

Abstract
Drug-likeness rules consider N-O single bonds as “structural alerts” which should not be present in a perspective drug candidate. In most cases this concern is correct, since it is known that N-hydroxy metabolites of branded drugs produce reactive species that cause serious side effects. However, this dangerous reactivity of the N-OH species generally takes place when the nitrogen atom is not comprised in a cyclic moiety. In fact, the same type of metabolic behavior should not be expected when the nitrogen atom is included in the ring of an aromatic heterocyclic scaffold. Nevertheless, heterocycles bearing endocyclic N-hydroxy portions have so far been poorly studied as chemical classes that may provide new therapeutic agents. This review provides an overview of N-OH-containing heterocycles with reported bioactivities that may be considered as therapeutically relevant and, therefore, may extend the chemical space available for the future development of novel pharmaceuticals. A systematic treatment of the various chemical classes belonging to this particular family of molecules is described along with a discussion of the biological activities associated to the most important examples.
Keywords: N-hydroxy-heterocycles, bioactivity, metabolism
1. Introduction
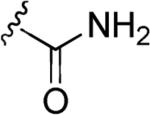
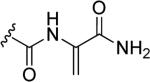
The presence of N-O single bonds is generally considered as a structural element which poses serious risks to the drug-likeness of the resulting molecule [1-3]. This warning derives from the general observation that most of the drugs bearing amino- or nitro-moieties generate reactive N-OH metabolites. These N-OH metabolites are usually generated by biochemical transformation via hydroxylation or oxidation of amines or by reduction of nitro groups present in the parent drugs and are considered as toxic metabolites. In fact, these reactive metabolites can either covalently bind to nucleic acids or interact with proteins and, therefore, be responsible for several side effects i.e. cellular necrosis, hypersensitivity, blood dyscrasia, fetotoxicity, hepatotoxicity etc. [4,5]. The high reactivity of these N-OH metabolites (1, Fig. 1) towards nucleophilic species is generally promoted by an initial conjugation with inorganic sulfate to produce corresponding O-sulfate ester (2), which further ionizes to generate the electrophilic nitrenium species (3). These cationic species covalently bind to nucleic acids and/or other nucleophilic sites of cellular components, to produce stable adduct which generate severe toxicity and, ultimately, may lead to the formation of malignant tumor (Fig. 1) [6].
Fig. 1.
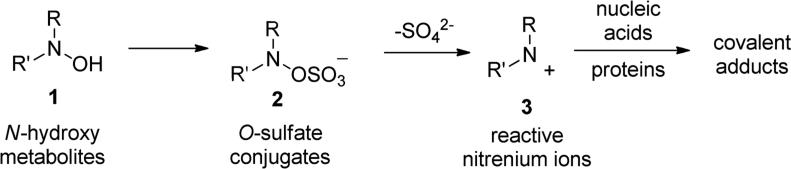
Schematic presentation of metabolic pathways involving primary N-hydroxy metabolites to electrophilic nitrenium species.
In details, primary N-hydroxy-substituted metabolites of drugs containing exocyclic and electron-rich nitrogen atoms are chemically unstable and susceptible to subsequent transformations, leading to the production of secondary metabolites in addition to nitrenium ions (3), such as: nitrones (4) if there are hydrogen atoms in α, nitroso- (5) and nitro- (6) metabolites if one of the two substituents is an hydrogen (R’ = H, Fig. 2).
Fig. 2.

Reactive secondary metabolites generated from primary N-hydroxy metabolites.
All these reactive secondary metabolites can cause severe toxicity and, for this reason in the past some drugs were withdrawn from the market. For example, Zimeldine (7, Fig. 3) a clinically effective antidepressant drug was withdrawn from the market due to unexpected incidences of the Guillain-Barré syndrome and other peripheral neuropathies [7], because of the formation of a N-hydroxy metabolite which is further metabolized to a nitrone derivative (90-95% of the total hydroxylamine metabolite) as shown in Fig. 3 [8].
Fig. 3.

Reactive secondary nitrone metabolite generated from primary N-hydroxy metabolite of Zimeldine.
In general, most of the drugs including aliphatic amines (primary, secondary and tertiary), alicyclic amines, aromatic amines (primary, secondary and tertiary), N-heterocycles are potentially susceptible to N-oxidation resulting to N-hydroxy metabolites. Thus, the knowledge of these metabolic transformations allows a rationale design of safer molecules either by changing the electron-density on these sensible portions, leading to the production of safer N-OH containing bioactive molecules, or by modifying the substituents present on the nitrogen atom. In any case, the design of new drugs generally avoids the introduction of N-OH structural portions for the abovementioned concerns.
Nowadays, a wide range of N-hydroxy derivatives included into acyclic portions have been identified to possess interesting pharmacological properties. Among them, chemistry of N-hydroxy derivatives such as hydroxamates (8), amidoximes (9), N-hydroxyurea (10) and aldoximes (11), have been explored in the production of various types of bioactive molecules (Fig. 4).
Fig. 4.
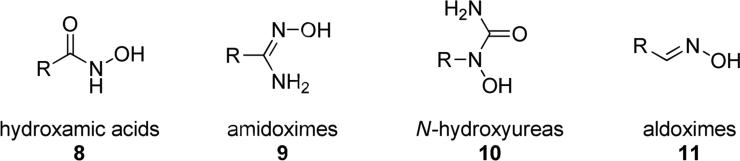
General representation of some biorelevant acyclic/exocyclic N-hydroxy-derivatives.
For example, some extensively explored hydroxamic acids have been identified to be able to inhibit histone deacetylase (HDAC) enzymes. Inhibition of HDACs is a clinically validated therapeutic strategy for treatment of various types of cancers [9-11]. Furthermore, some hydroxamates, N-hydroxyamidoximes, N-hydroxyureas displayed inhibitory activity against matrix metalloproteinases (MMPs), lysine-specific demethylase 1 (LSD1), human epidermal growth receptor 2 (HER2), TNF-α converting enzyme (TACE), inosine monophosphate dehydrogenase (IMPDH), and carbonic anhydrases (CA). All these effects were found to be potentially useful in the treatment of various pathologies, such as cancer, viral infections, rheumatoid arthritis and multiple sclerosis [12-20]. Furthermore, N-hydroxyamidoximes and N-hydroxyureas also showed antiprotozoal, antibacterial, anti-human immunodeficiency virus (HIV) and anti-leishmania activity [21,22]. In particular, several compounds belonging to these classes proved to be useful drugs in clinical or preclinical studies, such as the HDAC inhibitors suberoylanilide hydroxamic acid (SAHA, Vorinostat, 12 in Fig. 5) [23], m-carbocinnamic acid bishydroxamide (CBHA, 13) [24], Scriptaid (14) [25], the sulfonamide derivatives PXD101 (Belinostat, 15) [26] and Oxamflatin (16) [27], and the 3-substituted indole derivatives LBH589 (Panobinostat, 17) [28] and NVP-LAQ824 (Dacinostat, 18) [29] (Fig. 5).
Fig. 5.

Structures of N-hydroxy-bearing clinical and preclinical small molecules.
Finally, some aromatic aldoximes were developed as potent and selective estrogen receptor β (ERβ) agonists by both industrial [30-32] and academic groups [33,34].
The secret of success of these classes of exocyclic N-hydroxylated derivatives should be found in their metabolic stability. In fact, hydroxamates such as Vorinostat 12 (Fig. 5) are indeed metabolized by O-glucuronation or O-sulfation. However, the resulting conjugates are stable and are excreted as such without the generation of reactive intermediates, since the acyl-substituent on the nitrogen atom makes it quite electron-poor and, therefore, less prone to bear the positive charge of the nitrenium species, which are instead produced by electron-rich N-hydroxyamines (Fig. 2). For similar reasons, aromatic aldoximes are also stable towards this type of metabolism and, moreover, they are also quite resistant towards hydrolytic processes [35].
In spite of the success of the above-mentioned exocyclic (and electron-poor) N-hydroxy-derivatives, there is only a limited number of reports about heterocycles containing endocyclic N-hydroxy-portions, which should benefit from a comparable metabolic stability, since they are not prone to generate reactive intermediates, such as nitrenium ions and nitrones. There is a growing interest in expanding the chemical space of new potential therapeutic agents and N-hydroxy heterocycles may represent a newly emerging structural motif. Therefore, this review covers the scientific literature describing heterocycles containing endocyclic N-OH portions that proved to possess biorelevant properties. Fig. 6 gives a general overview of the various chemical classes which are systematically discussed herein. Chemical structures in red and green color are five-membered and six-membered heterocycles, respectively, which are shown in the inner yellow circle. Chemical structures in blue and black color respectively comprise five- and six-membered heterocyclic rings that are fused with other aromatic rings, and are placed in the outer black circle.
Fig. 6.

General structural overview of the types of endocyclic N-OH heterocyclic derivatives. Inner yellow circle: monocyclic 5-membered (red) and 6-membered (green) ring systems. Outer black circle: fused 5-membered (blue) and 6-membered (black) ring systems.
2. N-Hydroxy-substituted five-membered heterocycles
2.1. One endocyclic heteroatom
2.1.1. Pyrrolidinones, pyrrolidinethiones and pyrrolidinediones
N-Hydroxy-substituted pyrrolidinones, pyrrolidinethiones and pyrrolidindiones are heterocycles of great biopharmacological importance, because of their presence in numerous molecules showing interesting pharmacological applications. 3-Amino-1-hydroxypyrrolid-2-one, named as HA-966 (19 in Fig. 7), was first synthesized in 1959 and displayed various neuropharmacological properties. Chemically, it is similar to the cyclic form of the well-known amino acid γ-aminobutyric acid (GABA), which is an important inhibitory neurotransmitter in mammalian central nervous system. Bonta et al. reported that HA-966 19 showed various interesting properties, such as antitoxic effect against amphetamine-induced motor activity, selective central antitremor effect against tremor induced by chemical agents, antidepressant and anticonvulsant effects in mammals [36]. Further studies reported that it was one of the first substances endowed with moderately strong and selective action in antagonizing l-glutamate- or l-aspartate-evoked excitation of single neurons in the cerebral cortex. It reduced the excitatory effect of l-glutamate- or l-aspartate, while it had either no effect or weak effect on acetylcholine response. Hence, HA-966 reduced aminoacid-evoked responses stronger than that induced by acetylcholine [37]. The same authors reported a weak depressant action of HA-966 on chemically and synaptically excited neurons in cat cerebral cortex nucleus and potency of HA-966 relative to GABA was similar. Direct action of HA-966 on synaptic responses of central neurons may contribute to its complex effects and a blockade of excitatory amino acid receptors may contribute substantially to the depressant action on L-glutamate- and L-aspartate-induced excitation [38,39]. Moreover, selective action of HA-966 at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor complex was reported as maximum interaction at the highest concentration of 250 μM, and further increasing the concentration of HA-966 did not increase the degree of antagonism. In radioligand binding experiments, this compound inhibited strychnine-insensitive [3H]-glycine binding to rat cerebral cortex synaptic plasma membranes with an IC50 of 17.5 μM, and on increasing concentration up to 1 mM it caused minimal inhibition. These results demonstrated that the activity of HA-966 was due to the antagonism at the glycine modulatory site on the NMDA receptor complex [40-42]. Further studies of HA-966 suggested that (R)-(+)-enantiomer (19a, Fig. 7) was a selective inhibitor of glycine/NMDA receptor and this property was responsible for its anticonvulsant action in vivo. In contrast the (S)-(-)-enantiomer (19b, Fig. 7) showed only a weak interaction with glycine/NMDA receptor, but it displayed some significant muscle relaxation and sedative action. Nevertheless, the sedative/ataxic effect of racemic HA-966 was mainly attributable to the (S)-(-)-enantiomer which was 25-fold more potent than (R)-(+)-enantiomer [43]. Furthermore, the neuroprotective action of HA-966 was mainly due to its (R)-enantiomer 19a and it reduced NMDA-induced brain injury, whereas the other enantiomer was ineffective. Intravenously, racemic HA-966 and its (S)-enantiomer 19b prevented tonic extensor seizures from low-intensity electroshock with ED50 at 13.2 and 8.8 mg/kg, respectively, whereas the anticonvulsant effects of the (R)-enantiomer were much less potent (ED50 = 105.9 mg/kg) [44]. R-(+)-HA-966 19a significantly disinhibited both non-conditioned and conditioned suppressed behavior in rodents in preclinical models of anxiety, acting as anxiolytic without relevant side effects, while the S-(-) enantiomer 19b was devoid of any anxiolytic activity and it only produced behavioral sedation [45]. The enantiomer 19b also prevented in vivo restraint stress-induced dopamine utilization in brain regions in contrast to the lack of efficacy of its (R)-enantiomer, and it also suppressed fear-induced behaviors. At the dose of 3 mg/kg, 19b prevented locomotor sensitization in vivo, and at the highest dose of 5 mg/kg, it blocked acute cocaine-induced locomotion, but this treatment resulted in sedation of the animal, because this compound seems to act through the γ-hydroxybutyrate (γHB) binding site on the GABA receptor. This compound may thus represent a novel structural example of potential anxiolytic agents with γHB-like actions [46]. Continuous efforts were made aimed at identifying more potent analogues of HA-966, devoid of side effects and characterized by an improved brain penetration [47]. In particular Leeson et. al. reported a (3R, 4R)-cis-4-methyl derivative of HA-966 (20, also named L-687,414, Fig. 7), which was found to be a more potent glycine/NMDA receptor antagonist than HA-966,whereas its trans diastereoisomer was inactive [48]. The additional methyl group in 20, when compared to compounds 19a and 19b, may increase the hydrophobic contribution to the binding process to the glycine site of the receptor, but it may also affect the conformation of the five- membered ring. In addition to the above-mentioned (3R,4R)-derivative 20, the [3.2.l] bicyclic analogue 21 (Fig. 7) indicated that the energetically unfavorable conformation of the pyrrolidone ring, having the 3-amino group in a pseudoaxial situation, is essential for recognition by the glycine receptor. Hence, glycine receptor recognition requires the energetically less favoured 3-pseudoaxial conformation of the pyrrolidone ring, resulting in compound 20 being a more potent glycine/NMDA receptor antagonist than 19a [49]. Further development of di-substituted derivatives of racemic HA-966 by the same group demonstrated that only the cis orientation at 4-position allowed substitution and there was a strict tolerance to substituent size by the receptor. Ethyl derivative 22a (Fig. 7) displayed an IC50 value of 15 μM, which is equipotent to 19a (IC50 = 12.5 μM), but less active than 20 having the IC50 value of 1.4 μM, in the [3H]-glycine binding to rat cortical membrane assay. 4-Hydroxy-substituted derivative 22b (Fig. 7) proved to have a similar antagonist activity (IC50 = 1.3 μM) to the corresponding methyl-substituted analogue 20. Hence, the cis-4-methyl (20) and cis-4-hydroxy-substituted (22b) compounds exhibited similar in vitro anticonvulsant activities and good brain penetration levels, since they are not ionized at physiological pH. A smaller bicyclic analogue of compound 21 (IC50 = 19 μM) was also synthesized by reducing the length of the N-C5 ethylene bridge in 21 to a methylene portion, as in the [2.2.1]bicyclo-derivative 23 (Fig. 7), which proved to retain antagonist activity on the same receptor. However, the instability of 23 in aqueous solution did not allow an accurate determination of IC50 value [47]. N-hydroxy-2-pyrrolidinethione compound 24 (Fig. 7) when present in a zinc complex showed a good anti-microbial activity [50]. In addition, dioxopyrrolidine derivative 25 (Fig. 7) was an optically active derivative containing a sulfonylamino succinimide moiety, that showed some interesting inhibitory activity against the isoform 2 of matrix metalloprotease (MMP-2), an enzyme involved in tumor invasion. In vitro studies revealed that it exhibited a potent MMP-2 inhibitory activity, with an IC50 value of 0.0274 μM [51].
Fig. 7.

