

Abstract
Ag receptor stimulation of preactivated T cells causes rapid cell death in an IL-2– and Fas-dependent manner. This phenomenon, known as activation-induced cell death (AICD), plays a pivotal role in the removal of Ag-reactive T cells after initial expansion. In this study, we report a novel form of T cell apoptosis that is distinct from classic AICD. When peripheral T cells were activated with anti-CD3 and anti-CD28 Abs precoated onto plastic plates, CD4+CD25− and CD8 T cells initially expanded but underwent massive apoptosis after 4 d. Unlike classic AICD, this type of T cell apoptosis pathway requires engagement of CD28 and expression of p53, a tumor-suppressor gene. The most striking feature of this form of apoptosis was regulatory T cell resistance. Under the same stimulating conditions, CD4+CD25+ T cells grew continuously beyond 4 d. Consequently, when the entire CD4 population was cultured with plate-bound anti-CD3 plus anti-CD28 Ab, CD4+CD25+FoxP3+ regulatory T cells outgrew non-regulatory T cells and expanded >7000-fold after 11 d. The data presented herein demonstrate a novel process of Ag-induced T cell death by sustained TCR and CD28 engagement and represent a simple and efficient procedure for the expansion of regulatory T cells in vitro.
Peripheral T cells consist of several functionally distinct subpopulations (e.g., effector, regulatory, and memory T cells), and the distribution of these various subgroups plays a significant role in the control of immune responses. In particular, regulatory T cells (Tregs) have been intensely studied because of their important contribution to the control of immunological self-tolerance and the establishment of chronic infection by agents such as Leishmania major, HIV, and Mycobacterium tuberculosis (1, 2). These cells are characterized by the expression of the transcription factor FoxP3. On the cell surface, a majority of Tregs express CD4 and CD25. Under normal conditions, Tregs are present in the periphery and constitute ∼2–5% of the total peripheral T lymphocytes. FoxP3+ Tregs develop in the thymus (naturally arising Tregs [nTregs]) and in the periphery (inducible Tregs). Unlike thymic negative selection of self-Ag–reactive T cells, Tregs impose their immunosuppressive effect in a dominant manner on effector T cell activation (2). Tregs exert their suppressive effect via several mechanisms, including secretion of immunosuppressive cytokines (e.g., IL-10 and -35), alteration or elimination of APCs, and sequestration of cytokines (3–6). The presence of Tregs is essential for maintenance of immunological self-tolerance, because the loss of Tregs due to a mutation in FoxP3 causes lethal autoimmune disorders in humans and mice (7).
Activation-induced cell death (AICD) of T cells is also critical to immune regulation and is considered a major mechanism that controls the size of activated T cell populations after initial expansion (8). The mechanism of AICD has been studied extensively (9), and caspase activation and Fas (CD95) expression were shown to be required for AICD. IL-2 was also shown to be critical for AICD (10). IL-2R–mediated STAT5 activation induces the upregulation of Fas ligand (FasL) on the surface of T cells, leading to Fas-mediated cell death (11).
In addition to IL-2, the ligation of cell surface Ags can modulate AICD. For example, cross-linking of CD4 by anti-CD4 mAb or gp120 of the HIV envelope protein was shown to prime apoptosis of CD4+ T cells triggered by anti-CD3 stimulation (12, 13). Engagement of the costimulatory molecule CTLA-4 was shown to induce apoptosis (14). Although engagement of CD28 is known to enhance the production of IL-2, it was also shown to provide a survival signal for this form of AICD, rather than promoting apoptosis (15). This effect of CD28 is presumably due to enhanced Bcl-xL expression (16).
The previously studied form of AICD was demonstrated to be independent of p53, a proapoptotic tumor-suppressor gene (17). Activation of T cells from p53-deficient mice revealed that these T cells are equally sensitive to AICD. Although it is difficult to analyze autoimmunity in p53-deficient mice because of their high incidence of tumor development at an early age, p53-deficient mice showed exacerbated development of experimental autoimmune arthritis (18), experimental autoimmune encephalomyelitis (19), and streptozotocin-induced diabetes (20). Moreover, known downstream targets of p53, such as Bim, p21, and GADD45a, were shown to play critical roles in the maintenance of immunological tolerance, suggesting that p53 may also play a role in immune regulation (21–24).
In this study, we report a novel form of Ag receptor signal-induced apoptosis in CD4+CD25− and CD8+ T cells. This form of cell death is dependent on the engagement of CD28 and requires p53 and Fas expression in T cells. Most importantly, FoxP3+ nTregs are resistant to this form of apoptosis and undergo robust expansion after stimulation.
Materials and Methods
Mice
C57BL/6, BALB/c, CD28−/−, p53−/−, Faslpr, P21−/−, Bim−/−, and TNFR−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the Medical College of Georgia and Loyola University Chicago.
Abs and flow cytometry
Purified fluorochrome- and biotin-coupled mAbs specific for CD4 (RMA 4– 5, GK1.5), CD8 (53–6.7), CD25 (PC61), CD3-e (145-2C11), CD28 (37.51), and FoxP3 (FJK-16s) were purchased from eBioscience (San Diego, CA). Annexin V–PE, 7-aminoactinomycin D (7-AAD), anti-Fas (Jo2), anti-p53, and anti-FasL (MFL3) were purchased from BD Bio-sciences (San Jose, CA). Anti-MDM2, anti-p21, and anti-Bim were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–phopho-p53 was from Cell Signaling Technology (Beverly, MA). Anti–β-actin was from Sigma-Aldrich (St. Louis, MO). Anti-HSC70 was from Assay Designs (Ann Arbor, MI). Stained cells were analyzed on a FACS Calibur or FACS Canto flow cytometer (BD Biosciences).
Cell isolation and cell culture
Pooled lymph node and spleen cells were depleted of B cells and adherent cells by panning, stained with anti-CD4 (clone RMA4-5) and anti-CD25 (clone PC61, ebiosciences), followed by sorting using a FACSAria or MoFlo (DakoCytomation, Carpinteria, CA) into CD4+CD25− and CD4+ CD25+ populations. In some experiments (Fig. 3), the CD4+ T cells were purified by depletion of B cells, adherent cells, CD8+ T cells, and MHC class II+ cells by panning, as described previously (25). For isolation of dendritic cells (DCs), splenocytes were first enriched for CD11c+ cells using CD11c beads and auto MACS, followed by sorting of CD11c+CD8− cells using a FACSAria flow cytometer.
Figure 3.

Enrichment and expansion of functional nTregs from total CD4 cells by plate-bound Abs. A, Enrichment of Tregs by plate-bound stimulation of CD4 T cells. Total CD4 cells were stimulated with plate-bound anti-CD3 and anti-CD28 Abs for 11 d. Initial and expanded cells were analyzed for FoxP3 and CTLA4 expression. Numbers represent the percentage of positive cells in the corresponding quadrant. B, Fold increase in FoxP3+ and FoxP3− cells stimulated with plate-bound Abs. Cell numbers obtained for FoxP3+ and FoxP3− cells in the experiments shown in A were calculated and compared with the cell numbers of the initial population. C, In vitro suppression of T cell proliferation by freshly isolated nTregs or nTregs expanded by plate-bound Abs. A graded number of plate-bound expanded CD4+CD25+ (CD4+ plate-treated) and freshly sorted CD4+CD25+ T cells (CD4+CD25+ fresh) were tested for their ability to suppress anti-CD3–induced proliferation of CD4+CD25− T cells (2.5 × 104cells/well). Proliferation was measured by incorporation of 3H-thymidine after 3d of culture. D, Suppression of wasting diseases inRag1−/− mice by plate-bound expanded CD4+T cells. Rag1−/− mice were injected i.v. with 106 scurfy mouse CD4T cells (sf) with or without an equal number of plate-bound expanded CD4+ T cells [CD4(PB)]. At different time points after injection, the mice were analyzed for weight loss. Each line represents an individual mouse.
To measure the proliferation of CD4+CD25− or CD4+CD25+ T cells by 3H-thymidine incorporation, sorted cells (1500/well) were seeded in anti-CD3 and anti-CD28 (5 mg/ml each)-coated 96-well plates (flat-bottom) at 200 μl/well, and the cultures were pulsed with 0.5 μCi 3H-thymidine/well at the indicated times for the next 12 h. Incorporation of 3H-thymidine was determined as previously described (25).
For plate-bound activation, sorted CD4+CD25− or CD4+CD25+ T cells (105–3 × 105) were cultured in anti-CD3 and anti–CD28-coated (5 μg/ml each) 60-mm plates in 5 ml RPMI 1640 medium supplemented with 10% FCS (Atlanta Biologicals, Lawrenceville, GA), β-mercaptoethanol (50 μM), glutamine, sodium pyruvate (1 mM), and amino acids (Invitrogen, Carlsbad, CA) in the presence or absence of IL-2 (10 ng/ml), as indicated.
