

Abstract
Protein phosphatases regulate mRNA synthesis and processing by remodeling the carboxy-terminal domain (CTD) of RNA polymerase II (Pol2) to dynamically inscribe a Pol2 CTD code. Fission yeast Fcp1 (SpFcp1) is an essential 723-amino acid CTD phosphatase that preferentially hydrolyzes Ser2-PO4 of the YS2PTSPS repeat. The SpFcp1 catalytic domain (aa 140–580) is composed of a DxDxT acyl-phosphatase module (FCPH) and a BRCT module. Here we conducted a genetic analysis of SpFcp1, which shows that (i) phosphatase catalytic activity is required for vegetative growth of fission yeast; (ii) the flanking amino-terminal domain (aa 1–139) and its putative metal-binding motif C99H101Cys109C112 are essential; (iii) the carboxy-terminal domain (aa 581–723) is dispensable; (iv) a structurally disordered internal segment of the FCPH domain (aa 330–393) is dispensable; (v) lethal SpFcp1 mutations R271A and R299A are rescued by shortening the Pol2 CTD repeat array; and (vi) CTD Ser2-PO4 is not the only essential target of SpFcp1 in vivo. Recent studies highlight a second CTD code involving threonine phosphorylation of a repeat motif in transcription elongation factor Spt5. We find that Fcp1 can dephosphorylate Thr1-PO4 of the fission yeast Spt5 CTD nonamer repeat T1PAWNSGSK. We identify Arg271 as a governor of Pol2 versus Spt5 CTD substrate preference. Our findings implicate Fcp1 as a versatile sculptor of both the Pol2 and Spt5 CTD codes. Finally, we report a new 1.45 Å crystal structure of SpFcp1 with Mg2+ and AlF3 that mimics an associative phosphorane transition state of the enzyme-aspartyl-phosphate hydrolysis reaction.
Keywords: CTD code, Spt5, Ssu72
INTRODUCTION
The carboxyl-terminal domain (CTD) of the Rpb1 subunit of RNA polymerase II (Pol2) consists of tandemly repeated heptapeptides with the consensus sequence Y1S2P3T4S5P6S7. Phosphorylation of Tyr1, Ser2, Thr4, Ser5, and Ser7 and cis–trans isomerization of Pro3 and Pro6 inscribe a CTD code with up to 128n potential primary structures (where n is the number of heptads). The CTD code provides information about the state of the transcription machinery that is read by CTD receptor proteins that control transcription, modify chromatin structure, and catalyze or regulate RNA maturation (Corden 2013; Eick and Geyer 2013; Bentley 2014). The CTD undergoes waves of serine, threonine, and tyrosine phosphorylation and dephosphorylation during the transcription cycle. The instantaneous information content of the CTD reflects the balance between the activities of position-specific CTD kinases and CTD phosphatases that act as writers and erasers of the phosphorylation code.
The principal CTD serine phosphatase is Fcp1 (Chambers and Dahmus 1994; Chambers and Kane 1996; Archambault et al. 1997; Cho et al. 1999; Kobor et al. 1999). Fcp1 is conserved among eukarya and is essential for cell viability in budding and fission yeast (Archambault et al. 1997; Kimura et al. 2002) and for Drosophila development (Tombácz et al. 2009). A partial deficiency of human Fcp1 underlies an autosomal recessive developmental disorder (Varon et al. 2003).
Schizosaccharomyces pombe Fcp1 (SpFcp1), a 723-aa monomeric protein, is a prototype of the Fcp1 family. SpFcp1 catalyzes metal-dependent hydrolysis of either Ser2-PO4 or Ser5-PO4 CTD substrates, but displays a preference for Ser2-PO4 (Hausmann and Shuman 2002). The minimal effective CTD substrate for SpFcp1 is a single heptad of phasing SPSYS2PPT. The Tyr1 and Pro3 side chains that flank the Ser2-PO4 mark are important determinants of SpFcp1 activity (Hausmann et al. 2004). Deletion analysis has defined the central segment, SpFcp1-(140–580), as sufficient for phosphatase activity in vitro (Hausmann and Shuman 2003). Mutational analysis of SpFcp1 identified the essential active site constituents (Hausmann and Shuman 2002, 2003; Hausmann et al. 2004) and established SpFcp1 as a member of the DxDxT superfamily of phosphotransferases (Burroughs et al. 2006) that form a covalent enzyme-(aspartyl-Oδ-)-phosphate intermediate at the first Asp of the DxDxT motif (Fig. 1).
FIGURE 1.

Requirements for SpFcp1 activity in vivo. (A) Tertiary structure of SpFcp1(140–580) from pdb 3EF1. The core catalytic FCPH module sits at the base of the Y-shaped protein. The left prong of the Y is formed by a helical module inserted into the FCPH domain; the right prong is a BRCT domain (magenta). The active site (arrow) is demarcated by a Mg2+ ion (magenta sphere) and Asp170-BeF3 adduct (stick model). (B) Cartoon representations of the wild-type and mutated Fcp1 proteins. The FCPH and BRCT modules within the central catalytic domain are denoted as beige and magenta cylinders. The position of the signature DxDxT motif of the acyl-phosphatase superfamily is highlighted. The flanking N and C domains are horizontal lines. The column at right indicates whether the respective recombinant Fcp1 proteins have phosphatase activity in vitro. The column at left indicates whether the fcp1 alleles encoding the indicated proteins are functional in vivo in S. pombe. Lethal alleles are indicated by −. (C) Exponentially growing cultures of S. pombe strains with the indicated chromosomal fcp1 alleles were adjusted to A600 of 0.1 and aliquots of serial fivefold dilutions were spotted to YES agar and incubated at the indicated temperatures.
The crystal structure of SpFcp1-(140–580) revealed a Y-shaped globular protein in which the acyl-phosphatase (FCPH) domain is situated at the base of the Y (Fig. 1A; Ghosh et al. 2008). An α helical module inserted into the FCPH domain comprises the left arm of the Y. The right arm of the Y is a BRCT domain. The phosphatase active site is located within the FCPH domain at the base of a deep canyon, the sides of which are formed by the helical insert and BRCT modules (Fig. 1A). It is proposed that the CTD threads through the SpFcp1 canyon to access the active site (Ghosh et al. 2008).
Fcp1 is a pan-eukaryal CTD phosphatase that plays an essential role in Pol2 transcription. Its essentiality is likely linked to dephosphorylation of Pol2 (Fuda et al. 2012), but there are key unresolved questions, including the following: (i) Is Fcp1's essentiality in vivo tied to its action at a specific CTD phosphate mark, e.g., Ser2-PO4 versus Ser5-PO4? (ii) Do Fcp1 enzymes dephosphorylate other proteins? (iii) Does an Fcp1 catalytic domain suffice for function in vivo? (iv) Do the noncatalytic amino-terminal and carboxy-terminal segments of Fcp1 contribute to its essential function?
Here we address these issues genetically in the fission yeast S. pombe, taking advantage of a collection of biochemically characterized SpFcp1 mutants, including structure-guided mutations of “gatekeeper” residues that alter the Ser2/Ser5 substrate selectivity in vitro (Ghosh et al. 2008). We also exploit S. pombe Rpb1 mutants (Schneider et al. 2010; Schwer and Shuman 2011; Schwer et al. 2014) to illuminate genetic interactions between SpFcp1 activity and CTD composition. We conduct a new mutational analysis of the noncatalytic N domain of SpFcp1, which we show is essential in vivo. We report that SpFcp1 acts as a threonine phosphatase at the CTD of the fission yeast transcription elongation factor Spt5. In addition, we report a new atomic structure of SpFcp1 that illuminates the transition state of the aspartyl-phosphoenzyme hydrolysis reaction.