N-hydroxy-substituted pyrrolidinones, pyrrolidinethiones and pyrrolidindiones.
2.2. Two endocyclic heteroatoms
2.2.1. Imidazolidinones, imidazolidinediols and imidazolinethiones
N-Hydroxy-imidazoles and derivatives are important bioactive small organic molecules. NHydroxy imidazolidinone derivative 26 (Fig. 8) was found to exhibit affinity and efficacy similar to that of d-cycloserine, a representative partial agonist for the strychnine-insensitive glycine binding site of NMDA receptor. It was a potent ligand for strychnine-insensitive glycine binding site, with an IC50 value of 6.8 μM. It was reported that compound 26 possesses higher affinity than that of the partial agonist d-cycloserine, but it exhibits moderate affinity and an efficacy lower than the full agonist glycine, therefore it would be a useful tool in the exploration of the therapeutic potential of ligands interacting with glycine site of the NMDA receptor [52,53]. Compound 26 displayed an inhibitory activity against the interaction of glutamate with two receptors belonging to the same family of NMDA receptor, the α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate (AMPA) and the kainate receptors, which play important functions in pathogenesis of cancer, thus proving to be likely useful in treating cancer [54]. Similar kind of N-OH bearing small molecules, such as spiroimidazolidinone derivatives 1-hydroxy-7,7,9,9-tetramethyl-1,4,8-triazaspiro[4.5]decan-2-one 27a (Fig. 8) and (±)-1-hydroxy-3,7,7,9,9-pentamethyl-1,4,8-triazaspiro[4.5]decan-2-one 27b (Fig. 8) were reported by Vystorop et al. and ESR spectroscopy studies showed that these cyclic spirohydroxamic acids behave as NO donors in in vitro biological systems. The NO-donor activity of compounds 27a and 27b was found to be substantially higher in the presence of DMSO, compared to the aqueous medium. Compound 27b was a stronger NO-donor than 27a and it also exhibited a high antimetastatic activity on the B16-melanoma model, since NO-donor agents are known to display various pharmacological effects, including a cytotoxic action [55]. Nieto et al. presented a dimeric N-hydroxy-imidazole (28, Fig. 8), which is an analog of the trypanocidal agent 4,4’-bis(imidazolinylamino)diphenylamine. Compound 28 showed antiprotozoal activity in the submicromolar range, as shown by its IC50 values of 0.37 and 0.055 μM against Trypanosoma brucei rhodesiense and Plasmodium falciparum, respectively. Furthermore, compound 28 showed a significantly higher permeability through human blood-brain barrier, when compared to its non hydroxylated counterpart, as determined in vitro by transport assays through the human brain endothelial CMEC/D3 cell line. These results showed that the introduction of a hydroxyl group on the nitrogen atom of the imidazolyl ring enhanced the blood-brain barrier permeability of the resulting molecule (28). Furthermore, in the T. brucei STIB900 murine model of acute sleeping sickness, compound 28 was found to possess moderate activity at a dosage of 20 mg/kg by intraperitoneal administration and this modest effect might reflect an incomplete bioactivation of the molecule in vivo [56]. N-Hydroxy derivatives 29a-e (Fig. 8) displayed inhibitory effects on platelet aggregation in the range 28.9-54.2% at the concentration of 0.2 mg/mL, thus proving to be more efficient than aspirin, which showed a 24.6% inhibitory effect at the same concentration. Compound 29c displayed the most efficient inhibitory effect among the compounds of this series (54.2%), which was even higher than that of ticlopidine (44.6%) on platelet aggregation. A study of acute toxicity of these compounds showed that LD50 lay in the range 500-2500 mg/kg, thus proving that these compounds presented a low toxicity. Observation of the systemic arterial pressure variations after administration of these compounds suggested that compound 29e exhibited a combination of antihypertensive activity and 32.1% inhibition of platelet antiaggregation, which resulted in a high clinical priority for this compound, since both mechanisms synergistically contribute to the treatment of hypertonic disease [57]. Compound 30 (Fig. 8) bearing two OH groups on its two nitrogen atoms was found to be active against Bacillus cereus. The mechanism of action of 30 was hypothesized to involve a bidentate chelation of zinc ions of the active site of the Bacillus cereus phospholipase C. In fact, it did not exhibit significant inhibition of this enzyme at pH 7.3, whereas it turned out to be active at pH 9.5, in fact ionized N-hydroxyl function would be expected to bind better than a neutral hydroxyl group to an active site zinc ion [58]. 1-Hydroxy-4-(4-methoxyphenyl)-1H-imidazole-2(3H)-thione 31 (Fig. 8) was prepared and tested as tyrosinase inhibitor, useful for the treatment and prevention of hyperpigmentation. In enzymatic assays, this compound exhibited a potent tyrosinase inhibitory activity with an IC50 value of 3.7 μM, so that it might find application in cosmetic preparations for the prevention or treatment of signs of aging or dry skin [59].
Fig. 8.

N-Hydroxy-substituted imidazolidinones, imidazolidinediols and imidazolinethiones.
2.2.2. Thiazolidinones and thiazolidinethiones
Oxalatrunculin B 32 (Fig. 9), a natural product containing a N-hydroxylated thiazolidinone ring, was isolated from Red Sea sponge Negombata corticata. Actin polymerization inhibition assays and molecular modeling calculations demonstrated that oxalatrunculin B weakly bound to actin and exhibited significant antifungal and anticancer activity. Compound 32 displayed a significant antifungal activity against Saccharomyces cerevisiae with a MIC value of 56 μM. Cytotoxicity of oxalatrunculin B against several cancer cell lines suggested that compound 32 displayed nonspecific cytotoxicity against solid tumor cells and hematopoietic cancerous cells, possessing MIC value of 16.3 μM against hepatocellular carcinoma. The weak binding to actin and the stronger cytotoxic activity suggest that may exist a secondary target responsible for the different activities of 32 [60]. Austin et al. patented a series of N-hydroxy derivatives as industrial biocides. Out of these compounds, 3-hydroxy-4-methyl-thiazol-2(3H)thione 33 (Fig. 9) at the concentration of 25 ppm completely inhibited Escherichia coli proliferation and at 1 ppm completely inhibited viability of Staphylococcus aureus and Aspergillus niger [61]. A bivalent zinc complex of 33 at 1000 ppm inhibited the growth of bacteria and yeast such as Escherichia coli, Enterococcus faecalis and Candida albicans. This metal complex also reduced odor and inhibited the pH increase, hence it could be used as the absorbent layer in disposable articles for collecting body fluids [62]. Furthermore, the same author reported a similar derivative, p-chlorophenyl substituted compound 34 (Fig. 9), that showed an antimicrobial activity against Pseudomonas aeruginosa together with a low toxicity. This could be useful in a personal care formulation for cosmetic compositions containing compound 34 as an antimicrobial agent [63].
Fig. 9.

N-Hydroxy-substituted thiazolidinones and thiazolidinethiones.
3. N-Hydroxy-substituted six-membered heterocycles
3.1. One endocyclic heteroatom
3.1.1. Piperidines
N-Hydroxy-substituted six membered ring systems, i.e. piperidine derivatives, have great importance due to their numerous biological applications. N-hydroxy-substituted piperidines displayed broad spectrum of biological activities, such as: antioxidant properties for the treatment of cellular oxidative stress, or anxiolytic and antidepressant activities, or they can be used in arresting the development of cataracts or macular degeneration, or they possess anti-inflammatory activity in the liver, which might be useful in the treatment of hepatitis, as well as an anti-angiogenic activity [64-68]. Komarov et. al. reported the antioxidant properties of some N-hydroxy derivatives of 2,2,6,6-tetramethylpiperidines, such as compound 35 (Fig. 10). 35 showed in vitro effective antioxidant properties on membrane models of lipid peroxidation, and significantly it slowed down the rate of peroxidation at concentrations of 10−6-10−5 M. An increase in the length of the alkyl chain on the sulfur atom caused a reduction of the anti-oxidative effect of the resulting compound. The antioxidant effect observed with this compound is due to the oxidation to the corresponding nitroxyl radicals in aqueous medium. Moreover, compound 35 proved to be active in the treatment of ischemic and reperfusive injuries caused by lipid peroxidation in biomembranes. The anti-ischemic activity was demonstrated in models of long-term storage of isolate liver, as well as on models of ischemia of the liver, isolated ischemized heart, and ischemic shock [69]. N-hydroxytetramethylpiperidine derivatives 36, 37a-b and 38 (Fig. 10) showed interesting inhibitory activities against the cytokine Tumour Necrosis Factor α (TNFα) protein: 36, 37a-b and 38 exhibited IC50 values of 3.8, 1.0, 0.9 and 6.7 μM, respectively. Compound 38 also exhibited lipid peroxidation inhibitory activity of 91.4% at 10 μM and 87% at 1 μM. The less active compounds 36, 37a and 37b showed 64.4% 83.8% and 81.8% of lipid peroxidation inhibitory activity at 1 μM. Compound 38 alone efficiently exerted its cytotoxic action on both doxorubicin-resistant cells as well as sensitive cells, revealing that these type of inhibitors are able to bypass drug resistance. Further studies of compound 38 in combination with doxorubicin demonstrated that 38 displayed strong ability to reverse drug resistance in osteosarcoma, breast cancer and neuroblastoma [70,71].
Fig. 10.

N-Hydroxy-substituted piperidine derivatives.
3.2. Two endocyclic heteroatoms
3.2.2. Pyrazinones and piperazinones
Pyrazinones and piperazinones substituted with N-hydroxy groups have so far shown a significant biological importance, because of their emergence as subunits of various bioactive natural products as well as of other synthetic molecules. Natural product flutamide 39 in Fig. 11, a fully substituted 2,6-diketopirazine, which was isolated from fungus Delitschia confertaspora, exhibited inhibitory activity against cap-dependent transcriptase of influenza A and B viruses and it had no effect on the activities of other polymerases, hence it selectively inhibits the transcriptase of influenza viruses [72,73]. Both natural and synthetic flutamide [74,75] are endowed of inhibitory activity against influenza virus A transcription, with comparable IC50 values of 5.1 and 5.6 μM respectively, without showing any activity against other transcriptases. Compound 39 proved to efficiently inhibit the replication of influenza A and B viruses in cell culture. The selective antiviral properties of this natural product validate influenza virus endonuclease as a target for antiviral agents. A natural product known as sclerominol 40 (Fig. 11) was isolated from cultures of hypovirulent isolates of the fungal plant pathogen Sclerotinia minor, with a 1-hydroxy-2,6-pyrazinedione structure similar to flutamide. On the basis of the structural similarity with flutamide, sclerominol 40 was tested for the same biological activity, but this compound proved to be inactive on influenza virus cell lines. The role of this compound in the physiology of hypovirulent isolates of S. minor has not been determined yet [76]. Natural products NBRI16716A 41 and NBRI16716C 42 in Fig. 11 were isolated from the fermentation broth of Perisporiopsis melioloides and include in their structures several N-OH groups, in the endocyclic piperazine nitrogen atom and in the side chains. These compounds showed antitumor activities against human prostate cancer cells DU-145, and they inhibited the growth of these cells in their co-culture with human prostate stromal cells PrSCs without apparent cytotoxicity against PrSCs. Moreover, 41 showed efficient antitumor effect in xenograft models of DU-145 cells with PrSCs, without any effect on body weight of mice [77].
Fig. 11.