Polystyrene beads (4.5 μm) (Polysciences, Warrington, PA) were coated with anti-CD3 and anti-CD28 Abs according to the manufacturer's recommendations. In general, 0.1 ml beads were incubated with anti-CD3 (clone 145-2C11, 30 μg/ml) and anti-CD28 (clone 37-51, 30 μg/ml) in 1 ml 0.1 M borate buffer overnight at room temperature. At the end of the incubation, beads were blocked with 10 mg/ml BSA in 1 ml borate buffer, washed three times, and stored at 4°C in PBS until used.
For bead-bound stimulation, T cells were mixed with anti-CD3– and anti-CD28–coated beads at a 1:2 ratio and cultured in complete medium. The cells were stained with Annexin V and 7-AAD (BD Bioscience) or anti-CD4 and anti-FoxP3 Abs at the indicated time points. In some experiments, the cells were harvested at the indicated time points, and the total number of live cells was determined by trypan blue exclusion. In some experiments involving CD4+CD25− T cells stimulated with plate-bound Abs, the cultures were supplemented after 2 d with nothing, DMSO (Sigma-Aldrich), z-VAD-fmk, or z-DEVD-fmk (BD Biosciences), and cell viability was analyzed 4 d later.
Suppression assays
The suppressive capability of Tregs in vivo and in vitro was tested as described earlier (25). Briefly, for the in vitro suppression assays, graded numbers of Tregs were cultured with the indicated number of CD4+CD25− T cells in the presence of γ-irradiated (2000 rad) syngeneic splenocytes and anti-CD3 (0.5 μg/ml). After 72 h, the cultures were analyzed for proliferation by 3H-thymidine incorporation, as described above. To test the suppressive activity of Tregs in vivo, Rag1−/− mice were injected with 106 scurfy mouse CD4+ T cells with or without an equal number of Tregs. The recipient mice were monitored for weight loss.
Cell lysis and Western blot
For Western blot analysis, cells were lysed in SDS sample buffer (containing 2% SDS, 5% β-mercaptoethanol, 5% glycerol, and 62 mM Tris, pH 6.5), boiled, and frozen at −80°C until analysis. Lysates from an equal number of cells were loaded in each well, followed by electrophoresis and blotting onto a poly-vinylidene difluoridemembrane. Membranes were probed with Abs specific for p53, p21, MDM-2, β-actin, phospho-serine 18-p53, Bim, and HSC70, followed by the appropriate HRP-conjugated secondary Abs (Cell Signaling Technology) and enhanced chemiluminescence (GE Healthcare, Piscataway, NJ).
Caspase assay
Activation status of caspase 3 was determined using CaspGLOW assay kit (BioVision, Mountain View, CA). CD4+CD25− and CD4+CD25+ T cells were purified by a cell sorter (FACS Aria) and stimulated with plate-bound anti-CD3 and anti-CD28 Abs (coated at 5 μg/ml each) for 4 d. Cells were then stained to detect activated caspase 3, according to the conditions instructed by the manufacturer.
Results
Plate-bound anti-CD3/anti-CD28 Ab stimulation fails to support continuous growth of CD4+CD25− but promotes robust expansion of CD4+CD25+ T cells
The ability to grow a large number of Tregs is a critical step for applications using Tregs in the clinical setting. Stimulation of sorted nTregs by bead-coated anti-CD3 and anti-CD28 Abs has been used as an effective procedure to expand nTregs; however, the procedure also facilitates the expansion of effect or T cells, indicating that a small amount of contaminating nonregulatory T cells can take over the culture system. Indeed, successful expansion of Tregs by anti-CD3– and anti-CD28–coated beads requires >98% purity of the initial CD4+CD25+CD62L+ population (26).
While examining conditions that allow for effective expansion of CD4+CD25+ T cells, we discovered that plate-bound anti-CD3 and anti-CD28 Abs and stimulation with exogenous IL-2 for 5 d promoted robust growth of CD4+CD25+ T cells (Fig. 1A). The proliferation rates of CD4+CD25− and CD4+CD25+ cells were comparable for the first 3 d. However, after this time point, a stark difference in proliferation rates was observed between the two groups. Although CD4+CD25+ T cells continued to show increased uptake of 3H-thymidine, CD4+CD25− T cell proliferation decreased, as evidenced by the decline in 3H-thymidine uptake.
Figure 1.
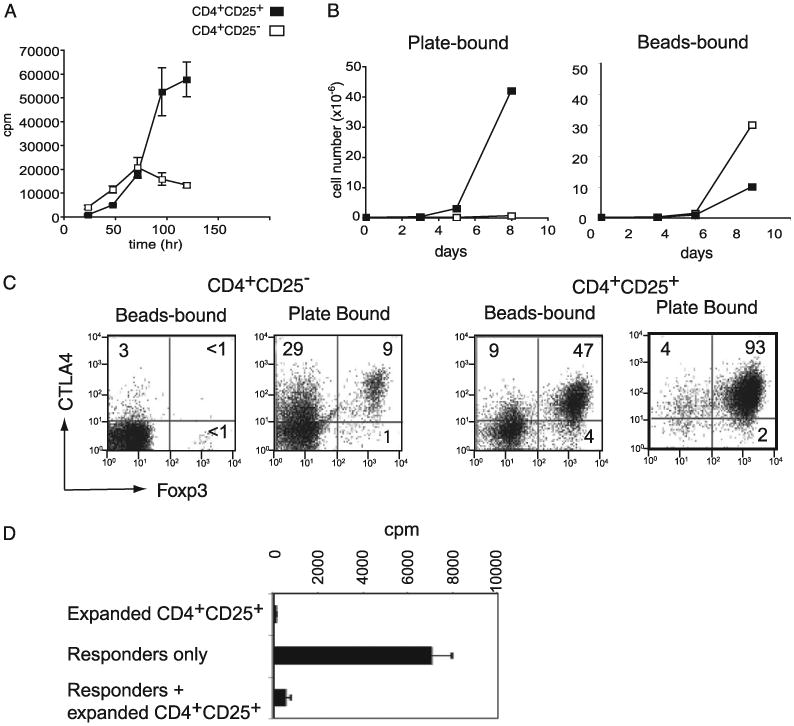
Plate-bound anti-CD3 stimulation of CD4 T cells induces selective expansion of Tregs. A, T cell proliferation stimulated by plate-bound Abs. CD4+CD25+ and CD4+CD25− T cells were stimulated with plate-bound anti-CD3 and anti-CD28 Abs. The proliferation of each sample was measured by incorporation of 3H-thymidine uptake for 12 h after the point indicated on the x-axis. B, Expansion of CD4+CD25− and CD4+CD25+ T cells stimulated with anti-CD3 and anti-CD28 Abs precoated onto polystyrene beads or polystyrene plates. The number of live cells was counted at the indicated points by trypan blue exclusion. C, FoxP3 and CTLA4 expression by T cells expanded by anti-CD3 and anti-CD28 stimulation. CD4+CD25− and CD4+CD25+ T cells were stimulated with bead-bound or plate-bound anti-CD3 and anti-CD28 Abs. The cells were harvested after 8 d of stimulation and stained for FoxP3 and CTLA4. The numbers represent the percentage of positive cells in the corresponding quadrant. D, Anergy and suppressive functions of CD4+ CD25+ T cells expanded by plate-bound Abs. Primary CD4+CD25− T cells (responder only) or plate-bound expanded CD4+CD25+ T cells (expanded CD4+ CD25+) were stimulated with anti-CD3 Ab plus APCs separately or together (responder + expanded CD4+CD25+). Cell proliferation was measured by 3H-thymidine uptake 72 h after the start of stimulation.
No increase in the proliferation of CD4+CD25− T cells from 4 to 5 d could have been due to the inability of the cells to proliferate or to the lack of nutrient. To rule out the possible complication of nutrient deprivation, we set up the culture of a small number of CD4+CD25− or CD4+CD25+ T cells (1 × 105 cells per plate) in 60-mm plates coated with anti-CD3 and anti-CD28 Abs. All of the cultures were supplemented with 10 ng/ml of IL-2. After 8 d of culture, the number of CD4+CD25+ T cells increased >400-fold, whereas the CD4+CD25− T cell number increased <5–6-fold (Fig. 1B). This difference was not due to the intrinsic inability of cells to continue growing in vitro or deterioration of the culture conditions, because cells stimulated with polystyrene beads coated with anti-CD3 and anti-CD28 Abs under the same culture conditions supported more robust growth of CD4+CD25− T cells compared with CD4+CD25+ T cells (Fig. 1B). Expansion of CD4+ CD25− and CD4+CD25+ T cells over 8 d via bead-bound stimulation was >250- and 90-fold, respectively.
