactiae subsp. equisimilis strain with Pshp2-luxAB on pLZ12-spec | 33; this study |
| NCTC 10999(pJC254) | Wild-type S. porcinus Collins et al. (ATCC 43138) strain with Pshp3-luxAB reporter on pJC254; Specr | 70; this study |
| Plasmids | ||
| p7INT | Shuttle-suicide vector that integrates at streptococcal bacteriophage T12 attB site; Ermr | 71 |
| pLZ12-spec | Shuttle vector encoding spectinomycin resistance; pWV01 origin; Specr | 60 |
| pJC254 | 227-bp upstream region of shp3 fused to luxAB genes and cloned in BamHI and EcoRI sites of pLZ12-spec | This study |
| pWAR368 | pET 15b expression vector with UA159 comR cloned into NdeI and BamHI restriction sites; Ampr | This study |
| pWAR193 | 947-bp upstream region of NZ131 speB cloned in front of luxAB in p7INT; Ermr | This study |
| pLC301 | 500-bp upstream region of S. pyogenes shp2 fused to luxAB genes and cloned in pLZ12-spec; Specr | This study |
Construction of plasmids.
Construction of derivative strains and luciferase reporters has been discussed in detail previously (20, 33, 59). Plasmid pLC301 (Pshp2-luxAB reporter for S. dysgalactiae) was obtained by PCR amplifying Pshp2-luxAB from plasmid pBL111 (20) using primers BL27 and BL43 (20) and cloning the fragment into the pLZ12-spec plasmid using BamHI and EcoRI restriction sites. pJC254 was constructed to monitor the expression of the S. porcinus shp3 gene. A 227-bp upstream region of the open reading frame was amplified using primers JC324 (CATGGGATCCTACAAGATATTTCGGACTCG) and JC325 (CAAATATTTCCAAACTTCATCTCGCTTCTCCTTTTACTTT), and this promoter product was fused to luxAB in a second PCR using JC324/BL27 (20). This product was inserted into BamHI and EcoRI sites of pLZ12-spec (60). Genotypes were confirmed by PCR and sequencing.
Purification of recombinant Rgg3 and ComR.
Details of His6-SUMO-Rgg3 purification scheme have been described previously (20, 54). The S. mutans UA159 comR gene was amplified using primers LW10:51 (GCGTGCATATGTTAAAAGATTTTGGGAA) and LW10:52 (GCGTGGGATCCTTATGTCCCGTTCTGAGAAT) and was cloned into the NdeI and BamHI sites of the pET 15b expression vector downstream of the His6 tag; the resulting vector, pWAR368, was electroporated in E. coli C41(DE3) cells. Expression of His6-comR was induced at an approximate optical density at 600 nm (OD600) of 0.6 with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 6 h at 30°C. Cells were pelleted and suspended in buffer A (phosphate-buffered saline [PBS] [pH 7.4], 20 mM imidazole, 10 mM β-mercaptoethanol) with Complete EDTA-free protease inhibitor (Roche). Cells were disrupted by sonication on ice, and cellular debris was removed by centrifugation at 45,000 × g for 20 min at 4°C. His6-ComR was then purified using a His-Trap-HP nickel column (GE Biosciences) and eluted with 250 mM imidazole. The purified protein was dialyzed in PBS followed by addition of glycerol to a final concentration of 20% glycerol. Aliquots were flash-frozen in a dry ice-ethanol bath and stored at −80°C.
HTS of the compound library.
