

ABSTRACT
Nitric oxide (NO) plays an important signaling role in all domains of life. Many bacteria contain a heme-nitric oxide/oxygen binding (H-NOX) protein that selectively binds NO. These H-NOX proteins often act as sensors that regulate histidine kinase (HK) activity, forming part of a bacterial two-component signaling system that also involves one or more response regulators. In several organisms, NO binding to the H-NOX protein governs bacterial biofilm formation; however, the source of NO exposure for these bacteria is unknown. In mammals, NO is generated by the enzyme nitric oxide synthase (NOS) and signals through binding the H-NOX domain of soluble guanylate cyclase. Recently, several bacterial NOS proteins have also been reported, but the corresponding bacteria do not also encode an H-NOX protein. Here, we report the first characterization of a bacterium that encodes both a NOS and H-NOX, thus resembling the mammalian system capable of both synthesizing and sensing NO. We characterized the NO signaling pathway of the marine alphaproteobacterium Silicibacter sp. strain TrichCH4B, determining that the NOS is activated by an algal symbiont, Trichodesmium erythraeum. NO signaling through a histidine kinase-response regulator two-component signaling pathway results in increased concentrations of cyclic diguanosine monophosphate, a key bacterial second messenger molecule that controls cellular adhesion and biofilm formation. Silicibacter sp. TrichCH4B biofilm formation, activated by T. erythraeum, may be an important mechanism for symbiosis between the two organisms, revealing that NO plays a previously unknown key role in bacterial communication and symbiosis.
IMPORTANCE
Bacterial nitric oxide (NO) signaling via heme-nitric oxide/oxygen binding (H-NOX) proteins regulates biofilm formation, playing an important role in protecting bacteria from oxidative stress and other environmental stresses. Biofilms are also an important part of symbiosis, allowing the organism to remain in a nutrient-rich environment. In this study, we show that in Silicibacter sp. strain TrichCH4B, NO mediates symbiosis with the alga Trichodesmium erythraeum, a major marine diazotroph. In addition, Silicibacter sp. TrichCH4B is the first characterized bacteria to harbor both the NOS and H-NOX proteins, making it uniquely capable of both synthesizing and sensing NO, analogous to mammalian NO signaling. Our study expands current understanding of the role of NO in bacterial signaling, providing a novel role for NO in bacterial communication and symbiosis.
INTRODUCTION
Nitric oxide (NO) serves dual biological roles as both a signaling molecule and a cytotoxin (1–3). NO is synthesized by nitric oxide synthase (NOS), which has been characterized extensively in mammals. As a gaseous signaling molecule, NO can diffuse freely across cellular membranes to neighboring cells. For instance, in mammalian signaling, nanomolar concentrations of NO are generated by nitric oxide synthase (NOS) in endothelial cells. This NO diffuses to neighboring smooth muscle cells, where NO activates soluble guanylate cyclase (sGC), leading to formation of the second messenger, cyclic GMP (cGMP), which increases vasodilation (4, 5). sGC senses NO via a heme cofactor that selectively binds NO, but not O2. The sGC heme domain is a member of the heme-nitric oxide/oxygen binding (H-NOX) protein family, which is also present in many bacteria, including a number of pathogens (6–8).
Besides its role in signaling, NO is also an important component in the host response to infection, acting as a cytotoxic antimicrobial agent when generated at localized micromolar concentrations (9, 10). H-NOX proteins are one of several bacterial NO sensors that mediate response to the gas, through conserved signaling mechanisms that regulate histidine kinases (HKs) or diguanylate cyclases (DGCs) contained within the same operon (8). H-NOX proteins in Legionella pneumophila and Shewanella woodyi inhibit biofilm formation by regulating the activity of a diguanylate cyclase/phosphodiesterase fusion protein that decreases levels of the bacterial second messenger cyclic diguanosine monophosphate (c-di-GMP) (11, 12). In Shewanella oneidensis, the H-NOX protein functions as a sensor protein for an HK, forming part of a bacterial two-component signaling pathway (13, 14). S. oneidensis contains a particularly complex NO-controlled multicomponent regulatory network, in which the HK activity is inhibited by the NO-bound H-NOX, and the HK has three cognate response regulators (RRs). Two of these RRs regulate biofilm formation by controlling c-di-GMP concentrations, while the third RR acts as a transcriptional regulator that controls the signaling network in a positive-feedback loop (14, 15). A similar signaling network is found in Vibrio cholerae, suggesting a broader role for H-NOX proteins as part of the bacterial defense mechanism to form a biofilm as protection against NO toxicity (14). In V. cholerae, the NO sensed by the H-NOX is likely produced by mammalian NOS (mNOS) activity from the host, but for the rest of these bacteria, the physiologically relevant source of NO for bacterial H-NOX signaling is unclear.
NOS catalyzes the conversion of arginine into NO and citrulline, consuming O2 and NADPH as cosubstrates (9, 16, 17). mNOS contains an oxygenase domain responsible for catalysis, and a reductase domain involved in electron transfer. mNOS forms a complex with Ca2+-calmodulin (CaM) that promotes electron transfer between the oxygenase and reductase domains. Electrons are transferred from NADPH via flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) bound within the reductase domain to the P450-type heme in the oxygenase domain. Through sequence searching of genome data banks, bacterial open reading frames coding for proteins with high sequence similarity to the oxygenase domain of mNOS have been discovered. These isolated bacterial oxygenase domains were initially characterized from Deinococcus radiodurans and Bacillus subtilis (18, 19) and subsequently from pathogens such as Bacillus anthracis and Staphylococcus aureus (20, 21). In the presence of a separate flavin-containing reductase protein, these bacterial NOS homologs were shown to have NOS activity. This bacterially derived NO has been proposed to protect against oxidative stress and antibiotics (18–20, 22). A full-length bacterial NOS containing a fused oxygenase and reductase domain in one polypeptide sequence was later discovered in Sorangium cellulosum (23). In contrast to mNOS, the reductase domain cofactors of S. cellulosum NOS (scNOS) are FAD and an iron-sulfur cluster (see Fig. S1 in the supplemental material). The function of NO generated from scNOS remains unclear, and the physiological inputs that stimulate NOS activity in these bacteria are unknown. H-NOX proteins are well-characterized NO sensors; however, to date, a bacterium carrying genes known to encode both the NOS and H-NOX proteins for a full NO signaling pathway had not been characterized.
Here, we report a novel NO signaling pathway in the alphaproteobacterium Silicibacter sp. strain TrichCH4B, which contains both a full-length NOS and an H-NOX NO sensor protein, resembling the mammalian NOS/sGC signaling system with NOS-derived NO binding to the H-NOX to activate downstream signaling. Additionally, the H-NOX of Silicibacter sp. TrichCH4B is encoded by a gene adjacent to a histidine kinase gene, suggesting involvement in a two-component signaling pathway. While bacterial NOS and H-NOX proteins have separately been shown to sense and defend against NO, in Silicibacter sp. TrichCH4B, NO acts as a signaling molecule that controls biofilm formation through a two-component phosphorelay system. Furthermore, the NO signaling pathway in Silicibacter sp. TrichCH4B is activated through interaction with its algal symbiont Trichodesmium erythraeum, indicating that NO plays a previously unknown role in bacterial communication.
RESULTS
SiliNOSox forms NO under single-turnover conditions.
The NOS of Silicibacter sp. strain TrichCH4B (SiliNOS) was identified as a putative nitric oxide synthase through sequence homology to the mammalian NOS oxygenase domain and to the full-length NOS in Sorangium cellulosum (scNOS) (23). The domain architecture of SiliNOS is similar to that of scNOS; SiliNOS contains an N-terminal reductase domain with NAD(P)H and FAD binding sites and a predicted 2Fe-2S cluster. Attempts to express full-length SiliNOS in an active form were unsuccessful, most likely due to improper assembly or instability of the 2Fe-2S cluster or heme cofactor. Therefore, a SiliNOS oxygenase domain (SiliNOSox) construct, designed based on alignment with both the mammalian and S. cellulosum NOS proteins, was expressed and purified from Escherichia coli.
The activity of SiliNOSox in the presence of oxygen, substrate, and other cofactors was investigated under single-turnover conditions using N-hydroxyarginine (NHA), the product of the first catalytic step of the NOS reaction, as a substrate. Following a protocol similar to one previously described for scNOSox (oxygenase domain of scNOS) (23), the heme cofactor of SiliNOSox was reduced from the ferric state to the ferrous state with a molar equivalent of dithionite, and the reduced SiliNOSox was incubated with NHA and cofactor tetrahydrobiopterin (H4B) or tetrahydrofolate (H4F) under anaerobic conditions. Aerobic buffer was introduced to provide the O2 required to initiate the reaction, and NO in the headspace was determined using a nitric oxide analyzer. NO was detected only in samples containing NHA, oxygen, and either H4B or H4F (Fig. 1), demonstrating that SiliNOSox is capable of synthesizing NO.