N-Hydroxy-substituted pyrazinone and piperazinone-based natural products.
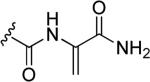
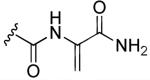
Synthetic small molecules deriving from natural product flutamide 39 were obtained by including various aromatic substituents in its structure and displayed interesting activities. Some of the most active compounds against cap-dependent endonuclease activity of influenza virus were obtained by replacing one of the isopropyl groups of flutamide with p-substituted phenyl groups, such as a p-fluorophenyl derivative 43a and a p-methoxyphenyl derivative 43b in Fig. 12. In fact, compounds 43a and 43b displayed IC50 values of 0.9 and 0.8 μM in vitro, respectively, vs. flutamide 39 which instead exhibited an IC50 of 5.1 μM in the same assay. Monoaryl-substituted compounds 43a and 43b appeared to be more potent in the cell-based antiviral assay too, but unfortunately they showed a certain degree of cytotoxicity at concentrations greater than 10 μM, whereas natural product flutamide 39 did not show any appreciable toxicity at a concentration of 100 μM. Replacement of both alkyl substituents of flutamide with two phenyl rings (44a-c, Fig. 12), caused only a slight improvement in the activity for compounds 44b and 44c (IC50 = 2.8 and 3.5 μM, respectively) compared to the value of flutamide, whereas compound 44a was nearly equipotent to flutamide (IC50 = 6.5 μM) [78]. Tomassini et al. studied the pharmacological activities of these chemically-modified analogs of flutamide. A structure-activity relationship analysis demonstrated that both N-hydroxy- and olefin groups in substituted 2,6-diketopirazine derivatives were required for activity. The aromatic substitution of the isopropyl side chains caused an increase in potency. Conversely, removal or transformation of N-hydroxy group resulted in a decrease of activity [73]. Marques et al. reported sulfonamide derivatives which displayed a good ability in inhibiting MMPs. These derivatives possess a new type of zinc-binding group at the enzyme catalytic center, and exhibited an inhibitory activity in the nanomolar range. The phenoxyphenly sulfonamide derivative 45 (Fig. 12) exhibited an efficient inhibitory activity against MMPs thanks to its ability to chelate the catalytic zinc ion of these enzymes. Compound 45 was screened against a broad spectrum of MMP isoforms and TACE and it showed a predominant inhibitory activity against MMP13 with an IC50 value of 9.5 nM, thus confirming that the 1-hydroxypiperazine-2,6-dione moiety may represent a valid potential substitute for the hydroxamate moiety as zinc binding group, typically present in most MMP inhibitors [79]. Other small heterocyclic molecules, such as pyrazinone derivative 46 in Fig. 12, were synthesized as artificial siderophores, as potential therapeutic agents for iron overloaded disease. Compounds 46 was reported as one of the first examples of microbial growth-promoters on Aureobacterium flavescens JG-9, showing the highest growth-promotion activity comparable to that of desferrioxamine B, which is a natural trihydroxamate siderophore [80].
Fig. 12.

N-Hydroxy-substituted pyrazinone and piperazinone-based synthetic compounds.
4. N-Hydroxy-substituted fused five-membered ring systems
4.1. One endocyclic heteroatom
4.1.1. Indoles
N-Hydroxyindoles are fascinating molecular entities, which have attracted considerable attention of medicinal chemists. In fact, N-hydroxyindoles were found to be an integral part of the structure of some complex natural products (47-48, Fig. 13 and Table 1). Recently discovered natural products birnbaumin A 47a and birnbaumin B 47b in Fig. 13, having N-hydroxyindole moieties in their structure, were isolated from extract of yellow fruit bodies of the tropical mushroom Leucocoprinus birnbaumii. However, no biological properties for these two natural compounds have been identified yet [81].
Fig. 13.

N-Hydroxyindole-containing natural products.
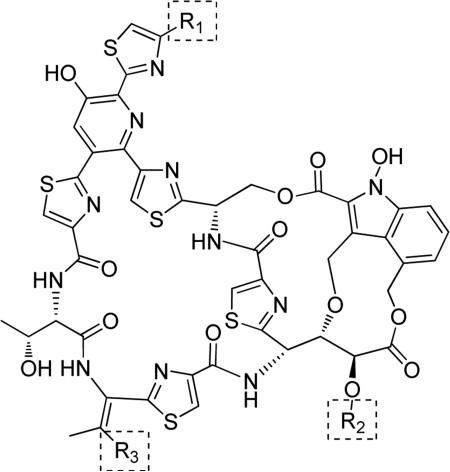
Table 1.
Natural products S-54832/A-I, nocathiacins and thiazomycins.
| ||||
|---|---|---|---|---|
| R1 | R2 | R3 | ||
| 48a | S-54832/A-I |
|
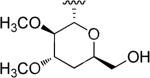
|
H |
| 48b | Nocathiacin I |
|
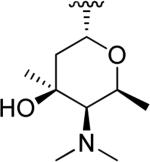
|
OCH3 |
| 48c | Nocathiacin III |
|
H | OCH3 |
| 48d | Nocathiacin IV |
|
|
OCH3 |
| 48e | Thiazomycin |
|
|
OCH3 |
| 48f | Thiazomycin A |
|

|
OCH3 |
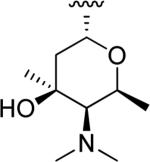
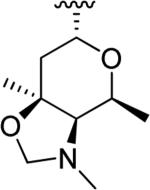
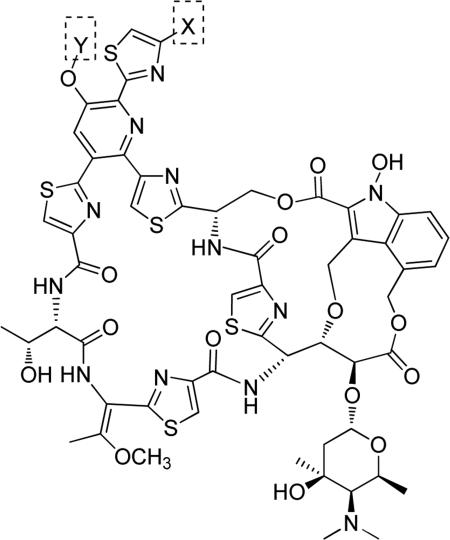
N-Hydroxyindole-bearing natural product S-54832/A-I 48a (Table 1), a thiazolyl peptide-based antibiotic, was isolated in 1984 from Micromonospora globosa cultures, and it exhibited excellent growth-inhibiting effect toward Gram-positive bacteria, including Staphylococci, Streptococci, Corynebacteria and Mycobacteria in vitro and against Staphilococcus pyogenes, pneumoniae and S. aureus in vivo [82,83]. N-Hydroxyindoles were also found in nocathiacins I 48b, III 48c and IV 48d in Table 1, a series of antibiotics that were independently isolated by two research groups from bacteria species Nocardia and the fungus Amicolaptosis and they closely resemble the structure of compound 48a [84-86]. These nocathiacins (48b-d) exhibited in vitro and in vivo antibacterial activity against Gram-positive bacteria including multi drug-resistant strains, Mycobacterium tuberculosis and Mycobacterium avium. Nocathiacin I 48b revealed a nanomolar potency against S. aureus strains and an excellent in vivo efficacy in a systemic S. aureus infection mice model. It was also active against Gram-positive anaerobes, revealing a clinical potential for these compounds to be used as antibiotics in humans [87,88]. Other natural products bearing N-hydroxyindole moiety are thiazomycin 48e and thiazomycin A 48f in Table 1. They are thiazole-rich potent antibacterial antibiotics that differ from nocathiacins for the oxazolidine ring as part of the amino-sugar moiety in contrast to the dimethyl amino group (R2 in Table 1). They were isolated from Amycolatopsis fastidiosa and were found extremely potent and selective against Gram-positive pathogens in vitro and in vivo with MIC range 0.002-0.064 μg/ml. The in vitro activity of thiazomycin A 48f was generally slightly lower than thiazomycin and nocathiacin I, suggesting a detrimental role of the methyl substitution in the sugar moiety. Thiazomycin 48e showed ED99 value of 0.15 mg/kg and was highly efficacious against Staphylococcus aureus via subcutaneous administration in mice. Furthermore, nocathiacin and thiazomycin antibiotics exhibited antibacterial activity by selective inhibition of protein synthesis through a direct interaction with the L11 protein and 23S rRNA of the bacterial 50S ribosome. Moreover, they did not show any cross-resistance to clinically used antibiotic classes, such as vancomycin, oxazolidinones and quinolones [89-91]. Similarly, thiazomycin A 48f selectively inhibited protein synthesis with IC50 value of 0.7 μg/ml and it did not inhibit any other macromolecules as RNA, DNA, peptidoglycan, and phospholipid of S. aureus [92]. In general, N-hydroxyindole scaffold-bearing natural products showed excellent activity against Gram-positive bacteria and could be further developed as anti-bacterial agents.
Due to the promising antibacterial activity of nocathiacins and to the presence of several functional groups in their structure suitable for chemical manipulation, some research groups reported the semi-synthetic derivatives of these natural products in order to improve aqueous solubility while maintaining the intrinsic biological activity [93,94]. Representative N-hydroxyindole scaffold-based semi-synthetic products 49a-c in Table 2 were obtained by the addition of amines to the dehydroalanine side chain of nocathiacin I, in particular primary, secondary and cyclic amines with different functional groups were inserted to create analogues of nocathiacin I. The resulting compounds were endowed of potent antibacterial activity in vitro on a panel of Gram-positive bacteria along with good in vivo efficacy in a lethal S. aureus systemic infection model and good solubility. Among the primary amine analogues, the ethylendiamine derivative 49a reached the best MIC values against methicillin-sensitive Staphylococcus aureus (MIC = 0.25 μg/mL), methicillin-sensitive Enterococcus faecalis (MIC = 0.5 μg/mL) and penicillin-resistant Streptococcus pneumoniae (MIC = 0.03 μg/mL), but it had overall decreased antibacterial activity compared to the parent compound nocathiacin I (MIC = 0.007, 0.03 and 0.001 μg/mL against S. aureus, E. faecalis and S. pneumonia, respectively). The 2-(methylamino)ethanol analogue 49b and the morpholine analogue 49c exhibited antibacterial profiles in vivo and in vitro comparable to nocathiacin I. These derivatives showed better water solubility in comparison to the parent compound, especially 49b possessed a solubility higher than 10 mg/mL, significantly improved compared to that of nocathiacin (0.34 mg/mL). Consequently, 49b was proposed as a lead candidate for further study and safety evaluation on the basis of its promising properties and activity [95]. A different amide substitution on thiazole ring gave compounds 50a and 50b (Table 2) that exhibited potent in vitro antibacterial activity against S. aureus, S. pneumoniae and E. faecalis. The N-methyl-piperazinyl compound 50a produced a significant reduction of kidney burden in a systemic S. aureus infection mouse model, with ED99 value of 0.28 mg/Kg comparable to that of vancomycin. Derivative 50b with a neutral urea side chain displayed excellent antibacterial activity both in vitro (MICs range 0.0038-0.25 μg/mL) and in vivo and it exhibited acceptable aqueous solubility, thus revealing to be one of the most potent water-soluble nocathiacin analogues synthesized to date [96]. Finally, O-substituted derivative of nocathiacin I 51 in Table 2 also showed potent antibacterial activity with MICs ranging from 0.06 to 0.25 g/mL against the same series of pathogens. Compound 51 retained the in vivo potency of nocathiacin I (PD50 = 0.26 mg/Kg). Moreover, mono-methylphosphonate 51 possessed very good aqueous solubility [97].
Table 2.
Semisynthetic nocathiacin I derivatives.
| |||||
|---|---|---|---|---|---|
| X | R1 | R2 | Y | ||
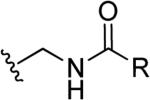
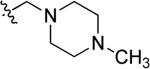
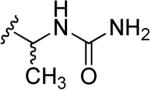
| 49 | a |
|
H | CH2CH2NHCH3 | H |
| b | CH2CH2OH | ||||
| c |
|
||||
| R | |||||
| 50 | a |
|
|
H | |
| b |
|
||||
| 51 |
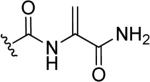
|
|
|||
Not only natural products and semi-synthetic compounds deriving from natural products showed excellent pharmacological properties, but recently also synthetic N-hydroxyindole heterocycles have acquired importance in the medicinal chemistry field. N-hydroxyindole-based compounds (NHIs) were reported as new and efficient inhibitors of the human isoform 5 of the enzyme lactate dehydrogenase (hLDH5), which is involved in the growth and survival of hypoxic tumours, with potential therapeutic applications of NHIs as anti-cancer agents [98,99]. N-hydroxyindole-based LDH inhibitors possess a hydroxyl group on the nitrogen atom at position 1, along with a carboxylic group at position 2 of the indole scaffold. The X-ray crystal structures of hLDH5 show that the enzymatic cavity is quite polar and rich in cationic residues such as arginines, because normally hosts the substrate pyruvate and the cofactor NADH, hence the OH/COOH moiety should bind in the catalytic binding site of the enzyme, as confirmed by molecular modeling studies. An extensive structure-activity relationship was made on the aryl-substituted NHIs and revealed that aryl substituents were generally beneficial for the activity, particularly if they were placed at position 5 and 6. Moreover, the presence of electron-withdrawing substituents in the aryl groups or of additional aryl portions generally improved inhibitory potency. Compound 52a (Fig. 14), in which the phenyl group was present in position 5 and compound 52b (Fig. 14) with the phenyl in position 6 showed 99% and 84% inhibitory activity of hLDH5, respectively, at 125 μM and less than 3% inhibition of the heart isoform hLDH1, thus revealing a good level of selectivity. The presence of the electron withdrawing group -CF3 at 4 position and a phenyl ring at 6 position in compound 52c (Fig. 14) caused a 87% inhibition of hLDH5 although against hLDH1 a minimal residual activity (11%) was observed. All these three compounds were competitive versus NADH and pyruvate, with Ki values in the low micromolar range (Ki's ranging from 4.7 to 35.4 μM). Cellular assays revealed that these NHIs were able to reduce lactate production in cancer cells and they possessed a strong antiproliferative activity, which was particularly pronounced under hypoxic conditions [100,101]. Further modification at 6 position with triazole ring in place of phenyl ring decreased the inhibitory potency noticeably, but when the triazole ring was moved to position 4 it produced a good hLDH5 inhibitory activity, as for compound 53 (Fig. 14) that selectively inhibited of 73% the enzymatic activity. A sharp preference for hLDH5 was also found with dimeric compound 54 in Fig. 14, in which two N-hydroxyindole portions at their 6 positions are linked through a 1,3-bis(triazole)-propane chain, with a 83% inhibitory activity, but it also displayed 32% inhibitory activity against hLDH1 [102]. In the case of compound 55 (Fig. 20), insertion of parachloro atom in the N-phenyl ring of N-methylsulfonamide portion at position 6 of the indole scaffold led to efficient inhibition levels versus both the cofactor (Ki = 6.6 μM) and the substrate (Ki = 5.6 μM) [103]. N-hydroxy derivative 56 (Fig. 14) substituted at 2 position with an urea moiety and at 3 position with an unsubstituted amide revealed inhibitory activity on Iκb kinase β (IKKβ) of 86% at 1 μM. Thanks to the IKKβ inhibition activity, compound 56 could be useful as a prophylactic and/or therapeutic agent for several kinds of diseases associated with IKKβ activity [104]. Somei et. al. developed a variety of N-hydroxy derivatives with promising pharmacological application and they found that N-hydroxytryptamines play an important role in inhibition of blood platelet aggregation, hence they have potential utility for the treatment of erectile dysfunction and cerebral infarction. Compounds 57a-b (Fig. 14) are tryptamine derivatives differing for the length of the acyl side chain and reached vascular relaxation effects about 66 and 79% respectively, compared to the α2-blocker yohimbine that was considered as a standard control (100%) [105]. Compound 58a-b (Fig. 14) based on 3-cyano-2-phenyl-substituted-N-hydroxyindole scaffold exhibited fungicidal activity on leaf rust on wheat plant against pathogen Puccinia recondita, possessing EC50 values of 60 and 50 ppm, respectively [106].
Fig. 14.

