

Abstract
Yhc1 and U1-C are essential subunits of the yeast and human U1 snRNP, respectively, that stabilize the duplex formed by U1 snRNA at the pre-mRNA 5′ splice site (5′SS). Mutational analysis of Yhc1, guided by the human U1 snRNP crystal structure, highlighted the importance of Val20 and Ser19 at the RNA interface. Though benign on its own, V20A was lethal in the absence of branchpoint-binding complex subunit Mud2 and caused a severe growth defect in the absence of U1 subunit Nam8. S19A caused a severe defect with mud2▵. Essential DEAD-box ATPase Prp28 was bypassed by mutations of Yhc1 Val20 and Ser19, consistent with destabilization of U1•5′SS interaction. We extended the genetic analysis to SmD3, which interacts with U1-C/Yhc1 in U1 snRNP, and to SmB, its neighbor in the Sm ring. Whereas mutations of the interface of SmD3, SmB, and U1-C/Yhc1 with U1-70K/Snp1, or deletion of the interacting Snp1 N-terminal peptide, had no growth effect, they elicited synthetic defects in the absence of U1 subunit Mud1. Mutagenesis of the RNA-binding triad of SmD3 (Ser-Asn-Arg) and SmB (His-Asn-Arg) provided insights to built-in redundancies of the Sm ring, whereby no individual side-chain was essential, but simultaneous mutations of Asn or Arg residues in SmD3 and SmB were lethal. Asn-to-Ala mutations SmB and SmD3 caused synthetic defects in the absence of Mud1 or Mud2. All three RNA site mutations of SmD3 were lethal in cells lacking the U2 snRNP subunit Lea1. Benign C-terminal truncations of SmD3 were dead in the absence of Mud2 or Lea1 and barely viable in the absence of Nam8 or Mud1. In contrast, SMD3-E35A uniquely suppressed the temperature-sensitivity of lea1▵.
Keywords: 5′, splice site, Prp28, Sm proteins, spliceosome assembly
INTRODUCTION
Pre-mRNA splicing initiates when the U1 snRNP engages the intron 5′ splice site (5′SS). Human U1 snRNP consists of a trimethylguanosine (TMG) capped 164-nt U1 snRNA, a seven-subunit Sm protein ring, and three U1-specific snRNP subunits: U1-70K, U1-A, and U1-C (van der Felz et al. 2012). The composition of the U1 snRNP is more complex in the budding yeast Saccharomyces cerevisiae with respect to the size of the U1 snRNA (568 nt) and the number of U1-specific protein subunits (10) (Fabrizio et al. 2009). Yeast Snp1, Mud1, and Yhc1 are the homologs of U1-70K, U1-A, and U1-C, respectively. The yeast U1 snRNP has seven additional U1-specific subunits: Prp39, Prp40, Snu71, Snu56, Luc7, Prp42, and Nam8. The conserved 5′ leader sequence of yeast and human U1 RNA—m2,2,7GpppAUACUUACCU—contains a hexanucleotide motif (underlined) that is complementary to the consensus yeast 5′SS (GUAUGU). The ACUUAC sequence pairs with the pre-mRNA to nucleate the U1•pre-mRNA complex. Bridging interactions between the yeast U1 snRNP and the yeast Msl5•Mud2 heterodimer engaged at the intron branchpoint stabilize a “commitment complex” and prepare a scaffold for recruitment of the U2 snRNP to the branchpoint (Abovich and Rosbash 1997). (Msl5 and Mud2 are the S. cerevisiae counterparts of human splicing factors SF1 and U2AF65.) The U1 snRNP is ultimately ejected from the pre-mRNA•U1•U2-containing spliceosome at the point when the U5•U4•U6 tri-snRNP complex joins en route to forming a pre-mRNA•U2•U5•U6 spliceosome. Dissociation of U1 snRNP is triggered by the DEAD-box ATPase Prp28 (Staley and Guthrie 1999; Chen et al. 2001; Jacewicz et al. 2014), acting to disrupt the short U1:5′SS RNA duplex or remodel protein–RNA contacts at the 5′SS (or both).
The human U1 snRNP has been the focus of intensive efforts at reconstitution and structure determination (Pomeranz Krummel et al. 2009; Weber et al. 2010; Kondo et al. 2015). These have culminated most recently in a 3.3 Å structure of a core subcomplex of human U1 snRNP containing the Sm ring, a truncated U1 snRNA, U1-C, an amino-terminal fragment of U1-70K, and a 5′SS-containing RNA oligonucleotide (Kondo et al. 2015). This structure affords deep insights, in atomic detail, to the internal protein–protein and protein–snRNA interactions that buttress the U1 snRNP and the mRNA interactions that enable splice site recognition.
The yeast U1 snRNP has been the focus of similarly intensive genetic analysis. Traditional and genome-wide approaches have highlighted a network of genetically buffered functions during early spliceosome assembly, embracing the U1-specific snRNP proteins Mud1, Nam8, and Yhc1, the U1 snRNA, the TMG cap, the Cbc2•Sto1 nuclear m7G cap-binding complex, the DEAD-box ATPase Prp28, and the Msl5•Mud2 branchpoint-binding complex (Liao et al. 1991; Abovich et al. 1994; Colot et al. 1996; Gottschalk et al. 1998; Hausmann et al. 2008; Wilmes et al. 2008; Costanzo et al. 2010; Chang et al. 2012; Qiu et al. 2012; Schwer et al. 2013; Schwer and Shuman 2014). This network is defined by the numerous instances in which null alleles of inessential players, or benign mutations in essential factors, elicit synthetic lethal and sick phenotypes when combined with other benign mutations in the splicing machinery.
The interpretation of yeast synthetic phenotypes can be elusive, especially when the atomic structures or biochemical activities of the genetically interacting yeast splicing factors are unknown. When structures are available, they can be exploited to program mutations with specific functional defects and then systematically test an allelic series for phenotypes per se and for synthetic genetic interactions with other spliceosome components or splicing factors. Toward this end, we have solved crystal structures of the yeast Msl5 • branchpoint RNA complex and the yeast Prp28•AMPPNP complex and have subjected these proteins to structure-guided mutagenesis (Jacewicz et al. 2014, 2015). However, there is (to our knowledge) no structure in hand for any component of the yeast U1 snRNP. Therefore, we rely on the human U1 snRNP structures to guide genetic dissection of the yeast U1 snRNP.
Our initial efforts focused on Yhc1, the essential yeast homolog of human U1-C (Tang et al. 1997). The conserved amino-terminal structure of U1-C/Yhc1 (Fig. 1A) consists of two β strands and two α helices (Fig. 2A) nucleated by a Cys-Cys-His-His zinc coordination complex, the importance of which was underscored by findings that alanine mutations of the zinc-binding residues Cys6, Cys9, and His30 in yeast Yhc1 are either lethal or elicit temperature-sensitive (ts) growth defects (Schwer and Shuman 2014). Although alanine mutations of conserved Yhc1 N domain residues Arg3, Tyr12, Thr14, His15, Arg21, Lys22, Asn29, Lys28, and Asp36 had no effect of yeast growth per se, several YHC1-Ala alleles displayed mutational synergies with other splicing components. For example, YHC1-R21A was synthetically lethal in the absence of Mud2 and sick in the absence of Nam8, Mud1, and Tgs1 (the enzyme that forms the TMG cap) or the presence of variant U1 snRNAs. YHC1 alleles K28A, Y12A, T14A, K22A, and H15A displayed a progressively narrower range of synergies. R21A and K28A bypassed the essentiality of Prp28, suggesting that they affected U1•5′SS complex stability (Schwer and Shuman 2014). The new atomic-resolution human U1 snRNP structure (Kondo et al. 2015) reveals that Arg21 makes an intramolecular salt bridge to Asp16 and an intersubunit salt bridge to SmD3 Asp38 (Fig. 2A). The U1-C equivalent of Lys28 is Arg28, which projects toward the uncapped 5′ triphosphate terminus of the U1 snRNA in the new crystal structure. Our speculation is that Lys28/Arg28 coordinates the 5′ cap guanosine nucleoside in the native U1 snRNP.
FIGURE 1.