FIG 1 .
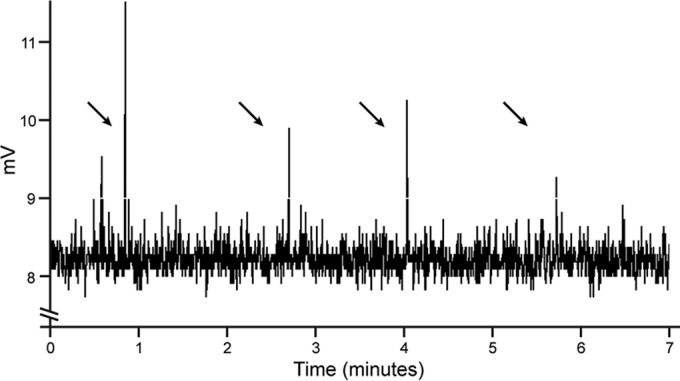
Single-turnover NO formation by SiliNOSox. Representative NO analyzer trace of a SiliNOSox single-turnover experiment as described in Materials and Methods. A 40-µl assay contained ferrous SiliNOSox (100 µM) with 500 µM NHA and 200 µM H4B in a sealed Reacti-vial, and the reaction was initiated by introducing aerobic buffer to provide the necessary oxygen for NOS activity. The reaction headspace was then injected into the NOA via a gas-tight syringe. Arrows indicate the times that reaction headspace was injected into the NOA.
SiliH-NOX binds nitric oxide but not oxygen.
NO generated from SiliNOS could interact with the Silicibacter sp. TrichCH4B H-NOX protein (SiliH-NOX). SiliH-NOX was expressed and purified from E. coli. Purified SiliH-NOX contains a ferrous heme that forms stable NO and CO complexes, displaying spectra nearly identical to those of H-NOX from S. oneidensis (SoH-NOX) (13) (see Fig. S2 in the supplemental material). Like SoH-NOX, SiliH-NOX has no measurable affinity to O2, indicating that SiliH-NOX likely functions as a selective NO sensor.
NO-bound SiliH-NOX inhibits SiliHK histidine kinase activity.
The gene encoding SiliH-NOX is located adjacent to a gene encoding a histidine kinase in the Silicibacter sp. TrichCH4B genome (SiliHK), suggesting that SiliH-NOX could function as a sensor for SiliHK. SiliHK is a hybrid histidine kinase, containing both a kinase domain and a receiver domain with an aspartic acid predicted to be the phosphoryl acceptor from the kinase domain (see Fig. S3 in the supplemental material). To test SiliHK autophosphorylation activity, SiliHK was incubated with γ-S-labeled ATP (ATPγS) as a substrate. After the ATPγS reaction was quenched by EDTA, the alkylating agent para-nitrobenzylmesylate (PNBM) was added, which alkylates thiophosphates and cysteines, and an antibody specific for the alkylated thiophosphate was then used to detect the resulting thiophosphate esters (24). SiliHK autophosphorylation activity was observed by immunoblotting (Fig. 2A).
FIG 2 .
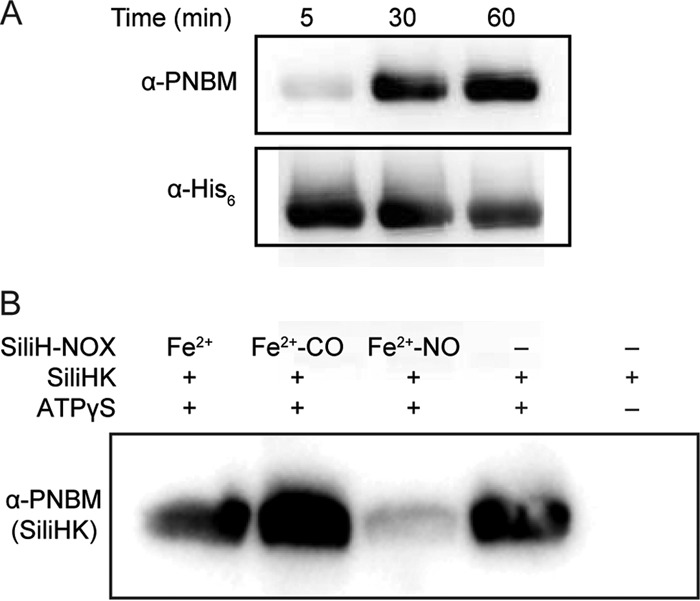
SiliHK autophosphorylation and regulation by SiliH-NOX. (A) Autophosphorylation of SiliHK. Kinase autophosphorylation assays were carried out with γ-S-labeled ATP (ATPγS), and aliquots were taken at 5, 30, and 60 min and analyzed by Western blotting as described in Materials and Methods. α-PNBM, anti-PNBM antibody. (B) Effect of SiliH-NOX on the kinase activity of SiliHK. Kinase assays containing 5 µM SiliHK were incubated for 30 min with 1 mM ATPγS in the presence of 30 µM SiliH-NOX in the ligation states indicated. Samples were analyzed by Western blotting as described in Materials and Methods.
Next, the effect of the SiliH-NOX ligation state on SiliHK autophosphorylation was investigated. Unliganded (Fe2+) and ferrous-carbonyl (Fe2+-CO) SiliH-NOX did not affect kinase activity. However, the SiliH-NOX ferrous-nitrosyl (Fe2+-NO) complex inhibited SiliHK autophosphorylation. At a fivefold excess of Fe2+-NO-labeled SiliH-NOX, SiliHK autophosphorylation was nearly completely inhibited (Fig. 2B), indicating that SiliHK is regulated by SiliH-NOX in an NO-dependent manner.
Phosphotransfer from SiliHK to SiliHpt and SiliDGC.
SiliHK is a hybrid histidine kinase; therefore, a histidine phosphotransfer protein (Hpt) is required to mediate phosphotransfer to its cognate response regulator (25). Using the SMART (simple modular architecture research tool) domain database (26, 27), searching the Silicibacter sp. TrichCH4B genome for stand-alone Hpt proteins resulted in only one gene, hereafter referred to as SiliHpt. Therefore, SiliHpt was cloned and then expressed and purified from E. coli.
Physiological cognate phosphotransfer partners are predicted to display fast (typically within 1 min) phosphotransfer kinetics in vitro (28, 29). In phosphotransfer assays with both SiliHpt and SiliHK, SiliHpt phosphorylation was monitored by immunoblotting for PNBM adducts as previously described (24). SiliHpt phosphorylation was observed within 1 min after assay initiation (Fig. 3A), consistent with the rapid transfer expected for an in vivo cognate pair (30).
FIG 3 .
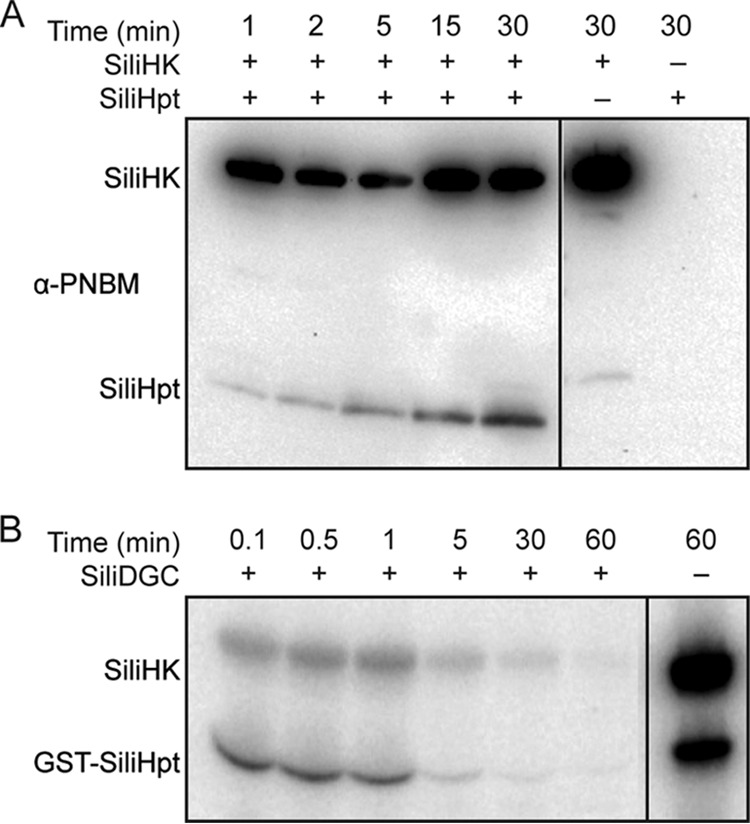
Phosphotransfer from SiliHK to its cognate partners. (A) Phosphotransfer between SiliHK and SiliHpt. SiliHK (5 µM) was mixed with SiliHpt (10 µM) in the presence of ATPγS (1 mM) for a 30-min time course as described in Materials and Methods. SiliHPT phosphorylation was observed within 1 min. Empty lanes were removed from the gel image, as indicated by the line. All lanes are part of the same gel, with the same exposure settings. (B) Phosphotransfer from SiliHK/SiliHpt to SiliDGC. Loss of SiliHK/SiliHpt phosphorylation was used to monitor phosphotransfer to a cognate response regulator, SiliDGC. SiliHK and SiliHpt were mixed with 5 µCi [γ-32P]ATP for 15 min and then desalted to remove excess ATP. SiliDGC (10 µM) was then added to the reaction mix for a 60-min time course as described in Materials and Methods. SiliDGC was the only RR from a panel of 12 orphan RRs to cause rapid loss of SiliHK/SiliHpt phosphorylation (between 1 and 5 min). Empty lanes were removed from the gel image, as indicated by the line. All lanes are part of the same gel, with the same exposure settings.