All HTS assays were assembled on a Tecan Freedom EVO 200 with an integrated Tecan Infinite F200 Pro plate reader fitted with appropriate polarized filters. Assays were performed in duplicate in black, flat-bottom 384-well plates (Greiner Bio-One 781-076) at room temperature. For Rgg3 assays, 0.1 µl of 10 mM compound in dimethyl sulfoxide (DMSO) (Prestwick Chemical Library) was added by using a pin tool (V&P Scientific) to test wells containing 30 µl of Rgg3 assay mix (1× PBS, 0.1 mg/ml bovine serum albumin [BSA], 0.01% Triton X-100, 5 mM dithiothreitol [DTT], 180 nM His6-Sumo-Rgg3, 10 nM FITC–SHP2-C8). Thirty-two negative-control wells per plate received 0.1 µl of DMSO, and 32 positive-control wells per plate received 0.1 µl of 1 mM unlabeled SHP2-C8 peptide. Following compound/control addition, plates were shaken at 1,500 rpm for 20 s on a Te-Shake plate shaker and then incubated at room temperature for 15 min. Fluorescence polarization values were measured at excitation and emission wavelengths of 485 (20) nm and 535 (25) nm, respectively, and percent inhibition was calculated for each sample using the following equation: %I = 100{1 − [(Px − μc+)/(μc− − μc+)]}, where %I is the percent inhibition of sample x, Px is the polarization value of sample x, μc+ is the mean polarization value of the positive controls per plate, and μc− is the mean polarization value of the negative controls per plate.
ComR was assayed as described above, with the exception that the ComR assay mix contained 1× PBS, 0.1 mg/ml BSA, 0.01% Triton X 100, 10 mM β-mercaptoethanol, 1 µM ComR, and 10 nM FITC-XIP. ComR positive-control wells contained the above buffer lacking ComR protein.
To assess the quality of the screening data, Z′ values (61) were calculated for each plate using the following equation: , where σc+ and σc− are the standard deviations of the positive- and negative-control polarization values, respectively, and μc+ and μc− are their mean values.
FP.
Fluorescence polarization (FP) assays were performed as described in detail earlier (21). Briefly, for the direct-FP assay, the concentration of N-terminally FITC-labeled synthetic peptides was kept constant at 10 nM for all reactions. Purified Rgg and ComR were serially diluted, ranging from 10 µM to 5 nM, and mixed with peptide in a final reaction volume of 50 µl in protein storage buffer (PBS [pH 7.4], 10 mM β-mercaptoethanol, and 20% [vol/vol] glycerol). For FP, the storage buffer was supplemented with 0.01% Triton X-100 and 0.1 mg/ml BSA. Polarization values were measured using a BioTek Synergy 2 plate reader, and the resulting millipolarization values were plotted for each protein concentration tested to assess protein-peptide interactions (62).
For competition-FP assays, 10 nM FITC–SHP2-C8 or FITC-XIP was incubated for 10 min with the concentration of Rgg or ComR corresponding to the Kd value, as determined from the direct-FP assay (160 nM His6-SUMO-Rgg3 or 1 µM ComR). Reactions were then titrated against serial dilutions of either unlabeled peptide or compounds identified from HTS. Millipolarization values were determined as described above. Plots in Fig. 2A show the means of at least three independent experiments. Kd values were determined by applying linear regression on dose-response curves using GraphPad Prism (version 6.01).
Bacterial growth curves and luminescence transcriptional reporter assays.
To test the effect of compounds on bacterial growth, S. pyogenes cultures were grown at 37°C in CDM to an OD600 of 0.1 and then provided with either 10 nM SHP2-C8 or with 10 nM SHP2-C8 and 10 µM quorum-sensing inhibitor (QSI) compound. OD600 was measured every 30 min for 8 h.
To assess the transcription-inhibiting activity of QSI compounds, luciferase reporter strains (Table 3) were used. This strain was grown to an exponential-growth-phase OD600 of 0.1 in CDM. One hundred microliters of this culture was then dispensed to a 96-well, clear-bottom plate, with each well containing either 10 nM SHP2-C8 or 10 nM SHP2-C8 with 10 µM of compound. Decanal, the aldehyde substrate required for luciferase, was provided as a 1% solution in mineral oil and was included in the plate in spaces outside the bioassay wells, as has been described previously (59, 63). The plate was lidded, sealed, and read in a Synergy 2 plate reader (BioTek) set to 37°C with continuous shaking to prevent cells from settling at the bottom of the plate. The OD600 and luminescence values (in counts per second [cps]) were monitored every 15 min for 8 h. The maximum cps/OD600 (relative luminescence) reached by each culture was plotted for Fig. 2B, C, D, and F and 4. Alternatively, luminescence measurements were taken in a Turner Biosystems Veritas microplate luminometer as previously described (59).