The majority of CD4+CD25+ T cells are FoxP3+ nTregs. Thus, the data presented above indicate the possibility that nTregs are resistant to plate-bound Ab-induced apoptosis. Indeed, CD4+CD25+ T cells that expanded for 8 d by stimulation with the plate-bound Ab were mostly FoxP3+ (93%) (Fig. 1C). In contrast, the same CD4+CD25+ T cells stimulated with bead-bound anti-CD3 and anti-CD28 Abs for 8 d were composed of only 47% FoxP3+ cells (Fig. 1C). These FoxP3+ cells expressed a higher level of CTLA4 than the FoxP3− T cells. Approximately 1% and 9%, respectively, of the CD4+ CD25− T cells stimulated with bead- or plate-bound Abs for 8 d were FoxP3+. We next examined whether these FoxP3+ T cells expanded from CD4+CD25+ T cells by plate-bound stimulation possessed Treg functions. Plate-bound expanded CD4+CD25+ T cells were anergic to stimulation by anti-CD3 and irradiated APCs in the absence of exogenous IL-2 (Fig. 1D). Moreover, CD4+CD25+ T cells expanded by plate-bound Ab stimulation demonstrated potent in vitro suppressive activity against the proliferation of freshly isolated CD4+CD25− naive T cells, indicating that these cells are bona fide Tregs.
Sustained anti-CD3/anti-CD28 stimulation is required for apoptosis of CD4+CD25− T cells
The data presented in Fig. 1 showed that CD4+CD25− T cell proliferation declined after 3 d of plate-bound stimulation. The decline in the proliferation of cells might have been caused by growth arrest or death. After 5 d of culture, most CD4+CD25− T cells stimulated with the plate-bound anti-CD3 and anti-CD28 appeared dead under microscope by trypan blue exclusion assay (data not shown). Indeed, when analyzed for viability on day 5 after stimulation with plate-bound anti-CD3 and anti-CD28 Abs, >71% of the cells were 7-AAD+(dead), and 7%were AnnexinV+, 7-AAD− (considered the early stage of apoptosis). In contrast, the same Abs bound to beads caused limited cell death of CD4+CD25− T cells (4% 7-AAD+). As predicted, based on their robust cell growth, CD4+CD25+ T cells stimulated with plate-bound or bead-bound Abs showed low levels of cell death (9% and 5%, respectively) (Fig. 2A).
Figure 2.
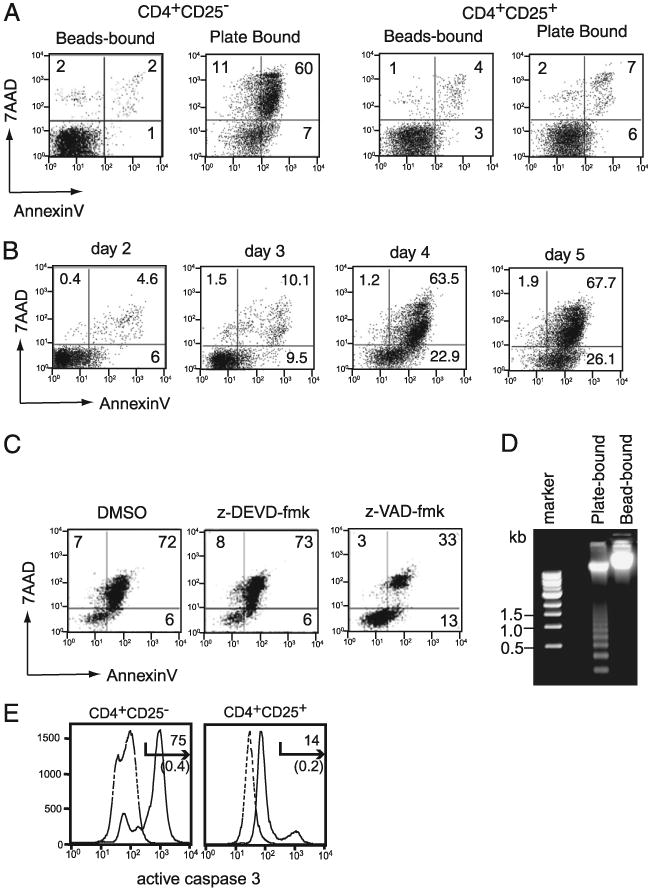
Plate-bound Ab stimulation causes apoptotic cell death in CD4+CD25− cells. A, CD4+ CD25− and CD4+CD25+ T cells were stimulated with bead-bound or plate-bound anti-CD3 plus anti-CD28 Abs for 5 d and analyzed for Annexin V and 7-AAD staining. The percentage of positive cells in each quadrant is indicated. B, CD4+CD25− T cells were stimulated with plate-bound anti-CD3 and anti-CD28 Abs in the presence of IL-2 (10 ng/ml). Cells were harvested at indicated time points and cultured on a new plate devoid of any anti-CD3 or anti-CD28 Abs. On day 5, all of the cells were analyzed for cell death by Annexin V and 7-AAD staining. The numbers represent the percentage of positive cells in the corresponding quadrant. C, Effect of the pan-caspase inhibitor (Z-VAD-fmk) and caspase 3-specific inhibitor (Z-DEVD-fmk) on cell death. CD4+CD25− T cells were stimulated with plate-bound anti-CD3 and anti-CD28 Abs. After 2 d, the indicated cultures were supplemented with DMSO, z-VAD-fmk, or no treatment (control). Cell death was determined by Annexin V and 7-AAD staining on day 4. D, DNA ladder analysis of CD4+ CD25− T cells stimulated for 5 d with plate-bound or bead-bound anti-CD3 plus anti-CD28 Abs. Total DNA extracted from CD4+CD25− T cells stimulated with plate-bound or bead-bound Abs were subjected to electrophoresis. The size of the marker DNA is indicated in the left lane. E, CD4+CD25− T cells were stimulated with plate-bound anti-CD3 and anti-CD28 Abs, as in A. After 4 d of stimulation, cells were analyzed for activation of caspase 3 by staining with CaspGLOW staining reagent. Solid lines represent data from stimulated cells, and dotted lines show the data for unstimulated cells. The numbers above the arrow represent the percentage of positive cells from stimulated samples, and the numbers in parentheses show the percentage of positive cells from unstimulated samples.
The data presented in Fig. 1 suggest that CD4+CD25− cell growth ceased after 3 d of culture with plate-bound anti-CD3 and anti-CD28. This observation led us to hypothesize that prolonged activation after 3 d is required for apoptosis of CD4+CD25− T cells in the plate-bound condition. To test this hypothesis, we removed CD4+CD25− T cells on days 2–5 after culturing with Ab-coated plates and cultured them in new plates lacking anti-CD3 and anti-CD28 Abs in the medium containing IL-2 for later analysis at day 5. After 4 d of continuous stimulation, the combination of plate-bound anti-CD3 plus anti-CD28 Ab stimulation induced cell death in >64% of the CD4+CD25− cells (7-AAD+), and >20% of the cells showed signs of apoptosis (Annexin V+, 7-AAD−). Cells removed from the stimulation plates after 2 or 3 d showed few signs of death (Fig. 2B), and more live cells were recovered on day 5 if stimulation was terminated on day 2 rather than on day 3 (Supplemental Fig. 1). The greatest cell number was observed in samples stimulated for 2 d, whereas when stimulation was sustained for 4 d, apoptotic/dead cell frequency increased (>80%), and live cell number decreased dramatically (Supplemental Fig. 2). These data explain why cell death is not observed in the method used routinely to expand T cells by stimulation with plate-bound anti-CD3/anti-CD28, because, in most cases, T cells are harvested after 2–3 d. CD8 T cells also underwent cell death by stimulation with plate-bound anti-CD3 and anti-CD28 Abs (data not shown). These results show that sustained CD3 and CD28 engagement over 3 d, provided on a flat surface, is detrimental to the growth of CD4+CD25− T cells.
The presence of Annexin V+, 7-AAD− cells suggests that cell death was due to apoptosis. Because the caspase family of proteases is integral to the induction of apoptosis under many situations, the pan-caspase inhibitor z-VAD-fmk or the caspase 3-specific inhibitor z-DEVD-fmk was added to the cultures after 2 d of activation, and the cells were analyzed for apoptosis after 4 d of culture. T cells treated with medium alone, DMSO (solvent for caspase inhibitors), or the caspase 3-specific inhibitor z-DEVD-fmk demonstrated 78% (not shown), 79%, and 81% dead cells (7-AAD+), respectively, whereas the addition of the pancaspase inhibitor z-VAD-fmk reduced the number of dead cells to 36%, suggesting that plate-bound Ab-induced cell death requires activation of caspases other than caspase 3 (Fig. 2C).