RESULTS
Phosphatase activity of SpFcp1 is essential in vivo
As it now stands, the genetics of S. pombe Fcp1 are limited to the following two points: (i) Deletion of the S. pombe fcp1+ gene is lethal (Kimura et al. 2002); and (ii) screening of a fission yeast ts mutant library for conditional loss of viability upon entering quiescence (by nitrogen starvation) yielded an fcp1-ts allele with a single R223K coding change (Sajiki et al. 2009). The latter finding reveals an intriguing phenotype, but is not mechanistically informative, insofar as the SpFcp1 R223K mutant is severely defective as a phosphatase in vitro (Hausmann and Shuman 2003) and the crystal structure teaches that Arg223 is not a catalytic residue, but plays a structural role in tethering the active site 170DxDxT loop to surrounding secondary structure elements (Ghosh et al. 2008). Our aim here was to query genetically the essential functions of SpFcp1. To do so, we established a complementation assay by allelic exchange at the chromosomal fcp1 locus. Wild-type and mutant fcp1 integration cassettes (marked with hygMX 3′ of the ORF) were exchanged by homologous recombination for one of the chromosomal fcp1+ alleles in a diploid strain of S. pombe. The correct integrations were verified by diagnostic PCR and by sequencing to ensure that the entire fcp1 sequence was incorporated at the fcp1 locus of hygromycin-resistant diploids. The diploids were sporulated at 30°C and large numbers (≥1000) of viable haploid progeny were scored for the presence of the hygMX gene linked to the fcp1 locus. Failure to recover any viable hygromycin-resistant haploids signified that the pertinent fcp1 allele was lethal. When viable haploids were obtained, we resequenced the fcp1 locus to confirm that the entire mutated ORF was encoded and that no unwanted changes or partial allelic exchanges (e.g., by crossing over within the fcp1 ORF) had occurred.
We used this assay to ask and answer two key questions: (i) Is the phosphatase activity of SpFcp1 essential for S. pombe viability? [YES]; and (ii) does catalytic activity suffice for SpFcp1 function in vivo? [NO]. The essentiality of phosphatase activity was established by the lethality of fcp1-D170A (Fig. 1B), which encodes a catalytically inert SpFcp1 protein (Hausmann and Shuman 2002). However, phosphatase activity was not sufficient for SpFcp1 function, given the lethality of fcp1-(140–580) (Fig. 1), which encodes the central FCPH+BRCT domain that has full phosphatase activity in vitro (Hausmann and Shuman 2003).
The noncatalytic N domain of SpFcp1 is essential in vivo
By singly deleting the amino-terminal and carboxy-terminal segments flanking the catalytic domain, we found that the N domain (aa 1–139) was essential for SpFcp1 function in vivo, whereas the C domain (aa 581–723) was dispensable (Fig. 1B,C). Indeed, the fcp1-(1–580) strain grew as well as wild-type yeast on YES agar medium at 18°C–34°C (Fig. 1B, scored as +++ growth based on colony size), while forming slightly smaller colonies at 37°C (Fig. 1C). In contrast, the equivalent N and C domains of S. cerevisiae Fcp1 are both dispensable, though their simultaneous deletion is lethal (Kobor et al. 2000).
The initial crystal structure of the SpFcp1 catalytic domain highlighted a disordered 70-aa segment within the FCPH module, from aa 327–396. Previously, we produced and purified an internal deletion mutant of the SpFcp1 catalytic domain, SpFcp1-(149–580)-Δ330–393, in which amino acids 330–393 were removed and the two flanking segments (aa 149–329 and 394–580) were fused. The purified recombinant SpFcp1-(149–580)-Δ330–393 protein retained full phosphatase activity in vitro for Ser2-PO4 and Ser5-PO4 CTD substrates (Ghosh et al. 2008). Here we tested the internally deleted fcp1-(Δ330–393) allele for complementation and found that it supported wild-type growth at all temperatures tested (Fig. 1B,C).
Mutational analysis of the N domain of SpFcp1
We conducted a phylogenetic-guided mutational analysis of the essential N domain of SpFcp1. The primary structures of the N domains of S. pombe, Candida albicans, and Aspergillus fumigatus Fcp1 are aligned in Figure 2A, with 52 positions of side chain identity/similarity indicated by a circle (•). Our initial efforts to delineate a minimally functional domain entailed testing complementation of fcp1Δ by alleles with serial amino-terminal deletions of 20, 40, or 60 amino acids. The fcp1-(21–723), fcp1-(41–723), and fcp1-(61–723) alleles were uniformly lethal, suggesting that the full N domain is necessary for SpFcp1 function in vivo.
FIGURE 2.
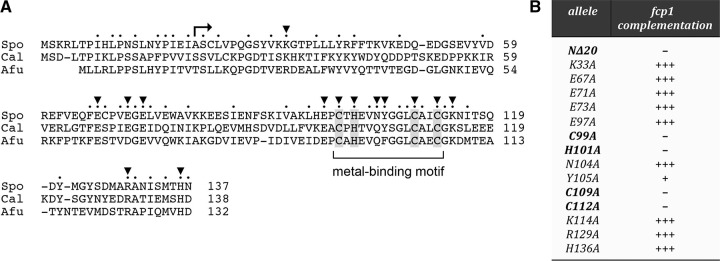
Mutational analysis of the essential N domain. (A) Alignment of the primary structures of the Fcp1 N domains of Schizosaccharomyces pombe (Spo), Candida albicans (Cal), and Aspergillus fumigatus (Afu). Positions of amino acid side chain identity/similarity in all three proteins are indicated by •. Conserved residues targeted for alanine scanning in SpFcp1 are denoted by ▾. A putative CHCC metal-binding motif is demarcated by a bracket. The lethal Δ20 truncation is denoted by . (B) The indicated fcp1 alleles were tested for complementation in vivo. Lethal alleles are denoted by −. +++ Indicates growth on YES agar indistinguishable from wild-type fcp1+. Y105A cells grew slowly (scored as +) and formed tiny colonies on YES agar.
To our inspection, the distinctive feature of the N domain is a 99CxHxxxxxxxCxxC112 motif (bracketed in Fig. 2A), which is widely conserved among Fcp1 homologs from diverse taxa (including human Fcp1), and is a likely metal-binding module. Our initial test of this idea entailed complementation of fcp1Δ by two double-alanine mutant alleles, C99A-H101A and C109A-C112A, that should compromise metal binding. Indeed we found that both of these alanine cluster mutations were lethal in vivo. We then tested complementation by single alanine mutants and found that C99A, H101A, C109A, and C112A were all lethal (Fig. 2B). We conclude that the putative metal-binding site is essential for SpFcp1 function in vivo.
In addition, we tested the effects of alanine changes at 10 other conserved basic, acidic, or polar residues on fcp1 complementation: Lys33, Glu67, Glu71, Glu73, Glu97, Asn104, Tyr105, Lys114, Arg129, and His136 (denoted by ▾ in Fig. 2A). Nine of the fcp1-Ala alleles supported wild-type fission yeast growth at all temperatures tested (scored as +++ growth in Fig. 2B). The lone outlier was the fcp1-Y105A strain, which was viable but sick, forming tiny colonies on rich agar medium (scored as + growth in Fig. 2B). Tyr105 is situated in the loop segment between the pairs of putative zinc ligands (Fig. 2A).
SpFcp1 phosphatase essentiality is not bypassed by eliminating its preferred CTD Ser2 target
SpFcp1 is four- to tenfold more active in vitro in dephosphorylating Ser2-PO4 versus Ser5-PO4 CTD substrates (Hausmann and Shuman 2002; Ghosh et al. 2008). A phospho-mimetic glutamate substitution for Ser2 in every consensus CTD heptad is lethal in vivo (Schwer and Shuman 2011). If it is the case that removal of the Ser2-PO4 mark is the singular essential function of SpFcp1 in vivo, then the lethality of the phosphatase-dead fcp1-D170A allele might be rescued if there were no Ser2-PO4 marks to erase. Putting this thought experiment to the test is feasible in fission yeast, because S. pombe is viable when all of the Ser2 positions in the consensus CTD heptad repeat array are replaced by alanine (Schwer and Shuman 2011). We found that fcp1-D170A remained lethal in the rpb1-CTD-S2A genetic background, signifying that Ser2-PO4 is not the sole essential target of SpFcp1.
