

Abstract
Enzymes can catalyze various reactions with high selectivity and are involved in many important biological processes. However, the general instability of enzymes against high temperature often limits their application. To address this, we synthesized a trehalose-based hydrogel in two steps from commercial starting materials with minimal purification procedures. Mono- and multi-functional trehalose monomers were cross-linked by redox-initiated radical polymerization to form a hydrogel. Phytase, an important enzyme utilized in animal feedstock, was employed to study the effectiveness of the trehalose hydrogel to stabilize proteins against heat. Addition of the phytase solution to the hydrogel resulted in enzyme internalization as confirmed by confocal microscopy. The phytase in the hydrogel retained 100% activity upon heating at 90 °C compared to 39% when the hydrogel was absent. The enzyme could also be recovered from the hydrogel. The trehalose hydrogel synthesis reported herein should be readily scalable for thermal stabilization of a wide variety of enzymes.
Introduction
Enzymes have well-defined three-dimensional structures formed by multiple noncovalent interactions such as hydrogen bonds, salt bridges, and hydrophobic interactions.1 At high temperatures, enzymes lose their original structure and denature to form insoluble aggregates that are no longer active.1-3 Because of their high efficiency and selectivity in catalyzing biological processes, enzymes are used for numerous industrial purposes.2, 4-6 However, this thermal instability of the proteins has negative impact on their applications in the pharmaceutical, food, and biotechnology industries. Many techniques such as chemical modification7, 8 and protein engineering9-12 have been developed to address this problem. Additionally, polymers have been used as conjugates or excipients to enhance thermostability of enzymes.13-17 Yet some of these approaches are too expensive for certain industrial and agricultural applications.
For industrial applications, polymeric hydrogels are especially attractive materials for enzyme stabilization. Enzyme immobilization by hydrogels has been extensively studied in the context of industrial enzyme stabilization, especially to organic solvents.18 Enzymes can be loaded onto hydrogels without the need of a conjugation reaction, which simplifies the synthesis and stabilization process. And unlike polymer excipients that are difficult to remove from the enzyme solution, the macroscopic hydrogels can be easily separated by filtration or centrifugation. Due to these advantages, hydrogels have been frequently used for stabilization of enzymes as well as other proteins.19-21 Herein, we propose a novel hydrogel system based on trehalose as an effective excipient for enhancing the stability of enzymes at elevated temperatures.
Trehalose is a non-reducing disaccharide that has been shown to stabilize proteins and cells against stresses such as heat,22-24 desiccation,25-27 and freezing.28-30 Some animals accumulate trehalose to significant levels in response to environmental stresses,31, 32 emphasizing the ability of trehalose to stabilize biological molecules. Moreover, trehalose is generally regarded as safe (GRAS)33 and is used in several pharmaceutical drugs as stabilizers.34 Our group has previously utilized trehalose-based linear polymers as excipients35 or conjugates36 to stabilize proteins and retain their activity against heat and lyophilization. We sought to develop trehalose-based material to stabilize enzymes against heat and focused on hydrogels for the advantages described above.
We chose to study stabilization of phytase because of its importance in the animal feed industry. Phytase is a phosphohydrolytic enzyme that catalyzes the conversion of phosphate in indigestible phytic acid to a highly digestible form.37-40 The conversion of phytic acid is essential for simple-stomached species such as swine, poultry, and fish to utilize this storage form of phosphate present in common feed grains such as corn, soy, and wheat.37 In 2011, phytase accounted for approximately 60% of the $550 million global feed enzyme market.41 Yet, the biggest challenge in the use of phytase in animal feeds is its low thermostability during steam heating of the pelleting process, during which the temperature between 70-90 °C is reached.37, 42 Despite previous efforts to enhance its heat stability,37, 42-44 a simple and cost-effective method is still of great interest. As described below, we have found that phytase retains 100% activity when heated to 90 °C in the presence of trehalose hydrogels.
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich and Fisher Scientific and were used without purification unless noted otherwise. All solvents for liquid chromatography mass spectrometry (LCMS) were purchased from VWR or Fisher Scientific in LCMS grade. Trehalose was purchased from The Healthy Essential Management Corporation (Houston, TX), and was azeotropically dried with ethanol and kept under vacuum until use. Phytase was provided by Phytex, LLC.