Cells undergoing apoptosis exhibit cleavage of nucleosomal DNA that appears as multimers and monomers of 200 bp by agarose gel electrophoresis. A significant level of DNA laddering appeared in CD4+CD25− cells treated with plate-bound anti-CD3/anti-CD28 Abs, whereas no laddering was observed in the DNA obtained from cells stimulated with bead-bound Ab (Fig. 2D). Seventy-five percent of CD4+CD25− T cells activated by plate-bound Abs contained activated caspase 3, whereas only 14% of CD4+CD25+ cells had activated caspase 3 (Fig. 2E). Moreover, CD4+CD25− T cells stimulated with plate-bound Abs showed a substantial level of DNA degradation (>50%), as indicated by the appearance of sub-G1/G0 DNA in the cell cycle analysis (N. Singh and M. Iwashima, unpublished data). Together, these data suggest that plate-bound anti-CD3 and anti-CD28 Abs induced a classic form of apoptotic cell death in CD4+CD25− T cells that requires activation of caspase family proteins.
Plate-bound Ab stimulation of total CD4+ T cells enriches for functional Treg populations
The data presented herein suggest that plate-bound Ab stimulation represents an effective tool for the expansion and enrichment of nTregs from unsorted T cell populations. To test this hypothesis, we determined the frequency of FoxP3+ cells expanded by plate-bound Ab stimulation of unsorted CD4+ T cells (Fig. 3A). In this experiment, we isolated CD4+ T cells from total splenocytes by the depletion of other cells, to avoid any unexpected effect from anti-CD4 Ab. After 11 d of culture with plate-bound Ab stimulation, >70% of the T cells were FoxP3+CTLA4+. Estimation of cell numbers showed that FoxP3+ T cells expanded >7000-fold, whereas FopxP32 cells in the same culture increased by ∼300-fold (Fig. 3B).
The cells expanded from total CD4+ T cells possessed potent regulatory functions in vitro (Fig. 3C). When graded doses of CD4+ T cells expanded by plate-bound Abs were mixed with naive CD4+CD25− T cells, they demonstrated a suppressive effect on T cell proliferation. The level of suppression by plate-bound Ab-expanded Tregs was comparable to freshly isolated CD4+ CD25+ Tregs. This suppressive activity was reversed by the addition of IL-2, but it was insensitive to anti–TGF-β inhibition, suggesting that the mode of suppression is the same as that induced by classic nTregs (data not shown).
We also tested the function of FoxP3+ cells derived from total CD4+ cells by plate-bound Ab in vivo using the scurfy mouse T cell-mediated autoimmune inflammatory bowel disease (IBD) model (25). Scurfy mice, which carry a mutation in the FoxP3 gene, lack nTregs; injection of mature T cells from these mice into Rag1−/− mice causes severe autoimmune IBD. Coinjection of nTregs prevents disease development in recipient mice (25). When CD4+ T cells expanded by plate-bound Abs from total CD4 T cells were coinjected with scurfy mouse T cells, induction of IBD, which was monitored by weight loss, was completely blocked (Fig. 3D). Thus, these FoxP3+ cells are potent Tregs and are functional in vitro and in vivo. Together, the data indicate that plate-bound anti-CD3/anti-CD28 stimulation enriches and expands functional Tregs without requiring fractionation of the CD4+ T cell population.
CD3 and CD28 are required for apoptosis of non-Tregs
Because the conditions used to induce T cell apoptosis by plate-bound Abs differ from those previously reported for AICD, we examined the apoptotic process in more detail and determined whether it occurred via the same mechanism as that previously reported for AICD. One characteristic feature of classic AICD is its dependence on IL-2 (10). Other published data indicate that CD28 stimulation provides a survival signal, presumably by inducing the expression of Bcl-xL (11).
To determine the role of IL-2 and CD28 in plate-bound Ab-induced apoptosis, we examined the effect of stimulation without CD28 engagement. When CD4+CD25− T cells were stimulated with plate-bound anti-CD3 Ab alone, only a marginal level (17%) of cell death was observed (Fig. 4A). This cell survival was not due to the lack of IL-2 production, because addition of exogenous IL-2 to the culture did not increase the level of cell death. As shown above, anti-CD3 plus anti-CD28 plate-coated Abs induced cell death in 75% and 74% of the cells in the presence or absence of exogenous IL-2, respectively. The live cell number also correlated with the percentage of cell death, and anti-CD3–stimulated samples with IL-2 showed the greatest live cell number after the culture (Supplemental Fig. 3). These data suggest that, unlike classic AICD, CD28 is required for plate-bound Ab-induced T cell apoptosis.
Figure 4.
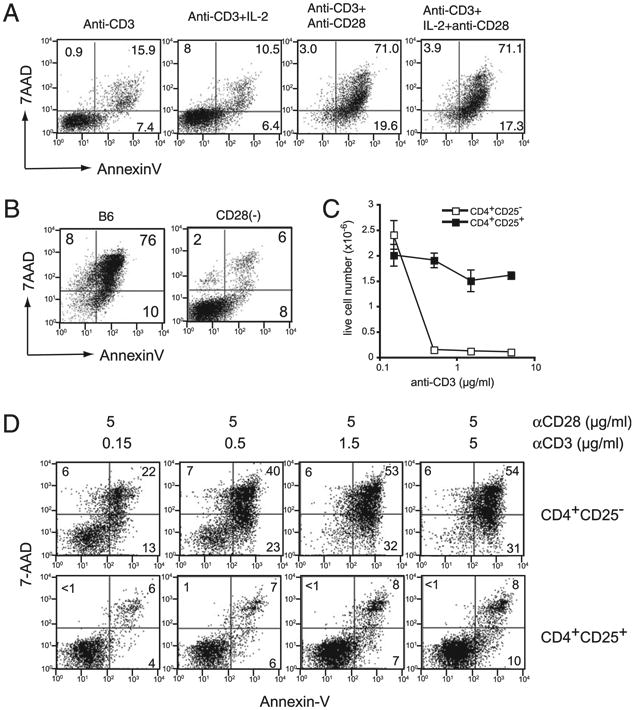
CD28 stimulation is required for CD4+ CD25− T cell apoptosis. A, The role of CD28 and IL-2 in cell death. CD4+CD25− T cells were stimulated with plate-bound anti-CD3 Ab alone; anti-CD3 Ab plus IL-2; anti-CD3 plus anti-CD28 Abs; or anti-CD3 Ab, anti-CD28 Ab, and IL-2 for 5 d and analyzed for Annexin V and 7-AAD staining. B, Plate-bound stimulation of CD28−/− mouse T cells. CD4+CD25− T cells from B6 or CD28−/− mice were stimulated with plate-bound anti-CD3 and anti-CD28 Abs plus IL-2 and analyzed 5 d later for Annexin V and 7-AAD staining. C and D, Effect of the anti-CD3 Ab concentration on cell death. CD4+CD25− and CD4+ CD25+ T cells from B6 mice were stimulated for 5 d with graded doses (coated on the plastic plates at concentrations of 0.15–5 μg/ml) of the anti-CD3 Ab, along with anti-CD28 Ab (5 μg/ml) and IL-2. Live cell number (C) and cell-staining data for Annexin V and 7-AAD (D) are shown.
To confirm these data, we examined the responses of CD4+ CD25− T cells from CD28-deficient and wild-type (WT) C57.BL/6 mice to plate-bound anti-CD3 plus anti-CD28 Abs stimulation for 5 d. Cells were analyzed for survival and apoptosis by Annexin V and 7-AAD staining. T cells from CD28-deficient mice were clearly resistant to plate-bound Ab-induced cell death, whereas C57.BL.6 mouse T cells underwent massive apoptosis (Fig. 4B). The resistance of CD28−/−CD4+CD25− T cells was not due to lack of activation, because their numbers increased >10-fold during the 5 d of culture (data not shown). The data obtained for CD28-deficient mouse T cells also suggest that plate-bound Ab causes death, not by an Fc portion effect, but through the engagement of CD28.
Although CD28 stimulation is required for apoptosis, it is not sufficient. Titration of the anti-CD3 coating Ab with fixed amounts of anti-CD28 coating Ab (5 μg/ml) revealed that a similar degree of CD4+CD25− T cell apoptosis occurred when anti-CD3 Ab concentration ranged between 0.5 and 5 μg/ml, whereas a significant reduction in death was observed when concentrations were 0.15 μg/ml (Fig. 4C, 4D). The number of live cells was 16 times greater in cultures stimulated with 0.15 μg/ml of anti-CD3 coating Ab compared with those stimulated with 0.5–5 μg/ml of anti-CD3 Ab. In contrast, CD4+CD25+ cultures did not exhibit a significant difference in apoptosis of live cell numbers when stimulated with 0.15–5 μg/ml of coating anti-CD3 Ab and a fixed amount of anti-CD28 (5 μg/ml) (Fig. 4C, 4D). Thus, anti-CD3 and anti-CD28 Abs are required for the induction of T cell death.