Ser5 is the only phospho-acceptor amino acid in the Pol2 CTD that is strictly essential for growth of fission yeast (Schwer and Shuman 2011). Replacing all of the Ser5 residues in the consensus heptads with alanine is lethal because the Ser5-PO4 mark is needed for recruitment of the fission yeast mRNA capping enzymes RNA triphosphatase and RNA guanylyltransferase to the Pol2 elongation complex (Pei et al. 2001; Doamekpor et al. 2014). The lethality of rpb1-CTD-S5A can be rescued by covalently fusing mammalian capping enzyme (MCE, a bifunctional RNA triphosphatase-guanylyltransferase) to the mutant Rpb1-S5A polypeptide (Schwer and Shuman 2011). Here we found that the phosphatase-defective fcp1-D170A allele remained lethal in the rpb1-CTD-S5A-MCE genetic background, from which we infer that Ser5-PO4 is not the sole essential target of SpFcp1.
SpFcp1 substrate ‘gatekeeper’ mutations R271A and R299A are lethal in vivo
Guided by the SpFcp1 crystal structure, we previously sought to identify determinants of substrate specificity by introducing mutations of amino acids that either mediate contacts between the FCPH core and the helical insert (e.g., Arg271) or line the surface of the canyon (e.g., Arg299, Trp305, Val313, Pro314, Trp516). Recombinant mutant SpFcp1-(140-580) proteins were assayed for phosphatase activity with tri-heptad CTD substrates phosphorylated uniquely at Ser2 or Ser5 of each heptad. By testing both substrates in parallel, we identified biased specificity phenotypes in the cases of the R271A, R299A, and W516S mutations, which suppressed Ser5 phosphatase activity but had relatively modest effects on Ser2 phosphatase activity (Ghosh et al. 2008). These mutations skewed the Ser2/Ser5 activity ratio such that R271A, R299A, and W516S had greater preferences for Ser2-PO4 than did wild-type Fcp1. We proposed that Arg271, Arg299, and Trp516 can act as gatekeepers of SpFcp1 substrate choice, i.e., they are permissive for Ser5-PO4 when present and restrictive when removed. In an initial attempt to gain a genetic correlate to the biochemical effects of an Ser2/Ser5 activity skew, we introduced the equivalent mutations into S. cerevisiae Fcp1 (R298A, R326A, and W529S) and tested them by plasmid shuffle for complementation of a lethal S. cerevisiae fcp1Δ mutant, finding that these three ScFcp1 mutations had no apparent effect on S. cerevisiae growth at any temperature tested (Ghosh et al. 2008). However, in light of the significant differences in the genetic requirements for Fcp1 domains in fission yeast versus budding yeast, and the differences in CTD structure–activity relations in these two yeast taxa, we elected to genetically test the effects of the SpFcp1 gatekeeper mutations in S. pombe.
The salient findings were that the R271A and R299A alleles were lethal in S. pombe, whereas a W516S strain was viable and grew normally at all temperatures (Fig. 3A). Several other mutations at the SpFcp1 canyon surface also had no impact on fission yeast growth: W305S, V313D, and P314D (Fig. 3A).
FIGURE 3.

Lethality of gatekeeper mutations R271A and R299A is rescued by CTD shortening. (A) Exponentially growing cultures of S. pombe strains with the indicated chromosomal fcp1 alleles were adjusted to A600 of 0.1 and aliquots of serial fivefold dilutions were spotted to YES agar and incubated at the indicated temperatures. The R271A and R99A alleles were lethal in an rpb1+ genetic background. (B) rpb1–natMX cassettes in which the Pol2 CTD was serially truncated from the C terminus to reduce the number of repeats to 26, 20, 18, 16, 13, or 12 were exchanged at the rpb1 locus of heterozygous fcp1+, fcp1-R271A–hygMX, and fcp1-R299A–hygMX diploid strains. hygR natR haploids with the indicated genotypes were recovered after sporulation, revealing that CTD truncation to ≤20 repeats rescued the lethality of R271A and R299A. Serial dilutions of exponentially growing yeast cultures as specified were spotted to YES agar and incubated at the indicated temperatures.
Shortening the CTD repeat array rescues lethality of fcp1-R271A and fcp1-R299A
The lethality of the fcp1-R271A and fcp1-R299A alleles was observed in an rpb1+ strain that expresses the native wild-type fission yeast Pol2 CTD, which consists of 25 YSPTSPS repeats connected to the body of the Rpb1 subunit via 4 degenerate repeats (the CTD “rump”) that deviate in size and/or sequence from the consensus heptad. Because the R271A and R299A SpFcp1 mutants are biochemically hypomorphs, we considered that they might regain biological activity if the dephosphorylation “workload” were lessened. To test this idea, we exploited a set of fission yeast strains with chromosomal rpb1 alleles encoding CTDs serially truncated at their carboxy-termini so as to comprise 26, 20, 18, 16, 13, or 12 repeats (i.e., the rump plus 22, 16, 14, 12, 9, or 8 consensus heptads) (Schneider et al. 2010). Whereas fcp1-R271A and fcp1-R299A remained lethal in the rpb1-26 background, shortening the CTD to 20 repeats or fewer permitted complementation by R271A and R299A (Fig. 3B). In a wild-type fcp1+ background, the rpb1-20, rpb1-18, and rpb1-16 strains grow as well as rpb1-26 at all temperatures; fcp1+ rpb1-13 and fcp1+ rpb1-12 strains are cold-sensitive (Schneider et al. 2010). Here we found that fcp1-R299A supported vigorous growth at 30°C, 34°C, and 37°C with a CTD having 20, 18, or 16 repeats, while displaying a tight cold-sensitive growth defect, seen as slow growth at 25°C and no growth at 20°C (Fig. 3B). fcp1-R271A had a more pronounced cold-sensitive defect in the context of 20, 18, or 16 repeats (e.g., no growth at 25°C) and displayed a serial improvement in growth at 30°C as the CTD was shortened from 20 to 16 repeats (Fig. 3B). Our inference from these results is that downward titration of SpFcp1 phosphatase activity by certain hypomorphic mutations can be compensated in vivo (at least at higher temperatures) by reducing CTD length, and hence the number of potential phosphorylation marks.
Synthetic genetic interaction of Fcp1 mutations with Ssu72 null and phosphatase-dead mutants
The CTD phosphatase Ssu72 is an agent of Ser5 dephosphorylation in the budding yeast Saccharomyces cerevisiae (Krishnamurthy et al. 2004; Hausmann et al. 2005). Ssu72 catalyzes phosphoryl transfer via a covalent enzyme-cysteinyl-S-phosphate intermediate (Zhang et al. 2011); mutation of the active site cysteine to serine or alanine abolishes phosphatase activity. The Ssu72 protein and its phosphatase activity are essential for the viability of S. cerevisiae (Krishnamurthy et al. 2004). In contrast, S. pombe Ssu72 is dispensable for growth of fission yeast (Schwer et al. 2012). We considered the possibility that partial functional overlap between SpFcp1 and Ssu72 might mask potential effects of hypomorphic SpFcp1 mutations on fission yeast growth. To test this idea, we introduced fcp1 alleles V313D, P314D, W305S, and 1-580 into ssu72+, ssu72Δ, and ssu72-C13S cells that expressed wild-type Ssu72, no Ssu72, and a catalytically dead Ssu72, respectively. Spot tests of growth on YES agar revealed mutational synergies of canyon surface mutations P314D and W305S (but not V313D) in the ssu72Δ and ssu72-C13S genetic backgrounds, evinced by inability to grow at 37°C and slow growth at 34°C (Fig. 4). Deletion of the SpFcp1 C domain elicited the same synthetic ts growth defect when Ssu72 was inactivated (Fig. 4).
FIGURE 4.

Synthetic genetic interaction of Fcp1 mutations with null mutants of Ssu72. Haploid fcp1–hygMX strains were mixed with ssu72+–kanMX (WT), ssu72::kanMX (Δ), or ssu72-(C13S)–kanMX strains (Schwer et al. 2012) of the opposite mating type on malt agar for mating and sporulation, after which hygR kanR haploids with the indicated genotypes were recovered. Exponentially growing cultures of the strains specified were adjusted to A600 of 0.1 and aliquots of serial fivefold dilutions were spotted to YES agar and incubated at the indicated temperatures.