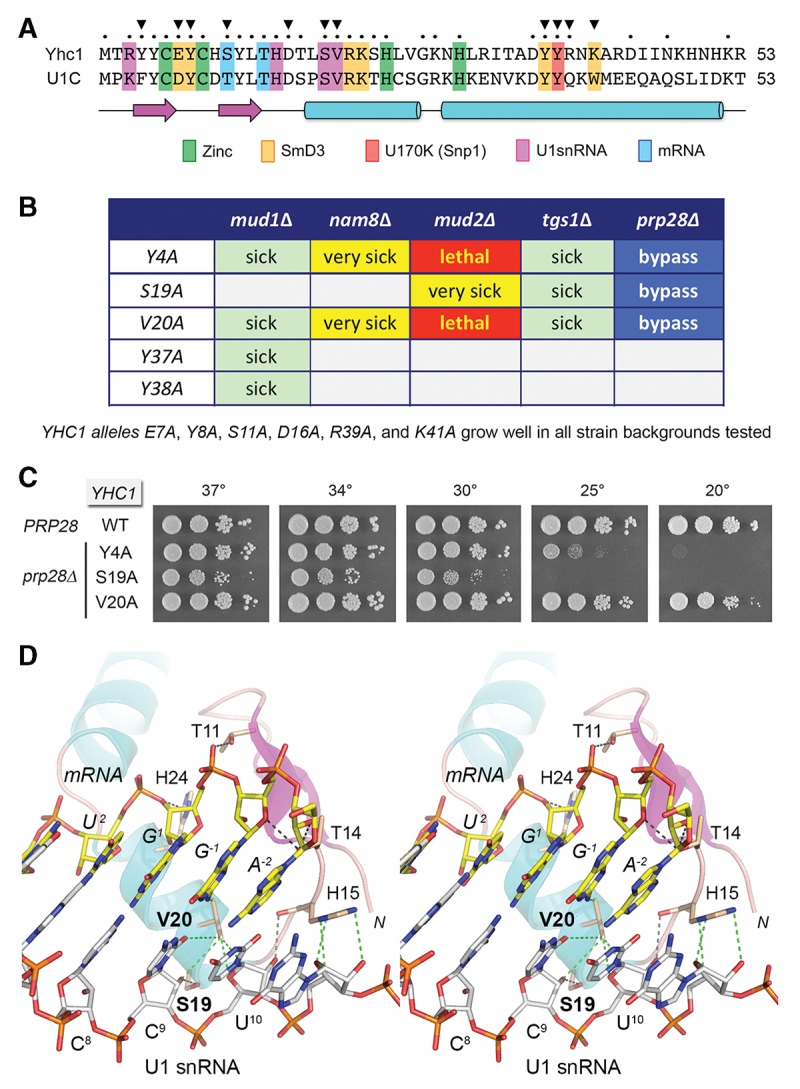
Structure-guided mutational analysis of Yhc1. (A) Alignment of the primary structures of the N-terminal domains of S. cerevisiae Yhc1 and human U1-C. Positions of side-chain identity/similarity are indicated by • above the alignment. The secondary structure elements are depicted below the alignment, with β strands as magenta arrows and α helices as cyan cylinders. U1-C/Yhc1 amino acids that coordinate zinc, make contacts to other U1 snRNP subunits, and contact the U1 snRNA or the mRNA 5′ splice site sequence are highlighted in color-coded boxes as indicated. (B) Summary of genetic interactions of the indicated YHC1-Ala alleles. (C) Prp28 bypass. Yeast prp28▵ yhc1▵ cells harboring the indicated YHC1 allele on a CEN LEU2 plasmid and either wild-type PRP28 (CEN HIS3) or an empty CEN HIS3 plasmid (prp28▵) were spot-tested for growth on YPD agar at the temperatures specified. (D) Stereo view of the human U1 snRNP structure highlighting the interactions of U1-C/Yhc1 (depicted as a cartoon trace with magenta β strands and cyan helices, with selected amino acids as stick models with beige carbons) with the RNA duplex formed by the U1 snRNA 5′ leader (5′-ACUUAC8C9U10, depicted as a stick model with gray carbons) and the mRNA 5′SS exon–intron junction (5′-A−2G−1/G1U2, depicted as a stick model with yellow carbons). Protein–RNA contacts are indicated by dashed lines: black for hydrogen bonds and green for van der Waals interactions.
FIGURE 2.
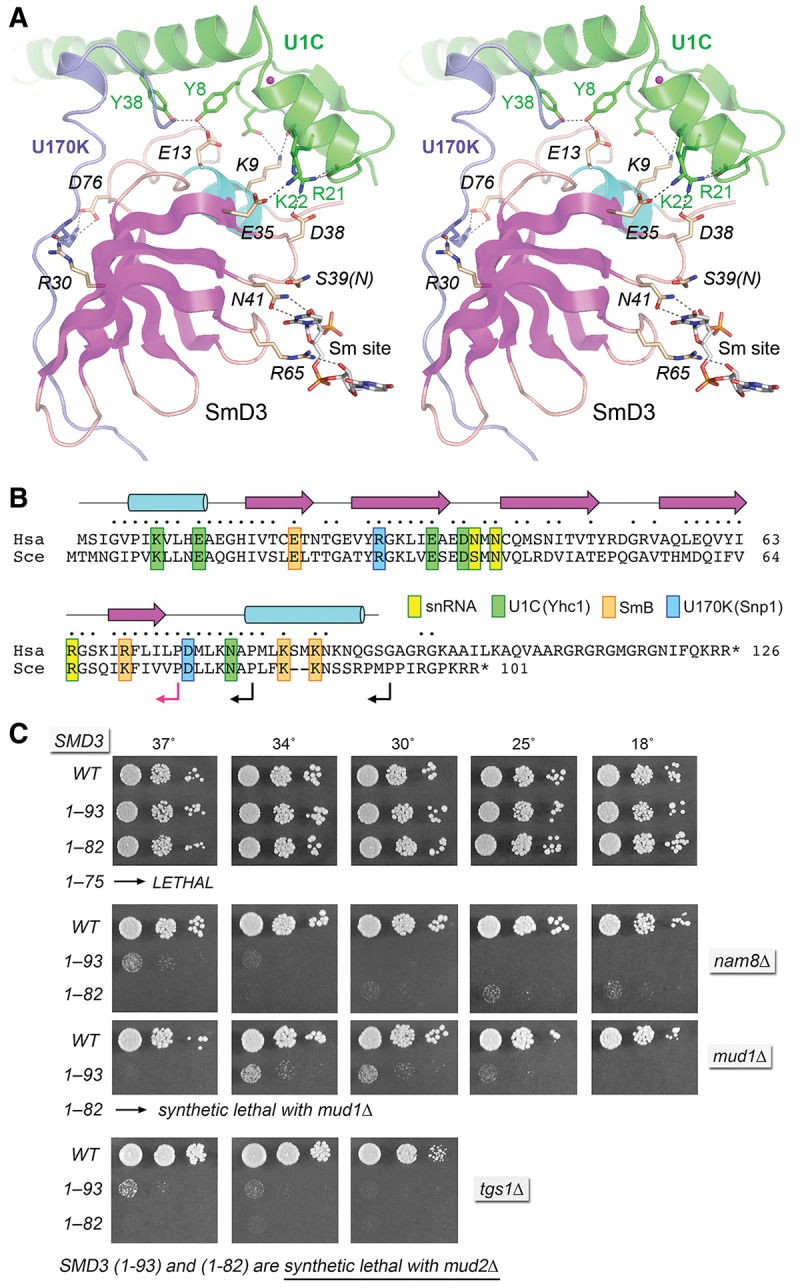
Structure-guided mutagenesis of SmD3. (A) Stereo view of the human U1 snRNP structure highlighting the fold of SmD3 (depicted as a cartoon trace with magenta β strands and cyan helices) and its interactions with neighboring subunits U1-C/Yhc1 (green) and U1-70K/Snp1 (blue) and with the Sm site in U1 snRNA. The zinc atom nucleating the U1-C/Yhc1 fold is depicted as a magenta sphere. Selected amino acids are shown as stick models and numbered according to their positions in the yeast polypeptides. Atomic contacts are indicated by dashed lines. (B) Alignment of the primary structures of the S. cerevisiae (Sce) and human (Hsa) SmD3. Positions of side-chain identity/similarity are indicated by • above the alignment. The secondary structure elements are depicted above the alignment, with β strands as magenta arrows and α helices as cyan cylinders. SmD3 amino acids that make contacts to other U1 snRNP subunits or contact the U1 snRNA are highlighted in color-coded boxes as indicated. Reverse arrowheads indicate the boundaries of the C terminal truncations of yeast SmD3; black and red arrowheads denote viable and lethal truncations, respectively. (C) The wild-type and truncated SMD3 alleles were tested for activity by plasmid shuffle in smd3▵, smd3▵ mud2▵, smd3▵ nam8▵, smd3▵ mud1▵, and smd3▵ tgs1▵ strains. The viable FOA-resistant smd3▵ strains bearing the indicated SMD3 alleles were spot-tested for growth on YPD agar at the temperatures specified. SMD3 alleles listed at the bottom of each panel failed to complement smd3▵ in the plasmid shuffle assay and were deemed lethal in that genetic background.
According to the 3.3 Å U1 snRNP structure, the U1-C N-domain makes a network of physical contacts with the U1:5′SS RNA duplex and the U1-70K and SmD3 protein subunits (Kondo et al. 2015). Our aim in the present study was to functionally interrogate these contacts via a new round of mutagenesis of yeast Yhc1. The results reveal the importance of interactions of the conserved Yhc1 Val20 and Ser19 side chains with the U1 5′ leader RNA and the pre-mRNA at the exon-5′SS junction.
Although the structure of the human Sm ring and its RNA interface has been appreciated for some time (Kambach et al. 1999; Weber et al. 2010; Leung et al. 2011), there has been little in the way of genetic structure–function analysis of the Sm proteins in yeast, beyond the early findings that (i) the SmB, SmD1, SmD2, SmD3, SmE, SmF, and SmG proteins are all essential for vegetative growth; (ii) the disordered carboxy-terminal tails of SmB, SmD1, and SmD3 are individually dispensable for vegetative growth; and (iii) simultaneous deletion of the tails of SmB and SmD1 is synthetically lethal (Bordonné 2000; Zhang et al. 2001). The uridine-rich Sm site of the snRNAs threads through the central hole of the Sm protein ring so that the individual nucleobases are engaged sequentially by the SmF, SmE, SmG, SmD3, SmB, SmD1, and SmD2 subunits (Kondo et al. 2015).
Our aim here was to initiate an in vivo mutational analysis of the yeast Sm protein ring, focusing on the SmD3 and SmB subunits and their atomic interactions with RNA and neighboring proteins, including Yhc1. Our results indicate that none of the SmD3 and SmB amino acids that mediate these contacts are essential per se. We define a network of genetically redundant constituents of the Sm ring that display diverse mutational synergies: within the Sm subunit, between neighboring Sm subunits, with Mud1, Nam8 and Mud2, and with the U2 snRNP subunit Lea1.