Plate-bound CD28 stimulation induces high levels of Fas/FasL coexpression in CD4+CD25− T cells
IL-2–induced FasL upregulation plays a critical role in classic AICD of T cells (27). The requirement for Fas/FasL interaction in AICD has been considered the main reason why mice deficient in Fas or FasL experience lymphoproliferative disorder as they age. TNF-αR also plays a role in AICD (28–30). Therefore, we examined whether the Fas/FasL interaction is involved in apoptosis induced by plate-bound anti-CD3 and anti-CD28 Abs. When CD4+CD25− T cells from Fas-deficient lpr mice were stimulated with plate-bound anti-CD3/anti-CD28 Abs in the presence of IL-2, very little apoptosis was observed, and the cells continued to grow. In contrast, TNF-αR–deficient mouse CD4+CD25− T cells underwent significant cell death, similar to WT mouse T cells (Fig. 5A). Thus, plate-bound Ab-induced apoptosis requires the expression of Fas, but not TNF-αR, in T cells.
Figure 5.

Fas is required for plate-bound Ab-induced apoptosis. A, CD4+CD25− T cells from C57.BL/6 (B6), Fas-deficient (lpr), or TNF-R–deficient (TNFR−/−) mice were stimulated with plate-bound anti-CD3 and anti-CD28 Abs. After 5 d, the cells were harvested and stained for Annexin V and 7-AAD, followed by analysis using a flow cytometer. B, CD4+CD25+ or CD4+ CD25− T cells from B6 mice were stimulated with plate-bound anti-CD3 Ab or anti-CD3 and anti-CD28 Abs for 3 d. At the end of the culture period, the cells were harvested and analyzed for Fas and FasL expression by flow cytometry. The percentage of cells in each rectangle is shown.
Based on these data, we examined the expression of Fas and FasL in T cells after stimulation with plate-bound anti-CD3 and anti-CD28 Abs for 3 d in the presence of IL-2. CD4+CD25− and CD4+ CD25+ cells expressed a similar level of Fas on their surface (>70%), but FasL expression differed between these two groups of cells after plate-bound Ab stimulation. More than 20% of the CD4+ CD25− cells expressed Fas and FasL, whereas only 8% of the CD4+CD25+ T cells expressed both. Moreover, the level of Fas and FasL expression differed substantially between these cells. CD4+ CD25− T cells categorized in the FasL− group demonstrated a much greater mean fluorescence intensity for Fas and FasL (76.1 and 8.1, respectively) compared with the corresponding CD4+ CD25+ cell group (48 and 6.8, respectively). Thus, the data indicate that there is a clear difference in the frequency and level of Fas/FasL expression in CD4+CD25− cells compared with CD4+CD25+ cells and that the appearance of double-positive cells correlates with the induction of apoptosis. Fas/FasL double-positive cells were not observed among CD4+CD25− cells when anti-CD28 Ab was not included during stimulation, suggesting that CD28 upregulates the surface expression of FasL in CD4+CD25− cells during the induction of apoptosis.
Requirement of p53 for plate-bound Ab-induced apoptosis
The tumor-suppressor molecule p53 is activated by cellular stressors or DNA damage, and it blocks cell cycle progression and/or causes apoptosis by inducing the expression of multiple target genes (31). For example, p21Waf1 and GADD45 are downstream targets of p53. Mice lacking the expression of these p53 target genes develop spontaneous autoimmune disorders, suggesting that the p53 pathway plays a role in immune regulation (22–24).
The classic form of AICD occurs independently of p53 (17). To determine whether plate-bound Ab-induced apoptosis is caused by the same apoptotic pathway, we examined whether p53 is involved in plate-bound Ab-induced apoptosis. Splenic CD4+CD25− T cells from p53−/− mice were stimulated with anti-CD3 and anti-CD28 Abs, and their responses were compared with those of WT C57.BL/6 mouse T cells. Although plate-bound anti-CD3 plus anti-CD28 Abs induced cell death in CD4+CD25− T cells from age-matched WT C57.BL/6 mice, CD4+CD25− T cells from p53−/− mice showed complete resistance to plate-bound Ab-induced cell death (Fig. 6A). As reported previously, these p53−/− T cells were sensitive to classic AICD and underwent apoptosis to the same degree as WT mouse T cells (Supplemental Fig. 4). Moreover, when cell numbers were compared during cell culture, CD4+CD25− T cells from p53−/− mice continued to grow substantially after 5 d of stimulation (Fig. 6B). The data clearly demonstrate that, unlike previous reports of AICD, the apoptotic cell death caused by plate-bound anti-CD3 and anti-CD28 Abs is dependent on p53. Because p53−/− T cells were able to continuously grow after 5 d with plate-bound Ab stimulation, CD4+CD25− WT T cell death was not due to deterioration of the culture conditions, such as exhaustion of nutrients/growth factors. Although CD4+CD25+ T cells from WT and p53−/− mice showed resistance to plate-bound anti-CD3 and anti-CD28 Abs, p53−/−CD4+CD25+ T cells grew ∼3-fold more than their WT counterparts (data not shown).
Figure 6.
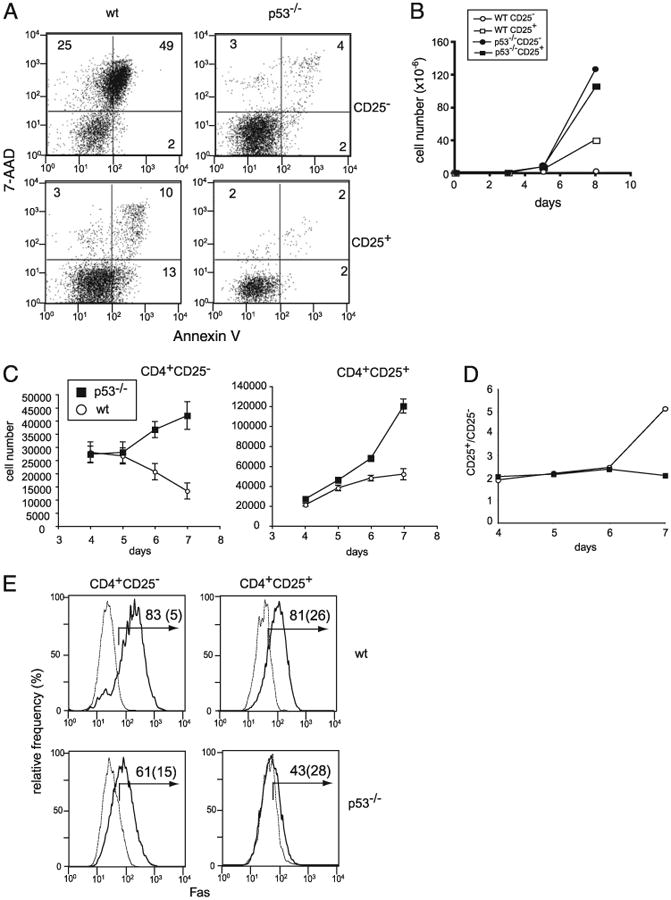
Apoptosis caused by plate-bound Abs is dependent on p53. A, CD4+CD25− (upper panels) and CD4+CD25− (lower panels) T cells from WT (left panels) and p53−/− C57.BL/6 (right panels) mice were stimulated with plate-bound anti-CD3 and anti-CD28 Abs for 5 d and analyzed for cell death by Annexin V and 7-AAD staining. B, Total number of live cells recovered on indicated days after stimulation of CD4+CD25− (circles) and CD4+ CD25+ (squares) T cells from WT B6 (open symbols) and p53−/− (filled symbols) mice. C, Allogenic DCs cause p53-dependent T cell death. Sorted CD4+CD25− and CD4+ CD25+ T cells from B6 and p53−/− (B6 background) mice were stimulated with sorted CD11c+ cells from BALB/c mice (2.5 × 103 T cells/well and 4 × 103 DCs per well in U-bot-tom 96-well plates in the presence of IL-2). At the indicated time points, cells were harvested, and viable cells were analyzed by analysis of trypan blue exclusion and FoxP3 expression. D, The live cell numbers of FoxP3− and FoxP3+ T cells in CD4+CD25+ cultures. The number of FoxP3+ cells was calculated based on total live cells and the percentage of FoxP3+ T cells present in the culture. At the start of the culture, WT and p53−/− CD4+CD25+ T cell populations contained 92% and 91% FoxP3+ cells, respectively. E, Fas expression by CD4+CD25− cells and CD4+CD25+ cells from WT C57.BL/6 (upper panels) and p53−/− mice (lower panels). Fas expression by un-stimulated cells (dotted lines) and cells stimulated with plate-bound anti-CD3 plus anti-CD28 Abs (solid lines) are shown. Numbers represent the percentage of Fas-positive cells for stimulated and unstimulated (parenthesized) cells.