SpFcp1 dephosphorylates Thr1 of the Spt5 CTD
Transcription elongation factor Spt5 is a large polypeptide, composed of multiple domain modules, that associates with the Pol2 transcription complex shortly after initiation and can exert negative and positive effects on transcription elongation (Hartzog and Fu 2013). Fission yeast Spt5 has a distinctive carboxy-terminal repeat domain (the “Spt5 CTD”), composed of 18 repeats of a nonapeptide motif (T1P2A3W4N5S6G7S8K9), that (i) binds the fission yeast RNA capping enzymes RNA triphosphatase and RNA guanylyltransferase (Pei and Shuman 2002), and (ii) is targeted for threonine phosphorylation by the Cdk9 kinase (Pei and Shuman 2002, 2003; Viladevall et al. 2009). The CTDs of fission yeast Pol2 and Spt5 play overlapping roles in recruiting the capping enzymes in vivo (Schneider et al. 2010). Unlike the capping enzyme·Pol2-CTD interactions, which stringently depend on the Ser5-PO4 mark, binding of fission yeast triphosphatase and guanylyltransferase to the Spt5 CTD is independent of Thr1 phosphorylation (Pei et al. 2001; Pei and Shuman 2002). Indeed, their binding to the Spt5 CTD is antagonized by Thr1 phosphorylation (Doamekpor et al. 2014, 2015). The phosphatase(s) responsible for removing the Thr1-PO4 mark from the Spt5 CTD have not been defined.
Here we evaluated the ability of purified recombinant SpFcp1-(140–580) to act as an Spt5 CTD phosphatase. The substrate used was a 22-aa long synthetic peptide (SGSKT1PPAWNSGSKT1PPAWNSGSK) in which both Thr1 residues were phosphorylated. The enzyme was assayed in parallel for Pol2 CTD Ser2 and Ser5 phosphatase activities, using 28-aa synthetic peptides, comprising four tandem YS2PPTSPS or YSPTS5PPS repeats. Increasing concentrations of SpFcp1 were reacted with 25 µM of the Pol2 CTD peptides or 50 µM Spt5 CTD peptide (to achieve the same concentration of 100 µM total phosphoamino acid) for 45 min at 37°C and release of inorganic phosphate was quantified colorimetrically. Product was divided by reaction time and the values were plotted as a function of Fcp1 concentration (Fig. 5A). Turnover numbers were calculated from the slopes of the titration curves (Fig. 5A). SpFcp1 was several-fold more active as a Pol2 CTD phosphatase at Ser2-PO4 (0.13 ± 0.005 s−1) than Ser5-PO4 (0.042 ± 0.002 s−1). The instructive finding was that SpFcp1 also hydrolyzed Spt5 CTD-Thr1-PO4 (0.016 ± 0.001 s−1), albeit one-third as well as it hydrolyzed Pol2 CTD-Ser5-PO4. Mutating the SpFcp1 active site nucleophile Asp170 to asparagine abolished CTD Thr1-PO4 hydrolysis (data not shown), affirming that the threonine phosphatase activity inhered to SpFcp1.
FIGURE 5.
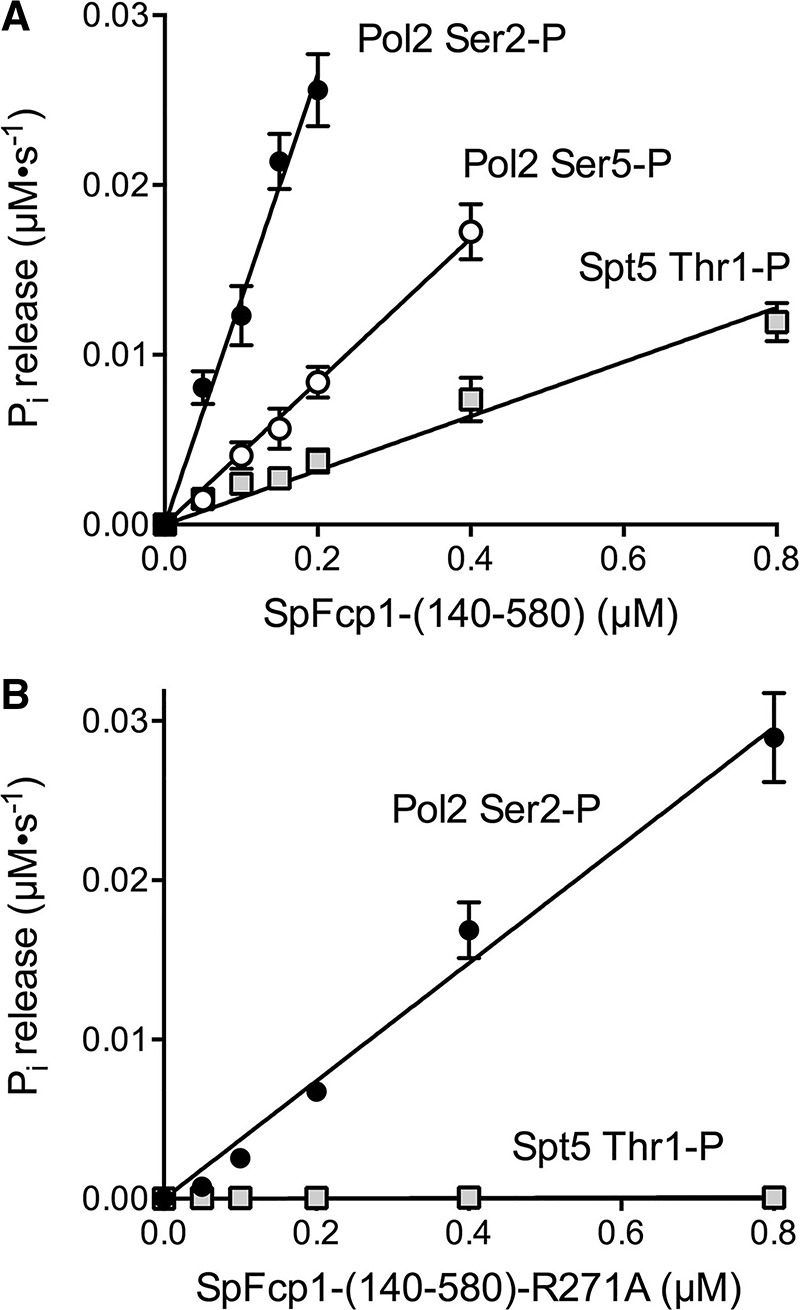
Spt5 CTD threonine phosphatase activity of SpFcp1. (A) Phosphatase activity of SpFcp1-(140–580) with the indicated Pol2 CTD Ser2-PO4, Ser5-PO4, or Spt5 CTD Thr1-PO4 peptide substrates was assayed as described in Materials and Methods. Phosphate release (normalized to reaction time) is plotted as a function of input SpFcp1-(140–580) concentration. (B) Phosphatase activity of SpFcp1-(140-580)-R271A with the Pol2 CTD Ser2-PO4 and Spt5 CTD Thr1-PO4 peptide substrates is plotted as a function of enzyme concentration. Each datum in the graphs is the average of three independent experiments (that were each performed in triplicate) ± SEM. The data were fit by linear regression in Prism.
Arg271 enables Spt5 CTD threonine phosphatase activity
In light of earlier findings that Arg271 influences Pol2 CTD substrate preference (Ghosh et al. 2008) we queried the impact of the R271A mutation on hydrolysis of Pol2 CTD-Ser2-PO4 versus Spt5 CTD-Thr1-PO4 (Fig. 5B). The turnover number of R271A as Ser2 phosphatase (0.037 ± 0.002 s−1) was about threefold lower than wild-type SpFcp1, a modest decrement. In contrast, the R271A protein was inert as an Spt5 CTD Thr1 phosphatase (Fig. 5B). These results fortify the role of Arg271 as a gatekeeper of substrate choice, whereby SpFcp1 acts on Spt5 as a CTD Thr1 phosphatase when Arg271 is present, but is limited to its Pol2 CTD Ser2 phosphatase activity when Arg271 is absent.