RESULTS
Synthetic genetic interactions of new Yhc1-Ala mutants with Mud2, Nam8, Mud1, and Tgs1
We performed a new alanine scan of amino acids in the yeast Yhc1 N-domain that are predicted, based on the human U1 snRNP structure, to contact the U1 snRNA (Ser19, Val20), the mRNA 5′ splice site (Ser11), U1-70K/Snp1 (Tyr38) (Fig. 2A), and SmD3 (Glu7, Tyr8, Tyr37, Lys41). We also targeted Tyr4, which internally contacts Arg21 in Yhc1, an important residue identified previously (Schwer and Shuman 2014). The 11 new YHC1-Ala alleles were placed on CEN HIS3 plasmids under the control of the native YHC1 promoter and tested by plasmid shuffle for complementation of a yhc1▵ p[CEN URA3 YHC1] strain. All of the YHC1-Ala strains grew as well as wild-type YHC1 on YPD agar (Supplemental Figs S1, S2).
To survey genetic interactions of the Yhc1-Ala mutants, we tested by plasmid shuffle for complementation of strains in which the genes encoding inessential splicing factors Mud2, Nam8, or Mud1 were deleted in the yhc1▵ p[CEN URA3 YHC1] background. Yeast mud2▵, nam8▵, and mud1▵ single mutants grow as well as wild-type yeast at all temperatures. We also tested for complementation of a yhc1▵ shuffle strain lacking the gene encoding the TMG capping enzyme Tgs1. Yeast tgs1▵ cells grow well at 30°C–37°C, but they fail to grow at 18°C–20°C. The spot tests for growth of the various double mutants are shown in Supplemental Figures S1, S2 and the results are summarized in Fig. 1B.
The genetics highlight the importance of Tyr4 and Val20 in Yhc1 activity in vivo when other components of the early spliceosome are perturbed. To wit, the YHC1 Y4A and V20A alleles were lethal in combination with mud2▵. They also caused strong growth defects in combination with nam8▵, whereby (i) the YHC1-V20A nam8▵ strain was sick at 37°C and 34°C and failed to thrive at ≤30°C (Supplemental Fig. S1), and (ii) the YHC1-Y4A nam8▵ strain grew well at 37°C and 34°C, but did not grow at ≤30°C (Supplemental Fig. S2). In the tgs1▵ background, Y4A and V20A caused cold-sensitive (cs) synthetic defects. In the mud1▵ background, V20A synergized to preclude growth at cold temperatures (Supplemental Fig. S1), whereas Y4A caused a ts defect at 37°C (Supplemental Fig. S2).
In the human U1 snRNP structure, Val20 makes multiple van der Waals contacts with the C9 and U10 nucleosides of the U1 snRNA 5′ leader sequence ACUUACC9U10 (Fig. 1D). It also makes a van der Waals contact to the mRNA G−1 base at the exon–intron junction. Tyr4 makes only intramolecular van der Waals contacts to the Arg21 and Asp16 side chains of U1-C/Yhc1.
YHC1-S19A displayed less drastic mutational synergies, being severely sick at all temperatures in the mud2▵ background and slow growing at 30°C and 25°C in the tgs1▵ background (Fig. 1B; Supplemental Fig. S1). Ser19 makes a hydrogen bond to the C9 ribose 2′-OH of the U1 snRNA 5′ leader (Fig. 1D).
YHC1 Y37A and Y38A synergized exclusively with mud1▵, resulting in failure to grow at 37°C and in the case of Y37A slow growth at 34°C (Fig. 1B; Supplemental Fig. S2). Tyr37 makes van der Waals contacts to a conserved Ala-Pro dipeptide in SmD3 (Ala80-Pro81 in human; Ala81-Pro82 in yeast SmD3). Tyr38 makes a van der Waals contact to U1-70K Phe13, the conserved counterpart of yeast Snp1 Phe16 (Fig. 8, below). Tyr38 also makes a hydrogen bond to U1-C/Yhc1 Tyr8, which in turn donates a hydrogen bond to a conserved glutamate in SmD3 (Fig. 2A). The six other YHC1-Ala alleles supported normal growth in the mud2▵, nam8▵, mud1▵, and tgs1▵ genetic backgrounds.
Yhc1 Val20 and Ser19 mutations bypass the essentiality of Prp28
Yeast Prp28 is an essential DEAD-box ATPase implicated in displacing the U1 snRNP from the 5′SS during the transition from a pre-mRNA•U1•U2 spliceosome to a pre-mRNA•U2•U5•U6 spliceosome. The essentiality of Prp28 for vegetative growth can be bypassed by mutations in the essential U1 snRNP subunits Yhc1, Prp42, and Snu71, and by specific U1 snRNA mutations located within and flanking the segment that base pairs with the intron 5′SS (Chen et al. 2001; Hage et al. 2009; Schwer et al. 2013; Schwer and Shuman 2014), the common thread being that such mutations are thought to weaken the U1•5′SS contacts and thereby alleviate the requirement for Prp28 during U1 snRNP ejection from the early spliceosome.
Here we queried whether any of the 11 new Yhc1 mutations might bypass Prp28. Three YHC1-Ala mutants did: Y4A, S19A, and V20A. The viable yhc1▵ prp28▵ cells bearing these bypass alleles were tested for growth on YPD agar (Fig. 1C). YHC1-Y4A prp28▵ cells grew as well as wild-type cells at 37°C, 34°C, and 30°C, as gauged by colony size, but they grew slowly at 25°C and did not thrive at 20°C. YHC1-S19A prp28▵ cells grew well at 37°C and 34°C, but were slow-growing at 30°C and failed to grow at 25°C or 20°C. The findings suggest that higher temperatures destabilize the weakened U1•5′SS interface in Y4A and S19A cells and thereby permit U1 snRNP ejection without the assistance of Prp28. However, when the U1:5′SS duplex pairing is more stable at lower temperatures, Prp28 is still required. Cold-sensitivity is a characteristic shared with other prp28 bypass mutations described to date (Chen et al. 2001; Hage et al. 2009; Schwer et al. 2013; Schwer and Shuman 2014).
In contrast, the new YHC1-V20A allele restored growth to prp28▵ cells at all temperatures tested (Fig. 1C). YHC1-V20A prp28▵ colony size was indistinguishable from wild-type at 37°C, 34°C, 30°C, and 25°C; colony size was slightly smaller than wild-type at 20°C (Fig. 1C) and 18°C (Supplemental Fig. S3). To extend these findings, we replaced Val20 with other hydrophobic amino acids: isoleucine, leucine, or phenylalanine. The V20I, V20L, and V20F strains were viable after plasmid shuffle and grew as well as wild-type YHC1 on YPD agar at all temperatures (Supplemental Fig. S3, top panel). The V20L and V20F mutations bypassed Prp28. V20L was less effective than V20A in this regard; V20L permitted growth of prp28▵ cells at 37°C, 34°C, and 30°C, but not at ≤25°C (Supplemental Fig. S3, bottom panel). The notable finding was that V20F was a better bypass suppressor than V20A, i.e., YHC1-V20F prp28▵ cells grew well at all temperatures and formed larger colonies at 18°C than did YHC1-V20A prp28▵ cells (Supplemental Fig. S3, bottom panel).
We considered the prospect that RNA contacts made by Yhc1 Ser19 and Val20 might be functionally redundant during vegetative growth, but this turned out not to be the case, insofar as a doubly mutated S19A-V20A strain was viable and grew as well as wild-type YHC1 at all temperatures (Supplemental Fig. S3, top panel). Moreover, the S19A-V20A allele bypassed Prp28 as effectively as the V20A single mutant (Supplemental Fig. S3, bottom panel).
Taken together, the mutational synergies and Prp28 bypass experiments underscore the significant contributions of Val20 and Ser19 interactions with the U1 leader/5′SS RNA duplex to early spliceosome assembly and stability.
Carboxy-terminal truncations of yeast SmD3 define a minimized functional domain
The 101-amino acid yeast SmD3 protein is homologous to the amino-terminal 102-amino acid segment of the 126-amino acid human SmD3 polypeptide, with 66 position of side-chain identity/similarity (Fig. 2B). The fold of human SmD3 in the U1 snRNP crystal of Weber et al. (2010) extends to position 94 and comprises a five-strand antiparallel β sheet of topology β5↑•β1↓•β2↑•β3↓•β4↑ flanked by two α helices (Fig. 2B). In the U1 snRNP structure of Kondo et al. (2015), the SmD3 fold extends to position 83 and does not include the distal α helix (Fig. 2A). We used the yeast/human alignment to guide serial carboxy-terminal truncations of yeast SmD3, at sites denoted by reverse arrows in Figure 2B. The wild-type and truncated alleles were placed on CEN LEU2 plasmids under the control of the native SMD3 promoter and tested by plasmid shuffle for complementation of a smd3▵ p[CEN URA3 SMD3] strain. The resulting SMD3-(1-93) and SMD3-(1-82) strains were viable after FOA selection and grew as well as wild-type SMD3 cells on YPD agar (Fig. 2C). The SMD3-(1-75) allele was lethal, i.e., it failed to complement growth of the shuffle test strain on FOA at any temperature tested. We conclude that (i) the carboxy-terminal 20 amino acids (embracing the distal α helix) are dispensable for yeast SmD3 function in vivo; and (ii) the segment from amino acids 76–82 is essential for yeast viability. We found that the steady-state levels of U1 and U2 snRNAs in SMD3-(1-93) and SMD3-(1-82) cells were similar to those of wild-type SMD3 cells, as gauged by primer-extension (Supplemental Fig. S4A).