Although p53−/− mice are not known to develop autoimmune disorders, previous studies described p53−/− mice as being more susceptible to autoimmune disorders in several mouse models (18–20). These studies indicate that there is a physiological phase that requires p53-mediated apoptosis to regulate the expansion of effector T cells in vivo. To determine whether natural Ag presentation causes differences between the growth of effector T cells obtained from WT and p53−/− T cells, we next examined the growth of effector and Tregs from p53−/− mice in response to alloantigens. We used DCs as the source of alloantigens, because a subpopulation of DCs (CD8+ and CD8−) was shown to induce T cell death in a Fas/FasL- and Ag-dependent manner (32, 33). Live CD11c+ DCs from BALB/C (H-2d) spleen were used as the antigenic source, and CD4+CD25− T cells from p53−/− and age/sex-matched C57.BL6 (H-2b) spleen were used as responders. The number of WT CD4+CD25− T cells increased slightly between days 4 and 6 after the start of stimulation, but the number of live cells decreased after day 5 (Fig. 6C). In contrast, CD4+CD25− T cells from p53−/− mice demonstrated continuous growth after day 4, and their numbers continued to increase after day 5 (Fig. 6C). CD4+CD25+ cells also displayed similar responses to live allogeneic DCs. On day 4, WT and p53−/− CD4+CD25+ T cell cultures demonstrated an increase in cell numbers and similar numbers of live cells. From this point onward, p53-deficient CD4+CD25+ T cells continued to expand, whereas WT CD4+CD25+ cultures showed a slight increase after day 4.
Because the difference between WT and p53−/− CD4+CD25+ cells was contradictory to that observed for plate-bound Ab stimulation, we determined the number of FoxP3+ and FoxP3− T cells in these cultures. At the beginning of culture, WT and p53−/− mouse CD4+CD25+ T cells contained similar numbers of FoxP3+ cells (92% and 93%, respectively; data not shown). At later time points, the CD4+CD25+ cultures from p53−/− mice demonstrated a significant increase in FoxP3− T cells, whereas the number of FoxP3− cells in the WT population remained the same from day 4 to 6 (Fig. 6D). Thus, the increased growth rate of CD4+ CD25+ T cells from p53−/− mice in response to allogenic WT DCs was caused by expansion of FoxP3− cells. The data suggest that CD4+CD25− and CD4+CD25+ T cells deficient in p53 continued to grow in response to allogenic APCs.
One of the known downstream effector apoptotic mechanisms is upregulation of Fas (34). CD4+CD25− T cells from WT and p53−/− mice expressed equivalent level of Fas on the surface in the resting stage (Fig. 6E). Four days after stimulation with plate-coated anti-CD3 and anti-CD28 Abs in the presence of IL-2, the surface Fas level was clearly elevated in the WT mouse T cells, whereas stimulated p53−/− mouse T cells showed a markedly lower level of Fas on the surface compared with the WT T cells. Thus, the data indicate that p53 contributes to cell death of CD4+CD25− cells via upregulation of Fas.
A role for the p53-dependent pathway in cell death caused by plate-bound Ab stimulation
Because p53 is required for plate-bound Ab-induced apoptosis, we examined the levels of p53 expression in plate-bound Ab-stimulated T cells versus bead-bound Ab-stimulated T cells. Cell lysates were prepared after 3 d of activation to obtain live cells at the stage just prior to apoptosis. Western blot analysis of total cell lysates isolated from CD4+CD25− T cells after plate-bound stimulation demonstrated an increased level of p53 protein compared with that in cells stimulated with bead-bound Abs (Fig. 7A). Plate-bound Ab-stimulated CD4+CD25+ T cells exhibited an increase in p53 expression, but the level was markedly less than that observed for plate-bound Ab-stimulated CD4+CD25− T cells.
Figure 7.
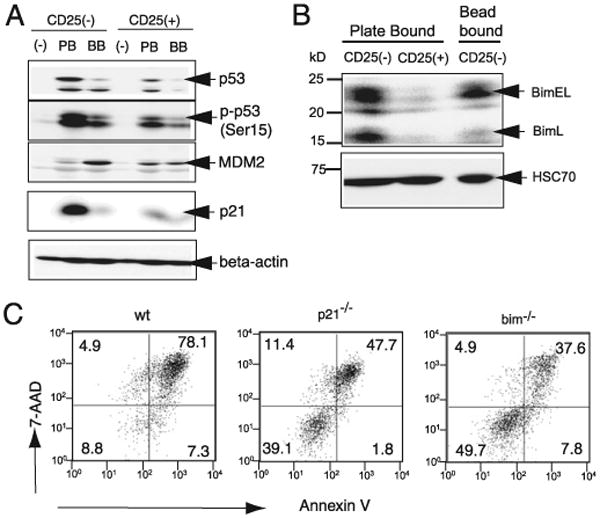
MDM2, p21, and Bim are differentially regulated between plate-bound Ab-stimulated CD4+CD25− and CD4+CD25+ T cells. A, Expression of p53, MDM2b, and p21 by CD4+CD25− and CD4+CD25+ T cells after plate-bound or bead-bound anti-CD3 and anti-CD28 Ab stimulation. After 3 d of activation, total cell lysate from the indicated cells was prepared and subjected to SDS-PAGE (0.75 × 106 cells/lane), followed by Western blotting and immunodetection of the indicated molecules. The specificity of the p53 band was confirmed by Western blotting of lysates from p53−/− T cells (data not shown). B, The indicated cells types were stimulated with plate- or bead-bound Abs; after 3 d, total cell lysate was analyzed for Bim by Western blotting, as in A. The lower panel shows the expression of the constitutively expressed protein HSC70. C, CD4+CD25− T cells from WT, p21−/−, and Bim−/− mice were stimulated by plate-bound anti-CD3 and anti-CD28 Abs for 5 d and analyzed for Annexin V and 7-AAD staining.
The protein level of p53 is controlled at the posttranslational level by MDM2, an E3 ubiquitin ligase that targets p53 for degradation (35). MDM2 expression is induced transcriptionally in a p53-dependent manner. Thus, regulation of p53 by MDM2 forms an autoinhibitory loop. Phosphorylation of Ser15 (Ser18 in humans) of p53 plays a critical role in p53 activation, in part by blocking the interaction between MDM2 and p53 (36). This phosphorylation is known to be mediated by a number of protein kinases, including the ATM family and DNA-dependent protein kinase.
Western blot analysis of cell lysates from plate-bound and bead-bound stimulated T cells revealed that p53 is heavily phosphorylated at the critical Ser15 residue in plate-bound Ab-treated CD4+ CD25− T cells, whereas bead-bound Ab-stimulated samples showed only a small increase in phospho-Ser15. Moreover, the data showed increased expression of MDM2 in bead-bound Ab-stimulated samples compared with plate-bound Ab-stimulated samples. Together, the data suggest that plate-bound Ab stimulation leads to increased p53 expression associated with a high level of Ser15 phosphorylation and lack of MDM2 expression in CD4+ CD25− T cells. In contrast, lysates from CD4+CD25+ T cells showed little, if any, differences between plate-bound and bead-bound Ab-stimulated samples. All samples demonstrated a marginal increase in p53 expression and phosphorylation compared with unstimulated samples, as well as a small increase in MDM2 expression.
Downstream of p53, the cyclin-dependent kinase inhibitor 1A (CDKN1A or P21) plays a critical role in cell cycle arrest. It was demonstrated that p21-deficient mice develop autoimmune disorders due to aggressive autoreactive T cells (22, 37). To further understand the mechanism by which plate-bound stimulation causes p53-dependent apoptosis, we analyzed the expression of p21 in stimulated CD4+CD25− and CD4+CD25+ T cells. CD4+ CD25− T cells showed a substantial increase in p21 expression after plate-bound Ab stimulation, whereas bead-bound Ab induced only a small increase in p21 expression (Fig. 7A). In CD4+CD25+ T cells, the level of p21 expression was comparable between plate-bound and bead-bound conditions and was similar to that detected for bead-bound Ab-stimulated CD4+CD25− T cells (Fig. 7A). Quantitative analysis of each band confirmed the results (Supplemental Fig. 5). Thus, the level of p21 expression in these samples correlated well with p53 expression and the induction of apoptosis.
Bim, another downstream target of p53, is involved in immune regulation, and double-knockout Bim and Fas mice exhibit severe autoimmune disorders (38). Bim was shown to regulate the negative selection of thymocytes, the death of superantigen-stimulated T cells, and the shutdown of immune response (39). Two isoforms of Bim, BimL, and BimEL, were shown to play a role in T cell apoptosis (40, 41). When the level of Bim expression was examined, we found barely detectable expression in plate-bound Ab-stimulated CD4+CD25+ T cells, whereas bead- and plate-bound Ab-stimulated CD4+CD25− T cells showed comparable levels of BimEL (Fig. 7B, Supplemental Fig. 6). However, BimL expression was much lower in bead-bound Ab-stimulated CD4+ CD25− T cells. Together, the data suggest that Bim could also play a role in plate-bound Ab-induced celled death in non-regulatory T cells.