Analytical techniques
NMR spectra were recorded on a Bruker DRX 500 MHz spectrometer. LCMS experiments were carried out on a Waters Acquity UPLC connected to a Waters LCT-Premier XE Time of Flight Instrument controlled by MassLynx 4.1 software. The mass spectrometer was equipped with a Multi-Mode Source operated in the electrospray mode. Trehalose samples were separated using an Acquity BEH C18 1.7 um column (2.1 × 50 mm) and were eluted with a gradient of 5 – 50% solvent B over 6 min (solvent A: water, solvent B: acetonitrile, both with 0.2% formic acid (vol/vol)). Mass spectra were recorded in the negative ion mode in the m/z range of 70–2000 with leucine enkephalin (Sigma L9133) as the lock mass standard. Preparatory reverse phase HPLC was carried out on a Shimadzu HPLC system equipped with a UV detector using a Luna 5 μm C18 100A column (preparatory: 5 μm, 250 × 21.2 mm) with monitoring at λ = 215 nm and 254 nm. A linear gradient solvent system (H2O : methanol = 70:30 to 50:50) was used as the mobile phase at a flow rate of 10 mL/min. Scanning electron microscopy (SEM) images were acquired on a FEI Nova Nano 230 SEM in the UCLA Molecular and Nano Archaeology (MNA) facility under a low vacuum of 50 Pa and high voltage of 5 or 2.5 kV with a spot size of 3.0. Fluorescence images of the hydrogels were acquired using a confocal laser scanning microscope (Leica SP2 1P-FCS, Leica) at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA. Diameter of phytase (PDB: 1DKL)45 was measured using Swiss-PdbViewer (Swiss Institute of Bioinformatics).46 Fluorescence measurements were made on a FlexStation II (Molecular Devices). Light absorbance for phytase activity assay was measured using a Biotek EPOCH microtiter plate reader.
One pot synthesis of trehalose monomers and cross-linkers
The one pot reaction for the monomers and cross-linkers was performed by modifying a previously reported literature procedure.35 Sodium hydroxide (NaOH, 4.44 g, 1.11×10-1 mol) was added to dimethyl sulfoxide (DMSO, 96 mL). After stirring for 5 min, trehalose (4.86 g, 1.42×10-2 mol) was added to the reaction. After all the trehalose was dissolved, 4-vinylbenzyl chloride (0.4 mL, 2.84×10-3 mol) was slowly added to the reaction and was stirred for 24 h at 25 °C. The crude product was then precipitated into 2 L of DCM to remove highly modified trehalose. The resulting solid was dried in vacuo and used for gelation without further purification.
Preparation of phytase-loaded trehalose hydrogel
The crude mixture (3.23 g) from the previous Williamson etherification was dissolved in H2O (3.23 mL) and then treated with tetramethylethylenediamine (TEMED, 16 μL, 1.07×10-4 mol). Next, 807 μL of 10 mg/mL aqueous stock solution of ammonium persulfate (APS, 8.07 mg, 3.54×10-5 mol) was added to initiate the gelation. The solution started gelling within 10 min at 25 °C. LCMS was used to quantify the extent of conversion, by comparing the relative amount of mono-substituted trehalose compared to unmodified trehalose before and after gelation. LCMS analysis showed that all cross-linkers had reacted after 24 h. After the gelation, the gel was washed with a Soxhlet extractor for 3 days with H2O to remove unreacted monomers. The hydrogel was lyophilized and then grinded into fine powder. 10 μL of phytase solutions of different concentrations were added to each dried gel to make phytase : hydrogel ratios of 1:1, 1:10, and 1:40 weight equivalents. The gels were incubated at 4 °C with the phytase solution for 24 h and lyophilized to yield a white powder for testing in the heat burden study.
Fluorescein isothiocyanate (FITC) labeling of phytase
Phytase (2 mg, 3.57×10-2 μmol) and FITC (0.3 mg, 7.71×10-1 μmol) were dissolved in 50 mM borate buffer, pH 8.5 (1 mL). The mixture was magnetically stirred at room temperature for an hour. Excess FITC was removed by repeated centrifugation through a 3,000 Da MWCO membrane using 0.5 mL centrifugal filtration tubes until no FITC was detected by UV-Vis in filtrate. Degree of labeling was 0.28 FITC per phytase as determined by UV absorbance.47
Release of phytase from trehalose hydrogel
FITC-labeled phytase (74 mg/mL) in 0.1 M sodium acetate buffer (pH 5.0, 10 μL) was added to 4 mg of trehalose hydrogel. The mixture was incubated at 4 °C for 24 h, and then lyophilized. To the gel was added 1000 μL buffer to initiate the passive diffusion of the phytase from the hydrogel. Aliquots (200 μL) were taken at respective time points and the samples were immediately replenished with fresh buffer. The concentrations of the time point aliquots were calculated from the fluorescence measured on a spectrofluorometer using a FITC-labeled phytase calibration curve.