Genetic interactions of SmD3 carboxy-terminal truncations
We surveyed genetic interactions of the two otherwise benign SmD3 carboxy-terminal truncations with mud2▵, nam8▵, mud1▵, and tgs1▵. The results (Fig. 2C) disclosed an informative hierarchy of synthetic mutational effects. SMD3-(1-93) and SMD3-(1-82) were lethal at all temperatures in the absence of Mud2, indicating that the essential contributions of the SmD3 carboxy-terminus to early spliceosome assembly/stability are buffered by the cross-intron bridging interactions of Mud2 (engaged with Msl5 at the branchpoint) with the U1 snRNP. SMD3-(1-93) was barely viable in the nam8▵, mud1▵, and tgs1▵ genetic backgrounds. SMD3-(1-82) was synthetically lethal with mud1▵ and barely viable in the nam8▵ and tgs1▵ backgrounds. These extreme synergies indicate that the SmD3 carboxy-terminus plays an important, albeit buffered, role in the function of the U1 snRNP.
Probing the interactions of SmD3 with Yhc1
SmD3 is the sole member of the Sm ring that interacts with U1-C/Yhc1. Therefore, we were acutely interested in the functional significance of the SmD3•U1-C atomic contacts revealed by the new U1 snRNP structure (Kondo et al. 2015). We reported previously that alanine mutation of Yhc1 Asp38, which we now see makes a salt bridge to Arg21 in U1-C/Yhc1 (Fig. 2A), mimicked YHC1-R21A with respect to bypass of prp28▵ and synthetic lethality with mud2▵ (Schwer and Shuman 2014). Here we performed an alanine scan of the other yeast SmD3 equivalents of human SmD3 residues that contact U1-C (Fig. 2A). These include (using the yeast SmD3 amino acid numbering): Lys9, which makes a hydrogen bond to the main-chain carbonyl of position 4 and a salt bridge to the carboxylate of residue 7 in U1-C/Yhc1; Glu13, which makes a hydrogen bond to the U1-C/Yhc1 Tyr8 hydroxyl; and Glu35, which makes a salt bridge to U1-C/Yhc1 Lys22 (Fig. 2A). Also, human SmD3 Asn79 (Asn80 in yeast SmD3) makes a hydrogen bond to U1-C Trp41 (corresponding to Lys41 in Yhc1). The K9A, D13A, E35A, and N80A mutations were introduced into the gene encoding full-length SmD3 and the alleles were tested by plasmid shuffle for smd3▵ complementation. SMD3 K9A, D13A, E35A, and N80A strains were viable after FOA selection and grew as well as wild-type SMD3 cells on YPD agar (Fig. 3, top panel; Fig. 4, top panel). K9A, D13A, and E35A displayed no mutational synergies with mud2▵ or nam8▵ (Fig. 3). Whereas K9A and E35A also displayed no synergies with mud1▵, the SMD3-E13A allele was severely sick at 37°C in the mud1▵ background (Fig. 3). The K9A, D13A, and E35A alleles did not bypass prp28▵. The SMD3-N80A allele caused no synthetic growth defects in the mud2▵, mud1▵, or nam8▵ backgrounds (Supplemental Fig. S5).
FIGURE 3.

Synthetic interactions of SmD3-Ala mutants. Yeast smd3▵ strains bearing the indicated SMD3-Ala alleles on a CEN LEU2 plasmid in an otherwise wild-type (top panel), mud2▵, mud1▵, or nam8▵ background as indicated were spot-tested for growth on YPD agar at the temperatures specified. Synthetic growth defects are denoted by •.
FIGURE 4.

Synthetic interactions of SmD3-Ala mutants. Yeast smd3▵ strains bearing the indicated SMD3-Ala alleles on a CEN LEU2 plasmid in an otherwise wild-type (top panel), mud2▵, mud1▵, nam8▵, or tgs1▵ background as indicated were spot-tested for growth on YPD agar at the temperatures specified. Synthetic growth defects are denoted by •.
Effects of mutating the SmD3 interface with SmB
The Sm ring is stabilized by main-chain and side-chain contacts between neighboring Sm protein subunits. SmB is the neighbor of SmD3 in the seven-subunit Sm ring. The 3.6 Å crystal structure of the human U4 snRNP core domain (Leung et al. 2011) highlighted an SmD3–SmB interface wherein (i) human SmD3 side-chain Lys84 and Lys87 (corresponding to yeast SmD3 Lys85 and Lys86) (Fig. 2B) make salt bridges to human SmB Asp44 (corresponding to yeast SmB Glu47) and Asp14 (equivalent to yeast SmB Asp17) and (ii) human SmD3 Arg69 (Lys70 in yeast SmD3) makes a salt bridge to Asp23 in human SmB (Asp27 in yeast SmB). To probe the importance of the SmD3 side of this interface, we mutated yeast SmD3 Lys70, Lys85, and Lys86 individually to alanine. The SMD3 K70A, K85A, and K86A strains were viable and grew normally on YPD agar (Fig. 4, top panel). The K70A, K85A, and K86A alleles engendered no mutational synergies with mud2▵ or nam8▵ (Supplemental Fig. S5). Yet they synergized uniquely with mud1▵, such that SMD3-K70A mud1▵ cells had a severe ts growth defect at 37°C and SMD3-K85A mud1▵ and SMD3-K86A mud1▵ cells grew slowly at 37°C (Supplemental Fig. S5).
Perturbing the interface of SmD3 with the snRNA Sm site
Each nucleotide of the U RNA Sm site is engaged by one of the subunits of the Sm protein ring. This entails a stereotypic set of contacts whereby the planar nucleobase is sandwiched by an arginine side chain from the β4–β5 loop (which makes a π-cation stack on the nucleobase) and a side chain from the β2–β3 loop, while an asparagine side chain of the β2–β3 loop makes hydrogen bonds to the nucleobase edge. Main-chain amides in the β2–β3 loop make additional hydrogen bonds to the nucleobase. In the case of the U1 snRNP (Kondo et al. 2015), the SmD3 subunit captures a uridine nucleotide of the Sm site. SmD3 side-chain Asn40 (Asn41 in yeast SmD3) makes bidentate hydrogen bonds from Nδ and Oδ to the O4 and N3 atoms of the uracil nucleobase (Fig. 2A). SmD3 Arg64 (Arg65 in yeast SmD3) makes the π-cation stack on the uracil and also makes a hydrogen bond to the ribose 2′-OH of the 5′-flanking uridine nucleoside (Fig. 2A). SmD3 Asn39 (Ser39 in yeast SmD3) completes the sandwich on the other face of the uracil. To interrogate the contributions of these contacts, we mutated yeast SmD3 residues Ser39, Asn41, and Arg65 to alanine. The S39A, N41A, and R65A strains were viable and grew normally on YPD agar (Fig. 4, top panel) and had normal steady-state levels of U1 and U2 snRNAs (Supplemental Fig. S4A). The S39A, N41A, and R65A alleles also supported normal growth in the nam8▵ genetic background (Fig. 4; Supplemental Fig. S5). The noteworthy findings were that these three mutations elicited distinct synergies with null mutations of other splicing factors. For example: SMD3-N41A mud2▵ was sick at 18°C–30°C and failed to grow at higher temperatures; SMD3-N41A mud1▵ failed to grow at 37°C; and SMD3-N41A tgs1▵ was barely viable (Fig. 4). In contrast, SMD3-S39A had no effect on the growth of the mud2▵, mud1▵ or tgs1▵ strains (Fig. 4). The SMD3-R65A allele was benign in the mud2▵ background and caused only slowed growth at 37°C in the mud1▵ background (Supplemental Fig. S5). These results implicate the nucleobase-specific contacts of the Asn41 side chain as the most important (albeit genetically buffered) for SmD3 function.
To evaluate whether there is functional redundancy of the Sm RNA-binding residues of SmD3, we constructed S39A-R65A and N41A-R65A double-mutants and found that they too grew normally on YPD agar (Fig. 3, top panel) and had normal steady-state levels of U1 and U2 snRNAs (Supplemental Fig. S4A). The double-mutants displayed an expanded set of mutational synergies vis-à-vis the single-mutants. For example, whereas S39A and R65A were benign with mud2▵, the S39A-R65A double-mutation caused a tight ts growth defect in the mud2▵ background (Fig. 3). N41A and R65A were benign with nam8▵, but N41A-R65A was ts in the nam8▵ background (Fig. 3).
Finally, we constructed a triple-mutant S39A-N41A-R65A that would efface all three of the SmD3 side-chains that contact the Sm RNA site. The S39A-N41A-R65A strain was very sick at 18°C–30°C and failed to thrive at higher temperatures (Supplemental Fig. S6). The same was true in nam8▵ and mud1▵ backgrounds. The S39A-N41A-R65A allele was synthetically lethal with mud2▵ and tgs1▵.