The data above indicate that p21 and Bim were involved in plate-bound Ab-induced apoptosis. Thus, we assessed the level of apoptosis in CD4+CD25− T cells from p21−/− or Bim−/− mice by stimulating them with plate-bound anti-CD3 plus anti-CD28 Abs (Fig. 7C). Although p21−/− or Bim−/− mouse CD4+CD25− T cells were not completely resistant to plate-bound Ab-induced apoptosis, a greater number of p21−/− and Bim−/− T cells survived (39.1% and 49.7%, respectively) compared with WT CD57. BL/6 mouse T cells (8.8%), suggesting that p21 and Bim may play roles in the induction of apoptosis by plate-bound stimulation.
Discussion
AICD is considered a major mechanism of control for the number of activated lymphocytes after the initial activation period. T cells undergo AICD in an IL-2–dependent and p53-independent manner in vitro. The data presented herein demonstrate the presence of another form of Ag-induced T cell death that occurs selectively in nonregulatory T cells. This apoptosis requires continuous Ag receptor and CD28 engagement over 3 d and is dependent on p53 and Fas. Under conditions provoking apoptosis in non-Tregs, nTregs proliferated effectively and expanded more then several thousand-fold. This apoptosis required high concentrations of anti-CD3 Ab and was observed only when stimulation was provided by the flat surface-coated Abs and not by bead-coated Abs. Thus, the effect is not caused by the nonspecific effect of a too high concentration of the Ab or by deteriorated culture conditions; rather, it is caused by a biologically programmed event. To distinguish this form of apoptosis from classic AICD, we designate it herein p53-induced CD28-dependent apoptosis (PICA).
A distinctive characteristic of PICA is the requirement for CD28. CD28 plays a pivotal role in Ag peptide-induced proliferation of naive T cells (42); increases the level of IL-2 mRNA via transcriptional and posttranscriptional mechanisms (43–45); upregulates the expression of the antiapoptotic protein Bcl-XL, which promotes cell survival (15, 46, 47); and elevates Ag receptor proximal events by strengthening immunological synapses (48, 49). Our data show that CD28 stimulation induces a higher level of FasL and Fas coexpression in CD4+CD25− T cells. This elevated level of FasL expression may exceed the threshold of cell death induced by homotypic Fas–FasL interactions on single cell membranes or cell–cell-mediated interactions between neighboring cells. Anti-CD3 plus anti-CD28–stimulated nTregs did not express high levels of FasL and exhibited lower levels of Fas on the cell surface, indicating that nTregs may be resistant to PICA because of an inability to reach the threshold of Fas–FasL interactions required for apoptosis. The mechanism whereby CD28 stimulation in non-Tregs superinduces FasL expression is not clear. PI3K is one of the well-known targets of CD28 signaling; however, the addition of PI3K inhibitors did not rescue the T cells from apoptosis (data not shown).
The data presented herein suggest that effector T cells possessing high-avidity TCR interactions with MHC over an extended period of time may lead to the deletion of T cells in a CD28-, Fas-, and p53-dependent manner. In this regard, it is interesting that a sub-population of DCs express FasL and caused cell death of T cells after 3 d of culture in a Fas-dependent manner (32, 33). Currently, we are testing whether this subpopulation of DCs is responsible for the growth inhibition by allogenic DCs and whether they support the growth of Tregs. It should be noted that because Fas– FasL interactions may take place on the T cell themselves, APCs that do not express FasL might also induce PICA by Ag and costimulatory molecule engagement.
p53 also plays an essential role in PICA. Because PICA requires sustained exposure to Ag, it is expected that PICA plays a role in conditions in which Ag is presented over a prolonged period of time, e.g., chronic infections, tumors, and self-Ags. Some previous studies suggested a potential role for p53 in immune regulation. It should be noted that lymphocytes from patients with rheumatoid arthritis express lower levels of p53 mRNA and protein compared with those from healthy controls (50). Impaired p53 activation was also reported for patients with multiple scleroses (51). In mice, p53 deficiency is associated with more severe Ag-induced arthritis and increased Ag-specific T cell responses (18–20). In the T cell-dependent type I diabetes model induced by a low dose of streptozotocin, p53-deficient mice underwent highly exacerbated diabetes compared with WT mice. These data strongly suggest that p53 is involved in immune regulation.
Our data demonstrate the presence of an Ag-induced form of T cell apoptosis that requires p53 and indicate that Tregs are resistant to this form of apoptosis. Furthermore, our data indicate that defects in p53 expression or function can impair activated effector T cell apoptosis and increase the relative frequency of Ag-reactive effector T cells compared with Tregs under certain conditions. Such enhanced T cell proliferation can lead to prolonged or elevated T cell responses against self- or nonself-Ags.
In addition to plate-bound Ab stimulation, p53-deficient T cells displayed robust and sustained expansion in response to live allogenic DCs. In response to allogenic stimulation, WT and p53−/− T cells showed similar expansion at early time points. At later time points of activation, WT T cell numbers decreased, whereas p53−/− T cells continued to proliferate. Thus, defective p53 expression could result in increased expansion of activated T cells, presumably due to a defect in PICA. It should be noted that although WT Tregs are resistant to PICA, p53−/− Tregs grow faster than WT Tregs in response to allogenic stimulation. Thus, one possible explanation for the lack of an autoimmune phenotype in p53−/− mice could be accelerated proliferation of Tregs, which effectively suppresses non-Tregs in vivo.
Although the requirement for p53 was clearly demonstrated, the mechanism by which p53 causes apoptosis in T cells remains to be determined. Expression of p53 was highly induced by plate-bound Ab stimulation, but not by bead-bound stimulation, and downstream effectors of p53, such as p21 and BimL, which were also highly expressed in plate-bound Ab-stimulated T cells. Induction of PICA negatively correlated with the level of MDM2, an inhibitor of p53 proteins that causes degradation of p53. Thus, a potential difference in the induction of apoptosis by plate-bound and bead-bound Abs may be due, in part, to the level of MDM2 or regulation of its activity.
Our data also suggest that p21, a downstream target of p53, was differentially expressed by T cells after plate-bound versus bead-bound Ab stimulation. Loss of p21 or Bim partially blocked PICA. Thus, p21 and Bim may play a role in PICA downstream of p53. However, the resistance of p21−/− or Bim−/− T cells to PICA is much less than that of p53−/− T cells. This finding suggests that other molecules downstream of p53 are involved in PICA. Loss of Fas significantly reduced the level of apoptosis caused by plate-bound Ab stimulation, suggesting that the Fas-dependent pathway is also critical for PICA. This pathway may represent another process that functions in combination with the above-mentioned p53-dependent pathway. Two recent reports using Fas and Bim double-knockout mice showed that these two molecules function coordinately to suppress T cell proliferation in vivo in response to viral infections (38). Although mice of the C57.BL/6 background carrying a single knockout of each gene suffer from mild forms of lymphadenopathy, double-knockout mice demonstrated severe lupus-like autoimmune and lymphoproliferative disorders. If PICA plays a role in the autoimmune disorders exhibited by Bim and Fas double-knockout mice, one would predict that the percentage of Tregs would be significantly reduced because of the lack of death among activated non-regulatory T cells. Indeed, this was found to be the case for these mice (38). The total number of Tregs did not change in these mice compared with WT mice, and nonregulatory T cells increased >10-fold in the p21−/−Bim−/− knockout mice. Together, these data suggest that the Fas and Bim pathways work in a synergistic manner with regard to the induction of lymphocyte apoptosis and that a physiological process requires both pathways for the prevention of autoimmunity.
A main difference between bead-bound Ab and plate-bound Ab stimulations relates to the physical constraint that the plate-bound stimulation imposes on T cells. We observed that with bead-bound Ab stimulation for 3–4 d, T cells detached themselves from the beads and proliferated elsewhere. This was even observed with the transwell culture system, in which stimulated T cells were placed in the transwell with beads (M. Yamamoto and M. Iwashima, unpublished data). A majority of activated T cells moved out of the transwell, although there were plenty of Ab-coated beads that were accessible to T cells. Thus, with bead-bound Ab stimulation, T cells seem to be able to avoid continuous stimulation and proliferate elsewhere. In contrast, with plate-bound Ab stimulation, cells are strongly and continuously attached to the surface of the plastic plate, because there is no surface without Abs; cells fall to the surface because of gravity and receive stimulating signal continuously. Together, the data suggest that continuous stimulation enforced by flat surface-coated Abs may be the cause of apoptosis on effector T cells.