Heat burden studies of phytase
To the dried hydrogel and phytase mixture, 53 wt % of H2O with respect to the phytase was added. The hydrogel was incubated at 4 °C for 24 h with gentle rocking to evenly distribute the solution. The hydrogel was then heated at 90 °C for 1 min, and diluted with 0.1 M sodium acetate buffer, pH 5, and incubated for at least 24 h prior to the activity assay.
Phytase activity assay
The control and heat treated hydrogels (10 uL) were first diluted in 10 mL of 0.2 M sodium citrate pH 5.5 buffer, and 0.5 mL aliquots of diluted sample were transferred to each of four reaction tubes (1 blank and 3 sample). To all sample tubes, 0.5 mL of 1% phytic acid solution (0.2 M sodium citrate buffer, pH 5.5) was added and the tubes were incubated at 37 °C for 15 minutes. The reactions were then quenched by the addition of 1.0 ml of 15% trichloroacetic acid, and 0.5 mL of phytic acid was added to the blank tubes. Samples (30 uL) were diluted ten-fold wih distilled water, and the diluted solutions (150 uL) were treated with 150 uL of 1:3:1 solution of 2.5% ammonium molybdate : 10% sulfuric acid : 10% ascorbic acid in a microtiter plate. The plate was incubated in a 50 °C water bath for 15 minutes, cooled at 4 °C for 15 minutes, and the 820 nm absorbance of individual wells were measured. Phytase activity (FTU) is defined as the amount of enzyme that catalyzes the release of 1.0 micromole of inorganic phosphate per minute from 1% phytic acid in pH 5.5 buffer at 37 °C.
Statistical analysis
One-tailed Student's t-test assuming unequal sample variance was used. Results were considered significantly different if p < 0.05.
Results and Discussion
Straightforward synthesis, commercially available starting materials, and simple purification steps are some of the most important factors in industrial-scale reactions.48 Thus, the hydrogel was synthesized in only two steps. First, Williamson etherification using 4-vinylbenzyl chloride and trehalose yielded a crude product mixture that was subsequently precipitated into DCM. The DCM wash contained mostly DMSO and some trehalose and mono- and di-substituted products, while the precipitate that was used for gelation consisted of unmodified trehalose and vinyl-substituted products (79 % mono-substituted, 16 % di-substituted, and 5 % tri-substituted) as measured by HPLC and LCMS (Figures S1 and S2 and Table S1). We envisioned that the multi-substituted products of the crude monomer reaction mixture could be used as cross-linkers to synthesize a trehalose-based hydrogel directly from the crude reaction mixture (Scheme 1). Due to the presence of cross-linkers, polymerization would yield a hydrogel rather than a linear polymer.
Scheme 1. Two-step synthesis of trehalose hydrogel.
The crude mixture was then polymerized by radical polymerization using a redox initiator pair, APS and TEMED. The crude mixture was dissolved in water with TEMED (Figure 1a). After the addition of APS, the solution started gelling within 10 min at 25 °C (Figure 1b). The resulting hydrogel had the same yellow color as the crude mixture. The hydrogel network remained intact after lyophilization and rehydration (Figure 1c and d). After 1 day, all of the di- and tri-substituted trehalose had reacted (Figure S3). The crude gel was washed with a Soxhlet extractor for 3 days to remove unreacted monomers, residual initiator and trehalose, yielding a colorless hydrogel. The purified trehalose hydrogel was grounded into a powder with a mortar and pestle for ease of handling and to increase the surface area for internalization of phytase (Figure 1e).
Figure 1.

(a) Trehalose-based monomer, cross-linker, and TEMED dissolved in water, (b) after adding APS, (c) after lyophilizing the hydrogel, (d) rehydration of lyophilized hydrogel, and (e) suspended in water after washing the hydrogel (grounded into fine powder before washing, and then rehydrated).