N-Hydroxyindole-based synthetic derivatives.
Fig. 20.

N-hydroxy-substituted acridone heterocycles.
4.1.2. Carbazoles and carbolines
N-hydroxy substituted carbazoles and carbolines are an important class of heterocycles, mainly present in natural products. Novel alkaloid coproverdine 59 (Fig. 15) possessing a N-hydroxycarbazole ring was isolated from marine organisms from New Zealand. Coproverdine displayed cytotoxic activity against a variety of murine and human tumor cell lines, with IC50 values ranging from 0.3 to 1.6 μM [107,108]. N-hydroxy pyrimidine-β-carboline alkaloid N-hydroxyannomontine 60 (Fig. 15) was isolated from the bark extract of Annona foetida, a plant of the Annonaceae family. The natural product 60 was characterized by antiparasitic activity against Leishmania braziliensis and Leishmania guyanensis, that are the main causes of leishmaniasis. It exhibited IC50 values of 437.5 and 252.7 μM against L. guyanensis and L. braziliensis, respectively [109]. Alkyl-guanidine-substituted β-carboline-based natural product opacaline B 61 (Fig. 15) was isolated from the New Zealand ascidian Pseudodistoma opacum and it exhibited a moderate antimalarial activity toward a chloroquine-resistant strain of Plasmodium falciparum, with an IC50 value of 4.5 μM (chloroquine was used as positive control, IC50 = 0.28 μM). Opacaline B was further screened against a wide range of parasites and it showed IC50 values of 27, 107 and 101 μM against Trypanosoma brucei rhodesiense, Trypanosoma cruzi and Leishmania donovani respectively, without being toxic toward the non-malignant L6 rat skeletal myoblast cell line [110].
Fig. 15.

N-Hydroxy-substituted carbazole-based natural products.
4.1.3. Other N-hydroxy-heterocycles
Among the N-hydroxy five-membered rings in tricyclic, tetracyclic or more complex natural products, we can mention omphalotins F-I 62a-d in Fig. 16 that are N-hydroxylated tricyclic tryptophan derivatives isolated by Liermann et. al. from mycelial extracts of the basidiomycete Omphalotus olearius. Compounds 62a, 62b, 62c and 62d exhibited strong and selective nematicidal activity against the plant pathogen Meloidogyne incognita, with LD50 values of 2, 1, 0.5-1 and 1 μg/mL respectively, while the positive control ivermectin possessed a comparable LD50 value of 2 μg/mL. None of these compounds showed antibacterial or antifungal activities, neither cytotoxic effects towards mouse leukaemia cells or human colon adenocarcinoma cells at concentrations up to 50 μg/mL [111]. Notamide A 63 (Fig. 16) is an indole alkaloid bearing the bicyclo [2.2.2] diazaoctane, obtained from a culture of Aspergillius sp., which is a marine-derived fungus isolated from the common mussel Mytilus edulis. It showed modest biological properties, exerting only moderate cytotoxicity against some tumor cell lines [112]. The same pyranoindole ring present in the structure of notamide A was also found in the natural product stephacidin B 64 in Fig. 16, a very complex dimeric alkaloid isolated from the fungus Aspergillus ochraceus. Natural product 64 demonstrated potent in vitro cytotoxicity against several human tumor cell lines (IC50 range 0.26-0.46 μM) with a good selectivity observed for the testosterone-dependent LNCaP cells (IC50 = 0.06 μM), and the growth inhibition effect was exerted by strongly inducing apoptosis in cell cultures [113,114].
Fig. 16.

N-hydroxy five-membered rings in complex systems.
4.2. Two endocyclic heteroatoms
4.2.1. Benzimidazoles and benzimidazolones
In literature, some N-hydroxybenzimidazole-based compounds exhibited promising pharmaceutical applications, for example they were patented as anti-infective agents that decreased resistance, virulence or growth of microbes or as compounds endowed with fungicidal and fungistatic activity [115-117]. It is noteworthy to mention N-hydroxy-benzoimidazolones 65a-c (Fig. 17) that showed in vitro potent inhibitory activity against human d-amino acid oxidase (DAAO), an enzyme that catalyzes the oxidation of d-amino acids to the corresponding imino acids and hydrogen peroxide. Compounds 65a-c displayed IC50 values of 0.6, 0.1 and 0.08 μM respectively, but they also exhibited similar inhibitory activity against porcine isoform of DAAO. The inhibitory potency was largely dependent on the size and position of substituents on the benzene ring. The two substituted compounds 65b and 65c were administered to mice (30 mg/kg) along with d-serine at the same dosage in order to assess their effects on d-serine plasma levels. It was demonstrated that activation of NMDA receptor by d-serine provides a new therapeutic approach for the treatment of schizophrenia, but unfortunately d-serine nephrotoxicity was provoked by hydrogen peroxide generated by DAAO-mediated d-serine metabolism. Pharmacokinetics studies revealed that compound 65b showed no effect on plasma d-serine due to the negligible oral bioavailability. Differently, the co-administration of 65c and d-serine increased the levels of d-serine, but only in a short period of time, because of the poor bioavailability of 65c. Metabolic stability tests of these compounds revealed that they were mainly metabolized by glucuronidation at the N-hydroxyl group, however this group was essential for the binding affinity to the DAAO active site. Therefore, new DAOO inhibitors should possess the N-OH group, but it would be necessary to optimize the presence of the surrounding substituents in order to improve the metabolic stability without compromise the activity [118]. N-hydroxybenzimidazole derivatives 66a-c (Fig. 17) were reported as potent inhibitors of LcrF, a multiple adaptational response transcription factor that regulates virulence in Yersinia pestis and Yersinia pseudotuberculosis. IC50 values of 3.9, 8.0 and 7.6 μM were reported for compounds 66a-c, respectively, in LcrF-DNA binding assay. Similarly, 66a-c were active also on ExsA, a multiple adaptational response transcription factor found in Pseudomonas aeruginosa with high homology with LcrF in the DNA binding domain, showing IC50 values of 3.5, 5.9 and 6.7 μM, respectively, in ExsA-DNA binding assay. In order to confirm their mechanism of action, these compounds were tested in an in vitro antimicrobial susceptibility test and they proved to be inactive against Y. pseudotuberculosis as well as S. aureus and E. coli bacteria, hence they did not target directly bacterial growth [119]. Similar N-hydroxybenzimidazoles 67a-c (Fig. 17) were developed as anti-virulence agents against infections caused by Pseudomonas aeruginosa and they showed IC50 values of 3.0, 13 and 7.5 μM, respectively, in ExsA-DNA binding assay and they effectively reduced the cytotoxicity of P. aeruginosa in an in vitro whole cell assay. These compounds demonstrated good metabolic stability from in vitro human liver microsomal assay [120]. Compound 68 (Fig. 17) belongs to the same class of N-hydroxybenzimidazoles, but it is substituted on the distal phenyl with a triazole ring. It could be useful in treatment of infections, because it targeted transcription factors of the AraC-XylS family and inhibition of these factors reduced the virulence of microbial cells. Specifically, compound 68 showed EC50 <10 μM against Pseudomonas aeruginosa [121].
Fig. 17.

N-hydroxy-substituted benzimidazoles and benzimidazolones.
5. N-Hydroxy-substituted fused six-membered ring systems
5.1. One endocyclic heteroatom
5.1.1. Quinolinones and isoquinolinones
In the group of N-hydroxy-quinolinones, compound 69a (Fig. 18), deriving from a HTS screening of the Pfizer compound collection, showed inhibitory activity against human kynurenine aminotransferase II (hKAT II), the primary enzyme in the brain responsible of catalyzing the formation of kynurenic acid from kynurenine, and this enzyme represents a potential target for the treatment of cognitive impairment associated with schizophrenia and other psychiatric disorders. Compound 69a showed selective inhibitory activity of hKAT II with IC50 value of 23 nM, while for the rat isoform rKAT II the IC50 value was of 263 nM. The R isomer 70 (Fig. 18) of 69a lost activity, displaying IC50 values of 219 nM for hKAT II and of 1170 nM for rKAT II. The methoxy derivative of the S isomer 69b (Fig. 18) showed a potent activity on the human isoform similar to 69a, but it was less selective (IC50 = 22 nM for hKAT II, IC50 = 137 nM for rKAT II). In vivo pharmacokinetic experiments revealed that after subcutaneous administration compound 69a displayed excellent CNS exposure in rat and good in vivo efficacy [122]. Derivatives 69c and 69d (Fig. 18) of the S isomer 69a were reported in a patent as the most potent hKAT II inhibitors of this chemical class, with IC50 values of 19 and 18 nM, respectively [123]. Compounds 71a-c (Fig. 18) showed potent antimicrobial activity against Enterobacter aerogenes, Serratia marcescens, Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, Lactobacillus casci, Lactobacillus plantarum, Leuconostoc dextranicum, and Streptococcus faecalis. Compounds 71a, 71b and 71c exhibited mean MIC values of 0.6, 0.6 and 0.2 μg/mL, respectively, on this panel of bacteria. In addition, chloro-substituted derivatives 71b and 71c weakly inhibited the growth of Candida albicans, with MICs of 20 μg/mL, and were 10-fold more effective than unsubstituted 71a (MIC = 200 μg/mL), even if they were less potent than the reference compound amphotericin B that exhibited MIC value of 0.6 μg/mL [124]. N-hydroxyquinolinone derivatives 72a-d (Fig. 18) were potent 5-lipoxygenase enzyme inhibitors, with IC50 values of 0.87, 0.34, 0.98 and 0.84 μM, respectively, and isoquinolinone derivative 73 (Fig. 18) exhibited IC50 value of 0.45 μM against the same enzyme. For compounds 72a-c and 73 the inhibition percentages against 12-lipoxygenase enzyme isolated from human platelets were evaluated and resulted of 97%, 98%, 97% and 99% at 100 μM, respectively. The same compounds 72a-c and 73 also displayed inhibitory activity against 15-lipoxygenase enzyme obtained from soybean, with inhibition percentages of 100%, 100%, 97% and 50% at 100 μM, respectively [125]. 1-Hydroxy-3,4-dihydroquinolin-2(1H)-one 74a and 1-hydroxy-6-chloro-3,4-dihydroquinolin-2(1H)-one 74b in Fig. 18 are derivatives of a natural benzoxazinone compound that has been shown to cause symptomatic improvements in patients with benign prostatic hyperplasia, chronic prostatitis and prostatodynia. Compounds 74a and b showed potent inhibitory effects on DU-145 prostate cancer cell growth, although they were not selective for this cell line because they inhibited MCF-7 cell growth at a concentration of 10 μg/mL [126].
Fig. 18.

N-hydroxy-substituted quinolinones and isoquinolinones.
5.1.2. Isoquinolinediones and benzo[de]isoquinolinediones
N-hydroxyimide compound 75a (Fig. 19) was initially reported as an inhibitor of the endonuclease dependent influenza polymerases, highlighting the absolute requirement of the N-hydroxy group for the activity [127]. Later, it was demonstrated that 75a and the methyl ester analogue 75b (Fig. 19) inhibited HIV-1 integrase and ribonuclease H and DNA polymerase functions of the HIV-1 reverse transcriptase. Interestingly, 75a and 75b complexed with magnesium ions were active, in particular these complexes required enolization of the ligands because magnesium cations were chelated by N-OH group and one of the adjacent OH groups of the corresponding tautomeric forms of 75a-b, and these complexes released superoxide anion [128]. The insertion of an alkyl chain in position 4 of the scaffold as in compound 75c (Fig. 19) did not improve the antiviral activity, but it conferred cytotoxicity to the compound. These inhibitors were only small prototype compounds with selectivity for two-metal ion enzymes, but they still need further optimization of substituents on the isoquinolinedione scaffold [129]. Representative 4-amide-substituted compound 75d (Fig. 19) possessed the same scaffold and was patented as a potent inhibitor against HIV integrase (IC50 = 3 nM), thus it was considered a promising anti-HIV agent useful in the treatment of HIV infection and acquired immune deficiency syndrome (AIDS) [130]. Anti-viral compounds 76a-b (Fig. 19) were characterized by the same scaffold with a substitution in position 7 of the fused phenyl ring. Compound 76a showed a good RNase H inhibitory activity (IC50 = 5.7 μM) and 76b showed noticeable overall integrase inhibitory activity (IC50 = 0.09 μM). Unfortunately, all the tested compounds possessing the 2-hydroxyisoquinoline-1,3(2H,4H)-dione scaffold exhibited high cellular cytotoxicity in cell cultures, which limits their applications as antiviral agents [131]. The 2-hydroxyisoquinolinedione 77 (Fig. 19) with a furan ring at the 6-position inhibited hepatitis C virus replication by targeting NS5B polymerase, which has a similar active site to that of HIV integrase and reverse transcriptase-associated ribonuclease H, showing IC50 value of 1.9 μM, and a similar value (1.3 μM) was obtained in a biochemical assay against recombinant NS5B [132]. Benzo[de]isoquinoline-1,3-dione derivatives 78 and 79 (Fig. 19) were developed as antibacterial agents, inhibiting selectively bacterial DNA gyrase and DNA topoisomerase IV. The inhibition of two different targets offer advantages, by lowering the possibility of bacterial resistance. These two potent derivatives were tested against a wide panel of bacteria and proved to be active against some Escherichia coli strains, Bacillus subtilis, Sthapylococcus aureus and pyogenes; in particular 78, in which the benzoisoquinoline ring was fused with a benzodioxin ring, showed MICs range of 0.13-8 μg/mL, whereas the pyrrolidine-substituted derivative 79 had MICs range of 0.06-2 μg/mL and it was nearly inactive against S. pyogenes. Moreover, it was confirmed that 79 had no any DNA intercalating or binding ability [133].
Fig. 19.