Crystal structures of the R271A phosphatase illuminate the Fcp1 reaction mechanism
SpFcp1 remodels the CTD by a two-step ping–pong reaction involving formation and hydrolysis of a phospho-enzyme intermediate. In the first step, the Asp170 Oδ acts as the nucleophile to attack the phosphorus atom of the CTD phosphoserine, resulting in formation of an acyl-phosphate intermediate and expulsion of the dephosphorylated CTD product. In the second step, a water nucleophile attacks the acyl-phosphate, resulting in formation of the inorganic phosphate product and expulsion of Asp170. Previous efforts to crystallize SpFcp1 bound to a CTD peptide were unsuccessful, conceivably because SpFcp1 can recognize either Ser2 or Ser5 as its target. Here we revisited the problem with the R271A mutant, which has enhanced selectivity for Ser2. We premixed purified recombinant SpFcp1-(149-580)-Δ330-393-(R271A) protein with AlCl3, NaF, MgCl2 and CTD peptide SPSYSPTSPS in an attempt to capture an enzyme-AlF3-peptide complex as a mimetic of the transition state of the first phosphoryl transfer step. A crystal grown by sitting drop vapor diffusion at pH 6.5 diffracted X-rays to 1.45 Å resolution. The structure was solved by molecular replacement and refined to Rwork/Rfree of 15.4/18.2 (see Table 1).
TABLE 1.
Crystallographic data and refinement statistics

The instructive crystallographic findings pertained to the occupancy of the active site by a single Mg2+ ion and a trigonal planar AlF3 adjacent to the Asp170 nucleophile. We detected no electron density for a CTD peptide on the face of the planar AlF3 opposite Asp170. Rather, this site was occupied by a water molecule that we propose corresponds to the nucleophile in the second hydrolysis step of the Fcp1 reaction pathway. The trigonal planar AlF3 complex captured in this new structure (Fig. 6, bottom panel) is a more faithful mimetic of the transition state geometry than the square planar AlF4− complex observed previously (Ghosh et al. 2008) and provides clearer insights to the reaction mechanism, as discussed in detail below.
FIGURE 6.
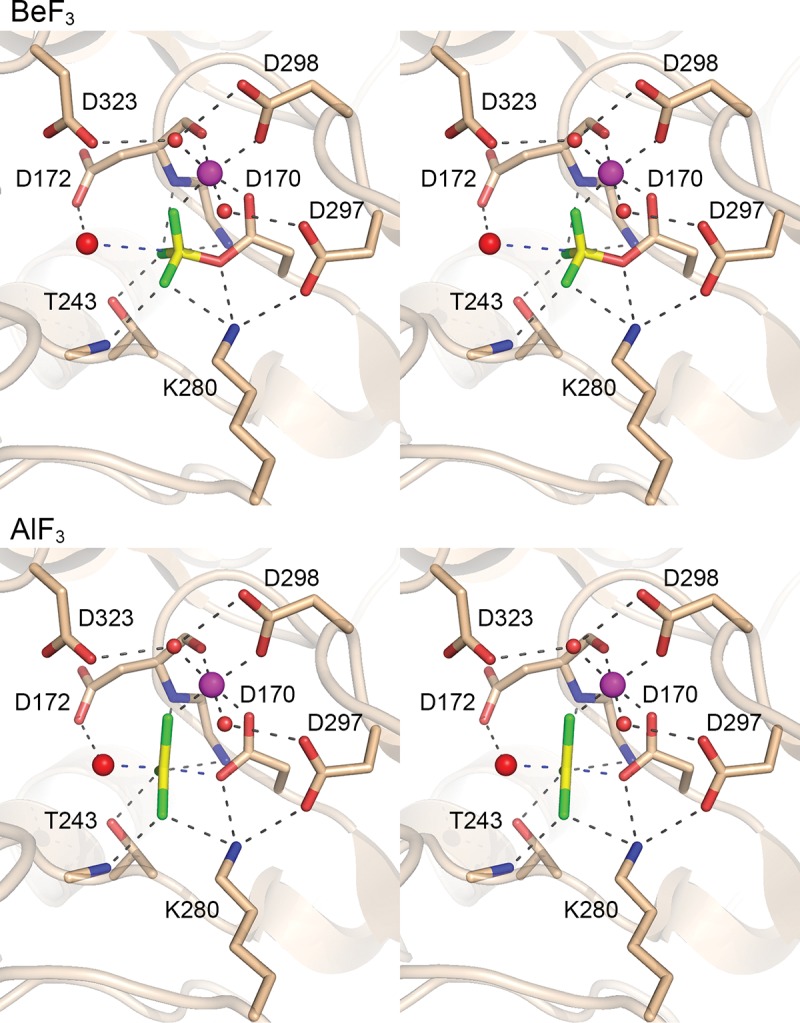
Fcp1 structure captured as a mimetic of the phosphorane transition state. (Top panel) Stereo view of the active site of Fcp1–BeF3 complex that mimics the Michaelis complex of the aspartyl-phosphate hydrolysis step. (Bottom panel) Stereo view of the active site of the Fcp1·AlF3 complex that mimics the transition state of the aspartyl-phosphate hydrolysis step. Selected side chains and main chain atoms are depicted in stick representation. Octahedrally coordinated Mg2+ ions are magenta spheres. Waters are red spheres. The fluoride atoms are colored green. The beryllium and aluminum atoms are colored yellow. Atomic contacts are indicated by dashed lines.
We also grew crystals from a mixture of SpFcp1-(149–580)-Δ330–393-(R271A), BeCl3, NaF, and MgCl2 that captured a covalent enzyme-aspartyl-BeF3 mimetic of the Michaelis complex of the aspartyl-phosphate hydrolysis reaction, the structure of which we solved at 1.85 Å resolution (Table 1). This complex, with tetrahedral geometry at the beryllium atom (Fig. 6, top panel), exemplifies the precursor of the transition state. Adjacent to the Asp170–BeF3 adduct is an octahedrally coordinated Mg2+ ion. The six metal ligands are as follows: the main-chain carbonyl of Asp172; Asp170 Oδ2; Asp298 Oδ1; a water coordinated by Asp297 Oδ1; another water coordinated by Asp298 Oδ2 and Asp323 Oδ1; and a fluorine atom of BeF3, which mimics a phosphate oxygen (Fig. 6, top panel). The enzyme forms an oxyanion hole around the BeF3, whereby the fluorine atoms (mimicking the nonbridging phosphate oxygens) are coordinated by Thr243 Oγ, Lys280 Nζ, and the main-chain amides of Leu171, Asp172, and Met244. Lys280 also contacts the bridging Asp170 Oδ1 oxygen. The putative water nucleophile for the hydrolysis step is poised 3.2 Å from the beryllium atom in an apical orientation to the bridging oxygen of Asp170 (water-Be-Oδ1 angle of 171°). The water nucleophile is coordinated by the Asp172 side chain of the DxD172xT motif, which acts as a general base catalyst of the hydrolysis reaction.
In the structure of the transition state analog, the planar AlF3 is at the center of a trigonal bi-pyramidal coordination complex with Asp170 Oδ at one apex and the attacking water at the other apical position (Fig. 6, bottom panel). The attacking water and the Asp170 Oδ leaving group subtend a 174° angle about the aluminum center. The aluminum atom is virtually equidistant from the leaving Asp170 Oδ (2.0 Å) and the attacking water (2.1 Å). Note that the distance from the water to the phosphorus-like center is shortened in the AlF3 transition state complex compared with the BeF3 Michaelis complex (3.2 Å), while the contact of the water nucleophile with the Asp172 general base is maintained. These structures provide evidence that Fcp1 catalyzes hydrolysis via an associative mechanism with a penta-coordinate phosphorane transition state. The octahedral magnesium complex is identical in the BeF3 and AlF3 structures, highlighting that the catalytic metal engages the same nonbridging phosphate oxygen in the transition as in the ground state. Lys280, Thr243, and the Leu171, Asp172, and Met244 main chain amides also contact the same nonbridging oxygens in the transition state as in the Michaelis complex.