Having determined that SiliHpt can serve as an intermediary between SiliHK and its cognate response regulator, we next searched for orphan response regulators within the Silicibacter sp. TrichCH4B genome using the SMART domain database. Orphan response regulators contain a receiver domain, but no histidine kinase in the same or nearby operons. Each of the 12 orphan response regulators discovered using this method was cloned, expressed, and purified from E. coli and then tested for phosphotransfer from SiliHK and SiliHpt (14, 31). SiliHK and SiliHpt were phosphorylated with [γ-32P]ATP prior to the addition of a response regulator. Rapid (within 1 min) loss of phosphorylation from both SiliHK and SiliHpt was observed for only one of the response regulators, SCH4B_1503 (Fig. 3B), whereas for the other response regulators, slow loss of phosphorylation from SiliHK/SiliHpt occurred over 15 to 30 min (see Fig. S4 in the supplemental material). Likely due to the instability of phosphoaspartate esters (32), a phosphorylation signal for SCH4B_1503 was not observed. The SCH4B_1503 protein is annotated to contain a GGDEF domain found in diguanylate cyclases (33), and hereafter will be referred to as SiliDGC.
Phosphorylation inhibits SiliDGC activity.
Diguanylate cyclases catalyze the formation of c-di-GMP from two molecules of GTP. Sequence alignment with characterized diguanylate cyclases confirmed the presence of the conserved GGDEF active site residues in SiliDGC. To confirm that SiliDGC is a functional diguanylate cyclase, SiliDGC was recombinantly expressed, purified from E. coli, and incubated with GTP. Formation of c-di-GMP was observed by high-performance liquid chromatography (HPLC), confirming that SiliDGC is an active diguanylate cyclase (see Fig. S5 in the supplemental material).
Phosphorylation of response regulator domains generally modulates the activity of the connected enzymatic or binding domains within the protein, so we then investigated the effect of response regulator phosphorylation on SiliDGC activity. When SiliDGC was incubated with SiliHK, SiliHpt, and ATP, the diguanylate cyclase activity decreased by ~40% (Fig. 4A). When either one of the phosphotransfer components (i.e., SiliHpt or ATP) was omitted, the activity was similar to that of SiliDGC alone. The addition of the SiliHK D386A receiver domain mutant, which is incapable of phosphotransfer, in place of wild-type SiliHK also did not significantly inhibit SiliDGC activity (Fig. 4A). A similar trend was observed when SiliDGC was incubated with beryllium-fluoride (BeFx), a known phosphorylation mimic (34) that is not subject to hydrolysis and thus more stable than phosphorylation. Titration of increasing BeFx concentrations resulted in decreasing levels of DGC activity, as SiliDGC activity decreased by ~80% in the presence of 0.5 mM BeFx (Fig. 4B). Taken together, these results indicate that phosphorylation of SiliDGC by SiliHK/SiliHpt decreases SiliDGC activity and therefore should decrease cellular c-di-GMP levels.
FIG 4 .
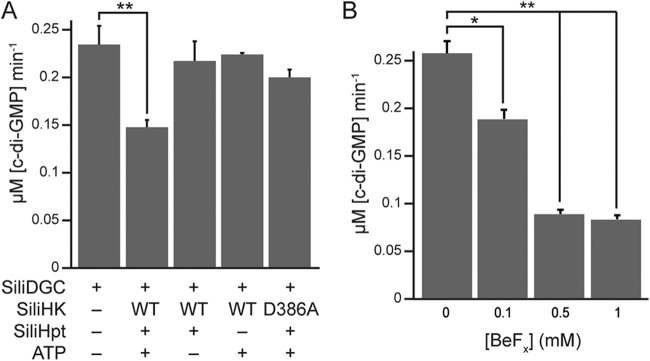
SiliDGC activity decreases in the presence of HK/HPT and ATP. (A) Phosphorylation of SiliDGC by SiliHK/SiliHpt inhibits SiliDGC activity. An assay containing 5 µM SiliDGC was incubated with some or all of the phosphotransfer components: SiliHK, SiliHpt, and ATP, and c-di-GMP formation was monitored by HPLC as described in Materials and Methods. A receiver domain mutant of the histidine kinase, SiliHK D386A, which is incapable of phosphotransfer to SiliHPT, was used as an additional control. Loss of SiliDGC activity was observed only with the addition of all of the necessary phosphotransfer components: SiliHK, SiliHpt, and ATP. WT, wild type. (B) SiliDGC is inhibited by a phosphorylation mimic. An assay mixture containing 5 µM SiliDGC was incubated with the phosphorylation mimic, beryllium-fluoride (BeFx) for 15 min before the reaction was initiated, and c-di-GMP formation was monitored by HPLC as described in Materials and Methods. Loss of SiliDGC activity was observed with increasing BeFx concentrations. Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
Silicibacter sp. TrichCH4B biofilm formation is induced by exogenous NO.
Cyclic di-GMP controls bacterial processes, such as motility, cellular aggregation, and biofilm formation (35). In particular, NO-mediated control of c-di-GMP levels has been shown to regulate bacterial biofilm formation (11, 12, 14). We tested whether NO induces a similar effect in Silicibacter sp. TrichCH4B, as SiliH-NOX inhibits SiliHK, thereby relieving the inhibitory effects of phosphorylation on SiliDGC. Thus, NO is expected to lead to an overall increase in c-di-GMP levels and biofilm formation (see Fig. 6). Static biofilm assays with Silicibacter sp. TrichCH4B were performed in an anaerobic chamber, with NO introduced via (Z)-1-[N-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NONOate) added to the growth medium at 0 to 200 µM concentrations. DETA-NONOate is a slow-release NO donor, with a half-life (t1/2) of 56 h at 25°C and pH 7 (36). Before biofilm formation was measured, cell density was measured by optical density at 600 nm (OD600), and the cells grew to similar densities under all conditions tested. NO addition resulted in a twofold increase in biofilm formation with 200 µM DETA-NONOate compared with no NO added, as measured by crystal violet staining (Fig. 5A).
FIG 6 .
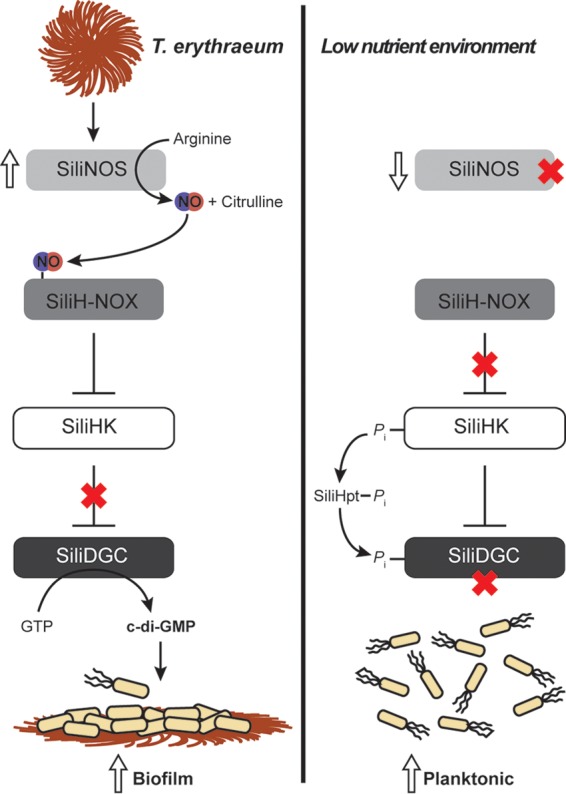
Silicibacter sp. TrichCH4B NO signaling. Summary of the NO signaling pathway in Silicibacter sp. TrichCH4B. (Left) SiliNOS is activated by a secreted T. erythraeum protein through an unknown mechanism. NO-bound SiliH-NOX inhibits SiliHK autophosphorylation activity. Loss of phosphorylation on SiliDGC leads to increased diguanylate cyclase activity, resulting in higher c-di-GMP levels and biofilm formation. (Right) In the absence of T. erythraeum, SiliNOS expression is reduced, which relieves inhibition of SiliHK autophosphorylation by SiliH-NOX. The resulting increase in SiliDGC phosphorylation via SiliHpt leads to decreased activity and lower c-di-GMP levels, and therefore less biofilm formation and more planktonic cells as a result.