Carboxy-terminal truncations of yeast SmB define a minimized functional domain
The amino-terminal half of the 196-aa S. cerevisiae SmB protein is homologous to the amino-terminal 91-amino acid segment of 231-amino acid human SmB modeled in the U1 snRNP crystal structure (Fig. 5B), but their carboxy-terminal primary structures diverge. S. cerevisiae SmB displays more extensive homology over its full length to the 165-aa Candida glabrata SmB, interrupted by a 26-amino acid insert near the carboxy-terminus of S. cerevisiae SmB that is missing from the C. glabrata protein (Fig. 5B). The amino-terminal SmB fold consists of a helix and a β sheet of the same topology as in SmD3 (Fig. 5A,B). S. cerevisiae and C. glabrata SmB have a longer β4–β5 loop than does human SmB, but the fungal and human proteins are otherwise conserved across their amino-termini, with 59 positions of amino acid identity/similarity in the three aligned polypeptides (Fig. 5B). We used the sequence alignment to guide carboxy-terminal deletions in S. cerevisiae SmB at sites denoted by reverse arrows in Figure 5B. The wild-type and truncated yeast SMB1 alleles were placed on CEN HIS3 plasmids under the control of the native SMB1 promoter and tested by plasmid shuffle for complementation of a smb1▵ p[CEN URA3 SMB1] strain. The SMB1 (1-180), (1-158), (1-138), (1-128), (1-118), and (1-101) strains grew as well as wild-type SMB1 cells on YPD agar at 20°C–37°C (Supplemental Fig. S7). [The steady-state levels of U1 and U2 snRNAs were similar in wild-type SMB1 and SMB1-(1-118) cells (Supplemental Fig. S4B).] The SMB1 (1-93) and (1-87) alleles were lethal. We conclude that (i) the carboxy-terminal 95 amino acids are dispensable in vivo, i.e., the core yeast SmB fold suffices for function; and (ii) the segment from amino acids 94–101 that includes the β5 strand is essential.
FIGURE 5.
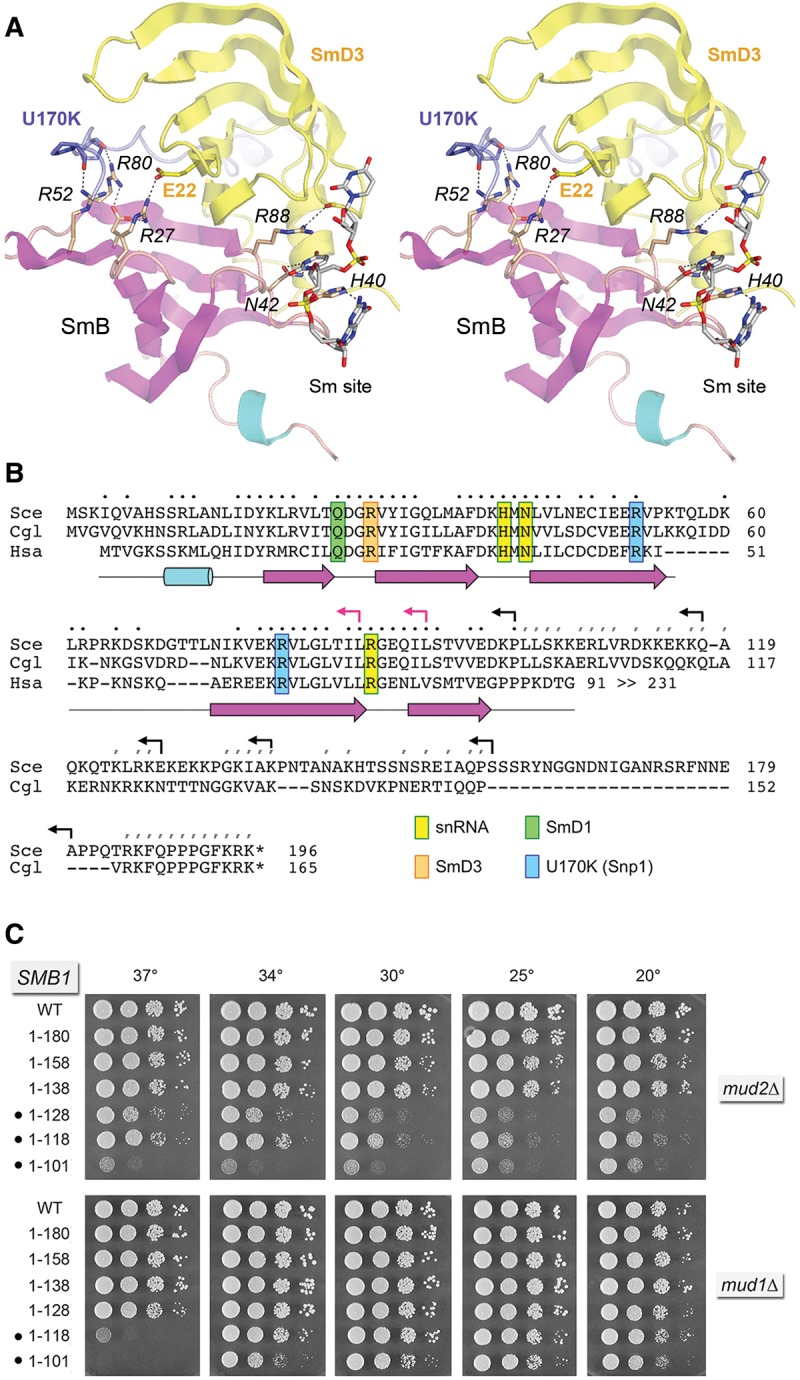
Structure-guided mutagenesis of SmB. (A) Stereo view of the human U1 snRNP structure highlighting the fold of SmB (depicted as a cartoon trace with magenta β strands and cyan helices) and its interactions with neighboring subunits SmD3 (yellow) and U1-70K/Snp1 (blue) and with the Sm site in U1 snRNA. Selected amino acids are shown as stick models and numbered according to their positions in the yeast polypeptides. Atomic contacts are indicated by dashed lines. (B) Alignment of the primary structures of the S. cerevisiae (Sce), Candida glabrata (Cgl), and human (Hsa) SmB. Positions of side-chain identity/similarity in the amino-terminal segments of all three proteins are indicated by • above the alignment. Positions of side chain identity/similarity between the carboxy-terminal segments of SceSmB and CglSmB are denoted by /. The secondary structure elements of the amino-terminal domain are depicted below the alignment, with β strands as magenta arrows and α helices as cyan cylinders. SmB amino acids that make contacts to other U1 snRNP subunits and contact the U1 snRNA are highlighted in color-coded boxes as indicated. Reverse arrowheads indicate the boundaries of the carboxy-terminal truncations of yeast SmB; black and red arrowheads denote viable and lethal truncations, respectively. (C) The wild-type and truncated SMB1 alleles were tested for activity by plasmid shuffle in smb1▵ mud2▵ and smb1▵ mud1▵ strains. The growth phenotypes of viable FOA-resistant smb1▵ strains bearing the indicated SMB1 alleles are shown. Synthetic defects are denoted by •.
Genetic interactions of SmB carboxy-terminal truncations
Whereas none of the viable SMB1-C▵ alleles displayed a synthetic growth phenotype in the absence of Nam8 (Supplemental Fig. S7), they synergized in distinct ways with mud2▵ and mud1▵ (Fig. 5C). The SMB1-(1-138) allele supported normal growth of mud2▵ cells, but further truncation of the carboxy-terminus to residue 128 or shorter resulted in synthetic sickness across the range of growth temperatures. In contrast, SMB1-(1-128) mud1▵ cells thrived, but trimming the SmB carboxy-terminus back to position 118 or 101 resulted in progressive failure to grow at 37°C (Fig. 5C). Our results indicate that (i) different segments of the SmB carboxyl domain interact genetically with different components of the early spliceosome; and (ii) deletions of the SmD3 and SmB carboxyl terminal tails have quite different genetic interaction profiles.
We evaluated the effects of simultaneously truncating the SmD3 and SmB carboxy-termini (Supplemental Fig. S8). The SmD3-(1-93) truncation, which was catastrophic in the absence of Mud2, Mud1, Nam8 and Tgs1 (Fig. 2C), had only a mild effect on growth of the SMB1-▵C allelic series, whereby it caused slowed growth at low temperatures when the SmB carboxy-terminus was retracted to positions 128, 118, and 101 (Supplemental Fig. S8).
Perturbing the interface of SmB with the snRNA Sm site
In the U1 snRNP structure, the SmB protein engages the uridine nucleotide of the Sm site immediately 3′ of the uridine bound by SmD3. SmB side chain Asn39 (Asn42 in yeast SmB) makes bidentate hydrogen bonds from Nδ and Oδ to the O4 and N3 atoms of the uracil nucleobase (Fig. 5A). SmB Arg73 (Arg88 in yeast SmB) makes the π-cation stack on the uracil and also makes a hydrogen bond to a main-chain carbonyl in the β2–β3 loop of the adjacent SmD3 subunit (Fig. 5A). SmB His37 (His40 in yeast SmB) completes the sandwich on the other face of the uracil and it also donates a hydrogen bond to the 3′ flanking guanine nucleobase (Fig. 5A). Here we mutated yeast SmB residues His40, Asn42, and Arg88 to alanine. The H40A, N42A, and R88A strains were viable, grew normally on YPD agar (Fig. 6, top panel), and had normal steady-state levels of U1 and U2 snRNAs (Supplemental Fig. S4B). The H40A, N42A, and R88A alleles also supported normal growth in the absence of Nam8 (Fig. 6). As with SmD3, the three Sm-binding site mutations in SmB elicited distinct synergies with null mutations of other splicing factors. For example: SMB1-N42A mud2▵ was sick at 34°C–37°C; SMB1-N42A mud1▵ grew slowly at 37°C; and SMB1-N42A tgs1▵ was constitutively sick (Fig. 6). In contrast, SMB1-H40A had no effect on the growth of the mud2▵, mud1▵, or tgs1▵ strains (Fig. 6). The SMB1-R88A allele was sick in the tgs1▵ background (Fig. 6). We also simultaneously changed His40 and Asn42 to alanine. SMB1-H40A-N42A cells grew well at 18°C–34°C, but slowly at 37°C. SMB1-H40A-N42A was synthetically lethal with tgs1▵ and barely viable only at low temperatures in the mud2▵ background (Fig. 6). These results implicate the nucleobase-specific contacts of the Asn42 side chain as the most important of the three RNA-binding side chains for SmB function. Indeed, the phenotypes and synergies of the RNA site mutations in SmB were quite concordant with those for SmD3.