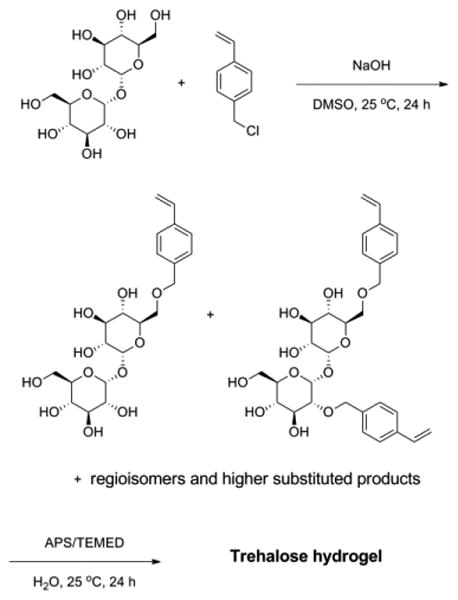
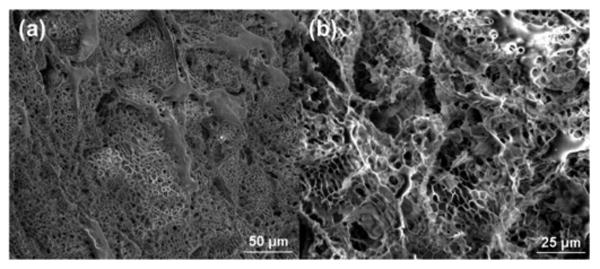
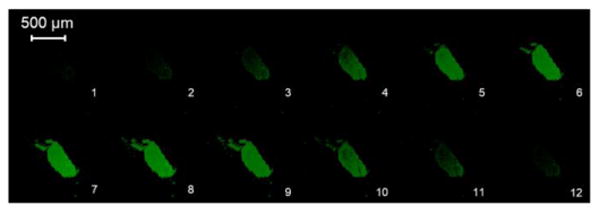
The purified hydrogel was characterized by a variable pressure SEM, as shown in Figure 2. The images revealed hydrogel architecture with micron-sized pores. Since phytase diameter is approximately 11.1 nm along the major axis as measured from the crystal structure (PDB: 1DKL),49 phytase was thus expected to be incorporated within the hydrogel. To test this hypothesis, we observed the hydrogel under a confocal microscope after incubation in fluorescein isothiocyanate (FITC)-labeled phytase solution followed by a brief wash in water (Figure 3). An even distribution of the fluorophore throughout the gel matrix demonstrated that the phytase was fully internalized into the hydrogel and not simply adsorbed on the hydrogel surface. Because of the pore size, we anticipated that the enzyme would be released from the hydrogel when diluted with water. Indeed, the release profile of FITC-labeled phytase from the hydrogel after lyophilization showed that 78% of the phytase was released in 6 hours (Figure 4). The release profile was similar to gel that has not been lyophilized (Figure S4). The results provide further evidence that the phytase is internalized inside the hydrogel and also demonstrate that the gel can be used to recover enzyme after loading.
Figure 2.
SEM images of trehalose hydrogel. (a) Images at 500× magnification and (b) at 1000× magnification.
Figure 3.
Confocal images of trehalose hydrogel incubated overnight in a solution containing FITC-labeled phytase and washed with deionized water. Numbers in the lower right corner indicate transaxial slice indices. Axial resolution = 2 μm.
Figure 4.
Release profile of FITC-labeled phytase from trehalose hydrogel after loading and lyophilization (n=6).
Currently in the animal feed industry, pelleting is the most common process for preparing animal feeds since it improves their efficiency and reduces nutrient excretion compared to mashed forms.39, 50 Typically temperatures reach 70 - 90 °C for a few minutes during pelleting. For phytase in particular, the dry ingredients including phytase are mixed in a pelleting mill conditioner, reaching a temperature of 80 - 90 °C for 35 – 45 sec, followed by extrusion to produce the desired pellets. Thus, phytase was loaded into the hydrogel and heated in a condition simulating the steam pelleting process (90 °C, 1 min). The phytase solution was added to three different weight equivalents (1, 10, and 40) of lyophilized trehalose hydrogel and incubated for 24 h. The sample was lyophilized again, 53 wt % of water was added to the phytase-loaded trehalose hydrogel, and the gel was incubated for another 24 h to replicate the moisture level of the steam heating process. The water is essential for the pelleting process, but it also expedites denaturation of phytase under the extreme heating.37, 42 The results showed that phytase heated in the presence of the hydrogel retained significantly higher activity for all weight equivalents tested. Even when only 1 weight equivalent of hydrogel was used, 81% activity was retained compared to the control that had not been heated, which was only 39% active, and 10 and 40 wt eq retained 100% enzyme activity (Figure 5). The average activity indicated that 10 weight equivalent of hydrogel to phytase was the optimal amount to completely retain the original phytase activity, while utilizing the minimal amount of hydrogel.