FIG 5 .

Silicibacter sp. strain TrichCH4B biological response to NO and Trichodesmium erythraeum. (A) Exogenous NO increases Silicibacter sp. TrichCH4B biofilm formation. Static biofilm assays were performed as described in Materials and Methods. With increasing amounts of NO, Silicibacter sp. TrichCH4B formed more biofilm, as quantified by crystal violet staining (OD570). Biofilm formation was normalized against growth (OD600). (B) Addition of T. erythraeum spent medium (TSM) increases Silicibacter sp. TrichCH4B NO formation. Silicibacter sp. TrichCH4B was grown anaerobically with TSM as described in Materials and Methods. To fully digest any proteins, TSM was also treated with proteinase K before the addition to Silicibacter sp. TrichCH4B. Only the TSM (high-molecular-weight [MW] fraction, or retentate, from a 5-kDa-MWCO membrane filter) showed stimulation of NO formation by Silicibacter sp. TrichCH4B. (C) TSM addition increases SiliNOS gene expression. Silicibacter sp. TrichCH4B was grown aerobically with various amounts of TSM, and cDNA from Silicibacter sp. TrichCH4B mRNA was prepared as described in Materials and Methods. Expression of the SiliNOS gene (ΔC(t)) was calculated using Bio-Rad CFX Manager software with rpoD as a reference gene. SiliNOS gene expression increased with the amount of TSM added. (D) TSM addition increases Silicibacter sp. TrichCH4B biofilm formation. Static biofilm assays were performed as described in Materials and Methods. Silicibacter sp. TrichCH4B biofilm formation increased with the amount of TSM added, as quantified by crystal violet staining. Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
Silicibacter sp. TrichCH4B NO formation.
To determine the factors that lead to NO synthesis and initiation of this novel bacterial signaling pathway, NO formation by Silicibacter sp. TrichCH4B was directly measured using a nitric oxide analyzer (Fig. 5B, None bar). The bacteria were grown anaerobically in sealed Hungate tubes, and before assaying the headspace for NO, a small volume of aerobic media was supplemented to provide sufficient oxygen for the O2-dependent NOS reaction. Under anaerobic growth conditions in seawater complete medium, Silicibacter sp. TrichCH4B produces NO. To confirm that the NO observed is produced by NOS, NO formation by two organisms that are closely related to Silicibacter sp. strain TrichCH4B, Silicibacter sp. strain TM1040 and Dinoroseobacter shibae FL-12 were also tested under the same conditions. These species do not contain a predicted NOS gene or contain genes that encode components of the NO-associated signaling pathway present in Silicibacter sp. TrichCH4B, and NO formation was not observed from either Silicibacter sp. TM1040 or D. shibae FL-12 (see Fig. S6 in the supplemental material).
T. erythraeum induces Silicibacter sp. TrichCH4B NO formation.
Having established that NO regulates Silicibacter sp. TrichCH4B aggregation and biofilm formation, we next focused on the signal(s) that induce NO synthesis in Silicibacter sp. TrichCH4B. To determine the factors regulating SiliNOS activity, we turned to the natural habitat of the organism.
Silicibacter sp. TrichCH4B was originally isolated from colonies of the T. erythraeum alga as a symbiont (37). Therefore, we hypothesized that T. erythraeum regulates SiliNOS activity, and the effect of T. erythraeum on NO production by Silicibacter sp. TrichCH4B was tested. Cultures of T. erythraeum IMS101 were grown, and the cells were filtered through a 0.2-µm membrane to remove the T. erythraeum cells from the spent medium, which was subsequently concentrated using a 5-kDa molecular-weight-cutoff (MWCO) membrane. The concentrated T. erythraeum spent medium (TSM) was added to Silicibacter sp. TrichCH4B cultures to test for stimulation of NOS activity. The high- and low-molecular-weight fractions of the concentrated medium (TSM, or retentate, and flowthrough of the 5-kDa-MWCO membrane, respectively) were added separately to Silicibacter sp. TrichCH4B cultures, and NO formation was measured using a nitric oxide analyzer. Cultures with the TSM, or high-molecular-weight fraction, exhibited an ~7-fold increase in NO formation over the samples with the flowthrough (low-molecular-weight fraction), or without any additions (Fig. 5B, None bar versus TSM bar). This demonstrates that T. erythraeum secretes a signaling agent captured in the TSM that induces NO production by SiliNOS.
To further characterize the stimulating factor of NO formation, TSM was treated with heat (95°C for 5 min) (data not shown) or protease (proteinase K). Silicibacter sp. TrichCH4B cultures with TSM under both treatments did not show any stimulated NO formation (Fig. 5B). Thus, the stimulant of Silicibacter sp. TrichCH4B NO formation appears to be a secreted protein from the T. erythaeum culture that needs to be properly folded to stimulate NO formation. We cannot rule out the possibility that the stimulant could be a small molecule that requires a protein carrier to deliver the molecule to Silicibacter sp. TrichCH4B. In this case, however, the protein carrier would mostly likely require a specific receptor on Silicibacter sp. TrichCH4B, since destroying the protein carrier by proteolysis abolishes stimulation of NO formation.
T. erythraeum induces SiliNOS gene expression.
To determine whether the stimulation of Silicibacter sp. TrichCH4B NO formation by T. erythraeum was regulated by gene expression or by direct activation of the SiliNOS enzyme, reverse transcription-quantitative PCR (RT-qPCR) was performed to examine the effect of TSM on SiliNOS gene expression. The addition of increasing amounts of TSM correlated with increasing levels of SiliNOS mRNA (Fig. 5C). The levels of expression of a control gene, rpoD encoding RNA polymerase, were unchanged under all conditions. The observed increase in SiliNOS gene expression correlated with the increase in NO formation by Silicibacter sp. TrichCH4B when grown with TSM (Fig. 5B), indicating that the stimulation of Silicibacter sp. TrichCH4B NO production occurs by inducing SiliNOS gene expression.
T. erythraeum spent medium induces Silicibacter sp. TrichCH4B biofilm formation.
Since TSM increases NO formation by Silicibacter sp. TrichCH4B and NO induces biofilm formation, we tested whether TSM addition also increases Silicibacter sp. TrichCH4B biofilm formation. Silicibacter sp. TrichCH4B biofilm formation was quantified using the crystal violet staining assay as described below. Addition of TSM leads to increased Silicibacter sp. TrichCH4B biofilm formation in a concentration-dependent manner, confirming that T. erythraeum secreted protein(s) activates the NO signaling pathway and increases biofilm formation in Silicibacter sp. TrichCH4B (Fig. 5D).
DISCUSSION
Silicibacter sp. strain TrichCH4B H-NOX signals through a conserved two-component signaling network.
H-NOX signaling has been shown to regulate bacterial motility through control of cellular c-di-GMP levels, either by direct regulation of a diguanylate cyclase (11) or through a two-component signaling network (12, 14). In Silicibacter sp. TrichCH4B, NO-bound SiliH-NOX inhibits SiliHK autophosphorylation (Fig. 2B), as is the case with all other characterized H-NOX-associated histidine kinases (8, 13). To identify the cognate response regulator for SiliHK, we relied on the principle that in vivo cognate interaction partners are expected to exhibit fast phosphotransfer kinetics in vitro, and only one orphan response regulator caused a rapid decrease in SiliHK/SiliHpt phosphorylation (see Fig. S4 in the supplemental material). This response regulator contains a GGDEF domain with diguanylate cyclase activity (SiliDGC) (Fig. S5), and phosphorylation of the SiliDGC receiver domain inhibits SiliDGC diguanylate cyclase activity (Fig. 4). However, inhibition of SiliHK by NO-bound SiliH-NOX relieves the SiliDGC inhibition, resulting in higher c-di-GMP levels than when SiliHK is fully active (Fig. 6).
In Shewanella oneidensis, NO-bound H-NOX inhibits the activity of its associated histidine kinase (13). In S. oneidensis, there are three cognate response regulators for the H-NOX-associated histidine kinase. One of the response regulators is an EAL-containing phosphodiesterase (PDE) that hydrolyzes c-di-GMP, and phosphorylation was shown to stimulate PDE activity. Kinase inhibition by NO-bound H-NOX results in lower PDE phosphorylation and activity and, therefore, higher c-di-GMP levels (14). A similar signaling network was confirmed in Vibrio cholerae (14). In the current study, the Silicibacter sp. TrichCH4B H-NOX/histidine kinase signaling network leads to the same overall response: NO signaling results in increased cellular c-di-GMP levels through relief of diguanylate cyclase inhibition (SiliDGC).