FIGURE 6.

Genetic interactions of SmB-Ala mutants. Yeast smb1▵ strains bearing the indicated SMB1-Ala allele on a CEN HIS3 plasmid in an otherwise wild-type (top panel), mud2▵, mud1▵, nam8▵, or tgs1▵ background as indicated were spot-tested for growth on YPD agar at the temperatures specified. Synthetic growth defects are denoted by •.
Effects of simultaneous Sm site mutations in SmD3 and SmB
The remarkable tolerance of the Sm sites of SmD3 and SmB to single and double alanine mutations of the trio of nucleotide-binding amino acids suggested that loosening of one RNA contact by a member of the Sm ring is not especially consequential in an otherwise wild-type genetic background. One simple explanation is that the Sm ring system has built-in redundancy, whereby the other six RNA-binding sites can pick up the slack when one site is mutated. This raises an important question of how many Sm-binding sites suffice for biological activity of the yeast Sm ring. We addressed this issue here by simultaneously introducing single alanine changes in lieu of RNA-binding amino acids of SmD3 and SmB: either synonymous changes of the equivalent contact residues (e.g., Ser39/His40, Asn41/Asn42, and Arg65/Arg88 in SmD3/SmB) or pairs of residues making different contacts. Among the synonymous double mutations, SMD3-N41A SMB1-N42A and SMD3-R65A SMB1-R88A were lethal (Fig. 7). This result shows that Sm ring function in vivo is fatally compromised when the sequential nucleotide-binding pockets of the SmD3 and SmB subunits lose their nucleobase hydrogen-bonding and π-cation stacking side chains. In contrast, simultaneous loss of the other nucleobase-sandwiching residues in the SMD3-S39A SMB1-H40A strain had no effect on cell growth (Fig. 7).
FIGURE 7.
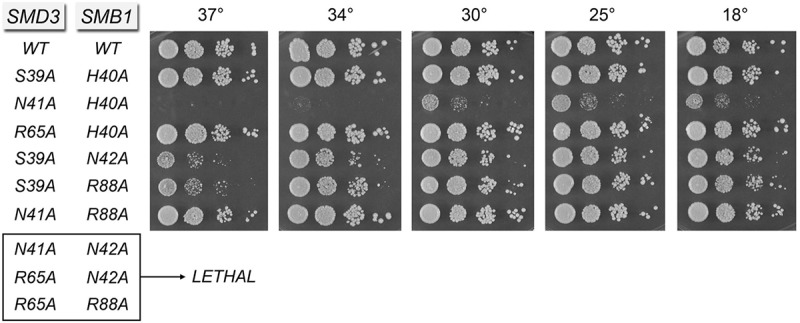
Effects of simultaneous Sm site mutations in SmD3 and SmB. SMD3 (WT) and SMD3-Ala alleles on CEN LEU2 plasmids were cotransformed with SMB1 (WT) and SMB1-Ala alleles on CEN HIS3 plasmids into a yeast smd3▵ smb1▵ p(CEN URA3 SMD3 SMB1) strain. The indicated viable FOA-resistant strains recovered after plasmid shuffle were spot tested for growth on YPD agar at the temperatures specified. SMD3 SMB1 allele pairs listed at the bottom failed to complement smd3▵ smb1▵ in the plasmid shuffle assay and were deemed synthetically lethal.
Among the nonsynonymous double mutants, SMD3-R65A SMB1-N42A was lethal whereas the “inverse” SMD3-N41A SMB1-R88A change had no effect on growth (Fig. 7). The SMD3-N41A SMB1-H40A mutant was very sick at 18°C–30°C and failed to thrive at 34°C–37°C, but the SMD3-R65A SMB1-H40A combination had no effect on growth (Fig. 7). Clearly, the effect of subtracting SmB His40 depends very much on the SmD3 mutation with which it is paired. In contrast, the corresponding S39A change in SmD3 caused only a modest ts phenotype when paired with N42A or R88A in SmB (Fig. 7).
Probing the interactions of SmD3 and SmB with U1–70K (Snp1)
In the U1 snRNP structure (Kondo et al. 2015), SmD3 and SmB interact with the amino-terminal peptide of the U1-specific subunit U1-70K, the homolog of yeast Snp1. The structure model includes U1-70K residues 2–59, which are aligned in Figure 8 to the corresponding amino-terminal segment of yeast Snp1, revealing 19 positions of side chain identity/similarity. Two amino acids in SmD3 make atomic contacts with a conserved arginine residue in U1-70K/Snp1. These are (using the yeast SmD3 and Snp1 amino acid numbering): Arg30, which makes a hydrogen bond to the Arg19 main-chain carbonyl in Snp1 (Arg16 in U1-70K); and Asp76, which makes a salt bridge to the Arg19 side chain in Snp1 (Fig. 2A). We found that mutating yeast SmD3 residues Arg30 and Asp76 to alanine had no effect on yeast growth on YPD agar (Fig. 3, top panel), from which we surmise that these constituents of the SmD3•Snp1 interface are inessential per se for U1 snRNP function in yeast.
FIGURE 8.

Genetic interactions of Snp1 amino-terminal truncations. (Top) Alignment of the amino-terminal amino acid sequences of the S. cerevisiae Snp1 and human U1-70K. Positions of side chain identity/similarity are indicated by • above the alignment. Arrowheads indicate the boundaries of the ▵N truncations of Snp1. (Bottom) Yeast snp1▵ strains bearing the indicated SNP1-▵N allele on a CEN HIS3 plasmid in an otherwise wild-type (top panel), mud2▵, mud1▵, nam8▵, or tgs1▵ background as indicated were spot-tested for growth on YPD agar at the temperatures specified. Synthetic growth defects are denoted by •.
The SMD3 R30A and D76A alleles elicited no mutational synergies with mud2▵ or nam8▵ (Fig. 3). Whereas D76A also thrived in the mud1▵ background, the SMD3-R30A mud1▵ strain was inviable at 37°C (Fig. 3). Thus, R30A resembled E13A in its distinctively narrow synthetic genetic interactions, limited to a ts growth defect in the absence of Mud1. The R30A and D76A alleles did not bypass prp28▵.
Two conserved amino acids in SmB make atomic contacts with the main-chain of U1-70K/Snp1. Human SmB side chains Arg49 and Arg65 (corresponding to yeast SmB Arg52 and Arg80) (Fig. 5A) donate hydrogen bonds from their terminal guanidinium nitrogens to the U1-70K Pro23 and Pro20 main-chain carbonyls, respectively (corresponding to residues 23 and 27 in yeast Snp1) (Fig. 5A). Mutating yeast SmB residues Arg52 and Asp80 to alanine had no effect on yeast growth (Fig. 6, top panel), suggesting that the SmB•Snp1 interface is also inessential per se for U1 snRNP function in yeast. The SMB1 R52A and R80A strains had apparently normal steady-state levels of U1 and U2 snRNAs (Supplemental Fig. S4B).
The SMB1 R52A and R80A alleles elicited no mutational synergies with mud2▵ or nam8▵ or tgs1▵ (Fig. 6). Rather, R52A and R80A synergized uniquely with mud1▵, causing a severe ts growth defect at 37°C (Fig. 6). It is noteworthy that alanine mutations of the SmD3•Snp1 interface (R30A) and the SmB•Snp1 interface (R52A, R80A) phenocopy each other with respect to their genetic interaction with Mud1.
Genetic interactions of Snp1 amino-terminal truncations
Deletions of the amino-terminal 21-amino acid peptide of yeast Snp1 that, in U1-70K, interacts with SmD3 and with U1-C had no effect on yeast growth at 18°C–37°C (Fig. 8). The N▵11, N▵14, and N▵21 SNP1 alleles were then surveyed for mutational synergies. N▵11 and N▵14 were uniquely synthetically ts in the mud1▵ background, but had no effect on growth in mud2▵, nam8▵, or tgs1▵ strains (Fig. 8). SNP1-N▵21 mud1▵ was barely viable at 18°C–25°C and failed to grow at 30°C–37°C. In contrast, SNP1-N▵21 mud2▵, SNP1-N▵21 nam8▵, and SNP1-N▵21 tgs1▵ cells thrived at 30°C–34°C, though they grew slowly at 37°C. Thus, the amino-terminal deletions of Snp1 phenocopy the predicted U1-70K/Snp1 interaction mutations of Yhc1 (Y38A), SmD3 (R30A), and SmB (R52A, R80A), with respect to specificity of synthetic interaction with Mud1.
Genetic interactions of SmD3 with U2 snRNP subunit Lea1
In the preceding sections, we focused on genetic interactions of Sm ring subunits with splicing factors involved in the earliest steps of spliceosome assembly, i.e., U1 snRNP subunits and Mud2. The U2, U4, and U5 snRNPs that act at later stages of the splicing pathway have the same Sm ring. The step immediately following commitment complex formation is the exchange of the U2 snRNP for Msl5•Mud2 at the branchpoint to form a pre-mRNA•U1•U2 complex. To address genetically whether and how our collection of SmD3 mutants might affect U2 snRNP function, we screened them for phenotypes in a yeast strain that lacks Lea1, which is an intrinsic subunit of the yeast U2 snRNP and a homolog of metazoan U2A′ (Caspary and Séraphin 1998; Caspary et al. 1999; Schwer et al. 2011).