N-hydroxy-isoquinolinediones and benzo[de]isoquinolinediones.
5.1.3. Acridone derivatives
Some acridone-based compounds are natural products, such as N-hydroxy-acridone alkaloid 80 (Fig. 20) isolated from the leaves of Sarcomelicope dogniensis [134]. Acridine-based drugs are well-known agents developed against malaria, and in particular floxacrine 81 (Fig. 20) exhibited in vitro and in vivo potent antimalarial activity (as racemic mixture). Floxacrine was able to bind heme, the by-product of parasite hemoglobin digestion, and to prevent heme crystallization, finally leading to parasite death, displaying IC50 value of 63 μM, comparable to the value of the reference anti-malarial drug chloroquine (IC50 = 56 μM) [135]. Unfortunately, its use would be limited only as a prophylactic agent, due to the requirement of high daily dosage, easy development of resistance to floxacrine and to the unwanted side effects produced by this molecule, such as chronic periarteritis and vascular cardiotoxicity. As a consequence, many floxacrine derivatives were synthesized to overcome the problems of the parent compound, but any small changes in inhibitor structure had negative effects on antimalarial activity, thus it was difficult to improve the activity-toxicity balance of the floxacrine derivatives [136-139]. Among the floxacrine analogues, N-hydroxy imine acridione derivative 82 (Fig. 20) was a potent antimalarial agent in both rodents and primates. In this structure, the replacement of the N-OH group with hydrogen did not completely remove the antimalarial activity, but the insertion of N-alkyl group caused a loss of activity. However, similarly to the parent compound floxacrine, 82 exerted some toxicity at higher doses [140].
5.2. Two endocyclic heteroatoms
5.2.1. Naphthyridinones
Compound 83 (Fig. 21) was a potent and selective RNase H inhibitor, showing IC50 value of 45 nM in biochemical assays, and it inhibited viral growth in vitro, without being cytotoxic [141]. A series of N-hydroxynaphthyridinone derivatives were patented as agents for the prophylaxis and treatment of infection by HIV, such as compound 84 (Fig. 21) that showed promising IC50 values in the low micromolar or nanomolar range in a RNase H-mediated RNA cleavage assay, in the integrase standard-transfer assay and against single-cycle HIV infectivity assay B, without being cytotoxic at this low concentrations [142]. Structurally similar representative derivative 85 (Fig. 21) showed inhibitory activity of hKAT II (IC50 = 52.7 nM), and therefore its potential use was for the treatment of cognitive deficits associated with schizophrenia and other neurodegenerative disorders [143].
Fig. 21.

N-hydroxy-substituted naphthyridinones.
5.2.2. Quinazolinediones and quinoxalinediones
N-hydroxyquinazoline-2,4-dione derivatives 86a-b (Fig. 22) were designed as Gly/NMDA and AMPA receptor ligands and their affinities were evaluated by measuring their ability to displace [3H]-glycine or [3H]-AMPA. Compound 86a showed the best affinity toward Gly/NMDA receptor (Ki = 0.20 μM), differently 86b substituted with a triazole ring showed a preferred binding to AMPA receptor (Ki = 0.25 μM), with a total selectivity for this receptor versus the Gly/NMDA receptor, that did not allow the presence of such a bulky substituent [144]. It is reasonable include in this group derivative 87, in which the pyrimidinone ring was fused with a thiophene heterocycle, instead of a phenyl ring (Fig. 22). Compound 87 was one of the most active and selective flap endonuclease-1 (an enzyme involved in DNA repair) inhibitors (IC50 = 35 nM) compared to a related endonuclease, xeroderma pigmentosum G (IC50 of about 8 μM). The N-hydroxy-urea moiety was essential for the inhibition of the target enzyme by coordinating the two divalent metal ions needed for catalysis [145]. Compound 88 (Fig. 22) was a representative inhibitor endowed with potent anti-microbial activity, displaying MIC values lower than 2 ppm against Candida albicans, Auresbasidium pulluans and Aspergillus niger [146]. Hydroxamate-substituted quinoxalinedione 89 (Fig. 22) was a bidentate zinc chelating agent and it showed anticoagulant properties due to the replacement of calcium ions with zinc ions in the serine protease domain of factor VII, thus causing an inhibitory effect on factor VII itself. Compounds exhibiting chelating binding affinity to zinc ions were considered useful for the treatment of coagulation-related diseased states [147].
Fig. 22.

N-hydroxy-substituted quinazolinediones and quinoxalinediones.
5.2.3. Benzoxazinones
2,4-Dihydroxy-1,4-benzoxazin-3-one (DIBOA) 90 (Fig. 23) is a cyclic hydroxamic acid possessing protective functions in plants against insects, bacteria and fungi, and it is abundant in sprouts of Gramineae family. In plants, it is stored in vacuoles as d-glycosides and the aglycone DIBOA is obtained after enzymatic hydrolysis. Compound 90 has been identified and quantified in grains and bakery products of rye and wheat. At present, uptake, metabolic fate and biological properties of this dietary compound in humans is still under study. Compound 90 was firstly isolated from commercial pollen extract, Cernitin T-60, and it proved to reduce the growth of a prostate cancer cell line [148]. Compound 90 and its methoxy derivative 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIMBOA) 91 (Fig. 23), which is the dominant benzoxazinoid compound found in wheat, exerted mutagenic activity against human liver cell line HepG2 and 91 caused stronger effects than the parent compound 90 [149]. Moreover, the cytotoxic effects of DIBOA were also observed on different cancer cell lines, such as DU-145 (prostate cancer) and MCF-7 (breast cancer) cells [126]. Derivatives of DIBOA such as compound 92 (Fig. 23) which bears a chlorine atom in position 6 and an ethyl chain in position 2 showed phytotoxic activity with IC50 value less of 1 μM against Allium cepa (onion) and Lepidium sativum (watercress) and it could be used for the development of a new generation of plant protection products of natural origin [150]. Halogenated benzoxazinone DIBOA derivative 93 (Fig. 23) was patented as a suitable agent for agricultural weed control. It showed phytotoxic activity, causing dose-dependent inhibitions of Allium cepa, Triticum aestivum (wheat), Lepidium sativum, Lycopersicon esculentum (tomato), Lactuca sativa (lettuce), Avena fatua (wild oats), Echinochloa crus-galli (barnyard grass) and Lolium rigidum (rigid ryegrass) [151]. Insertion of butyl group in place of the OH group of DIBOA produced compound 94 (Fig. 23) that possessed a potent phytotoxic activity making it useful as herbicide. The group in position 2 modulated the solubility and increased the phytotoxic activity compared to the parent compound. It was found that compound 94 inhibited growth of A. fatua with IC50 value of 131 μM and E. crus-galli with IC50 value of 190 μM [152].
Fig. 23.

N-hydroxy-substituted benzoxazinones.
5.2.4. Other N-hydroxy heterocycles
N-hydroxy-containing natural products 95 and 96 (Fig. 24) are bispyrroloiminoquinone oximes isolated from the South African latrunculid sponge Tsitsikamma favus. Compounds 95 and 96 possessed interesting pharmacological properties, because they exhibited cytotoxicity against human colon tumor cell line HCT-116, with IC50 values of 128.2 and 16.5 μM, respectively, moreover they showed DNA intercalating ability [153]. Some studies demonstrated the weak oncogenic activity of 3-hydroxyxanthine 97 (Fig. 24), that may be due to its metabolization by xanthine oxidase that lead to the formation of oncogenic metabolites [154,155].
Fig. 24.