Figure 5.
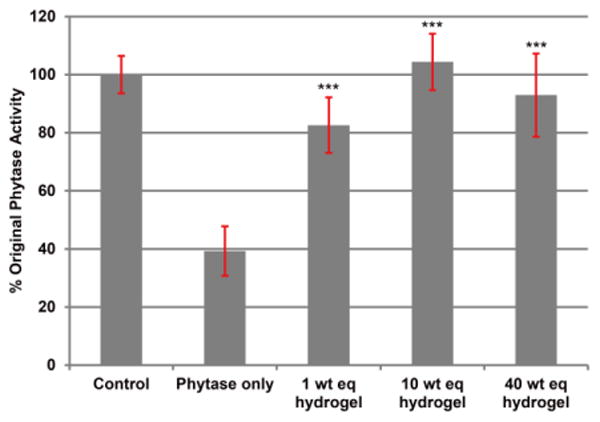
Activity of phytase after heating with different weight equivalents of trehalose hydrogel. All the samples except the control were heated for 1 min at 90 °C with 53 wt % of water (n=3). *** = p < 0.005 relative to phytase only.
The results demonstrate that the trehalose hydrogel can stabilize phytase against extreme heat conditions. The trehalose hydrogel may be suitable for industrial-scale applications as the synthesis only requires two steps and involves minimal purification that can be easily adapted to a large scale. Specifically, the proposed method uses chromatography-free purification, easily accessible starting materials, protecting group-free chemistry, and a minimal number of steps.48
Another advantage of hydrogel formulation is its ease of removal. The release results demonstrate that the protein of interest can be removed from the hydrogel. The release occurred over several hours with ∼80% release at 6 hours. However, this is with passive diffusion. One can anticipate that by rinsing or pushing water through the system, or with the agitation that occurs in the gastrointestinal track in the case of phytase-loaded hydrogel, the enzyme would be released faster. Since the hydrogel is not soluble in water or organic solvents, it can be separated from the mixture by simple filtration or centrifugation. This is a potential advantage of the system since the hydrogel could be added and then removed from the protein after stress if so desired.
In addition, despite much research on the genetic engineering of enzymes for improving their thermal stability, multiple optimization iterations or enzyme-specific mutation strategies are usually required, accompanied with a higher cost.51 Thus, the strategy described herein may be more flexible and cost effective than genetic engineering techniques. Since our group has already demonstrated that linear trehalose polymers stabilize various proteins against heating,35, 36 the trehalose-based hydrogel hereby described may be readily applicable to thermal stabilization of a wide variety of industrially important enzymes and proteins.
Conclusions
We have detailed the synthesis of a trehalose hydrogel for thermal stabilization of phytase as a model enzyme. This hydrogel can be prepared via simple synthesis and purification steps, which are important considerations in industrial processes. The resulting trehalose hydrogel fully preserved the activity of phytase under temperatures relevant in the pelleting procedure for animal feed preparation. Currently, many enzymes in animal feeds lose the majority of their activity during this steam pelleting process. As demonstrated by the stabilization of phytase in this report, the trehalose hydrogel is a promising material for stabilizing various enzymes and proteins against high-temperature processes.
Supplementary Material
Acknowledgments
This work was funded by the National Science Foundation (CHE-1112550) and a research grant from Phytex, LLC. (20131391). J.L. thanks the NIH Biotechnology Training Fellowship (5T32 GM675557) for funding. E.-W.L. thanks the 2014 Cram Fellowship for funding. The authors thank Dr. Greg Khitrov (UCLA Molecular Instrumentation Center) for assistance with the MS experiments. The LCMS was supported by the National Center for Research Resources (grant number S10RR025631). Confocal laser scanning microscope was supported by NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and a NSF Major Research Instrumentation grant (CHE-0722519).