DISCUSSION
The present study provides insights to the catalytic mechanism, substrate repertoire, and in vivo structure–activity relations of the essential fission yeast CTD phosphatase Fcp1. The chemistry of the aspartyl-phosphate hydrolysis step of the Fcp1 pathway was clarified by comparison of new crystal structures of the catalytic domain, as an aspartyl–BeF3 “Michaelis complex” (1.85 Å) and an AlF3 “transition state” (1.45 Å). Beyond affording higher atomic resolution than previous 2.1 Å SpFcp1 structures, the new AlF3 structure captures the “correct” trigonal bi-pyramidal geometry of the transition state, rather than the square bi-pyramidal configuration of the earlier AlF4– complex (Ghosh et al. 2008). The short bond distances from the aluminum center in AlF3 to the apically positioned attacking water and aspartyl-Oδ leaving group weigh in favor of an associative mechanism of phosphoryl transfer by Fcp1, as has been observed for other members of the acyl-phosphatase superfamily (Wang et al. 2002; Lahiri et al. 2003; Lu et al. 2008; Daughtry et al. 2013). The AlF3 structure highlights the relative “quietness” of the active site during progression from Michaelis complex to hydrolytic transition state, whereby none of the enzymic or Mg2+ contacts to the scissile phosphate in the ground state are remodeled in the transition state. The outstanding structural issue for Fcp1 is its interaction with its phosphorylated CTD substrate(s); such complexes have been refractory thus far to co-crystallization.
With regard to substrate repertoire, we show here that SpFcp1 is able to dephosphorylate Thr1-PO4 of the Spt5 CTD. To our knowledge, SpFcp1 is the first instance of a fission yeast Spt5 CTD threonine phosphatase. The strict essentiality of such an activity in fission yeast may be open to question in light of the findings that an spt5-CTD-T1E mutant, in which all Thr1 positions are replaced by the phosphomimetic glutamate, is viable at all temperatures (Schneider et al. 2010). The Thr1-PO4 mark (installed by Cdk9) is thought to be important for the positive transcription elongation activity of fission yeast Spt5, insofar as changing all Spt5 CTD Thr1 positions to alanine sensitized S. pombe cells to growth inhibition by 5-azauracil, whereas changing Thr1 to a phosphomimetic glutamate did not (Schneider et al. 2010). On the other hand, Thr1 phosphorylation antagonizes the interaction of the Spt5 CTD with the S. pombe mRNA capping enzymes (Doamekpor et al. 2014, 2015). Genetic evidence in fission yeast indicates that the unphosphorylated Spt5 CTD and the Ser5-phosphorylated Pol2 CTD collaborate in vivo to recruit the capping apparatus to the Pol2 elongation complex. The Fcp1 Spt5 threonine phosphatase could aid this process by ensuring availability of unphosphorylated Spt5 CTD.
Although the structural biology and biochemistry of SpFcp1 are well advanced, little was known previously about the genetics of SpFcp1, beyond its essentiality for fission yeast viability (Kimura et al. 2002). By implementing a complementation assay with biochemically characterized mutants, we show that (i) the phosphatase catalytic activity is necessary (full-length D170A is lethal), but not sufficient (catalytic domain alone is lethal), for Fcp1 function in vivo; and (ii) the N domain flanking the catalytic domain is essential in vivo, but the flanking C domain is dispensable. In contrast, the N and C domains of S. cerevisiae Fcp1 are both dispensable, though their simultaneous deletion is lethal (Kobor et al. 2000). It is posited that the N and C domains of S. cerevisiae Fcp1 make functionally redundant interactions with one or more components of the Pol2 elongation complex. Much attention has been paid to an acidic carboxy-terminal peptide motif in S. cerevisiae and human Fcp1 that binds TFIIF (Archambault et al. 1997; Kamada et al. 2003; Nguyen et al. 2003). Human Fcp1 has a second peptide motif proximal to its BRCT domain that interacts independently with the same docking site on TFIIF that binds the carboxy-terminal Fcp1 motif (Yang et al. 2009). In the S. pombe Fcp1 crystal structure (Ghosh et al. 2008), the equivalent peptide makes extensive intramolecular hydrophobic contacts that would preclude its binding to TFIIF in the mode reported for a synthetic peptide derived from human Fcp1. Moreover, the relevance of TFIIF binding to S. pombe Fcp1 is unclear, because available evidence points to a direct interaction of SpFcp1 with Pol2 as a likely mode of SpFcp1 binding to the elongation complex (Kimura et al. 2002; Suh et al. 2005).
By alanine scanning of the SpFcp1 N domain, we identify a putative CHCC metal-binding module as uniquely essential for Fcp1 function in vivo. It is conceivable that the N domain and metal-binding module target Fcp1 to Pol2 and/or the Pol2 transcription complex at the proper time and place, to either recycle phosphorylated Pol IIo to unphosphorylated Pol IIa for a new round of transcription initiation or to modulate the CTD phosphorylation state of the Pol2 transcription complex as it traverses a gene. Structure determination of the SpFcp1 N domain, and a search for interacting partners, will be critical to understanding its function.
Our studies uncovered two novel genetic interactions of Fcp1 mutants in fission yeast. First, we found that the R271A and R299A alleles, which were lethal in the presence of full-length Pol2 CTD, were revived when the CTD heptad array was shortened. We surmise that the impact of certain mutations that reduce SpFcp1 CTD phosphatase activity or alter its substrate preference can be compensated in vivo by reducing the phosphatase workload, in this case by having fewer CTD phosphorylation marks to contend with when the CTD repeat array is truncated. Second, we saw that certain fcp1 alleles, that elicited no growth defect by themselves (e.g., missense changes along the canyon surface of a deletion of the C domain), were synthetically sick when the inessential Ssu72 phosphatase was either deleted or its active site cysteine was mutated. These results suggest that Ssu72 shares some of the workload of Fcp1 in fission yeast, so that it can buffer the effects of hypomorphic SpFcp1 mutations. In the case of budding yeast, where Ssu72 is essential for viability, it was reported that the growth defect of ssu72-ts mutants at 35°C was suppressed by overexpression of Fcp1, but not vice versa (Ganem et al. 2003).
Finally, we provide here genetic evidence that neither the Ser2-PO4 mark nor the Ser5-PO4 mark on the Pol2 CTD is a sole essential substrate for SpFcp1 in vivo, insofar as the lethality of catalytically dead SpFcp1 could not be bypassed by individually precluding Ser2 or Ser5 phosphorylations. Whereas it is likely that SpFcp1 is needed to erase Ser2-PO4 and Ser5-PO4 marks in fission yeast, based on its competence to do so in vitro, we do not rule out the prospect that SpFcp1 might dephosphorylate Ser7-PO4, Thr4-PO4 or Tyr1-PO4 on the fission yeast Pol2 CTD (Sakurai and Ishihama 2002) or that its essential activity extends to other phospho-protein substrates. Two recent studies have invoked a Pol2 CTD Thr4 phosphatase function for Fcp1 in S. cerevisiae and chicken DT40 cells (Allepuz-Fuster et al. 2014; Hsin et al. 2014).
MATERIALS AND METHODS
Allelic exchange at the S. pombe chromosomal fcp1 locus
We constructed a series of pKS-based plasmids carrying an fcp1 CTD integration cassette marked with hygMX. The cassettes consisted of the following elements, proceeding from 5′ to 3′: (i) a 670-bp segment of genomic DNA 5′ of the fcp1+ start codon; (ii) an open reading frame encoding the wild-type SpFcp1 phosphatase or mutated versions thereof as specified; (iii) a 269-bp segment including termination/poly(A) signals from the nmt1+ gene; (iv) a hygMX gene conferring resistance to hygromycin; and (v) a 703-bp segment of genomic DNA 3′ of the fcp1+ stop codon. The integration cassettes were linearized and transformed into a diploid S. pombe strain. Hygromycin-resistant transformants were selected and diagnostic PCR and/or Southern blotting was used to confirm correct integrations at one of the fcp1+ loci. The fcp1–hygMX allele was amplified by PCR and sequenced to verify that the desired ORFs were present. The heterozygous fcp1+ fcp1–hygMX diploids were then sporulated and subjected to random spore analysis. Spores (∼5000) were plated to YES agar medium (to gauge the number of viable offspring) and to YES medium containing 300 µg/mL hygromycin. A finding that no viable hygromycin-resistant haploids were recovered after 7 d at 30°C was deemed to indicate lethality of a given fcp1 mutant allele. Viable fcp1–hygMX haploid strains formed colonies on selective agar at frequencies consistent with random segregation. To gauge the effect of the fcp1 mutations on vegetative growth, cultures of haploid S. pombe fcp1–hygMX strains were grown in liquid medium at 30°C until A600 reached 0.6–0.9. The cultures were adjusted to a final A600 of 0.1 and aliquots (3 µL) of serial fivefold dilutions were spotted on YES agar. The plates were incubated at 18°C or 20°C, 25°C, 30°C, 34°C, and 37°C.