Titration curves of top hits obtained from HTS in Fig. 2C were prepared by doing the experiment described above with serially diluted compounds provided as 1% of the final volume. The maximum cps/OD600 (relative luminescence) reached was recorded for each sample and plotted as cps/OD600 (relative luminescence) versus compound concentration to calculate the 50% inhibitory concentration (IC50 [concentration resulting in 50% of activity seen without inhibitor]) of each compound. To evaluate the effect of QSI compounds over RopB activity, PspeB-luxAB reporter strain was grown in C medium to an exponential-growth-phase OD600 of 0.1 and tested as described above.
Biofilm assays.
Bacterial strains were grown overnight in THY medium at 30°C and then back-diluted 1:100 into fresh CDM and grown at 37°C until they reached late exponential phase. Bacteria were back-diluted into tubes of fresh CDM containing 10 nM SHP2-C8 and 2-fold dilutions of cyclosporine A or valspodar, at concentrations ranging from 5 to 0.156 µM. NZ131 strains were back-diluted to an OD600 of 0.02, and GA19681 and MGAS5005 strains were back-diluted to an OD600 of 0.005. Tubes containing 0.1% DMSO were used as controls. Bacteria were incubated for approximately 1 h at 37°C until they reached an OD600 of ~0.1 for NZ131 strains and an OD600 of ~0.02 for GA19681 and MGAS5005 and then plated in duplicate in cell culture-treated 24 well polystyrene plates. Plates were then incubated at 37°C with 5% CO2 for 20 h to promote biofilm growth. Medium was aspirated, wells were washed once with 0.9% NaCl, and biomass was dry-fixed overnight. Biofilms were stained with 0.2% crystal violet solution, washed three times with a solution containing 0.9% NaCl and 10% ethanol, and quantified by measurement of absorbance at λ595 by an area scan of the wells in a Synergy 2 plate reader. Experiments were performed at least three times per condition.
ACKNOWLEDGMENTS
Support for this work was provided by NIH grant R01-AI091779, the Burroughs Wellcome Fund Investigators in the pathogenesis of Infectious Diseases, the UI Center for Drug Discovery, and the Chicago Biomedical Consortium High Throughput Screening grant program with support from the Searle Funds at the Chicago Community Trust.
We thank Laura C. Cook, Jennifer Chang, and Lauren Mashburn-Warren for the use of their strains, Andrew Jin for help with growth curve experiments, and Gregory Thatcher and Jason Hickok for their suggestions.
Footnotes
Citation Aggarwal C, Jimenez JC, Lee H, Chlipala GE, Ratia K, Federle MJ. 2015. Identification of quorum-sensing inhibitors disrupting Rgg-SHP signaling in streptococci. mBio 6(3):e00393-15. doi:10.1128/mBio.00393-15.