Footnotes
Conflict of interest: This study was partially funded by Phytex, LLC.
Electronic Supplementary Information (ESI) available: LCMS chromatogram, hydrated gel release profile, and mass spectrometry data. See DOI: 10.1039/b000000x/
Notes and references
- 1.Somero GN. Annu Rev Physiol. 1995;57:43–68. doi: 10.1146/annurev.ph.57.030195.000355. [DOI] [PubMed] [Google Scholar]
- 2.Rader AJ, Hespenheide BM, Kuhn LA, Thorpe MF. Proc Natl Acad Sci U S A. 2002;99:3540–3545. doi: 10.1073/pnas.062492699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fágáin CÓ. BBA-Protein Struct M. 1995;1252:1–14. doi: 10.1016/0167-4838(95)00133-f. [DOI] [PubMed] [Google Scholar]
- 4.Ravindran V, Son JH. Recent Pat Food Nutr Agric. 2011;3:102–109. doi: 10.2174/2212798411103020102. [DOI] [PubMed] [Google Scholar]
- 5.Samejima H, Kimura K, Ado Y. Biochimie. 1980;62:299–315. doi: 10.1016/s0300-9084(80)80159-3. [DOI] [PubMed] [Google Scholar]
- 6.Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Nature. 2001;409:258–268. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- 7.DeSantis G, Jones JB. Curr Opin Biotechnol. 1999;10:324–330. doi: 10.1016/S0958-1669(99)80059-7. [DOI] [PubMed] [Google Scholar]
- 8.Ryan O, Smyth MR, Fagain CO. Enzyme Microb Tech. 1994;16:501–505. doi: 10.1016/0141-0229(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 9.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJH, Denheijer M, Kluijtmans LAJ, Vandenheuvel LP, Rozen R. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 10.Matthews BW, Nicholson H, Becktel WJ. Proc Natl Acad Sci U S A. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar S, Tsai CJ, Nussinov R. Protein Eng. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- 12.Imanaka T, Shibazaki M, Takagi M. Nature. 1986;324:695–697. doi: 10.1038/324695a0. [DOI] [PubMed] [Google Scholar]
- 13.Gaertner HF, Puigserver AJ. Enzyme Microb Tech. 1992;14:150–155. doi: 10.1016/0141-0229(92)90174-m. [DOI] [PubMed] [Google Scholar]
- 14.Longo MA, Combes D. J Chem Technol Biot. 1999;74:25–32. [Google Scholar]
- 15.Yang Z, Domach M, Auger R, Yang FX, Russell AJ. Enzyme Microb Tech. 1996;18:82–89. [Google Scholar]
- 16.Kazan D, Erarslan A. Appl Biochem Biotech. 1997;62:1–13. doi: 10.1007/BF02787979. [DOI] [PubMed] [Google Scholar]
- 17.Tomita S, Nagasaki Y, Shiraki K. Biotechnol Bioeng. 2012;109:2543–2552. doi: 10.1002/bit.24531. [DOI] [PubMed] [Google Scholar]
- 18.Sheldon RA. Adv Synth Catal. 2007;349:1289–1307. [Google Scholar]
- 19.Leobandung W, Ichikawa H, Fukumori Y, Peppas NA. J Controlled Release. 2002;80:357–363. doi: 10.1016/s0168-3659(02)00028-7. [DOI] [PubMed] [Google Scholar]
- 20.Akiyoshi K, Sasaki Y, Sunamoto J. Bioconjug Chem. 1999;10:321–324. doi: 10.1021/bc9801272. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Yang Z, Gao Y, Ge W, Wang L, Xu B. Soft Matter. 2008;4:550–553. doi: 10.1039/b715439a. [DOI] [PubMed] [Google Scholar]
- 22.Lippert K, Galinski E. Appl Microbiol Biotechnol. 1992;37:61–65. [Google Scholar]
- 23.Kaushik JK, Bhat R. J Biol Chem. 2003;278:26458–26465. doi: 10.1074/jbc.M300815200. [DOI] [PubMed] [Google Scholar]
- 24.Baptista RP, Pedersen S, Cabrita GJ, Otzen DE, Cabral JM, Melo EP. Biopolymers. 2008;89:538–547. doi: 10.1002/bip.20926. [DOI] [PubMed] [Google Scholar]