Cyclic di-GMP is an important bacterial second messenger, controlling various processes such as motility and biofilm formation (35). Typically, higher c-di-GMP levels result in increased biofilm formation, consistent with the phenotype exhibited by Silicibacter sp. TrichCH4B (Fig. 5A and D), as well as S. oneidensis and V. cholerae (14). In addition to controlling a two-component signaling circuit, NO-bound H-NOX has been shown in Legionella pneumophila and Shewanella woodyi to directly control activity of an adjacent GGDEF and/or EAL-containing protein (11, 12). Thus, NO/H-NOX control of bacterial biofilm through c-di-GMP regulation may be a universal mechanism governing bacterial communal behavior.
Silicibacter sp. TrichCH4B: first characterized bacterium with NOS-dependent H-NOX pathway.
Silicibacter sp. TrichCH4B is unique among the aforementioned bacteria in that, in addition to the NO-sensing H-NOX, the Silicibacter sp. TrichCH4B genome also encodes a full-length nitric oxide synthase (SiliNOS). SiliNOS shares the same domain architecture as the previously characterized NOS from S. cellulosum (42% sequence identity) (23) (see Fig. S1 in the supplemental material). SiliNOS has a putative N-terminal reductase domain that contains a 2Fe-2S cluster as well as FAD and NAD(P)H binding sites and a predicted NOS oxygenase domain at the C terminus. In contrast, mammalian NOS reductase domains are encoded C terminally to the oxygenase domain and resemble cytochrome c reductase and other cytochrome P450 reductases, with FAD, FMN, and NADPH binding subdomains. Many bacterial NOS proteins (e.g., from Bacillus, Staphylococcus, and Geobacillus species) contain only the oxygenase domain without a fused reductase domain. In those organisms, an independent reductase protein is required for NO synthesis (18, 22). Thus, the oxygenase domain appears to be the common ancestor for bacterial and mammalian NOS, and variants have evolved to include different reductase domains or remain without a fused reductase.
To date, NO signaling in NOS-containing bacteria is not well understood. Proposed functions for bacterial NOS proteins include protection against oxidative stress and antibiotics (20, 38–40), but in most bacteria, the biological role of NO synthesized by bacterial NOS is unknown. As mentioned above, bacterial H-NOX proteins regulate bacterial biofilm formation. For these H-NOX-containing bacteria, the source of NO is likely from the host immune system or from nitrate reduction in the environment, such as surrounding soil (8).
In mammals, NO from NOS activates production of the mammalian second messenger cGMP by sGC. In Silicibacter sp. TrichCH4B, NO leads to increased levels of the bacterial second messenger c-di-GMP. Silicibacter sp. TrichCH4B is the first characterized bacterium with a complete NO signaling system, in which the bacterial H-NOX responds to NO synthesized by the bacteria, instead of NO generated in the environment. The clear parallels between mammalian and Silicibacter sp. TrichCH4B NO signaling pathways suggest a bacterial evolutionary origin for mammalian NO signaling. Although Silicibacter sp. TrichCH4B is the only organism so far with characterized NOS and H-NOX proteins, further bacterial genome sequencing will likely reveal additional organisms with similar NO signaling pathways.
Silicibacter sp. TrichCH4B NO signaling is induced by Trichodesmium erythraeum.
Silicibacter sp. TrichCH4B was originally isolated from colonies of the alga Trichodesmium, a filamentous cyanobacterium. The genus Trichodesmium has gained much scientific interest as one of the major diazotrophic (dinitrogen-fixing) species in the ocean (41) and is capable of doing so in a low-nutrient marine environment. Thus, Trichodesmium alga colonies provide a nutrient-rich environment that supports a variety of bacteria. The symbiotic interactions between Trichodesmium and associated bacteria are of great interest, as these bacteria are essential to the ecology of algae and dinitrogen fixation as well as maintaining a balanced ocean ecosystem (42–44).
Previous studies have shown that the bacteria, including Silicibacter sp. TrichCH4B, provide Trichodesmium with essential minerals and metals (such as iron), indicating a symbiotic relationship (44, 45). However, the specific signaling mechanisms between Trichodesmium and associated bacteria have not been explored. Here, we found that SiliNOS gene expression and NO production by Silicibacter sp. TrichCH4B is stimulated by the presence of concentrated spent medium from a growing T. erythraeum culture (TSM) (Fig. 5B). Accordingly, Silicibacter sp. TrichCH4B biofilm also increases with the addition of TSM (Fig. 5D). Protease and heat treatments of TSM removed any stimulation of Silicibacter sp. TrichCH4B NO production, suggesting that activation of Silicibacter sp. TrichCH4B NO production requires a secreted protein (Fig. 5B). Current efforts are directed at identification of this T. erythraeum signaling protein.
Other bacteria in the Roseobacter clade, which includes Silicibacter sp. TrichCH4B, are symbiotic with dinoflagellates and practice a “swim or stick” lifestyle, in which they switch from sessile to motile phases depending upon the presence of their algal symbiont (46, 47). Silicibacter sp. TrichCH4B and T. erythraeum appear to have a similar mechanism for symbiosis—when Silicibacter sp. TrichCH4B is in the vicinity of T. erythraeum, it senses a T. erythraeum secreted signaling protein. This protein could bind to an extracellular receptor in Silicibacter sp. TrichCH4B, or be translocated into the cytosol, which then induces SiliNOS gene expression and thus, increased NO formation. NO activates the H-NOX signaling pathway, leading to higher levels of cellular c-di-GMP and biofilm formation, most likely on the T. erythraeum colonies, poising the two species for nutrient exchange and improved symbiotic growth and production processes (Fig. 6). The Silicibacter sp. TrichCH4B NO signaling pathway may be key to the symbiosis between the two organisms, revealing a new role for NO in bacterial signaling and communication.
MATERIALS AND METHODS
SiliNOSox, SiliH-NOX, SiliHK, and SiliHpt expression and purification.
Plasmids containing the desired genes were transformed into E. coli BL21(DE3) cells. Expression cultures were grown at 37°C, induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of ~0.6, and grown at 18°C for 20 to 22 h. Cells were pelleted and resuspended in lysis buffer (for SiliNOSox, 50 mM sodium phosphate [pH 8.0], 150 mM NaCl, 10% [vol/vol] glycerol, 10 mM imidazole, 1 mM Pefabloc, 1 mM benzamidine; for all others, 50 mM diethanolamine [DEA] [pH 8.0], 300 mM NaCl, 10% [vol/vol] glycerol, 10 mM imidazole, 1 mM Pefabloc, 1 mM benzamidine) and lysed by passage through a high-pressure homogenizer (Avestin). Cell debris was removed by centrifugation at 100,000 × g for 30 min using an Optima XL-100K ultracentrifuge with an Ti-45 rotor (Beckman). The supernatant was loaded onto ~5-ml nickel resin (nickel-nitrilotriacetic acid [Ni-NTA] superflow from Qiagen) preequilibrated with wash buffer (for SiliNOSox, 50 mM sodium phosphate [pH 8.0], 150 mM NaCl, 10% [vol/vol] glycerol, 10 mM imidazole; for all others, 50 mM DEA [pH 8.0], 300 mM NaCl, 10% [vol/vol] glycerol, 10 mM imidazole). The resin was washed with 20 column volumes of wash buffer, and protein was eluted with 200 mM imidazole in wash buffer. Proteins were exchanged into storage buffer (wash buffer without imidazole) by dialysis, flash frozen in liquid nitrogen, and stored at −80°C.
Glutathione S-transferase (GST)-SiliHpt, SiliDGC, and other response regulators (RRs).
For expression, plasmids containing the desired genes were transformed into E. coli BL21(DE3)/pLysS cells (Life Technologies). Expression cultures were grown at 37°C, induced with 1 mM IPTG at an OD600 of ~0.6, and grown at 20°C for 20 to 22 h. Cell pellets were resuspended in lysis buffer (50 mM DEA [pH 8.0], 300 mM NaCl, 10% [vol/vol] glycerol, 1 mM Pefabloc, 1 mM benzamidine). Supernatant was prepared in the same manner as described above and loaded onto ~5 ml glutathione resin (GE Healthcare) preequilibrated with wash buffer (50 mM DEA [pH 8.0], 300 mM NaCl, 10% [vol/vol] glycerol). The resin was then washed with 10 column volumes of wash buffer, and protein was eluted with 10 mM reduced glutathione in wash buffer. Proteins were exchanged into storage buffer (wash buffer without glutathione) by dialysis, flash frozen in liquid nitrogen, and stored at −80°C.
UV-Vis spectroscopy.