N-hydroxy six-membered rings in heterocyclic systems.
6. Conclusions
The presence of many N-hydroxylated heterocycles among pharmaceutical active derivatives attests the growing interest toward this varied chemical class of N-hydroxy-substituted compounds as building blocks. Anti-cancer and anti-microbial properties are the principal biological activities demonstrated by N-hydroxy-substituted compounds, suggesting that they may be therapeutically useful for some types of cancer and several other infection diseases. In some cases the role of the NOH group was clear, representing a pharmacophoric moiety necessary for the biological activity of the whole molecule on the related target, while in some compounds the need for this group was not adequately investigated and evaluated. The insertion of a N-OH group in a molecule significantly modifies some of its properties, for example it can increase solubility or modify the acid-base behavior of the compound. Importantly, the N-OH moiety affects the binding of the molecule to the target protein, increasing the possibility to establish some hydrogen bonds with the protein residues in the active site or with surrounding water molecules. Certainly, as previously discussed in the present review, one of the most represented classes is the N-hydroxyindole series which includes both synthetic and natural compounds, although these chemical entities are quite rare in nature. In order to obtain functionalized N-hydroxyindoles some synthetic methods for the construction of this kind of scaffold have been developed [156-159]. However, this scaffold is unstable if not adequately supported by some substitutions, such as electron withdrawing groups or resonance stabilizing substituents on the indole nucleus, or substituents in position 2 that can hydrogen-bond to the hydroxyl group.
The present review summarizes the main biologically active N-OH containing heterocycles along with a discussion of their medicinal importance, pointing out the increasing use of this structural motif in medicinal chemistry in view of their applications in several fields. It is concluded that N-OH-substituted bioactive molecules exhibit a wide spectrum of biological activities and this review would help and encourage further developments and modifications of N-OH containing compounds, highlighting the potentialities of this multifaceted chemical class.
Heterocycles containing endocyclic N-hydroxy portions are metabolically stable molecular entities
We present several chemical classes of bioactive N-hydroxy heterocycles, according to the central scaffold of the molecules
The main applications in medicinal chemistry of the compounds discussed in the present review are as anticancer and antibacterial agents
Acknowledgements
We are grateful to NIH (R01M098453) (CG) and European Union Seventh Framework Programme grant (PIIF-GA-2011-299026) (RR) for financial support. We would like to thank Prof. Filippo Minutolo (University of Pisa) for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edwards PJ, Sturino C. Managing the liabilities arising from structural alerts: a safe philosophy for medicinal chemists. Curr. Med. Chem. 2011;18:3116–3135. doi: 10.2174/092986711796391714. [DOI] [PubMed] [Google Scholar]
- 2.Smith GF. Designing drugs to avoid toxicity. Prog. Med. Chem. 2011;50:1–47. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 3.Banoglu E. Current status of the cytosolic sulfotransferases in the metabolic activation of promutagens and procarcinogens. Curr. Drug Metab. 2000;1:1–30. doi: 10.2174/1389200003339234. [DOI] [PubMed] [Google Scholar]
- 4.Gillette JR. Commentary. A perspective on the role of chemically reactive metabolites of foreign compounds in toxicity I. Correlation of changes in covalent binding of reactivity metabolites with changes in the incidence and severity of toxicity. Biochem. Pharmacol. 1974;23:2785–2794. doi: 10.1016/0006-2952(74)90052-5. [DOI] [PubMed] [Google Scholar]
- 5.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 6.Jollow DJ, Kocsis JJ, Snyder R, Vainio H. Biological Reactive Intermediates: Formation, Toxicity and Inactivation. Plenum Press; New York: 1977. [Google Scholar]
- 7.Fagius J, Osterman PO, Sidén A, Wiholm BE. Guillain-Barré syndrome following zimeldine treatment. J. Neurol. Neurosurg. Psychiatry. 1985;48:65–69. doi: 10.1136/jnnp.48.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cashman JR, Yang ZC, Högberg T. Oxidation of N-hydroxynorzimeldine to a stable nitrone by hepatic monooxygenases. Chem. Res. Toxicol. 1990;3:428–432. doi: 10.1021/tx00017a007. [DOI] [PubMed] [Google Scholar]
- 9.Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs. 2014;25:140–149. doi: 10.1097/CAD.0000000000000040. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Lei J, Shan Y, Yang H, Song M, Ma Y. Recent progress in the development of histone deacetylase inhibitors as anti-cancer agents. Mini Rev. Med. Chem. 2013;13:1999–2013. doi: 10.2174/13895575113136660102. [DOI] [PubMed] [Google Scholar]
- 11.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 12.Hugenberg V, Riemann B, Hermann S, Schober O, Schäfers M, Szardenings K, Lebedev A, Gangadharmath U, Kolb H, Walsh J, Zhang W, Kopka K, Wagner S. Inverse 1,2,3-triazole-1-yl-ethyl substituted hydroxamates as highly potent matrix metalloproteinase inhibitors: (radio)synthesis, in vitro and first in vivo evaluation. J. Med. Chem. 2013;56:6858–6870. doi: 10.1021/jm4006753. [DOI] [PubMed] [Google Scholar]
- 13.Campestre C, Tortorella P, Agamennone M, Preziuso S, Biasone A, Nuti E, Rossello A, Gallina C. Peptidyl 3-substituted 1-hydroxyureas as isosteric analogues of succinylhydroxamate MMP inhibitors. Eur. J. Med. Chem. 2008;43:1008–1014. doi: 10.1016/j.ejmech.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Hazeldine S, Pachaiyappan B, Steinbergs N, Nowotarski S, Hanson AS, Casero RA, Jr, Woster PM. Low molecular weight amidoximes that act as potent inhibitors of lysine-specific demethylase 1. J. Med. Chem. 2012;55:7378–7391. doi: 10.1021/jm3002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sengupta P, Puri CS, Chokshi HA, Sheth CK, Midha AS, Chittur TR, Thennati R, Murumkar PR, Yadav MR. Synthesis, preliminary biological evaluation and molecular modeling of some new heterocyclic inhibitors of TACE. Eur. J. Med. Chem. 2011;46:5549–5555. doi: 10.1016/j.ejmech.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Su M, Chen Y, Li J, Lu W. The design and synthesis of a new class of RTK/HDAC dual-targeted inhibitors. Molecules. 2013;18:6491–6503. doi: 10.3390/molecules18066491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Petrelli R, Gao G, Wilson DJ, McLean GT, Jayaram HN, Sham YY, Pankiewicz KW. Dual inhibitors of inosine monophosphate dehydrogenase and histone deacetylase based on a cinnamic hydroxamic acid core structure. Bioorg Med Chem. 2010;18:5950–5964. doi: 10.1016/j.bmc.2010.06.081. [DOI] [PubMed] [Google Scholar]
- 18.Marques SM, Nuti E, Rossello A, Supuran CT, Tuccinardi T, Martinelli A, Santos MA. Dual inhibitors of matrix metalloproteinases and carbonic anhydrases: iminodiacetyl-based hydroxamate-benzenesulfonamide conjugates. J. Med. Chem. 2008;51:7968–779. doi: 10.1021/jm800964f. [DOI] [PubMed] [Google Scholar]
- 19.Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med. Chem. 2011;3:1165–1180. doi: 10.4155/fmc.11.69. [DOI] [PubMed] [Google Scholar]
- 20.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 21.Bertrand P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010;45:2095–2116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 22.Katritzky AR, Huang L, Chahar M, Sakhuja R, Hall CD. The chemistry of N-hydroxyamidoximes, N-aminoamidoximes, and hydrazidines. Chem. Rev. 2012;112:1633–1649. doi: 10.1021/cr200076q. [DOI] [PubMed] [Google Scholar]
- 23.Kelly WK, O' Conor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilidehydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coffey DC, Kutko MC, Glick RD, Butler LM, Heller G, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001;61:3591–3594. [PubMed] [Google Scholar]
- 25.Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, Davidson NE. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res. Treat. 2003;81:177–186. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- 26.Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS, Jeffers M. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol. Cancer Ther. 2006;5:2086–2095. doi: 10.1158/1535-7163.MCT-06-0111. [DOI] [PubMed] [Google Scholar]
- 27.Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene. 1999;18:2461–2470. doi: 10.1038/sj.onc.1202564. [DOI] [PubMed] [Google Scholar]
- 28.Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, Lebbé C, Assaf C, Squier M, Williams D, Marshood M, Tai F, Prince HM. Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur. J. Cancer. 2013;49:386–394. doi: 10.1016/j.ejca.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, Mitsiades N, Shringarpure R, LeBlanc R, Chauhan D, Munshi NC, Schlossman R, Richardson P, Griffin J, Anderson KC. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–2622. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 30.Edsall RJ, Jr., Harris HA, Manas ES, Mewshaw RE. ERβ Ligands. Part 1: The discovery of ERβ selective ligands which embrace the 4-hydroxy-biphenyl template. Bioorg. Med. Chem. 2003;11:3457–3474. doi: 10.1016/s0968-0896(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 31.Yang C, Edsall RJ, Jr., Harris HA, Zhang X, Manas ES, Mewshaw RE. ERβ Ligands. Part 2: Synthesis and structure-activity relationships of a series of 4-hydroxy-biphenylcarbaldehyde oxime derivatives. Bioorg. Med. Chem. 2004;12:2553–2570. doi: 10.1016/j.bmc.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 32.Mewshaw RE, Bowen SM, Harris HA, Xu ZB, Manas ES, Cohn ST. ERβ Ligands. Part 5: Synthesis and structure-activity relationships of a series of 4’-hydroxyphenyl-aryl-carbaldehyde oxime derivatives. Bioorg. Med. Chem. Lett. 2007;17:902–906. doi: 10.1016/j.bmcl.2006.11.066. [DOI] [PubMed] [Google Scholar]
- 33.Minutolo F, Bertini S, Granchi C, Marchitiello T, Prota G, Rapposelli S, Tuccinardi T, Martinelli A, Gunther JR, Carlson KE, Katzenellenbogen JA, Macchia M. Structural evolutions of salicylaldoximes as selective agonists for estrogen receptor β. J. Med. Chem. 2009;52:858–867. doi: 10.1021/jm801458t. [DOI] [PubMed] [Google Scholar]
- 34.Bertini S, De Cupertinis A, Granchi C, Bargagli B, Tuccinardi T, Martinelli A, Macchia M, Gunther JR, Carlson KE, Katzenellenbogen JA, Minutolo F. Selective and potent agonists for estrogen receptor beta derived from molecular refinements of salicylaldoximes. Eur. J. Med. Chem. 2011;46:2453–2462. doi: 10.1016/j.ejmech.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonta IL, De Vos CJ, Grijsen H, Hillen FC, Noach EL, Sim AW. 1-Hydroxy-3-amino-pyrrolidone-2(HA-966): a new GABA-like compound, with potential use in extrapyramidal diseases. Br. J. Pharmac. 1971;43:514–535. doi: 10.1111/j.1476-5381.1971.tb07182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies J, Watkins JC. Is 1-hydroxy-3-aminopyrrolidone-2 (HA-966) a selective excitatory amino-acid antagonist? Nat. New Biol. 1972;238:61–63. doi: 10.1038/newbio238061a0. [DOI] [PubMed] [Google Scholar]
- 38.Davies J, Watkins JC. Microelectrophoretic studies on the depressant action of HA-966 on chemically and synaptically excited neurones in the cat cerebral cortex and cuneate nucleus. Brain Res. 1973;59:31l–322. doi: 10.1016/0006-8993(73)90269-2. [DOI] [PubMed] [Google Scholar]
- 39.Davies J, Watkins JC. Antagonism of synaptic and amino acid induced excitation in the cuneate nucleus of the cat by HA-966. Neuropharmacology. 1973;12:637–640. doi: 10.1016/0028-3908(73)90116-0. [DOI] [PubMed] [Google Scholar]
- 40.Evans RH, Francis AA, Watkins JC. Mg+2 like selective antagonism of excitatory amino acid-induced responses by α,ε-diaminopimelic acid, D-α-aminoadipate and HA-966 in isolated spinal cord of frog and immature rat. Brain Res. 1978;148:536–542. doi: 10.1016/0006-8993(78)90744-8. [DOI] [PubMed] [Google Scholar]
- 41.Biscoe TJ, Davies J, Drav A, Evans RH, Martin MR, Watkins JC. d-α-aminoadipate, α-ε-diaminopimelic acid and HA-966 as antagonists of amino acid-induced and synaptic excitation of mammalian spinal neurones in vivo. Brain Res. 1978;148:543–548. doi: 10.1016/0006-8993(78)90745-x. [DOI] [PubMed] [Google Scholar]
- 42.Foster AC, Kemp JA. HA-966 antagonizes N-methyl-d-aspartate receptors through a selective interaction with the glycine modulatory site. J. Neurosci. 1989;9:2191–2196. doi: 10.1523/JNEUROSCI.09-06-02191.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh L, Donald AE, Foster AC, Hutson PH, Iversen LL, Iversen SD, Kemp JA, Leeson PD, Marshall GR, Oles RJ, Priestley T, Thorn L, Tricklebank MD, Vass CA, Williams BJ. Enantiomers of HA-966 (3-amino-1-hydroxypyrrolid-2-one) exhibit distinct central nervous system effects: (+)-HA-966 is a selective glycine/N-methyl-d-aspartate receptor antagonist, but (-)-HA-966 is a potent gamma-butyrolactone-like sedative. Proc. Natl. Acad. Sci. USA. 1990;87:347–351. doi: 10.1073/pnas.87.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vartanian MG, Taylor CP. Different stereoselectivity of the enantiomers of HA-966 (3-amino-1-hydroxy-2-pyrrolidinone) for neuroprotective and anticonvulsant actions in vivo. Neurosci. Lett. 1991;133:109–112. doi: 10.1016/0304-3940(91)90069-6. [DOI] [PubMed] [Google Scholar]
- 45.Dunn RW, Flanagan DM, Martin LL, Kerman LL, Woods AT, Camacho F, Wilmot CA, Cornfeldt ML, Effland RC, Wood PL, Roy C. Stereoselective R-(+) enantiomer of HA-966 displays anxiolytic effects in rodents. Eur. J. Pharmacol. 1992;214:207–214. doi: 10.1016/0014-2999(92)90120-s. [DOI] [PubMed] [Google Scholar]
- 46.Morrow BA, Lee EJ, Taylor JR, Elsworth JD, Nye HE, Roth RH. (S)-(-)-HA-966, a γ-hydroxybutyrate-like agent, prevents enhanced mesocorticolimbic dopamine metabolism and behavioral correlates of restraint stress, conditioned fear and cocaine sensitization. J. Pharmacol. Exp. Ther. 1997;283:712–721. [PubMed] [Google Scholar]
- 47.Leeson PD, Williams BJ, Rowley M, Moore KW, Baker R, Kemp JA, Priestley T, Foster AC, Donald AE. Derivatives of l-hydroxy-3-aminopyrrolidin-2-one (HA-966). Partial agonists at the glycine site of the NMDA receptor. Bioorg. Med. Chem. Lett. 1993;3:71–76. [Google Scholar]