PICA provides a potential clinical approach for the treatment of autoimmune disorders. Our data suggest that nTregs can expand as much as 7000-fold within 11 d in vitro. These cells outgrow nonregulatory T cells in culture. We were able to obtain functional nTregs from total CD4 T cells that were as potent as purified CD4+ CD25+ T cells. These data suggest that peripheral CD4 T cells can be used as a source of cells for ex vivo expansion of Tregs obtained from patients. Expanded Tregs may be useful for controlling immune responses to self- as well as nonself-Ags (such as transplanted organs).
Supplementary Material
Acknowledgments
The authors thank Christine Seroogy and Kathleen Mishler for critical reading of the manuscript.
This work was supported by National Institutes of Health Grants R01AI055022 and R21AG030953 (to M.I.) and the Medical College of Georgia Intramural Scientist Training Program (to N.S). Y.S. was supported by a fellowship from the Arthritis Foundation.
Abbreviations used in this paper
- 7-AAD
7-aminoactinomycin D
- AICD
activation-induced cell death
- DC
dendritic cell
- FasL
Fas ligand
- IBD
inflammatory bowel disease
- nTreg
naturally occurring T regulatory cell
- PICA
p53-induced CD28-dependent apoptosis
- Treg
T regulatory cell
- WT
wild-type
Footnotes
Disclosures: N. Singh and M. Iwashima have a patent application pending related to the contents of this manuscript.
References
- 1.Belkaid Y. Role of Foxp3-positive regulatory T cells during infection. Eur J Immunol. 2008;38:918–921. doi: 10.1002/eji.200738120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 5.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+ CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 6.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochs HD, Ziegler SF, Torgerson TR. FOXP3 acts as a rheostat of the immune response. Immunol Rev. 2005;203:156–164. doi: 10.1111/j.0105-2896.2005.00231.x. [DOI] [PubMed] [Google Scholar]
- 8.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–542. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- 9.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 10.Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Bárdos T, Shao Q, Tschopp J, Mikecz K, Glant TT, Finnegan A. IL-4 potentiates activated T cell apoptosis via an IL-2-dependent mechanism. J Immunol. 2003;170:3495–3503. doi: 10.4049/jimmunol.170.7.3495. [DOI] [PubMed] [Google Scholar]
- 12.Banda NK, Bernier J, Kurahara DK, Kurrle R, Haigwood N, Sekaly RP, Finkel TH. Crosslinking CD4 by human immunodeficiency virus gp120 primes T cells for activation-induced apoptosis. J Exp Med. 1992;176:1099–1106. doi: 10.1084/jem.176.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newell MK, Haughn LJ, Maroun CR, Julius MH. Death of mature T cells by separate ligation of CD4 and the T-cell receptor for antigen. Nature. 1990;347:286–289. doi: 10.1038/347286a0. [DOI] [PubMed] [Google Scholar]
- 14.Scheipers P, Reiser H. Fas-independent death of activated CD4(+) T lymphocytes induced by CTLA-4 crosslinking. Proc Natl Acad Sci USA. 1998;95:10083–10088. doi: 10.1073/pnas.95.17.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radvanyi LG, Shi Y, Vaziri H, Sharma A, Dhala R, Mills GB, Miller RG. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J Immunol. 1996;156:1788–1798. [PubMed] [Google Scholar]
- 16.Okkenhaug K, Wu L, Garza KM, La Rose J, Khoo W, Odermatt B, Mak TW, Ohashi PS, Rottapel R. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat Immunol. 2001;2:325–332. doi: 10.1038/86327. [DOI] [PubMed] [Google Scholar]
- 17.Boehme SA, Lenardo MJ. TCR-mediated death of mature T lymphocytes occurs in the absence of p53. J Immunol. 1996;156:4075–4078. [PubMed] [Google Scholar]
- 18.Simelyte E, Rosengren S, Boyle DL, Corr M, Green DR, Firestein GS. Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum. 2005;52:1876–1884. doi: 10.1002/art.21099. [DOI] [PubMed] [Google Scholar]
- 19.Okuda Y, Okuda M, Bernard CC. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;135:29–37. doi: 10.1016/s0165-5728(02)00428-9. [DOI] [PubMed] [Google Scholar]
- 20.Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, Chen YH. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005;54:1423–1428. doi: 10.2337/diabetes.54.5.1423. [DOI] [PubMed] [Google Scholar]
- 21.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 22.Balomenos D, Martín-Caballero J, García MI, Prieto I, Flores JM, Serrano M, Martínez-A C. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat Med. 2000;6:171–176. doi: 10.1038/72272. [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Tran E, Zhao Y, Huang Y, Flavell R, Lu B. Gadd45 β and Gadd45 γ are critical for regulating autoimmunity. J Exp Med. 2005;202:1341–1347. doi: 10.1084/jem.20051359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salvador JM, Hollander MC, Nguyen AT, Kopp JB, Barisoni L, Moore JK, Ashwell JD, Fornace AJ., Jr Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity. 2002;16:499–508. doi: 10.1016/s1074-7613(02)00302-3. [DOI] [PubMed] [Google Scholar]
- 25.Singh N, Seki Y, Takami M, Baban B, Chandler PR, Khosravi D, Zheng X, Takezaki M, Lee JR, Mellor AL, et al. Enrichment of regulatory CD4(+) CD25(+) T cells by inhibition of phospholipase D signaling. Nat Methods. 2006;3:629–636. doi: 10.1038/nmeth903. [DOI] [PubMed] [Google Scholar]
- 26.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 28.Speiser DE, Sebzda E, Ohteki T, Bachmann MF, Pfeffer K, Mak TW, Ohashi PS. Tumor necrosis factor receptor p55 mediates deletion of peripheral cytotoxic T lymphocytes in vivo. Eur J Immunol. 1996;26:3055–3060. doi: 10.1002/eji.1830261235. [DOI] [PubMed] [Google Scholar]
- 29.Sytwu HK, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 30.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 31.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 32.Lu L, Qian S, Hershberger PA, Rudert WA, Lynch DH, Thomson AW. Fas ligand (CD95L) and B7 expression on dendritic cells provide counter-regulatory signals for T cell survival and proliferation. J Immunol. 1997;158:5676–5684. [PubMed] [Google Scholar]
- 33.Süss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med. 1996;183:1789–1796. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caspari T. How to activate p53. Curr Biol. 2000;10:R315–R317. doi: 10.1016/s0960-9822(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 37.Arias CF, Ballesteros-Tato A, García MI, Martín-Caballero J, Flores JM, Martínez-A C, Balomenos D. p21CIP1/WAF1 controls proliferation of activated/memory T cells and affects homeostasis and memory T cell responses. J Immunol. 2007;178:2296–2306. doi: 10.4049/jimmunol.178.4.2296. [DOI] [PubMed] [Google Scholar]
- 38.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Zheng SJ, Chen YH. TRAIL, Bim, and thymic-negative selection. Immunol Res. 2003;28:295–301. doi: 10.1385/IR:28:3:295. [DOI] [PubMed] [Google Scholar]
- 40.Chen D, Zhou Q. Caspase cleavage of BimEL triggers a positive feedback amplification of apoptotic signaling. Proc Natl Acad Sci USA. 2004;101:1235–1240. doi: 10.1073/pnas.0308050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandalova E, Wei CH, Masucci MG, Levitsky V. Regulation of expression of Bcl-2 protein family member Bim by T cell receptor triggering. Proc Natl Acad Sci USA. 2004;101:3011–3016. doi: 10.1073/pnas.0400005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 43.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 44.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 45.Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 46.Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157:636–642. [PubMed] [Google Scholar]
- 47.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 48.Bromley SK, Iaboni A, Davis SJ, Whitty A, Green JM, Shaw AS, Weiss A, Dustin ML. The immunological synapse and CD28-CD80 interactions. Nat Immunol. 2001;2:1159–1166. doi: 10.1038/ni737. [DOI] [PubMed] [Google Scholar]
- 49.Wetzel SA, McKeithan TW, Parker DC. Live-cell dynamics and the role of costimulation in immunological synapse formation. J Immunol. 2002;169:6092–6101. doi: 10.4049/jimmunol.169.11.6092. [DOI] [PubMed] [Google Scholar]
- 50.Maas K, Westfall M, Pietenpol J, Olsen NJ, Aune T. Reduced p53 in peripheral blood mononuclear cells from patients with rheumatoid arthritis is associated with loss of radiation-induced apoptosis. Arthritis Rheum. 2005;52:1047–1057. doi: 10.1002/art.20931. [DOI] [PubMed] [Google Scholar]
- 51.Deng X, Ljunggren-Rose A, Maas K, Sriram S. Defective ATM-p53-mediated apoptotic pathway in multiple sclerosis. Ann Neurol. 2005;58:577–584. doi: 10.1002/ana.20600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.