UV-visible (UV-Vis) spectra were collected on a Varian Cary 300 Bio UV-Vis spectrophotometer. SiliNOSox was reduced to the ferrous (Fe2+) state by the addition of 1 mM Na2S2O4 for 20 min at 25°C and desalted using a PD-10 column (GE Healthcare) equilibrated with deoxygenated buffer (50 mM sodium phosphate [pH 8.0], 150 mM NaCl, 10% [vol/vol] glycerol). The protein was placed in a sealed anaerobic cuvette, and spectra were measured against a baseline of buffer from 200 to 700 nm. Fe2+, Fe2+-NO, and Fe2+-CO SiliH-NOX were prepared as previously described (13), and spectra were collected in the same manner as for SiliNOSox. Fe2+-O2 SiliH-NOX was not observed upon exposing the cuvette to oxygen or upon adding aerobic buffer to the protein sample.
SiliNOSox single-turnover assay.
NO formation was measured using an NO analyzer (NOA) (Sievers model 270; GE Analytical Instruments) as previously described (23). Briefly, reactions were carried out at room temperature in sealed Reacti-vials. A 40-µl final assay mix contained 500 µM N-hydroxyarginine, 0 or 200 µM H4B or H4F, and 100 µM reduced SiliNOSox (Fe2+) in 100 mM HEPES (pH 7.4). Reduced SiliNOSox was prepared as described above. Reactions were initiated with 40 µl of aerobic buffer. NO formation was measured in 30-s intervals by sampling 500 µl of Reacti-vial headspace using a gas-tight syringe (Hamilton) and injecting it into the NOA reaction vessel. Each reaction was sampled four times.
SiliHK autophosphorylation assay.
The kinase activity of SiliHK was assayed using γ-S-labeled ATP (ATPγS) as previously described (24). Briefly, 5 µM SiliHK was mixed with 5 mM MgCl2 and 1 mM ATPγS in 50 mM DEA (pH 8.0), 150 mM NaCl, and 5% (vol/vol) glycerol in 20-µl reaction mixtures. Reactions were quenched with 2.5 µl of 500 mM EDTA, and 1.5 µl of 50 mM para-nitrobenzylmesylate (PNBM) was added to alkylate the thiophosphate and incubated for 1 h at room temperature. Proteins were separated on 10-20% Tris-glycine SDS-polyacrylamide gels (Life Technologies), then transferred to nitrocellulose membranes (Whatman), and blocked with 5% (wt/vol) nonfat dry milk in phosphate-buffered saline (pH 8.0) with 0.5% (vol/vol) Tween 20 (PBST). Primary antibody specific for the alkylated thiophosphate ester (anti-PNBM) (monoclonal antibody 51-8; Epitomics) was added at a 1:5,000 dilution and incubated overnight at 4°C. The blot was then washed three times for 10 min each time with PBST at room temperature. Secondary antibody, goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP) (Pierce) was then added at a 1:1,000 dilution and incubated at room temperature for 1 h. The blot was then washed again three times for 10 min each time with PBST at room temperature. The blot was developed using Luminata Classico Western HRP substrate (Millipore) and imaged using a Chemidoc MP imager (Bio-Rad).
SiliH-NOX/SiliHK activity assay.
SiliHK (5 µM) was incubated with 30 µM Fe2+, Fe2+-NO, or Fe2+-CO SiliH-NOX, and 5 mM MgCl2. The reaction buffer was the same as for the SiliHK autophosphorylation assay. Reactions were initiated with 1 mM ATPγS. The reaction was quenched at specified times, and data were collected as described above for the SiliHK autophosphorylation assay.
SiliHK/SiliHpt phosphotransfer assay.
SiliHK (5 µM) was incubated with 10 µM SiliHpt and 5 mM MgCl2. Reactions were initiated with 1 mM ATPγS. The reaction buffer was the same as for the SiliHK autophosphorylation assay. The reaction was quenched at specified times, and data were collected as described above for the SiliHK autophosphorylation assay.
Phosphotransfer profiling of orphan response regulators.
Twelve orphan response regulators identified through the SMART database (26, 27) were tested for phosphotransfer from SiliHK/SiliHpt. SiliHK (15 µM) and GST-SiliHpt (5 µM) were preincubated with 1 mM ATP, 5 µCi [γ-32P]ATP, and 5 mM MgCl2 for 15 min and then desalted over a PD-10 column (GE Healthcare) to remove excess ATP. Each individual RR (10 µM) was then added to a separate reaction mix. At endpoints, reactions were quenched with 6× SDS loading dye, and the proteins were separated by SDS-PAGE. The gels were dried overnight on a slab gel dryer (Hoeffer Scientific Instruments), and dried gels were exposed overnight (16 to 20 h) on a Kodak phosphorimaging plate. Data were collected on a Typhoon phosphorimager at 100-µm resolution.
Diguanylate cyclase assay.
Purified SiliDGC protein was incubated in 50 mM DEA (pH 8.0), 150 mM NaCl, and 5% (vol/vol) glycerol with 10 mM MgCl2, and reactions were initiated by the addition of 0.5 mM GTP. An internal HPLC standard, 0.5 mM tryptophan, was also included. Aliquots (10 µl) were quenched at different time points by the addition of 25 µl trifluoroacetic acid (2% [vol/vol] in water). Quenched reaction volumes were adjusted to 100 µl, and the samples were filtered through a 0.2-µm spin filter and analyzed by HPLC on a Nova-Pak C18 column (3.9 by 150 mm) (4 µm) at a flow rate of 1 ml/min using the following gradient at the time indicated: from 0 to 6 min, 100% 20 mM ammonium acetate; from 6 to 7.5 min, 95% 20 mM ammonium acetate and 5% (vol/vol) acetonitrile; from 7.5 to 8.4 min, 85% 20 mM ammonium acetate and 15% (vol/vol) acetonitrile; from 8.4 to 9 min, 5% 20 mM ammonium acetate and 95% (vol/vol) acetonitrile; from 9.1 to 13.5 min, 100% 20 mM ammonium acetate. c-di-GMP concentration was calculated from peak integration and a standard curve of c-di-GMP. The c-di-GMP standard was synthesized enzymatically as previously described (14).
To examine the effects of phosphorylation on SiliDGC activity, phosphotransfer partners were included in the assay, SiliHK (15 µM) and SiliHpt (15 µM) were prephosphorylated with 0.5 mM ATP for 15 min. SiliDGC was then added to the reaction mixture and incubated for 15 min before initiating reactions by the addition of 0.5 mM GTP. SiliDGC activity assays in the presence of beryllium-fluoride were performed as previously described (14, 34). All experiments were performed in triplicate.
Crystal violet biofilm assay.
Biofilm assays with DETA-NONOate were performed in an anaerobic glove bag (Coy Laboratory Products) in 12-well polystyrene plates. Silicibacter sp. strain TrichCH4B was grown aerobically in seawater complete (SWC) medium at 30°C overnight. Anaerobic SWC medium was inoculated with 100-fold-diluted overnight cultures. Freshly prepared DETA-NONOate (Cayman Chemicals) in 10 mM NaOH was added at various concentrations. Cells were grown statically at 25°C, and a 3-ml reference culture was grown for OD measurements and normalization. Biofilm formation was quantified after 20 h and normalized to growth (OD600). Crystal violet staining was performed in the same manner as previously described (14, 48). Measurement from individual wells were averaged and normalized to growth (OD600).
Biofilm assays with concentrated T. erythraeum spent medium (TSM) were performed aerobically (to provide the necessary O2 for NOS activity) in the same manner as described above. Reported results are from the averages of three 12-well experiments on separate days.
Trichodesmium erythraeum growth and spent medium concentration.
A nonaxenic culture of Trichodesmium erythraeum IMS101 (obtained from D. Hutchins, University of Southern California) was grown at 25°C with a 12-h light/12-h dark cycle (70 to 100 μE m−2 s−1) in a modified Aquil* medium (49). The modified medium is composed of synthetic ocean water (SOW) with 10 µM NaH2PO4·H2O, 10 µM EDTA, 1 µM FeCl3·6H2O, 79.7 nM ZnSO4·7H2O, 121 nM MnCl2·4H2O, 50.3 nM CoCl2·6H2O, 100 nM Na2MoO4·2H2O, 297 nM thiamine, 2.25 nM biotin, and 0.37 nM cyanocobalamin. Cultures were diluted into fresh medium (volume increased by 2 to 5 times each transfer) every 7 days. Spent medium from 14 to 28 days of T. erythraeum culture growth was collected by filtration through a 0.2-µm filter and then concentrated 1,000 times by tangential-flow filtration with a 5-kDa MWCO membrane (Millipore). Further concentration was performed using a Macrosep Advance centrifugal device (Pall) to obtain a final protein concentration of ~1 mg/ml.
Heat treatment of the concentrated spent medium was performed by heating the solution for 5 min at 95°C. Protease treatment of the concentrated spent medium was performed by adding proteinase K (final concentration of 0.5 mg/ml) (Qiagen) according to the manufacturer’s instructions.