- 25.Guo N, Puhlev I, Brown DR, Mansbridge J, Levine F. Nat Biotechnol. 2000;18:168–171. doi: 10.1038/72616. [DOI] [PubMed] [Google Scholar]
- 26.Hengherr S, Heyer AG, Kohler HR, Schill RO. FEBS J. 2008;275:281–288. doi: 10.1111/j.1742-4658.2007.06198.x. [DOI] [PubMed] [Google Scholar]
- 27.Crowe JH, Crowe LM, Chapman D. Science. 1984;223:701–703. doi: 10.1126/science.223.4637.701. [DOI] [PubMed] [Google Scholar]
- 28.Beattie GM, Crowe JH, Lopez AD, Cirulli V, Ricordi C, Hayek A. Diabetes. 1997;46:519–523. doi: 10.2337/diab.46.3.519. [DOI] [PubMed] [Google Scholar]
- 29.Sundaramurthi P, Suryanarayanan R. J Phys Chem Lett. 2009;1:510–514. [Google Scholar]
- 30.Duong T, Barrangou R, Russell WM, Klaenhammer TR. Appl Environ Microbiol. 2006;72:1218–1225. doi: 10.1128/AEM.72.2.1218-1225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westh P, Ramløv H. J Exp Zool. 1991;258:303–311. [Google Scholar]
- 32.Madin KAC, Crowe JH. J Exp Zool. 1975;193:335–342. [Google Scholar]
- 33.Jain NK, Roy I. Protein Sci. 2009;18:24–36. doi: 10.1002/pro.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohtake S, Wang YJ. J Pharm Sci. 2011;100:2020–2053. doi: 10.1002/jps.22458. [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Lin EW, Lau UY, Hedrick JL, Bat E, Maynard HD. Biomacromolecules. 2013;14:2561–2569. doi: 10.1021/bm4003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mancini RJ, Lee J, Maynard HD. J Am Chem Soc. 2012;134:8474–8479. doi: 10.1021/ja2120234. [DOI] [PubMed] [Google Scholar]
- 37.Lei XG, Weaver JD, Mullaney E, Ullah AH, Azain MJ. Annu Rev Anim Biosci. 2013;1:283–309. doi: 10.1146/annurev-animal-031412-103717. [DOI] [PubMed] [Google Scholar]
- 38.Kuhn I, Partanen K. J Anim Sci. 2012;90:194–196. doi: 10.2527/jas.53902. [DOI] [PubMed] [Google Scholar]
- 39.Nahm KH. Crit Rev Env Sci Technol. 2002;32:1–16. [Google Scholar]
- 40.Silversides FG, Scott TA, Bedford MR. Poult Sci. 2004;83:985–989. doi: 10.1093/ps/83.6.985. [DOI] [PubMed] [Google Scholar]
- 41.Adeola O, Cowieson AJ. J Anim Sci. 2011;89:3189–3218. doi: 10.2527/jas.2010-3715. [DOI] [PubMed] [Google Scholar]
- 42.Slominski BA, Davie T, Nyachoti MC, Jones O. Livestock Sci. 2007;109:244–246. [Google Scholar]
- 43.Hughes KP, Soares JH., Jr Aquacult Nutr. 1998;4:133–140. [Google Scholar]
- 44.Cao L, Wang W, Yang C, Yang Y, Diana J, Yakupitiyage A, Luo Z, Li D. Enzyme Microb Technol. 2007;40:497–507. [Google Scholar]
- 45.Lim D, Golovan S, Forsberg CW, Jia Z. Nat Struct Biol. 2000;7:108–113. doi: 10.1038/72371. [DOI] [PubMed] [Google Scholar]
- 46.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 47.Schreiber AB, Haimovich J. Methods Enzymol. 1983;93:147–155. doi: 10.1016/s0076-6879(83)93039-2. [DOI] [PubMed] [Google Scholar]
- 48.Kuttruff CA, Eastgate MD, Baran PS. Nat Prod Rep. 2014;31:419–432. doi: 10.1039/c3np70090a. [DOI] [PubMed] [Google Scholar]
- 49.Oakley AJ. Biochem Biophys Res Commun. 2010;397:745–749. doi: 10.1016/j.bbrc.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 50.Thomas M, Van der Poel A. Anim Feed Sci Tech. 1996;61:89–112. [Google Scholar]
- 51.Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD. Science. 2007;315:804–807. doi: 10.1126/science.1137016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.