- 48.Tricklebank MD, Bristow LJ, Hutson PH, Leeson PD, Rowley M, Saywell K, Singh L, Tattersall FD, Thorn L, Williams BJ. The anticonvulsant and behavioural profile of L-687,414, a partial agonist acting at the glycine modulatory site on the N-methyl-d-aspartate (NMDA) receptor complex. Br. J. Pharmacol. 1994;113:729–736. doi: 10.1111/j.1476-5381.1994.tb17054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leeson PD, Williams BJ, Baker R, Ladduwahetty T, Moore KW, Rowley M. Effects of five-membered ring conformation on bioreceptor recognition: identification of 3R-amino-1-hydroxy-4R-methylpyrrolidin-2-one (L-687,414) as a potent glycine/N-methyl-d-aspartate receptor antagonist. J. Chem. Soc. Chem. Commun. 1990;22:1578–1580. [Google Scholar]
- 50.Payne JD. Microbicidal metal complexes of cyclic thiohydroxamic acids or 1-hydroxy-2-pyrrolidinethiones with enhanced stability and activity. 1998 WO9826665. [Google Scholar]
- 51.Watanabe F, Araki Y, Fujii Y. Sulfonylaminosuccinimides and matrix metalloprotease inhibitors containing them. 2002 JP2002105073. [Google Scholar]
- 52.Cordi A, Lacoste JM, Audinot V, Millan M. Design, synthesis and structure-activity relationship of novel strychnine-insensitive glycine receptor ligands. Bioorg. Med. Chem. Lett. 1999;9:1409–1414. doi: 10.1016/s0960-894x(99)00194-8. [DOI] [PubMed] [Google Scholar]
- 53.Cordi A, Lacoste JM, Millan M, Audinot V. Preparation of 3-hydroxy-4-imidazolidinone as a NMDA receptor agonist. 1997 EP780379. [Google Scholar]
- 54.Ikonomidou H. New use of glutamate antagonists for the treatment of cancer. 2000 EP1002535. [Google Scholar]
- 55.Vystorop IV, Konovalova NP, Nelyubina Yu.V., Varfolomeev VN, Fedorov BS, Sashenkova TE, Berseneva EN, Lyssenko KA, Kostyanovsky RG. Cyclic hydroxamic acids derived from α-amino acids 1. Regioselective synthesis, structure, NO-donor and antimetastatic activities of spirobicyclic hydroxamic acids derived from glycine and dl-alanine. Russ. Chem. Bull. 2010;59:127–135. [Google Scholar]
- 56.Nieto L, Mascaraque A, Miller F, Glacial F, Martínez C. Ríos, Kaiser M, Brun R, Dardonville C. Synthesis and antiprotozoal activity of N-alkoxy analogues of the trypanocidal lead compound 4,4’-bis(imidazolinylamino)diphenylamine with improved human blood-brain barrier permeability. J. Med. Chem. 2011;54:485–494. doi: 10.1021/jm101335q. [DOI] [PubMed] [Google Scholar]
- 57.Tikhonov A.Ya., Mazhukin DG, Grigor'eva LN, Khlestkin VK, Voinova NN, Syropyatov B.Ya., Shirinkina SS, Volodarsky LB. Synthesis and inhibitory effect on platelet aggregation and antihypertensive activity of 1-hydroxy-2,5-dihydro-1H-imidazole-2-carboxylic acid 3-oxides, 1,3-dihydroxyimidazolidine-2-carboxylic acids, and 1,4-dihydroxy-2,3-piperazinediones. Arch. Pharm. Pharm. Med. Chem. 1999;332:305–308. doi: 10.1002/(sici)1521-4184(19999)332:9<305::aid-ardp305>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 58.Martin SF, Follows BC, Hergenrother PJ, Franklin CL. A Novel class of zinc-binding inhibitors for the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. J. Org. Chem. 2000;65:4509–4514. doi: 10.1021/jo9915731. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki I, Erdelmeier I, Yadan JC, Pelisson I. Preparation of imidazole derivatives as tyrosinase inhibitors for therapeutic or cosmetic use. 2008 WO2008006824. [Google Scholar]
- 60.Ahmed SA, Odde S, Daga PR, Bowling JJ, Mesbah MK, Youssef DT, Khalif SI, Doerksen RJ, Hamann MT. Latrunculin with a highly oxidized thiazolidinone ring: structure assignment and actin docking. Organic Letters. 2007;9:4773–4776. doi: 10.1021/ol7020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Austin P. Preparation of thiazolethiones and imidazolidinethiones as industrial biocides. 1987 EP249328. [Google Scholar]
- 62.Payne JD. Non-woven material comprising cyclic thiohydroxamic acid. 1999 WO9945975. [Google Scholar]
- 63.Austin PW, Singer M. Antimicrobial agents for cosmetic compositions. 1992 EP498636. [Google Scholar]
- 64.Napolitano E, Basagni S, Trasciatti S. Preparation of cyclic hydroxylamine derivatives as antioxidants. 2011 WO2011076930. [Google Scholar]
- 65.Parsons P, Trasciatti S, Rosini S, Annis M. Preparation of cyclic hydroxylamine as psychoactive agents. 2004 WO2004111021. [Google Scholar]
- 66.Matier WL, Patil G. Esterified N-hydroxypiperidine compounds for the amelioration of the development of cataracts and other opthalmic diseases, preparation thereof, and compositions containing them. 2003 WO2003096991. [Google Scholar]
- 67.Matier WL, Patil G. Hydroxylamines and derivatives for treatment of inflammatory conditions of the liver. 2007 US20070197593. [Google Scholar]
- 68.Matier WL, Patil G. Hydroxylamines and derivatives as anti-angiogenic agents. 2007 WO2007092741. [Google Scholar]
- 69.Komarov PG, Morgunov AA, Bilenko MV, Zhdanov RI. Anti-ischemic effect of 1-hydroxy derivatives of nitroxyl bioantioxidants. Bioact. Spin Labels. 1992:491–507. [Google Scholar]
- 70.Patil G. Preparation of hydroxytetramethylpiperidine derivatives and analogs for use in drug resistance reversal in neoplastic diseases. 2008 WO2008103613. [Google Scholar]
- 71.Patil G, Mousa SA. Preparation of hydroxytetramethylpiperidine derivatives and analogs for use in drug resistance reversal in neoplastic diseases. 2008 WO2008101195. [Google Scholar]
- 72.Pelaez F, Polishook JD, Valldosera M, Guarro J. A new species of Delitschia from west Africa. Mycotaxon. 1994;50:115–122. [Google Scholar]
- 73.Tomassini JE, Davies ME, Hastings JC, Lingham R, Mojena M, Raghoobar SL, Singh SB, Tkacz JS, Goetz MA. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996;40:1189–1193. doi: 10.1128/aac.40.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hensens OD, Goetz MA, Liesch JM, Zink DL, Raghoobar SL, Helms GL, Singh SB. Isolation and Structure of Flutimide, a Novel Endonuclease Inhibitor of Influenza Virus. Tet. Lett. 1995;36:2005–2008. [Google Scholar]
- 75.Singh SB. Total synthesis of Flutimide, a Novel Endonuclease Inhibitor of Influenza Virus. Tet. Lett. 1995;36:2009–2012. [Google Scholar]
- 76.Savard ME, Melzer MS, Boland GJ, Bensimon C, Blackwell BA. A new 1-hydroxy-2,6-pyrazinedione associated with hypovirulent isolates of Sclerotinia minor. J. Nat. Prod. 2003;66:306–309. doi: 10.1021/np020445w. [DOI] [PubMed] [Google Scholar]
- 77.Kawada M, Someno T, Inoue H, Ohba S, Masuda T, Kato T, Ikeda D. NBRI16716A, a new antitumor compound against human prostate cancer cells, produced by Perisporiopsis melioloides Mer-f16716. J. Antibiot. 2010;63:319–323. doi: 10.1038/ja.2010.42. [DOI] [PubMed] [Google Scholar]
- 78.Singh SB, Tomassini JE. Synthesis of natural flutimide and analogous fully substituted pyrazine-2,6-diones, endonuclease inhibitors of influenza virus. J. Org. Chem. 2001;66:5504–5516. doi: 10.1021/jo015665d. [DOI] [PubMed] [Google Scholar]
- 79.Marques SM, Tuccinardi T, Nuti E, Santamaria S, André V, Rossello A, Martinelli A, Santos MA. Novel 1-hydroxypiperazine-2,6-diones as new leads in the inhibition of metalloproteinases. J. Med. Chem. 2011;54:8289–8298. doi: 10.1021/jm200593b. [DOI] [PubMed] [Google Scholar]
- 80.Saito R, Ghosh KK, Harada K, Katoh A. Microbial growth-promotion activity of 3-hydroxymonoazine- and N-hydroxydiazine-type heterocycles. Yakugaku Zasshi. 2002;122:703–705. doi: 10.1248/yakushi.122.703. [DOI] [PubMed] [Google Scholar]
- 81.Bartsch A, Bross M, Spiteller P, Spiteller M, Steglich W. Birnbaumin A and B: two unusual 1-hydroxyindole pigments from the “flower pot parasol” Leucocoprinus birnbaumii. Angew. Chem. Int. Ed. 2005;44:2957–2959. doi: 10.1002/anie.200500082. [DOI] [PubMed] [Google Scholar]
- 82.Keller-Juslen C, Kuhn M, King HD. Antibiotics, pharmaceutical compositions and method of use. 1984 US4478831. [Google Scholar]
- 83.Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide antibiotics. Chem. Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 84.Sasaki T, Otani T, Matsumoto H, Unemi N, Hamada M, Takeuchi T, Hori M. MJ347-81F4 A & B, novel antibiotics from Amycolatopsis sp.: taxonomic characteristics, fermentation, and antimicrobial activity. J. Antibiot. 1998;51:715–721. doi: 10.7164/antibiotics.51.715. [DOI] [PubMed] [Google Scholar]
- 85.Li W, Left JE, Ax HA, Gustavson DR, Brown DM, Turner L, Brown K, Clark J, Yang H, Fung-Tomc J, Lam KS. Nocathiacins, new thiazolyl peptide antibiotics from Nocardia sp. I. Taxonomy, fermentation and biological activities. J. Antibiot. 2003;56:226–231. doi: 10.7164/antibiotics.56.226. [DOI] [PubMed] [Google Scholar]
- 86.Leet JE, Li W, Ax HA, Matson JA, Huang S, Huang R, Cantone JL, Drexler D, Dalterio RA, Lam KS. Nocathiacins, new thiazolyl peptide antibiotics from Nocardia sp. II. Isolation, characterization, and structure determination. J. Antibiot. 2003;56:232–242. doi: 10.7164/antibiotics.56.232. [DOI] [PubMed] [Google Scholar]
- 87.Pucci MJ, Bronson JJ, Barrett JF, DenBleyker KL, Discotto LF, Fung-Tomc JC, Ueda Y. Antimicrobial evaluation of nocathiacins, a thiazole peptide class of antibiotics. Antimicrob. Agents Chemother. 2004;48:3697–3701. doi: 10.1128/AAC.48.10.3697-3701.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leet JE, Ax HA, Gustavson DR, Brown DM, Turner L, Brown K, Li W, Lam KS. Nocathiacin antibiotics. 2000 WO2000003722. [Google Scholar]
- 89.Jayasuriya H, Herath K, Ondeyka JG, Zhang C, Zink DL, Brower M, Gailliot FP, Greene J, Birdsall G, Venugopal J, Ushio M, Burgess B, Russotti G, Walker A, Hesse M, Seeley A, Junker B, Connors N, Salazar O, Genilloud O, Liu K, Masurekar P, Barrett JF, Singh SB. Isolation and structure elucidation of thiazomycin- a potent thiazolyl peptide antibiotic from Amycolatopsis fastidiosa. J. Antibiot. 2007;60:554–564. doi: 10.1038/ja.2007.70. [DOI] [PubMed] [Google Scholar]
- 90.Singh SB, Occi J, Jayasuriya H, Herath K, Motyl M, Dorso K, Gill C, Hickey E, Overbye KM, Barrett JF, Masurekar P. Antibacterial evaluations of thiazomycin- a potent thiazolyl peptide antibiotic from Amycolatopsis fastidiosa. J. Antibiot. 2007;60:565–571. doi: 10.1038/ja.2007.71. [DOI] [PubMed] [Google Scholar]
- 91.Zhang C, Herath K, Jayasuriya H, Ondeyka JG, Zink DL, Occi J, Birdsall G, Venugopal J, Ushio M, Burgess B, Masurekar P, Barrett JF, Singh SB. Thiazomycins, thiazolyl peptide antibiotics from Amycolatopsis fastidiosa. J. Nat. Prod. 2009;72:841–847. doi: 10.1021/np800783b. [DOI] [PubMed] [Google Scholar]
- 92.Zhang C, Zink DL, Ushio M, Burgess B, Onishi R, Masurekar P, Barrett JF, Singh SB. Isolation, structure, and antibacterial activity of thiazomycin A, a potent thiazolyl peptide antibiotic from Amycolatopsis fastidiosa. Bioorg. Med. Chem. 2008;16:8818–8823. doi: 10.1016/j.bmc.2008.08.079. [DOI] [PubMed] [Google Scholar]
- 93.Naidu BN, Sorenson ME, Bronson JJ, Pucci MJ, Clark JM, Ueda Y. Synthesis, in vitro, and in vivo antibacterial activity of nocathiacin I thiol-Michael adducts. Bioorg. Med. Chem. Lett. 2005;15:2069–2072. doi: 10.1016/j.bmcl.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 94.Naidu BN, Sorenson ME, Matiskella JD, Li W, Sausker JB, Zhang Y, Connolly TP, Lam KS, Bronson JJ, Pucci MJ, Yang H, Ueda Y. Synthesis and antibacterial activity of nocathiacin I analogues. Bioorg. Med. Chem. Lett. 2006;16:3545–3549. doi: 10.1016/j.bmcl.2006.03.079. [DOI] [PubMed] [Google Scholar]
- 95.Naidu BN, Sorenson ME, Zhang Y, Kim OK, Matiskella JD, Wichtowski JA, Connolly TP, Li W, Lam KS, Bronson JJ, Pucci MJ, Clark JM, Ueda Y. Nocathiacin I analogues: synthesis, in vitro and in vivo biological activity of novel semi-synthetic thiazolyl peptide antibiotics. Bioorg. Med. Chem. Lett. 2004;14:5573–5577. doi: 10.1016/j.bmcl.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 96.Xu L, Farthing AK, Dropinski JF, Meinke PT, McCallum C, Hickey E, Liu K. Synthesis and antibacterial activity of novel water-soluble nocathiacin analogs. Bioorg. Med. Chem. Lett. 2013;23:366–369. doi: 10.1016/j.bmcl.2012.10.065. [DOI] [PubMed] [Google Scholar]
- 97.Naidu BN, Sorenson ME, Hudyma T, Zheng X, Zhang Y, Bronson JJ, Pucci MJ, Clark JM, Ueda Y. Synthesis and antibacterial activity of O-substituted nocathiacin I derivatives. Bioorg. Med. Chem. Lett. 2004;14:3743–3746. doi: 10.1016/j.bmcl.2004.04.102. [DOI] [PubMed] [Google Scholar]
- 98.Minutolo F, Macchia M, Granchi C, Roy S, Giannaccini G, Lucacchini A. N-Hydroxyazole derivatives as lactate dehydrogenase inhibitors and their preparation and use for the treatment of diseases. 2011 WO2011054525. [Google Scholar]
- 99.Minutolo F, Macchia M, Granchi C, Di Bussolo V, Giannaccini G, Lucacchini A, Hergenrother PJ, Calvaresi EC. Preparation of indole derivatives as inhibitors of enzyme lactate dehydrogenase (LDH) 2013 WO2013092753. [Google Scholar]
- 100.Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G, Lucacchini A, Funel N, León LG, Giovannetti E, Peters GJ, Palchaudhuri R, Calvaresi EC, Hergenrother PJ, Minutolo F. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 2011;54:1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- 101.Granchi C, Roy S, De Simone A, Salvetti I, Tuccinardi T, Martinelli A, Macchia M, Lanza M, Betti L, Giannaccini G, Lucacchini A, Giovannetti E, Sciarrillo R, Peters GJ, Minutolo F. N-Hydroxyindole-based inhibitors of lactate dehydrogenase against cancer cell proliferation. Eur. J. Med. Chem. 2011;46:5398–5407. doi: 10.1016/j.ejmech.2011.08.046. [DOI] [PubMed] [Google Scholar]
- 102.Granchi C, Roy S, Del Fiandra C, Tuccinardi T, Lanza M, Betti L, Giannaccini G, Lucacchini A, Martinelli A, Macchia M, Minutolo F. Triazole-substituted N-hydroxyindol-2-carboxylates as inhibitors of isoform 5 of human lactate dehydrogenase (hLDH5) Med. Chem. Commun. 2011;2:638–643. doi: 10.1016/j.bmcl.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 103.Granchi C, Roy S, Mottinelli M, Nardini E, Campinoti F, Tuccinardi T, Lanza M, Betti L, Giannaccini G, Lucacchini A, Martinelli A, Macchia M, Minutolo F. Synthesis of sulfonamide-containing N-hydroxyindole-2-carboxylates as inhibitors of human lactate dehydrogenase-isoform 5. Bioorg. Med. Chem. Lett. 2011;21:7331–7336. doi: 10.1016/j.bmcl.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 104.Enomoto H, Kawashima K, Kudou K, Yamamoto M, Murai M, Inaba T, Ishizaka N. Preparation of novel indole derivatives having inhibitory activity on Iκb kinase β. 2008 WO2008087933. [Google Scholar]