Recombinant SpFcp1
DNAs encoding SpFcp1(140–580) and SpFcp1Δ330–393 (Ghosh et al. 2008) were inserted into pSMT3 (Mossessova and Lima 2000) to generate plasmids encoding His6Smt3-SpFcp1 fusion proteins under the control of a bacteriophage T7 promoter. Mutation R271A was introduced into the His6Smt3-SpFcp1Δ330–393 plasmid. The plasmids inserts were sequenced completely to exclude the acquisition of unwanted coding changes during amplification and cloning. The plasmids were transformed into E. coli BL21 (DE3) CodonPlus RIL (Novagen). Four-liter bacterial cultures were grown at 37°C in Super Broth (Teknova) supplemented with 50 μg/mL kanamycin and 100 µg/mL chloramphenicol in baffled flasks until A600 reached 2.0. The temperature was then reduced to 18°C, isopropyl-β-D-thiogalactoside (IPTG) was added to 0.5 mM, and the cultures were incubated overnight. Cells were harvested by centrifugation at 14,000g and resuspended in buffer containing 20 mM Tris–HCl (pH 8.0), 500 mM NaCl, 20 mM imidazole, 0.1% IGEPAL, 20% sucrose, 1 mM β-mercaptoethanol (BME). Cells were disrupted by sonication and debris was removed by centrifugation at 45,000g. The supernatants were applied to 10 mL Ni-NTA agarose columns that had been equilibrated with buffer A (50 mM Tris–HCl at pH 8.0, 15 mM imidazole, 500 mM NaCl, 1 mM BME). The columns were washed with buffer A and the His6Smt3-SpFcp1 proteins were eluted with buffer B (20 mM Tris–HCl at pH 8.0, 350 mM NaCl, 150 mM imidazole, 1 mM BME). The His6Smt3 tags were removed by overnight digestion at 4°C with the Smt3-specific protease Ulp1 (Mossessova and Lima 2000) at a 2000:1 ratio of SpFcp1:Ulp1. The tag-free Fcp1 proteins were then separated from His6Smt3 by Superdex 200 gel filtration in buffer containing 20 mM Tris–HCl at pH 8.0, 350 mM NaCl, 1 mM BME. Peak fractions were pooled and concentrated to 20–25 mg/mL in 20 mM Tris–HCl at pH 8.0, 50 mM NaCl, 5 mM DTT.
Crystallization and structure determination
Aluminum fluoride complex: A solution comprising 300 µM SpFcp1Δ330-393-(R271A) mutant protein with 600 µM AlCl3, 5 mM NaF, 5 mM MgCl2, and 500 µM CTD peptide SPSYSPTSPS was incubated for 1 h on ice prior to crystallization by sitting drop vapor diffusion against 35% PEG-550MME, 100 mM MES (pH 6.5). Beryllium fluoride complex: A solution of 300 µM SpFcp1Δ330–393, 600 µM BeCl2, 5 mM NaF, and 5 mM MgCl2 was incubated for 1 h on ice prior to crystallization by sitting drop vapor diffusion against 40% PEG-400, 100 mM MES (pH 6.5). The crystals were flash frozen in liquid nitrogen. Diffraction data at 1.45 Å resolution for the AlF3 complex and 1.85 Å resolution for the BeF3 complex were collected at the Advanced Photon Source (Argonne) at NE-CAT beamline 24-ID (Table 1). Data reduction was achieved with SCALEPACK (Otwinowski and Minor 1997) and CCP4 (CCP4 1994). Both crystals were in space group P212121 with a single Fcp1 protomer in the asymmetric unit. The structures were solved by molecular replacement using as a search model a prior Fcp1 polypeptide structure (pdb 3EF0) from which nonprotein ligands, metal, and waters were omitted. Atomic models were built into the electron density with Coot (Emsley and Cowtan 2004), and refined with Refmac and Phenix (Adams et al. 2010). Waters were added during refinements in Phenix and checked manually. Model quality and geometry were validated in MolProbity (Table 1; Chen et al. 2010).
CTD phosphatase assay
The Pol2 CTD substrates were a 28-aa synthetic peptide in which all Ser5 positions were phosphorylated (YSPTS5PPSYSPTS5PPSYSPTS5P PSYSPTS5PPS) or a 28-aa synthetic peptide in which all Ser2 positions were phosphorylated (YS2PPTSPSYS2PPTSPSYS2PPTSPSYS2P PTSPS). The Spt5 CTD substrate was a 22-aa synthetic peptide in which Thr1 positions were phosphorylated (SGSKT1P PAWNSGSKT1PPAWNSGSK). The content of Ser-PO4 (for the Pol2 CTD peptides) and Thr1-PO4 (for the Spt5 CTD peptide) was determined by measuring the release of inorganic phosphate after incubating the phosphopeptides with 0.5 unit of alkaline phosphatase (CIP, New England Biolabs) at 37°C for 2 h. CTD phosphatase reaction mixtures (25 µL) containing 50 mM Tris–acetate (pH 5.5), 10 mM MgCl2, 25 µM Pol2 CTD or 50 µM Spt5 CTD phosphopeptide substrates, and 0.05 to 0.8 µM SpFcp1-(140–580) proteins were incubated at 37°C for 45 min. The reactions were quenched by adding 100 μL malachite green reagent (BIOMOL green; BIOMOL Research Laboratories). The samples were incubated at room temperature for 30 min before measuring A620 with a SPECTRAmax Plus (Molecular Device, Inc.) spectrophotometer. Inorganic phosphate (Pi) was quantified by interpolating the absorbance values to a standard curve derived from KH2PO4 diluted in reaction buffer. Pi release (normalized to reaction time) was plotted as a function of input SpFcp1 concentration. Error bars denote the SEM calculated from three independent experiments that were each performed in triplicate. The titration data were fit by linear regression in Prism to derive the specific activity values.
DATA DEPOSITION
Structure factors and coordinates have been deposited in the RCSB Protein Data Base with accession codes 4XPZ (AlF3 complex) and 4XQ0 (BeF3 complex).