REFERENCES
- 1.Waters CM, Bassler BL. 2005. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol 21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 2.Antunes LC, Ferreira RB, Buckner MM, Finlay BB. 2010. Quorum sensing in bacterial virulence. Microbiology 156:2271–2282. doi: 10.1099/mic.0.038794-0. [DOI] [PubMed] [Google Scholar]
- 3.Rutherford ST, Bassler BL. 2012. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2:012427. doi: 10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romero M, Acuña L, Otero A. 2012. Patents on quorum quenching: interfering with bacterial communication as a strategy to fight infections. Recent Pat Biotechnol 6:2–12. doi: 10.2174/187220812799789208. [DOI] [PubMed] [Google Scholar]
- 5.LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 77:73–111. doi: 10.1128/MMBR.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nizet V. 2007. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J Allergy Clin Immunol 120:13–22. doi: 10.1016/j.jaci.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 8.DeMuri GP, Wald ER. 2014. The group A streptococcal carrier state reviewed: still an enigma. J Pediatr Infect Dis Soc 3:336–342. doi: 10.1093/jpids/piu030. [DOI] [PubMed] [Google Scholar]
- 9.Stollerman GH. 1991. Rheumatogenic streptococci and autoimmunity. Clin Immunol Immunopathol 61:131–142. doi: 10.1016/S0090-1229(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 10.Stollerman GH. 2001. Rheumatic fever in the 21st century. Clin Infect Dis 33:806–814. doi: 10.1086/322665. [DOI] [PubMed] [Google Scholar]
- 11.Dillon HC, Derrick CW, Dillon MS. 1974. M-antigens common to pyoderma and acute glomerulonephritis. J Infect Dis 130:257–267. doi: 10.1093/infdis/130.3.257. [DOI] [PubMed] [Google Scholar]
- 12.Silva F. 1998. Acute postinfectious glomerulonephritis and glomerulonephritis complicating persistent bacterial infection. Hepinstall’s pathology of the kidney, 5th ed., p 389–453. Lippincott Raven Publishers, Philadelphia, PA. [Google Scholar]
- 13.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 14.Shulman ST, Bisno AL, Clegg HW, Gerber MA, Kaplan EL, Lee G, Martin JM, Van Beneden C. 2012. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis 55:1279–1282. doi: 10.1093/cid/cis847. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan EL, Johnson DR. 2001. Unexplained reduced microbiological efficacy of intramuscular benzathine penicillin G and of oral penicillin V in eradication of group A streptococci from children with acute pharyngitis. Pediatrics 108:1180–1186. doi: 10.1542/peds.108.5.1180. [DOI] [PubMed] [Google Scholar]
- 16.Pichichero ME, Casey JR, Mayes T, Francis AB, Marsocci SM, Murphy AM, Hoeger W. 2000. Penicillin failure in streptococcal tonsillopharyngitis: causes and remedies. Pediatr Infect Dis J 19:917–923. doi: 10.1097/00006454-200009000-00035. [DOI] [PubMed] [Google Scholar]
- 17.Pichichero ME, Casey JR. 2007. Systematic review of factors contributing to penicillin treatment failure in Streptococcus pyogenes pharyngitis. Otolaryngol Head Neck Surg 137:851–857. doi: 10.1016/j.otohns.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 18.Martin JM, Green M, Barbadora KA, Wald ER. 2002. Erythromycin-resistant group A streptococci in schoolchildren in Pittsburgh. N Engl J Med 346:1200–1206. doi: 10.1056/NEJMoa013169. [DOI] [PubMed] [Google Scholar]