Silicibacter sp. strain TrichCH4B NO formation.
Overnight cultures of Silicibacter sp. TrichCH4B were inoculated into 10 ml aerobic SWC medium in sealed Hungate tubes. When examining the effects of T. erythraeum on NO formation by Silicibacter sp. TrichCH4B, TSM was added to the cultures in specified amounts. Cultures were grown with shaking for 20 h at 30°C. Ten minutes before measuring NO, 1 ml of aerobic SWC medium was delivered via syringe into the Hungate tubes to provide sufficient oxygen for the NOS reaction. Headspace (100 µl) was injected into an NO analyzer, and peaks were integrated using the NOAnalysis software for relative quantification. All experiments were performed in triplicate.
Silicibacter sp. TrichCH4B RNA preparation and quantitative PCR.
Cultures of Silicibacter sp. TrichCH4B (5 ml) were grown in the presence of various amounts of TSM. Cells were harvested after 12 h of growth. Total RNA was extracted using an RNeasy purification kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized from RNA with a SuperScript III reverse transcriptase kit according to the manufacturer’s instructions (Life Technologies). Control reaction mixtures lacking reverse transcriptase were included to confirm the absence of contaminating genomic DNA. Quantitative PCR (qPCR) amplification of the SiliNOS gene was performed using SYBR green (Life Technologies) with SiliNOS qPCR primers (see Table S1 in the supplemental material) on a C1000 thermal cycler with a CFX96 real-time system (Bio-Rad). A two-step amplification procedure was used: 10 min at 95°C, followed by 40 cycles, with 1 cycle consisting of 15 s at 95°C and 30 s at 55°C. Results were analyzed using the Bio-Rad CFX Manager software. Results were normalized relative to the level of expression of the housekeeping gene rpoD (37). The relative expression values represent the means ± standard deviations of triplicate samples from three independent experiments.
SUPPLEMENTAL MATERIAL
Mammalian and bacterial nitric oxide synthase domain architecture. Comparison of mammalian and bacterial nitric oxide synthases. The N and C termini and relative positions of the identified NOS domains are indicated. The heme/oxygenase domains are shown in gray, and the reductase domains are shown in white. Representative organisms with the stand-alone bacterial NOS oxygenase domain are indicated. S. cellulosum and Silicibacter sp. TrichCH4B are the only two known organisms with genes that encode a NOS with an N-terminal reductase domain containing a 2Fe-2S cluster. Download
SiliH-NOX ligation states. UV-Vis spectra of SiliH-NOX in different oxidation and ligation states are shown. The wavelengths of maximum absorption (λmax) follow: 411 nm for Fe3+, 426 nm for Fe2+, 399 nm for Fe2+-NO, and 420 nm for Fe2+-CO. Download
Silicibacter sp. TrichCH4B two-component phosphorelay signaling system. SiliHK is a hybrid histidine kinase, which has a receiver domain containing an aspartate phosphoryl acceptor. A separate Hpt protein is required as an intermediary for phosphotransfer from the receiver domain of SiliHK to the receiver domain of the response regulator SiliDGC. Download
Phosphotransfer from SiliHK/SiliHpt to orphan response regulators. SCH4B_3211, SCH4B_1525, and SCH4B_3426 are three representative orphan response regulators that did not cause rapid loss of SiliHK/SiliHpt phosphorylation. In contrast, SCH4B_1503 (SiliDGC), the cognate phosphotransfer partner of SiliHK/SiliHpt, caused complete loss of SiliHK/SiliHpt phosphorylation within 5 min. Control reactions with no response regulator added or with the E. coli response regulator OmpR also did not cause a rapid loss in SiliHK/SiliHpt phosphorylation. Download
SiliDGC activity assays. SiliDGC (10 µM) was mixed with 0.5 mM GTP, and aliquots were quenched at 1-min, 5-min, 30-min, 1-h, and 2-h time points as described in Materials and Methods. Near complete conversion of GTP to c-di-GMP was observed through the 2-h time course. Download
NO formation by Silicibacter sp. TrichCH4B compared with Roseobacter species. Silicibacter sp. TrichCH4B, Silicibacter sp. TM1040, and Dinoroseobacter shibae FL-12 were tested for NO formation using the NOA as described in Materials and Methods. Only Silicibacter sp. TrichCH4B forms NO. Seawater complete medium was also tested as a control. Download
Primers and plasmids used in this study
ACKNOWLEDGMENTS
We thank Katherine Barbeau and Shane Hogle from the Scripps Institution of Oceanography for providing Silicibacter sp. TrichCH4B, Silicibacter sp. TM1040, and D. shibae cultures and David Hutchins and Feixue Fu from the University of Southern California for providing T. erythraeum cultures and assistance with culture conditions.
Footnotes
Citation Rao M, Smith BC, Marletta MA. 2015. Nitric oxide mediates biofilm formation and symbiosis in Silicibacter sp. strain TrichCH4B. mBio 6(3):e00206-15. doi:10.1128/mBio.00206-15.
Contributor Information
George O’Toole, Geisel School of Medicine at Dartmouth.
Edward G. Ruby, University of Wisconsin–Madison.
REFERENCES
- 1.Dinerman JL, Lowenstein CJ, Snyder SH. 1993. Molecular mechanisms of nitric oxide regulation—potential relevance to cardiovascular disease. Circ Res 73:217–222. doi: 10.1161/01.RES.73.2.217. [DOI] [PubMed] [Google Scholar]
- 2.Moncada S, Palmer RM, Higgs EA. 1991. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43:109–142. [PubMed] [Google Scholar]
- 3.Kerwin JF, Lancaster JR, Feldman PL. 1995. Nitric oxide: a new paradigm for second messengers. J Med Chem 38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 4.Ignarro LJ, Cirino G, Casini A, Napoli C. 1999. Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol 34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Bredt DS, Snyder SH. 1992. Nitric oxide, a novel neuronal messenger. Neuron 8:3–11. doi: 10.1016/0896-6273(92)90104-L. [DOI] [PubMed] [Google Scholar]
- 6.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. 2004. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci U S A 101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karow DS, Pan D, Tran R, Pellicena P, Presley A, Mathies RA, Marletta MA. 2004. Spectroscopic characterization of the soluble guanylate cyclase-like heme domains from Vibrio cholerae and Thermoanaerobacter tengcongensis. Biochemistry 43:10203–10211. doi: 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- 8.Plate L, Marletta MA. 2013. Nitric oxide-sensing H-NOX proteins govern bacterial communal behavior. Trends Biochem Sci 38:566–575. doi: 10.1016/j.tibs.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alderton WK, Cooper CE, Knowles RG. 2001. Nitric oxide synthases: structure, function and inhibition. Biochem J 357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macmicking J, Xie QW, Nathan C. 1997. Nitric oxide and macrophage function. Annu Rev Immunol 15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 11.Carlson HK, Vance RE, Marletta MA. 2010. H-NOX regulation of c-di-GMP metabolism and biofilm formation in Legionella pneumophila. Mol Microbiol 77:930–942. doi: 10.1111/j.1365-2958.2010.07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N, Xu Y, Hossain S, Huang N, Coursolle D, Gralnick JA, Boon EM. 2012. Nitric oxide regulation of cyclic di-GMP synthesis and hydrolysis in Shewanella woodyi. Biochemistry 51:2087–2099. doi: 10.1021/bi201753f. [DOI] [PubMed] [Google Scholar]
- 13.Price MS, Chao LY, Marletta MA. 2007. Shewanella oneidensis MR-1 H-NOX regulation of a histidine kinase by nitric oxide. Biochemistry 46:13677–13683. doi: 10.1021/bi7019035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plate L, Marletta MA. 2012. Nitric oxide modulates bacterial biofilm formation through a multicomponent cyclic-di-GMP signaling network. Mol Cell 46:449–460. doi: 10.1016/j.molcel.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plate L, Marletta MA. 2013. Phosphorylation-dependent derepression by the response regulator HnoC in the Shewanella oneidensis nitric oxide signaling network. Proc Natl Acad Sci U S A 110:E4648–E4657. doi: 10.1073/pnas.1318128110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurshman AR, Marletta MA. 2002. Reactions catalyzed by the heme domain of inducible nitric oxide synthase: evidence for the involvement of tetrahydrobiopterin in electron transfer. Biochemistry 41:3439–3456. doi: 10.1021/bi012002h. [DOI] [PubMed] [Google Scholar]
- 17.Marletta MA, Hurshman AR, Rusche KM. 1998. Catalysis by nitric oxide synthase. Curr Opin Chem Biol 2:656–663. doi: 10.1016/S1367-5931(98)80098-7. [DOI] [PubMed] [Google Scholar]