ACKNOWLEDGMENTS
Use of the Advanced Photon Source (APS) is supported by the US Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the APS on the NE-CAT beamlines are supported by award GM103403 from the NCRR at the National Institutes of Health (NIH). A.G. was supported in part by a Human Frontier Science Fellowship. C.D.L. is an Investigator of the Howard Hughes Medical Institute. S.S. is an American Cancer Society Research Professor. This work was supported by the Office of Extramural Research of the NIH, grants GM052470 (S.S. and B.S.) and GM061906 (C.D.L.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.050286.115.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allepuz-Fuster P, Martínez-Fernández V, Garrido-Godino AI, Alonso-Aguado S, Hanes SD, Navarro F, Calvo O 2014. Rpb4/7 facilitates RNA polymerase II CTD dephosphorylation. Nucleic Acids Res 42: 13674–13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault J, Chambers RS, Kobor MS, Ho Y, Cartier M, Bolotin D, Andrews B, Kane CM, Greenblatt J 1997. An essential component of a C-terminal domain phosphatase that interacts with transcription factor IIF in Saccharomyces cerevisiae. Proc Natl Acad Sci 94: 14300–14305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L 2006. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol 361: 1003–1034. [DOI] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Chambers RS, Dahmus ME 1994. Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J Biol Chem 269: 26243–26248. [PubMed] [Google Scholar]
- Chambers RS, Kane CM 1996. Purification and characterization of an RNA polymerase II phosphatase from yeast. J Biol Chem 271: 24498–24504. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kim TK, Mancebo H, Lane WS, Flores O, Reinberg D 1999. A protein phosphatase functions to recycle RNA polymerase II. Genes Dev 13: 1540–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL 2013. RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughtry KD, Huang H, Malashkevich V, Patskovsky Y, Liu W, Ramagopal U, Sauder JM, Burley SK, Almo SC, Dunaway-Mariano D, et al. 2013. Structural basis for the divergence of substrate specificity and biological function within HAD phosphatases in lipopolysaccharide and sialic acid biosynthesis. Biochemistry 52: 5372–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD 2014. How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes. Genes Dev 28: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Schwer B, Sanchez AM, Shuman S, Lima CD 2015. Fission yeast RNA triphosphatase reads an Spt5 CTD code. RNA 21: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D, Geyer M 2013. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113: 8456–8490. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fuda NJ, Buckley MS, Wei W, Core LJ, Waters CT, Reinberg D, Lis JT 2012. Fcp1 dephosphorylation of the RNA polymerase II C-terminal domain is required for efficient transcription of heat shock genes. Mol Cell Biol 32: 3428–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem C, Devaux F, Torchet C, Jacq C, Quevillon-Cheruel S, Labesse G, Facca C, Faye G 2003. Ssu72 is a phosphatase essential for transcription termination of snoRNAs and specific mRNAs in yeast. EMBO J 22: 1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shuman S, Lima CD 2008. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol Cell 32: 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzog GA, Fu J 2013. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim Biophys Acta 1829: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann S, Shuman S 2002. Characterization of the CTD phosphatase Fcp1 from fission yeast: preferential dephosphorylation of serine 2 versus serine 5. J Biol Chem 277: 21213–21220. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Shuman S 2003. Defining the active site of Schizosaccharomyces pombe C-terminal domain phosphatase Fcp1. J Biol Chem 278: 13627–13632. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Erdjument-Bromage H, Shuman S 2004. Schizosaccharomyces pombe carboxyl-terminal domain (CTD) phosphatase Fcp1: distributive mechanism, minimal CTD substrate, and active site mapping. J Biol Chem 279: 10892–10900. [DOI] [PubMed] [Google Scholar]
- Hausmann S, Koiwa H, Krishnamurthy S, Hampsey M, Shuman S 2005. Different strategies for carboxyl-terminal domain (CTD) recognition by Serine5-specific CTD phosphatases. J Biol Chem 280: 37681–37688. [DOI] [PubMed] [Google Scholar]
- Hsin JP, Xiang K, Manley JL 2014. Function and control of RNA polymerase II C-terminal domain phosphorylation in vertebrate transcription and RNA processing. Mol Cell Biol 34: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada K, Roeder RG, Burley SK 2003. Molecular mechanism of recruitment of TFIIF-associating RNA polymerase C-terminal domain phosphatase (FCP1) by transcription factor IIF. Proc Natl Acad Sci 100: 2296–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Suzuki H, Ishihama A 2002. Formation of a carboxy-terminal domain phosphatase (Fcp1)/TFIIF/RNA polymerase II (pol II) complex in Schizosaccharomyces pombe involves direct interaction between Fcp1 and the Rpb4 subunit of pol II. Mol Cell Biol 22: 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobor MS, Archambault J, Lester W, Holstege FC, Gileadi O, Jansma DB, Jennings EG, Kouyoumdjian F, Davidson AR, Young RA, et al. 1999. An unusual eukaryotic protein phosphatase required for transcription by RNA polymerase II and CTD dephosphorylation in S. cerevisiae. Mol Cell 4: 55–62. [DOI] [PubMed] [Google Scholar]
- Kobor MS, Simon LD, Omichinski J, Zhong G, Archambault J, Greenblatt J 2000. A motif shared by TFIIF and TFIIB mediates their interaction with the RNA polymerase II carboxy-terminal domain phosphatase Fcp1p in Saccharomyces cerevisiae. Mol Cell Biol 20: 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S, He X, Reyes-Reyes M, Moore C, Hampsey M 2004. Ssu72 is an RNA polymerase II CTD phosphatase. Mol Cell 14: 387–394. [DOI] [PubMed] [Google Scholar]
- Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN 2003. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 299: 2067–2071. [DOI] [PubMed] [Google Scholar]
- Lu Z, Dunaway-Mariano D, Allen KN 2008. The catalytic scaffold of the haloalkanoic acid dehalogenase enzyme superfamily acts as a mold for the trigonal bipyramidal transition state. Proc Natl Acad Sci 105: 5687–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Lima CD 2000. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell 5: 865–876. [DOI] [PubMed] [Google Scholar]
- Nguyen BD, Abbott KL, Potempa K, Kobor MS, Archambault J, Greenblatt J, Legault P, Omichinski JG 2003. NMR structure of a complex containing the TFIIF subunit RAP74 and the RNA polymerase II carboxyl-terminal domain phosphatase FCP1. Proc Natl Acad Sci 100: 5688–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S 2002. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J Biol Chem 277: 19639–19648. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S 2003. Characterization of the Schizosaccharomyces pombe Cdk9/Pch1 protein kinase: Spt5 phosphorylation, autophosphorylation, and mutational analysis. J Biol Chem 278: 43346–43356. [DOI] [PubMed] [Google Scholar]
- Pei Y, Hausmann S, Ho CK, Schwer B, Shuman S 2001. The length, phosphorylation state, and primary structure of the RNA polymerase II carboxyl-terminal domain dictate interactions with mRNA capping enzymes. J Biol Chem 276: 28075–28082. [DOI] [PubMed] [Google Scholar]
- Sajiki K, Hatanaka M, Nakamura T, Takeda K, Shimanuki M, Yoshida T, Hanyu Y, Hayashi T, Nakaseko Y, Yanagida M 2009. Genetic control of cellular quiescence in S. pombe. J Cell Sci 122: 1418–1429. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Ishihama A 2002. Level of the RNA polymerase II in the fission yeast stays constant but phosphorylation of its carboxyl terminal domain varies depending on the phase and rate of cell growth. Genes Cells 7: 273–284. [DOI] [PubMed] [Google Scholar]
- Schneider S, Pei Y, Shuman S, Schwer B 2010. Separable functions of the fission yeast Spt5 carboxyl-terminal domain (CTD) in capping enzyme binding and transcription elongation overlap with those of the RNA polymerase II CTD. Mol Cell Biol 30: 2353–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell 43: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Sanchez AM, Shuman S 2012. Punctuation and syntax of the RNA polymerase II CTD code in fission yeast. Proc Natl Acad Sci 109: 18024–18029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Bitton DA, Sanchez AM, Bähler J, Shuman S 2014. Individual letters of the RNA polymerase II CTD code govern distinct gene expression programs in fission yeast. Proc Natl Acad Sci 111: 4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh MH, Ye P, Zhang M, Hausmann S, Shuman S, Gnatt AL, Fu J 2005. Fcp1 directly recognizes the C-terminal domain (CTD) and interacts with a site on RNA polymerase II distinct from the CTD. Proc Natl Acad Sci 102: 17314–17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombácz I, Schauer T, Juhász I, Komonyi O, Boros I 2009. The RNA Pol II CTD phosphatase Fcp1 is essential for normal development in Drosophila melanogaster. Gene 446: 58–67. [DOI] [PubMed] [Google Scholar]
- Varon R, Gooding R, Steglich C, Marns L, Tang H, Angelicheva D, Yong KK, Ambrugger P, Reinhold A, Morar B, et al. 2003. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet 35: 185–189. [DOI] [PubMed] [Google Scholar]
- Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokar KM, Schwer B, Leatherwood JK, Fisher RPl 2009. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell 33: 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cho HS, Kim R, Jancarik J, Yokota H, Nguyen HH, Grigoriev IV, Wemmer DE, Kim SH 2002. Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J Mol Biol 319: 421–431. [DOI] [PubMed] [Google Scholar]
- Yang A, Abbott KL, Desjardins A, De Lello P, Omichinski JG, Legault P 2009. NMR structure of a complex formed by the carboxyl-terminal domain of human RAP74 and a phosphorylated peptide from the central domain of the FCP1 phosphatase. Biochemistry 48: 1964–1974. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang M, Zhang Y 2011. Crystal structure of Ssu72, an essential eukaryotic phosphatase specific for the carboxy-terminal domain of RNA polymerase II, in complex with a transition state analog. Biochem J 434: 435–444. [DOI] [PubMed] [Google Scholar]