- 19.Robinson DA, Sutcliffe JA, Tewodros W, Manoharan A, Bessen DE. 2006. Evolution and global dissemination of macrolide-resistant group A streptococci. Antimicrob Agents Chemother 50:2903–2911. doi: 10.1128/AAC.00325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang JC, Lasarre B, Jimenez JC, Aggarwal C, Federle MJ. 2011. Two group A Streptococcal Peptide Pheromones act through Opposing Rgg regulators to control biofilm development. PLoS Pathog 7:e1002190. doi: 10.1371/journal.ppat.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aggarwal C, Jimenez JC, Nanavati D, Federle MJ. 2014. Multiple length peptide-pheromone variants produced by Streptococcus pyogenes directly bind Rgg proteins to confer transcriptional regulation. J Biol Chem 289:22427–22436. doi: 10.1074/jbc.M114.583989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Declerck N, Bouillaut L, Chaix D, Rugani N, Slamti L, Hoh F, Lereclus D, Arold ST. 2007. Structure of PlcR: insights into virulence regulation and evolution of quorum sensing in Gram-positive bacteria. Proc Natl Acad Sci U S A 104:18490–18495. doi: 10.1073/pnas.0704501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rocha-Estrada J, Aceves-Diez AE, Guarneros G, de la Torre M. 2010. The RNPP family of quorum-sensing proteins in Gram-positive bacteria. Appl Microbiol Biotechnol 87:913–923. doi: 10.1007/s00253-010-2651-y. [DOI] [PubMed] [Google Scholar]
- 24.Shi K, Brown CK, Gu ZY, Kozlowicz BK, Dunny GM, Ohlendorf DH, Earhart CA. 2005. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc Natl Acad Sci U S A 102:18596–18601. doi: 10.1073/pnas.0506163102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zouhir S, Perchat S, Nicaise M, Perez J, Guimaraes B, Lereclus D, Nessler S. 2013. Peptide-binding dependent conformational changes regulate the transcriptional activity of the quorum-sensor NprR. Nucleic Acids Res 41:7920–7933. doi: 10.1093/nar/gkt546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parashar V, Mirouze N, Dubnau DA, Neiditch MB. 2011. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol 9:e1000589. doi: 10.1371/journal.pbio.1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perego M, Higgins CF, Pearce SR, Gallagher MP, Hoch JA. 1991. The oligopeptide transport system of Bacillus subtilis plays a role in the initiation of sporulation. Mol Microbiol 5:173–185. doi: 10.1111/j.1365-2958.1991.tb01838.x. [DOI] [PubMed] [Google Scholar]
- 28.Linton KJ, Higgins CF. 2007. Structure and function of ABC transporters: the ATP switch provides flexible control. Pflugers Arch 453:555–567. doi: 10.1007/s00424-006-0126-x. [DOI] [PubMed] [Google Scholar]
- 29.Fleuchot B, Gitton C, Guillot A, Vidic J, Nicolas P, Besset C, Fontaine L, Hols P, Leblond-Bourget N, Monnet V, Gardan R. 2011. Rgg proteins associated with internalized small hydrophobic peptides: a new quorum-sensing mechanism in streptococci. Mol Microbiol 80:1102–1119. doi: 10.1111/j.1365-2958.2011.07633.x. [DOI] [PubMed] [Google Scholar]
- 30.Donlan RM, Costerton JW. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fux CA, Costerton JW, Stewart PS, Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol 13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Kania RE, Lamers GE, Vonk MJ, Huy PT, Hiemstra PS, Bloemberg GV, Grote JJ. 2007. Demonstration of bacterial cells and glycocalyx in biofilms on human tonsils. Arch Otolaryngol Head Neck Surg 133:115–121. doi: 10.1001/archotol.133.2.115. [DOI] [PubMed] [Google Scholar]
- 33.Cook LC, LaSarre B, Federle MJ. 2013. Interspecies communication among commensal and pathogenic streptococci. mBio 4:e00382-13. doi: 10.1128/mBio.00382-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleuchot B, Guillot A, Mézange C, Besset C, Chambellon E, Monnet V, Gardan R. 2013. Rgg-associated SHP signaling peptides mediate cross-talk in streptococci. PLoS One 8:e66042. doi: 10.1371/journal.pone.0066042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mashburn-Warren L, Morrison DA, Federle MJ. 2010. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol Microbiol 78:589–606. doi: 10.1111/j.1365-2958.2010.