Yeast lea1▵ cells thrive at 18°C–34°C but not at 37°C. The SMD3-Ala mutants R30A, D76A, N80A, K85A, and R86A caused no growth defects in the lea1▵ strain (Fig. 9). The SMD3-K9A lea1▵ and SMD3-E13A lea1▵ strains were slower growing at 34°C. In contrast, the S39A, N41A, and R65A mutations of the Sm RNA-binding site and the K70A mutation of the SmD3–SmB interface were synthetically lethal with lea1▵ (Fig. 9). The carboxy-terminal truncation mutants SMD3-(1–93) and SMD3-(1-82) were also synthetically lethal with lea1▵ (Fig. 9). Thus, the critical contributions of SmD3's RNA-binding site and carboxy-terminal tail to U2 snRNP function are genetically redundant to those of Lea1.
FIGURE 9.
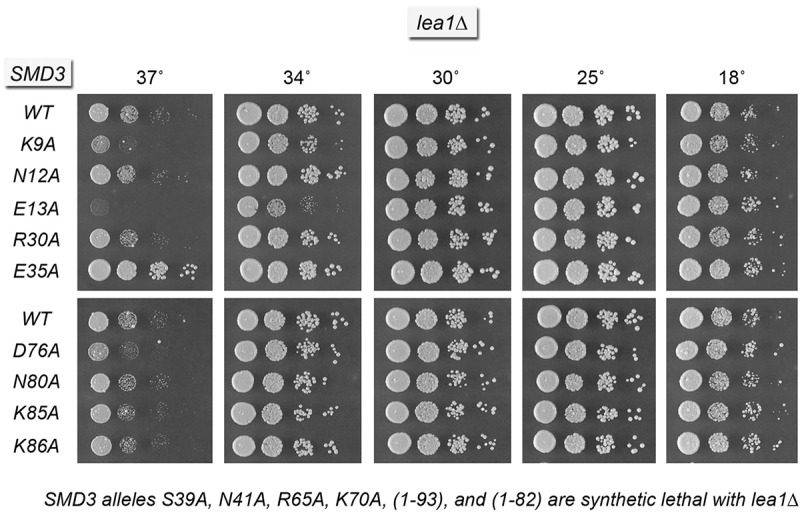
Genetic interactions of SmD3 with U2 snRNP subunit Lea1. Yeast lea1▵ strains bearing the indicated SMD3 alleles on a CEN LEU2 plasmid were spot-tested for growth on YPD agar at the temperatures specified.
A most remarkable finding was that of the SMD3-E35A allele uniquely suppressed the temperature-sensitivity of lea1▵, allowing for seemingly normal growth at 37°C (Fig. 9). We conclude that hypomorphic SmD3 mutations exert distinctive and allele-specific effects, negative and positive, on the functions of the U1 and U2 snRNPs.
DISCUSSION
Structure-guided genetic insights to U1-C/Yhc1 function
The present study marries the structural biology of the human U1 snRNP to the powerful genetics of budding yeast to gain new insights to structure–function relationships for the essential U1-C/Yhc1 subunit. The results, here and previously (Schwer and Shuman 2014), of a cumulative alanine scan of 24 conserved amino acids of the amino-terminal domain of Yhc1 reveal that, with the exception of three constituents of the zinc-binding cluster, none of the conserved residues are essential for yeast growth under standard laboratory conditions. This includes the yeast Yhc1 counterparts of all of the U1-C amino acid side chains that contact the U1:5′SS RNA duplex in the human U1 snRNP crystal structure (Ser11, Thr14, His15, Ser19, Val20, His24) (Fig. 1D). Yet, for Yhc1, as for so many components of the early yeast spliceosome, the effects of perturbing protein–RNA and protein–protein interactions are masked in an otherwise wild-type genetic background, because of built-in functional redundancy of the yeast splicing machine, especially the U1 snRNP.
The highly instructive findings here pertain to the key role of neighboring Yhc1 residues Ser19 and Val20 at the RNA interface. Val20 makes van der Waals contacts to the conserved C9 and U10 nucleosides of the U1 snRNA leader; Ser19 makes a hydrogen bond to the C9 ribose 2′-OH. Subtraction of Val20 is lethal in the absence of the Mud2 subunit of the Msl5•Mud2 intron branchpoint-binding complex and causes a severe growth defect in the absence of the U1 snRNP subunit Nam8. Loss of the Ser19 hydrogen bond caused a severe growth defect in the absence of Mud2. Bridging interactions between U1 snRNP at the 5′SS and Msl5•Mud2 at the branchpoint are important to establish and stabilize the early spliceosome. Thus, the dire impact in vivo of weakening the Yhc1-U1 RNA interface is felt only when other stabilizing factors are removed. Consistent with this interpretation, we find that subtracting Val20 or Ser19 bypasses the essentiality of Prp28.
Initial studies of the Prp28 bypass phenomenon by mutations of Yhc1 Leu13 were interpreted to mean that Leu13 makes specific contact to stabilize the U1 snRNA:5′SS duplex (Chen et al. 2001). However, the U1 snRNP structure clarifies that Leu13 does not make atomic contacts to RNA; Leu13 is 4.7 Å away from the G1 ribose of the 5′SS RNA. Because the first Prp28 bypass mutation of Yhc1 was mapped to L13F, one could envision that the added bulk of the phenylalanine might create a steric clash with the mRNA 5′SS. However, this does not hold for the second bypass mutation identified as L13S, or for the finding that 11 other substitutions for Leu13 also bypassed Prp28 (Chen et al. 2001). We construe that the Leu13 mutations bypass Prp28 via indirect effects on the conformation of the Yhc1 globular N-domain. Subsequent studies have shown that alanine mutations of other Yhc1 residues more remote from the RNA interface can also bypass Prp28, for example: Arg21, an important residue that mediates Yhc1 interaction with SmD3 (Schwer and Shuman 2014); and, as shown here, Tyr4. In the U1 snRNP structure, the Phe4 equivalent of Tyr4 makes direct contact to Arg21. Thus, we speculate that R21A and Y4A (which share the property of being synthetically lethal with mud2▵) bypass Prp28 via a common route involving weakening the Yhc1•SmD3 interface.
YHC1 V20F and V20A are the best Prp28 bypass suppressors in our collection, i.e., they permit growth of prp28▵ cells at all temperatures tested. In contrast, S19A bypass of Prp28 is cold-sensitive. It is conceivable that this difference in bypass strength reflects the nature, number, and nucleotide positions of the RNA contacts made by Val20 and Ser19. The nucleotide position of Yhc1's RNA contacts with the U1 snRNA:5′SS duplex may well be relevant to Prp28 bypass, insofar as alanine mutations of RNA-interaction residues Ser11, Thr14, and His15 do not bypass prp28▵. In this vein, it is worth noting that among the mutations of the U1 snRNA 5′ leader that have been shown to bypass Prp28, the strongest bypass was achieved by singly deleting the U10 nucleotide (Schwer et al. 2013), this being one of the leader positions contacted by Val20.
Structure–function analysis of yeast Sm proteins SmD3 and SmB
We extended the structure-guided mutagenesis to two of the subunits of the yeast Sm ring: SmD3, which interacts closely with U1-C/Yhc1 in the U1 snRNP; and SmB, the neighbor of SmD3. We began by defining minimum functional domains of SmD3 and SmB by serial carboxy-terminal truncations. Although it has been appreciated that their carboxy-terminal tails are dispensable for vegetative growth (Zhang et al. 2001), the domain margins had not been refined, nor had genetic interactions of the Sm protein truncations with non-Sm splicing factors been interrogated. Underscoring the theme of redundancy in the yeast U1 snRNP, we show that otherwise benign SMD3-C▵ mutants are barely viable in the absence of U1 snRNP subunits Nam8 or Mud1 and dead in the absence of Mud2. In contrast, benign SMB1-C▵ mutants are unaffected by the absence of Nam8 and have relatively modest synthetic growth defects in the absence of Mud1 or Mud2. We surmise that the carboxy-terminal tails of these Sm ring subunits play distinct (though genetically buffered) roles in U1 snRNP function.
In the U1 snRNP, SmD3, SmB, and U1-C/Yhc1 interact with the amino-terminus of U1-70K/Snp1. Replacing the side chains at the respective U1-70K/Snp1 interfaces with alanine had no effect on yeast vegetative growth, nor did deletion of the interacting Snp1 amino-terminal peptide. Genetic interaction tests revealed synergy with mud1▵ as a shared feature of perturbing protein contacts to the Snp1 amino-terminus. In other words, Mud1 masks functional deficits incurred by these perturbations.
Our genetic analysis of the Sm RNA-binding sites of SmD3 and SmB provides novel insights to the built-in redundancies of the Sm ring, whereby no individual side-chain of the conserved trio in the RNA-binding pocket is essential for yeast growth, but simultaneous mutations of the RNA-binding sites in the adjacent SmD3 and SmB ring subunits are lethal. This result is proof-of-principle that six of seven intact RNA-binding sites in the Sm ring can suffice for in vivo function, but five sites may not. Of course, this conclusion applies narrowly to SmD3 and SmB. It is conceivable that one of the other five Sm protein RNA sites is essential per se, or that yeast might survive if a different pair of RNA sites within the ring was disabled. Addressing this issue will require genetic interrogation of the RNA sites of all seven yeast Sm proteins, alone and in all combinations.
Benign mutations of the RNA sites of SmD3 and SmB elicited synthetic phenotypes with Mud1 and Mud2, suggesting that the Sm mutations affect U1 snRNP function and early spliceosome assembly. The intra-U1 genetic interactions were specifically with Mud1, i.e., there was little or no phenotype of the RNA-binding site mutations in the absence of Nam8. There are no available structures to help intuit how Mud1 and Nam8 fit into the yeast U1 snRNP. The genetics point toward distinct contributions of Nam8 and Mud1 to U1 snRNP function in vivo.