- 18.Adak S, Bilwes AM, Panda K, Hosfield D, Aulak KS, McDonald JF, Tainer JA, Getzoff ED, Crane BR, Stuehr DJ. 2002. Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc Natl Acad Sci U S A 99:107–112. doi: 10.1073/pnas.012470099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pant K, Bilwes AM, Adak S, Stuehr DJ, Crane BR. 2002. Structure of a nitric oxide synthase heme protein from Bacillus subtilis. Biochemistry 41:11071–11079. doi: 10.1021/bi0263715. [DOI] [PubMed] [Google Scholar]
- 20.Shatalin K, Gusarov I, Avetissova E, Shatalina Y, McQuade LE, Lippard SJ, Nudler E. 2008. Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages. Proc Natl Acad Sci U S A 105:1009–1013. doi: 10.1073/pnas.0710950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357:1225–1240. doi: 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 22.Crane BR, Sudhamsu J, Patel BA. 2010. Bacterial nitric oxide synthases. Annu Rev Biochem 79:445–470. doi: 10.1146/annurev-biochem-062608-103436. [DOI] [PubMed] [Google Scholar]
- 23.Agapie T, Suseno S, Woodward JJ, Stoll S, Britt RD, Marletta MA. 2009. NO formation by a catalytically self-sufficient bacterial nitric oxide synthase from Sorangium cellulosum. Proc Natl Acad Sci U S A 106:16221–16226. doi: 10.1073/pnas.0908443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlson HK, Plate L, Price MS, Allen JJ, Shokat KM, Marletta MA. 2010. Use of a semisynthetic epitope to probe histidine kinase activity and regulation. Anal Biochem 397:139–143. doi: 10.1016/j.ab.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West AH, Stock AM. 2001. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci 26:369–376. doi: 10.1016/S0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 26.Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A 95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Letunic I, Doerks T, Bork P. 2012. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laub MT, Goulian M. 2007. Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 29.Podgornaia AI, Laub MT. 2013. Determinants of specificity in two-component signal transduction. Curr Opin Microbiol 16:156–162. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Laub MT, Biondi EG, Skerker JM. 2007. Phosphotransfer profiling: systematic mapping of two-component signal transduction pathways and phosphorelays. Methods Enzymol 423:531–548. doi: 10.1016/S0076-6879(07)23026-5. [DOI] [PubMed] [Google Scholar]
- 31.Laub MT, Biondi EG, Skerker JM. 2007. Phosphotransfer profiling: systematic mapping of two-component signal transduction pathways and phosphorelays. Methods Enzymol 423:531–548. doi: 10.1016/S0076-6879(07)23026-5. [DOI] [PubMed] [Google Scholar]
- 32.Attwood PV, Besant PG, Piggott MJ. 2011. Focus on phosphoaspartate and phosphoglutamate. Amino Acids 40:1035–1051. doi: 10.1007/s00726-010-0738-5. [DOI] [PubMed] [Google Scholar]
- 33.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in Bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol 187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan D, Cho HS, Hastings CA, Igo MM, Lee SY, Pelton JG, Stewart V, Wemmer DE, Kustu S. 1999. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc Natl Acad Sci U S A 96:14789–14794. doi: 10.1073/pnas.96.26.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 36.Hrabie JAA, Klose JRR, Wink DAA, Keefer LKK. 1993. New nitric oxide-releasing zwitterions derived from polyamines. J Org Chem 58:1472–1476. doi: 10.1021/jo00058a030. [DOI] [Google Scholar]
- 37.Roe KL. 2012. Microbial iron cycling on Trichodesmium colonies: laboratory culture studies of Trichodesmium and associated model organisms. University of California, San Diego, CA. [Google Scholar]
- 38.Gusarov I, Shatalin K, Starodubtseva M, Nudler E. 2009. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325:1380–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gusarov I, Nudler E. 2005. NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc Natl Acad Sci U S A 102:13855–13860. doi: 10.1073/pnas.0504307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson EG, Sparks JP, Dzikovski B, Crane BR, Gibson DM, Loria R. 2008. Plant-pathogenic Streptomyces species produce nitric oxide synthase-derived nitric oxide in response to host signals. Chem Biol 15:43–50. doi: 10.1016/j.chembiol.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 41.Bergman B, Sandh G, Lin S, Larsson J, Carpenter EJ. 2013. Trichodesmium—a widespread marine cyanobacterium with unusual nitrogen fixation properties. FEMS Microbiol Rev 37:286–302. doi: 10.1111/j.1574-6976.2012.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janson S, Bergman B, Carpenter EJ, Giovannoni SJ, Vergin K. 1999. Genetic analysis of natural populations of the marine diazotrophic cyanobacterium Trichodesmium. FEMS Microbiol Ecol 30:57–65. doi: 10.1111/j.1574-6941.1999.tb00635.x. [DOI] [Google Scholar]
- 43.Hmelo L, Van Mooy B, Mincer T. 2012. Characterization of bacterial epibionts on the cyanobacterium Trichodesmium. Aquat Microb Ecol 67:1–14. doi: 10.3354/ame01571. [DOI] [Google Scholar]
- 44.Thompson AW, Zehr JP. 2013. Cellular interactions: lessons from the nitrogen-fixing cyanobacteria. J Phycol 49:1024–1035. doi: 10.1111/jpy.12117. [DOI] [PubMed] [Google Scholar]
- 45.Roe KL, Barbeau K, Mann EL, Haygood MG. 2012. Acquisition of iron by Trichodesmium and associated bacteria in culture. Environ Microbiol 14:1681–1695. doi: 10.1111/j.1462-2920.2011.02653.x. [DOI] [PubMed] [Google Scholar]
- 46.Sule P, Belas R. 2013. A novel inducer of Roseobacter motility is also a disruptor of algal symbiosis. J Bacteriol 195:637–646. doi: 10.1128/JB.01777-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller TR, Belas R. 2006. Motility is involved in Silicibacter sp. TM1040 interaction with dinoflagellates. Environ Microbiol 8:1648–1659. doi: 10.1111/j.1462-2920.2006.01071.x. [DOI] [PubMed] [Google Scholar]
- 48.Lassak J, Henche A-L, Binnenkade L, Thormann KM. 2010. ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl Environ Microbiol 76:3263–3274. doi: 10.1128/AEM.00512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sunda WG, Price NM, Morel FMM. 2005. Trace metal ion buffers and their use in culture studies, p 35–63. In Andersen RA (ed), Algal culturing techniques. Academic Press, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mammalian and bacterial nitric oxide synthase domain architecture. Comparison of mammalian and bacterial nitric oxide synthases. The N and C termini and relative positions of the identified NOS domains are indicated. The heme/oxygenase domains are shown in gray, and the reductase domains are shown in white. Representative organisms with the stand-alone bacterial NOS oxygenase domain are indicated. S. cellulosum and Silicibacter sp. TrichCH4B are the only two known organisms with genes that encode a NOS with an N-terminal reductase domain containing a 2Fe-2S cluster. Download
SiliH-NOX ligation states. UV-Vis spectra of SiliH-NOX in different oxidation and ligation states are shown. The wavelengths of maximum absorption (λmax) follow: 411 nm for Fe3+, 426 nm for Fe2+, 399 nm for Fe2+-NO, and 420 nm for Fe2+-CO. Download
Silicibacter sp. TrichCH4B two-component phosphorelay signaling system. SiliHK is a hybrid histidine kinase, which has a receiver domain containing an aspartate phosphoryl acceptor. A separate Hpt protein is required as an intermediary for phosphotransfer from the receiver domain of SiliHK to the receiver domain of the response regulator SiliDGC. Download
Phosphotransfer from SiliHK/SiliHpt to orphan response regulators. SCH4B_3211, SCH4B_1525, and SCH4B_3426 are three representative orphan response regulators that did not cause rapid loss of SiliHK/SiliHpt phosphorylation. In contrast, SCH4B_1503 (SiliDGC), the cognate phosphotransfer partner of SiliHK/SiliHpt, caused complete loss of SiliHK/SiliHpt phosphorylation within 5 min. Control reactions with no response regulator added or with the E. coli response regulator OmpR also did not cause a rapid loss in SiliHK/SiliHpt phosphorylation. Download
SiliDGC activity assays. SiliDGC (10 µM) was mixed with 0.5 mM GTP, and aliquots were quenched at 1-min, 5-min, 30-min, 1-h, and 2-h time points as described in Materials and Methods. Near complete conversion of GTP to c-di-GMP was observed through the 2-h time course. Download
NO formation by Silicibacter sp. TrichCH4B compared with Roseobacter species. Silicibacter sp. TrichCH4B, Silicibacter sp. TM1040, and Dinoroseobacter shibae FL-12 were tested for NO formation using the NOA as described in Materials and Methods. Only Silicibacter sp. TrichCH4B forms NO. Seawater complete medium was also tested as a control. Download
Primers and plasmids used in this study