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fontaine L, Boutry C, de Frahan MH, Delplace B, Fremaux C, Horvath P, Boyaval P, Hols P. 2010. A novel pheromone quorum-sensing system controls the development of natural competence in Streptococcus thermophilus and Streptococcus salivarius. J Bacteriol 192:1444–1454. doi: 10.1128/JB.01251-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mashburn-Warren L, Morrison DA, Federle MJ. 2012. The cryptic competence pathway in Streptococcus pyogenes is controlled by a peptide pheromone. J Bacteriol 194:4589–4600. doi: 10.1128/JB.00830-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rüegger A, Kuhn M, Lichti H, Loosli HR, Huguenin R, Quiquerez C, von Wartburg A. 1976. Cyclosporin A, a peptide metabolite from Trichoderma polysporum (link ex Pers.) Rifai, with a remarkable immunosuppressive activity. Helv Chim Acta 59:1075–1092. doi: 10.1002/hlca.19760590412. [DOI] [PubMed] [Google Scholar]
- 39.Matsuda S, Koyasu S. 2000. Mechanisms of action of cyclosporine. Immunopharmacology 47:119–125. doi: 10.1016/S0162-3109(00)00192-2. [DOI] [PubMed] [Google Scholar]
- 40.Steiner JP, Connolly MA, Valentine HL, Hamilton GS, Dawson TM, Hester L, Snyder SH. 1997. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat Med 3:421–428. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- 41.Wei L, Steiner JP, Hamilton GS, Wu YQ. 2004. Synthesis and neurotrophic activity of nonimmunosuppressant cyclosporin A derivatives. Bioorg Med Chem Lett 14:4549–4551. doi: 10.1016/j.bmcl.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 42.Jonsson B, Nilsson K, Nygren P, Larsson R. 1992. SDZ PSC-833—a novel potent in vitro chemosensitizer in multiple myeloma. Anti Cancer Drugs 3:641–646. doi: 10.1097/00001813-199212000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Loughman JA, Caparon M. 2006. Regulation of SpeB in Streptococcus pyogenes by pH and NaCl: a model for in vivo gene expression. J Bacteriol 188:399–408. doi: 10.1128/JB.188.2.399-408.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. 2008. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol 6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjarnsholt T, Ciofu O, Molin S, Givskov M, Høiby N. 2013. Applying insights from biofilm biology to drug development—can a new approach be developed? Nat Rev Drug Discov 12:791–808. doi: 10.1038/nrd4000. [DOI] [PubMed] [Google Scholar]
- 46.Allen RC, Popat R, Diggle SP, Brown SP. 2014. Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol 12:300–308. doi: 10.1038/nrmicro3232. [DOI] [PubMed] [Google Scholar]
- 47.Dreyfuss M, Härri E, Hofmann H, Kobel H, Pache W, Tscherter H. 1976. Cyclosporin A and C. Eur J Appl Microbiol 3:125–133. doi: 10.1007/BF00928431. [DOI] [Google Scholar]
- 48.Borel JF, Feurer C, Gubler HU, Stähelin H. 1994. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions 43:179–186. doi: 10.1007/BF01986686. [DOI] [PubMed] [Google Scholar]
- 49.Krönke M, Leonard WJ, Depper JM, Arya SK, Wong-Staal F, Gallo RC, Waldmann TA, Greene WC. 1984. Cyclosporin A inhibits T-cell growth factor gene expression at the level of mRNA transcription. Proc Natl Acad Sci U S A 81:5214–5218. doi: 10.1073/pnas.81.16.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. 1984. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 51.Herold KC, Lancki DW, Moldwin RL, Fitch FW. 1986. Immunosuppressive effects of cyclosporin A on cloned T cells. J Immunol 136:1315–1321. [PubMed] [Google Scholar]
- 52.Gaveriaux C. 1991. SDZ PSC 833, a non-immunosuppressive cyclosporin analog, is a very potent multidrug-resistance modifier. J Cell Pharmacol 2:225–234. [Google Scholar]
- 53.Archinal-Mattheis A, Rzepka RW, Watanabe T, Kokubu N, Itoh Y, Combates NJ, Bair KW, Cohen D. 1995. Analysis of the interactions of SDZ PSC 833 ([3′-keto-Bmt1]-Val2]-cyclosporine), a multidrug resistance modulator, with P-glycoprotein. Oncol Res 7:603–610. [PubMed] [Google Scholar]