Finally, although our main focus here was U1 snRNP-centric with respect to genetic interactions of SmD3 and SmB, we did extend the analysis of SmD3 to its role in U2 snRNP function, by unveiling negative and positive mutational synergies with the U2 snRNP subunit Lea1. We expect our allelic series will be useful for probing Sm ring function in the context of snRNPs that act at steps of the splicing pathway downstream from U1 and U2.
MATERIALS AND METHODS
Yhc1 expression plasmids and mutants. pRS413-YHC1 (CEN HIS3) plasmids bearing wild-type and mutated YHC1 genes under the control of the native YHC1 promoter have been described (Schwer and Shuman 2014). New missense mutations Y4A, E7A, Y8A, S11A, D16A, S19A, V20A, Y37A, Y38A, R39A, and K41A were introduced into YHC1 by two-stage PCR overlap extension with mutagenic primers. The PCR products were digested and then inserted into the pRS413-YHC1 expression plasmid. We used similar strategies to generate pRS415- or pRS413-based expression plasmids harboring wild-type and mutated SMD3 (nucleotides −445 to +552), SMB1 (nucleotides −500 to +875), and SNP1 (nucleotides −400 to +1190) genes. All genes in the resulting p415-SMD3 (CEN LEU2), p413-SMB1 (CEN HIS3), and p413-SNP1 (CEN HIS3) plasmids were sequenced completely to confirm that no unwanted changes were acquired during amplification and cloning.
Yeast strains and tests of function in vivo. Strains yhc1▵ [p316-YHC1], smd3▵ [p316-SMD3], and smd3▵ prp28▵ [p316-PRP28-SMD3] and assays to test the effects of YHC1 and SMD3 mutations in various genetic backgrounds have been described (Schwer and Shuman 2014). To develop plasmid shuffle assays to test the effects of SMB1 and SNP1 mutations in various genetic backgrounds, we first generated smb▵ [p316-SMB1] and snp1▵ [p316-SNP1] haploid cells by sporulation and dissection of heterozygous SMB1 smb1▵::kanMX and SNP1 snp1::kanMX diploids (Open Biosystems) that had been transfected with URA3 SMB1 (nucleotides −500 to +875) or URA3 SNP1 (nucleotides −400 to +1190) plasmids, respectively. smb1▵ [p316-SMB1] and snp1▵ [p316-SNP1] cells were resistant to G418 and unable to grow on medium containing 0.75 mg/mL 5-fluoroorotic acid (FOA). To assay the function of, for example, wild-type and mutated SMB1 alleles, smb1▵ [p316-SMB1] cells were transfected with CEN HIS3 SMB1 plasmids. Individual His+ transformants were selected and streaked on agar medium containing FOA. The plates were incubated at 20°C, 30°C, or 37°C and mutants that failed to form macroscopic colonies at any temperatures after 8 d were deemed lethal. Individual FOA-resistant colonies with viable SMB1 alleles were grown to mid-log phase in YPD broth and adjusted to the same A600 values. Aliquots (3 µL) of serial 10-fold dilutions were spotted to YPD agar plates, which were then incubated at temperatures ranging from 18°C to 37°C. We also developed plasmid shuffle assays to test mutational effects on SmB1 and Snp1 function in mud2▵, nam8▵, mud1▵, and tgs1▵ cells, and on SmD3 function in lea1▵ cells, using standard genetic manipulations of mating, sporulation, and dissection.
To investigate genetic interactions of SMD3 and SMB1, we first generated heterozygous smd3▵ SMD3 smb1▵ SMB1 diploids by crossing smd3▵::hygMX [p316-SMD3] cells with smb1▵::kanMX [p316-SMB1] cells of the opposite mating type, selecting diploids on YPD medium containing hygromycin plus G418, and plating them to FOA-containing medium. The heterozygous diploids were then transfected with a CEN URA3 SMD3 SMB1 plasmid (p316-SMD3-SMB1), in which the SMD3 gene (nucleotides −445 to +552) is arranged in a head-to-head configuration with the SMB1 gene (nucleotides −500 to +875). Ura+ heterozygous diploids were subjected to sporulation and tetrad dissection, after which haploid smd3▵ smb1▵ [p316-SMD3-SMB1] progeny were recovered. These cells were unable to grow on FOA medium, but the double-deletion strains could be complemented by cotransformation with p[CEN HIS3 SMB1] plus p[CEN LEU2 SMD3].
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are most grateful to Prof. Kiyoshi Nagai for sharing the human U1 snRNP crystal structure in advance of publication. This work was supported by the Office of Extramural Research of the National Institutes of Health (NIH) grants GM52470 (S.S.) and GM102961 (B.S.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.050583.115.
REFERENCES
- Abovich N, Rosbash M 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. [DOI] [PubMed] [Google Scholar]
- Abovich N, Liao XC, Rosbash M 1994. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev 8: 843–854. [DOI] [PubMed] [Google Scholar]
- Bordonné R 2000. Functional characterization of nuclear localization signals in yeast Sm proteins. Mol Cell Biol 20: 7943–7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary F, Séraphin B 1998. The yeast U2A′/U2B complex is required for pre-spliceosome formation. EMBO J 17: 6348–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary F, Shevchenko A, Wilm M, Séraphin B 1999. Partial purification of the yeast U2 snRNP reveals novel yeast pre-mRNA splicing factor required for pre-spliceosome assembly. EMBO J 18: 3463–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Schwer B, Shuman S 2012. Structure-function analysis and genetic interactions of the yeast branchpoint binding protein Msl5. Nucleic Acids Res 40: 4539–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Stands L, Staley JP, Jackups RR Jr, Latus LJ, Chang TH 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell 7: 227–232. [DOI] [PubMed] [Google Scholar]
- Colot HV, Stutz F, Rosbash M 1996. The yeast splicing factor Mud13p is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes Dev 10: 1699–708. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. 2010. The genetic landscape of a cell. Science 327: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Lührmann R 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Tang J, Puig O, Salgado J, Neubauer G, Colot HV, Mann M, Séraphin B, Rosbash M, Lührmann R, et al. 1998. A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA 4: 374–393. [PMC free article] [PubMed] [Google Scholar]
- Hage R, Tung L, Du H, Stands L, Rosbash M, Chang TH 2009. A targeted bypass screen identifies Ynl187p, Prp42p, Snu71p, and Cbp80p for stable U1 snRNP/pre-mRNA interaction. Mol Cell Biol 29: 3941–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann S, Zheng S, Costanzo M, Brost RL, Garcin D, Boone C, Shuman S, Schwer B 2008. Genetic and biochemical analysis of yeast and human cap trimethylguanosine synthase: functional overlap of TMG caps, snRNP components, pre-mRNA splicing factors, and RNA decay pathways. J Biol Chem 283: 31706–31718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz A, Schwer B, Smith P, Shuman S 2014. Crystal structure, mutational analysis and RNA-dependent ATPase activity of the yeast DEAD-box pre-mRNA splicing factor Prp28. Nucleic Acids Res 42: 12885–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz A, Chico L, Smith P, Schwer B, Shuman S 2015. Structural basis for recognition of intron branchpoint RNA by yeast Msl5 and selective effects of interfacial mutations on splicing of yeast pre-mRNAs. RNA 21: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach C, Walke S, Yound R, Avis JM, de la Fortelle E, Raker VA, Lührmann R, Li J, Nagai K 1999. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96: 375–387. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Oubridge C, van Roon AM, Nagai K 2015. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKW, Nagai K, Li J 2011. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature 473: 536–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao XC, Tang J, Rosbash M 1991. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: implications for snRNP protein function in pre-mRNA splicing. Genes Dev 7: 419–428. [DOI] [PubMed] [Google Scholar]
- Pomeranz Krummel DA, Oubridge C, Leung AKW, Li J, Nagai K 2009. Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution. Nature 458: 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu ZR, Chico L, Chang J, Shuman S, Schwer B 2012. Genetic interactions of hypomorphic mutations in the m7G cap binding pocket of yeast nuclear cap binding complex: an essential role for Cbc2 in meiosis via splicing of MER3 pre-mRNA. RNA 18: 1996–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S 2014. Structure-function analysis of the Yhc1 subunit of yeast U1 snRNP and genetic interactions of Yhc1 with Mud2, Nam8, Mud1, Tgs1, U1 snRNA, SmD3 and Prp28. Nucleic Acids Res 42: 4697–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Erdjument-Bromage H, Shuman S 2011. Composition of yeast snRNPs and snoRNPs in the absence of trimethylguanosine caps reveals nuclear cap binding protein as a gained U1 component implicated in the cold-sensitivity of tgs1▵ cells. Nucleic Acids Res 39: 6715–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Chang J, Shuman S 2013. Structure-function analysis of the 5′ end of yeast U1 snRNA highlights genetic interactions with the Msl5•Mud2 branchpoint binding complex and other spliceosome assembly factors. Nucleic Acids Res 41: 7485–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell 3: 55–64. [DOI] [PubMed] [Google Scholar]
- Tang J, Abovich N, Fleming MJ, Séraphin B, Rosbash M 1997. Identification and characterization of a yeast homolog of U1 snRNP-specific protein C. EMBO J 16: 4082–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Feltz C, Anthony K, Brilot A, Pomeranz Krummel DA 2012. Architecture of the spliceosome. Biochemistry 51: 3321–3333. [DOI] [PubMed] [Google Scholar]
- Weber G, Trowitzsch S, Kastner B, Lührmann R, Wahl MC 2010. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J 29: 4172–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, et al. 2008. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell 32: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Abovich N, Rosbash M 2001. A biochemical function for the Sm complex. Mol Cell 7: 319–329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.