- 105.Yamada K, Tanaka Y, Somei M. Synthesis of Nb-acyltryptamines and their 1-hydroxytryptamine derivatives as new α2-blockers heterocycles. Heterocycles. 2009;79:635–645. [Google Scholar]
- 106.Schleigh WR, Welter TR. 1-Hydroxyindole fungicides. 1992 EP470665. [Google Scholar]
- 107.Urban S, Blunt JW, Munro MHG. Coproverdine, a novel, cytotoxic marine alkaloid from a new zealand ascidian. J. Nat. Prod. 2002;65:1371–1373. doi: 10.1021/np010594z. [DOI] [PubMed] [Google Scholar]
- 108.Munro MHG, Blunt JW, Urban S, Garcia Gravalos D. Antitumor carbazoles, and coproverdine isolation. 2002 WO2002048107. [Google Scholar]
- 109.Costa EV, Pinheiro ML, Xavier CM, Silva JR, Amaral AC, Souza AD, Barison A, Campos FR, Ferreira AG, Machado GM, Leon LL. A pyrimidine-β-carboline and other alkaloids from Annona foetida with antileishmanial activity. J. Nat. Prod. 2006;69:292–294. doi: 10.1021/np050422s. [DOI] [PubMed] [Google Scholar]
- 110.Chan ST, Pearce AN, Page MJ, Kaiser M, Copp BR. Antimalarial β-carbolines from the new zealand ascidian Pseudodistoma opacum. J. Nat. Prod. 2011;74:1972–1979. doi: 10.1021/np200509g. [DOI] [PubMed] [Google Scholar]
- 111.Liermann JC, Opatz T, Kolshorn H, Antelo L, Hof C, Anke H. Omphalotins E-I, five oxidatively modified nematicidal cyclopeptides from Omphalotus olearius. Eur. J. Org. Chem. 2009;8:1256–1262. [Google Scholar]
- 112.Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Notoamides A-D: prenylated indole alkaloids isolated from a marine-derived fungus, Aspergillus sp. Angew. Chem. Int. Ed. 2007;46:2254–2256. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]
- 113.Cutrone JQ, Krampitz KD, Shu Y-Z, Chang L-P, Lowe SE. Stephacidin antitumor antibiotics. 2001 US6291461. [Google Scholar]
- 114.Qian-Cutrone J, Huang S, Shu Y-Z, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. Stephacidin A and B: two structurally novel, selective inhibitors of the testosterone-dependent prostate LNCaP cells. J. Am. Chem. Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 115.Levy SB, Alekshun MN, Podlogar BL, Ohemeng K, Verma AK, Warchol T, Bhatia B, Bowser T, Grier M. Substituted benzoimidazole compounds as transcription factor-modulating compounds useful as anti-infectives. 2004 WO2004041209. [Google Scholar]
- 116.Alekshun MN, Amoo V, Kim OK, Verma AK. Benzimidazole derivative transcription factor-modulating compounds for use as antiinfective agents. 2006 WO2006076009. [Google Scholar]
- 117.Velikorodov AV, Kovalev VB, Degtyarev OV. Monohydrochlorides and sodium salts of tautomeric 5(6)-alkoxycarbonylamino derivatives of 2-aryl-1-hydroxybenzimidazole 3-oxide with high antifungal activity and synthesis method thereof. 2011 RU2427574. [Google Scholar]
- 118.Berry JF, Ferraris DV, Duvall B, Hin N, Rais R, Alt J, Thomas AG, Rojas C, Hashimoto K, Slusher BS, Tsukamoto T. Synthesis and SAR of 1-hydroxy-1H-benzo[d]imidazol-2(3H)-ones as inhibitors of d-amino acid oxidase. ACS Med. Chem. Lett. 2012;3:839–843. doi: 10.1021/ml300212a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim OK, Garrity-Ryan LK, Bartlett VJ, Grier MC, Verma AK, Medjanis G, Donatelli JE, Macone AB, Tanaka SK, Levy SB, Alekshun MN. N-Hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J. Med. Chem. 2009;52:5626–5634. doi: 10.1021/jm9006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grier MC, Garrity-Ryan LK, Bartlett VJ, Klausner KA, Donovan PJ, Dudley C, Alekshun MN, Tanaka SK, Draper MP, Levy SB, Kim OK. N-Hydroxybenzimidazole inhibitors of ExsA MAR transcription factor in Pseudomonas aeruginosa: In vitro anti-virulence activity and metabolic stability. Bioorg. Med. Chem. Lett. 2010;20:3380–3383. doi: 10.1016/j.bmcl.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 121.Kim OK. Preparation of benzoimidazole compounds as transcription factor modulating compounds to treat infections. 2008 WO2008130368. [Google Scholar]
- 122.Dounay AB, Anderson M, Bechle BM, Campbell BM, Claffey MM, Evdokimov A, Evrard E, Fonseca KR, Gan X, Ghosh S, Hayward MM, Horner W, Kim JY, McAllister LA, Pandit J, Paradis V, Parikh VD, Reese MR, Rong S, Salafia MA, Schuyten K, Strick CA, Tuttle JB, Valentine J, Wang H, Zawadzke LE, Verhoest PR. Discovery of brain-penetrant, irreversible kynurenine aminotransferase II inhibitors for schizophrenia. ACS Med. Chem. Lett. 2012;3:187–192. doi: 10.1021/ml200204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dounay AB, Helal CJ, Tuttle JB, Verhoest PR. Preparation of quinoline compounds as KATII inhibitors for treatment of nervous system disorders and other diseases. 2012 WO2012073146. [Google Scholar]
- 124.Davis AL, Hulme KL, Wilson GT, McCord TJ. In vitro antimicrobial activity of some cyclic hydroxamic acids and related lactams. Antimicrob. Agents Chemother. 1978;13:542–544. doi: 10.1128/aac.13.3.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gunn BP, Summers JB., Jr. Preparation of quinolinone and isoquinolinone derivatives as lipoxygenase inhibitors. 1987 EP240859. [Google Scholar]
- 126.Roberts KP, Iyer RA, Prasad G, Liu LT, Lind RE, Hanna PE. Cyclic hydroxamic acid inhibitors of prostate cancer cell growth: selectivity and structure activity relationships. Prostate. 1998;34:92–99. doi: 10.1002/(sici)1097-0045(19980201)34:2<92::aid-pros3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 127.Parkes KE, Ermert P, Fässler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2003;46:1153–1164. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 128.Billamboz M, Bailly F, Lion C, Touati N, Vezin H, Calmels C, Andréola ML, Christ F, Debyser Z, Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J. Med. Chem. 2011;54:1812–1824. doi: 10.1021/jm1014692. [DOI] [PubMed] [Google Scholar]
- 129.Billamboz M, Bailly F, Lion C, Calmels C, Andréola M-L, Witvrouw M, Christ F, Debyser Z, De Luca L, Chimirri A, Cotelle P. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: influence of the alkylation of position 4. Eur. J. Med Chem. 2011;46:535–546. doi: 10.1016/j.ejmech.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 130.Bailly F, Billamboz M, Christ F, Cotelle P, Debyser Z, Lion C, Suchaud V. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones and related compounds as HIV integrase inhibitors and their preparation and use for the treatment of HIV infection and AIDS. 2012 WO2012085003. [Google Scholar]
- 131.Billamboz M, Bailly F, Barreca ML, De Luca L, Mouscadet JF, Calmels C, Andréola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J. Med. Chem. 2008;51:7717–7730. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- 132.Chen YL, Tang J, Kesler MJ, Sham YY, Vince R, Geraghty RJ, Wang Z. The design, synthesis and biological evaluations of C-6 or C-7 substituted 2-hydroxyisoquinoline-1,3-diones as inhibitors of hepatitis C virus. Bioorg. Med. Chem. 2012;20:467–479. doi: 10.1016/j.bmc.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 133.Amegadzie AK, Carey ME, Domagala JM, Huang L, Micetich RG, Singh R, Stier MA, Vaisburg A, Sanchez JP. Preparation of isoquinolones as antibacterial agents. 1998 WO9819648. [Google Scholar]
- 134.Mitaku S, Skaltsounis AL, Tillequin F, Koch M, Pusset J, Sevenet T. Plants from New Caledonia. Part 144. New alkaloids from Sarcomelicope dogniensis. Product Lett. 1995;7:219–225. [Google Scholar]
- 135.Biagini GA, Fisher N, Berry N, Stocks PA, Meunier B, Williams DP, Bonar-Law R, Bray PG, Owen A, O'Neill PM, Ward SA. Acridinediones: selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol. Pharmacol. 2008;73:1347–1355. doi: 10.1124/mol.108.045120. [DOI] [PubMed] [Google Scholar]
- 136.Raether W, Fink E. Antimalarial activity of Floxacrine (HOE 991) I. Studies on blood schizontocidal action of Floxacrine against Plasmodium berghei, P. vinckei and P. cynomolgi. Ann. Trop. Med. Parasitol. 1979;73:505–526. [PubMed] [Google Scholar]
- 137.Schmidt LH. Antimalarial properties of floxacrine, a dihydroacridinedione derivative. Antimicrob. Agents Chemother. 1979;16:475–485. doi: 10.1128/aac.16.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Raether W, Enders B, Hofmann J, Schwannecke U, Seidenath H, Hänel H, Uphoff M. Antimalarial activity of new floxacrine-related acridinedione derivatives: studies on blood schizontocidal action of potential candidates against P. berghei in mice and P. falciparum in vivo and in vitro. Parasitology Res. 1989;75:619–626. doi: 10.1007/BF00930959. [DOI] [PubMed] [Google Scholar]
- 139.Dorn A, Scovill JP, Ellis WY, Matile H, Ridley RG, Vennerstrom JL. Short report: floxacrine analog WR 243251 inhibits hematin polymerization. Am. J. Trop. Med. Hyg. 2001;65:19–20. doi: 10.4269/ajtmh.2001.65.19. [DOI] [PubMed] [Google Scholar]
- 140.Kesten SJ, Degnan MJ, Hung J, McNamara DJ, Ortwine DF, Uhlendorf SE, Werbel LM. Synthesis and antimalarial properties of 1-imino derivatives of 7-chloro-3-substituted-3,4-dihydro-1,9(2H,10H)-acridinediones and related structures. J. Med. Chem. 1992;35:3429–3447. doi: 10.1021/jm00097a001. [DOI] [PubMed] [Google Scholar]
- 141.Williams PD, Staas DD, Venkatraman S, Loughran HM, Ruzek RD, Booth TM, Lyle TA, Wai JS, Vacca JP, Feuston BP, Ecto LT, Flynn JA, DiStefano DJ, Hazuda DJ, Bahnck CM, Himmelberger AL, Dornadula G, Hrin RC, Stillmock KA, Witmer MV, Miller MD, Grobler JA. Potent and selective HIV-1 ribonuclease H inhibitors based on a 1-hydroxy-1,8-naphthyridin-2(1H)-one scaffold”. Bioorg. Med. Chem. Lett. 2010;20:6754–6757. doi: 10.1016/j.bmcl.2010.08.135. [DOI] [PubMed] [Google Scholar]
- 142.Williams PD, Venkatraman S, Langford HM, Kim B, Booth TM, Grobler JA, Staas D, Ruzek RD, Embrey MW, Wiscount CM, Lyle TA. Preparation of 1-hydroxynaphthyridin-2(1H)-one derivatives as anti-HIV agents. 2008 WO2008010964. [Google Scholar]
- 143.Claffey MM, Dounay AB, Gan X, Hayward MM, Rong S, Tuttle JB, Verhoest PR. Preparation of bicyclic and tricyclic compounds as KAT II inhibitors for treating cognitive and other disorders. 2010 WO2010146488. [Google Scholar]
- 144.Colotta V, Catarzi D, Varano F, Calabri FR, Filacchioni G, Costagli C, Galli A. 3-Hydroxy-quinazoline-2,4-dione as a useful scaffold to obtain selective Gly/NMDA and AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 2004;14:2345–2349. doi: 10.1016/j.bmcl.2004.01.109. [DOI] [PubMed] [Google Scholar]
- 145.Tumey LN, Bom D, Huck B, Gleason E, Wang J, Silver D, Brunden K, Boozer S, Rundlett S, Sherf B, Murphy S, Dent T, Leventhal C, Bailey A, Harrington J, Bennani YL. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg. Med. Chem. Lett. 2005;15:277–281. doi: 10.1016/j.bmcl.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 146.Hani R, Berkowitz PT, Peterson JL. N-hydroxyquinazolinone and 2-N-hydroxythiourea benzoate compounds as biocides. 1993 WO9306830. [Google Scholar]
- 147.Petersen LC, Olsen OH, Richter SL, Jakobsen P. Factor VII-binding reagent. 1997 WO9749684. [Google Scholar]
- 148.Zhang X, Habib FK, Ross M, Burger U, Lewenstein A, Rose K, Jaton JC. Isolation and characterization of a cyclic hydroxamic acid from a pollen extract, which inhibits cancerous cell growth in vitro. J. Med. Chem. 1995;38:735–738. doi: 10.1021/jm00004a019. [DOI] [PubMed] [Google Scholar]
- 149.Buchmann CA, Nersesyan A, Kopp B, Schauberger D, Darroudi F, Grummt T, Krupitza G, Kundi M, Schulte-Hermann R, Knasmueller S. Dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIMBOA) and 2,4-dihydroxy-1,4-benzoxazin-3-one (DIBOA), two naturally occurring benzoxazinones contained in sprouts of Gramineae are potent aneugens in human-derived liver cells (HepG2) Cancer Lett. 2007;246:290–299. doi: 10.1016/j.canlet.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 150.Macias Dominguez FA, Gonzalez Molinillo JM, Varela Montoya RM, Arroyo Garcia E, Marin Mateos D, Chinchilla Salcedo N. Halogenated derivatives of 2H-1,4-benzoxazin-3(4H)-one with C-2 alkyl chains or N-4 acyl residues as herbicides useful for weed control. 2010 WO2010133724. [Google Scholar]
- 151.Macias Dominguez FA, Gonzalez Molinillo JM, Varela Montoya RM, Chinchilla Salcedo N, De Siqueira JM, Marin Mateos D. Phytotoxic halogenated derivatives of benzoxazinones, especially of 4-hydroxy-2H-1,4-benzoxazin-3(4H)-one. 2008 WO2008012385. [Google Scholar]
- 152.Macias Dominguez FA, Gonzalez Molinillo JM, Marin Mateos D, Varela Montoya RM, Chinchilla Salcedo N, Arroyo Garcia E. Derivatives of 2-alkyl-2H-1,4-benzoxazin-3(4H)-one and 2-alkoxycarbonyl-2H-1,4-benzoxazin-3(4H)-one, with phytotoxic activity, and their preparation and use as herbicides. 2009 WO2009010606. [Google Scholar]
- 153.Antunes EM, Beukes DR, Kelly M, Samaai T, Barrows LR, Marshall KM, Sincich C, Davies-Coleman MT. Cytotoxic pyrroloiminoquinones from four new species of South African latrunculid sponges. J. Nat. Prod. 2004;67:1268–1276. doi: 10.1021/np034084b. [DOI] [PubMed] [Google Scholar]
- 154.Lee TC, Teller MN, Budinger JM, Klötzer W, Brown GB. Chemical reactivities and oncogenicities of a series of N-hydroxyheterocycles. Chem. Biol. Interact. 1979;25:369–372. doi: 10.1016/0009-2797(79)90059-0. [DOI] [PubMed] [Google Scholar]
- 155.Teller MN, Giner-Sorolla A, Stöhrer G, Budinger JM, Brown GB. Oncogenicity of purine 3-oxide and unsubstituted purine in rats. Cancer Res. 1978;38:2229–2232. [PubMed] [Google Scholar]
- 156.Nicolaou KC, Lee SH, Estrada AA, Zak M. Construction of substituted N-hydroxyindoles: synthesis of a nocathiacin I model system. Angew. Chem. Int. Ed. Engl. 2005;44:3736–3740. doi: 10.1002/anie.200500724. [DOI] [PubMed] [Google Scholar]
- 157.Nicolaou KC, Estrada AA, Lee SH, Freestone GC. Synthesis of highly substituted N-hydroxyindoles through 1,5-addition of carbon nucleophiles to in situ generated unsaturated nitrones. Angew. Chem. Int. Ed. Engl. 2006;45:5364–5368. doi: 10.1002/anie.200601808. [DOI] [PubMed] [Google Scholar]
- 158.Nicolaou KC, Estrada AA, Freestone GC, Lee SH, Alvarez-Mico X. New synthetic technology for the construction of N-hydroxyindoles and synthesis of nocathiacin I model systems. Tetrahedron. 2007;63:6088–6114. doi: 10.1016/j.tet.2007.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Somei M. 1-Hydroxyindoles. Heterocycles. 1999;50:1157–1211. [Google Scholar]