- 54.Lasarre B, Aggarwal C, Federle MJ. 2013. Antagonistic Rgg regulators mediate quorum sensing via competitive DNA binding in Streptococcus pyogenes. mBio 3:e00333-12. doi: 10.1128/mBio.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts AL, Connolly KL, Kirse DJ, Evans AK, Poehling KA, Peters TR, Reid SD. 2012. Detection of group A Streptococcus in tonsils from pediatric patients reveals high rate of asymptomatic streptococcal carriage. BMC Pediatr 12:3. doi: 10.1186/1471-2431-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Connolly KL, Roberts AL, Holder RC, Reid SD. 2011. Dispersal of group A streptococcal biofilms by the cysteine protease SpeB leads to increased disease severity in a murine model. PLoS One 6:e18984. doi: 10.1371/journal.pone.0018984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howell-Adams B, Seifert HS. 2000. Molecular models accounting for the gene conversion reactions mediating gonococcal pilin antigenic variation. Mol Microbiol 37:1146–1158. doi: 10.1046/j.1365-2958.2000.02067.x. [DOI] [PubMed] [Google Scholar]
- 58.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 59.LaSarre B, Chang JC, Federle MJ. 2013. Redundant group a streptococcus signaling peptides exhibit unique activation potentials. J Bacteriol 195:4310–4318. doi: 10.1128/JB.00684-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Husmann LK, Scott JR, Lindahl G, Stenberg L. 1995. Expression of the Arp protein, a member of the M protein family, is not sufficient to inhibit phagocytosis of Streptococcus pyogenes. Infect Immun 63:345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J-H, Chung TDY, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screening 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 62.Rossi AM, Taylor CW. 2011. Analysis of protein-ligand interactions by fluorescence polarization. Nat Protoc 6:365–387. doi: 10.1038/nprot.2011.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bachmann H, Kleerebezem M, van Hylckama Vlieg JE. 2008. High-throughput identification and validation of in situ-expressed genes of Lactococcus lactis. Appl Environ Microbiol 74:4727–4736. doi: 10.1128/AEM.00297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McShan WM, Ferretti JJ, Karasawa T, Suvorov AN, Lin S, Qin B, Jia H, Kenton S, Najar F, Wu H, Scott J, Roe BA, Savic DJ. 2008. Genome sequence of a nephritogenic and highly transformable M49 strain of Streptococcus pyogenes. J Bacteriol 190:7773–7785. doi: 10.1128/JB.00672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Degnan BA, Fontaine MC, Doebereiner AH, Lee JJ, Mastroeni P, Dougan G, Goodacre JA, Kehoe MA. 2000. Characterization of an isogenic mutant of Streptococcus pyogenes Manfredo lacking the ability to make streptococcal acid glycoprotein. Infect Immun 68:2441–2448. doi: 10.1128/IAI.68.5.2441-2448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci U S A 99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ajdić D, McShan WM, McLaughlin RE, Savić G, Chang J, Carson MB, Primeaux C, et al. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A 99(2):14434–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lancefield RC, McCarty M, Everly WN. 1975. Multiple mouse-protective antibodies directed against group B streptococci. Special reference to antibodies effective against protein antigens. J Exp Med 142:165–179. doi: 10.1084/jem.142.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O’Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc Natl Acad Sci U S A 102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bentley RW, Leigh JA, Collins MD. 1991. Intrageneric structure of Streptococcus based on comparative analysis of small-subunit rRNA sequences. Int J Syst Bacteriol 41:487–494. doi: 10.1099/00207713-41-4-487. [DOI] [PubMed] [Google Scholar]
- 71.McShan WM, McLaughlin RE, Nordstrand A, Ferretti JJ. 1998. Vectors containing streptococcal bacteriophage integrases for site-specific gene insertion. Methods Cell Sci:51–57. doi: 10.1023/A:1009773309163. [DOI] [Google Scholar]