

Abstract
The OLR1 gene encodes the oxidized low-density lipoprotein receptor (LOX-1), which is responsible for the cellular uptake of oxidized LDL (Ox-LDL), foam cell formation in atheroma plaques and atherosclerotic plaque rupture. Alternative splicing (AS) of OLR1 exon 5 generates two protein isoforms with antagonistic functions in Ox-LDL uptake. Previous work identified six single nucleotide polymorphisms (SNPs) in linkage disequilibrium that influence the inclusion levels of OLR1 exon 5 and correlate with the risk of cardiovascular disease. Here we use minigenes to recapitulate the effects of two allelic series (Low- and High-Risk) on OLR1 AS and identify one SNP in intron 4 (rs3736234) as the main contributor to the differences in exon 5 inclusion, while the other SNPs in the allelic series attenuate the drastic effects of this key SNP. Bioinformatic, proteomic, mutational and functional high-throughput analyses allowed us to define regulatory sequence motifs and identify SR protein family members (SRSF1, SRSF2) and HMGA1 as factors involved in the regulation of OLR1 AS. Our results suggest that antagonism between SRSF1 and SRSF2/HMGA1, and differential recognition of their regulatory motifs depending on the identity of the rs3736234 polymorphism, influence OLR1 exon 5 inclusion and the efficiency of Ox-LDL uptake, with potential implications for atherosclerosis and coronary disease.
Keywords: alternative splicing, OLR1, SNP, SR proteins, RRM, linkage disequilibrium, coronary disease
INTRODUCTION
The process of intron removal from mRNA precursors (pre-mRNA splicing) is mediated by the spliceosome, one of the most complex ribonucleoprotein machineries within the cell (Wahl et al. 2009), and represents an essential and versatile step for the regulation of gene expression in multicellular organisms (Jangi and Sharp 2014). Alternative patterns of intron removal (alternative splicing, AS) expand the coding potential of the genome (Nilsen and Graveley 2010) and alterations of cis and trans-acting elements involved in splice site recognition are frequently associated with the establishment and progression of disease states (Wang and Cooper 2007; Cooper et al. 2009, Tazi et al. 2009).
A major hallmark of the initiation of atherosclerotic lesions is the uptake and accumulation of low-density lipoproteins (LDLs and Ox-LDLs), resulting in foam cell formation in atheroma plaques (Vohra et al. 2006). Oxidized LDL (Ox-LDL) is considered a key initial event in atherosclerosis and also in atherosclerotic plaque rupture by promoting lipid accumulation, pro-inflammatory responses, and apoptotic cell death (Mehta et al. 2006; Chen et al. 2007). OLR1 encodes the scavenger lectin-like oxidized low-density lipoprotein receptor (LOX-1), which is responsible for the uptake of Ox-LDL (Aoyama et al. 1999; Ohki et al. 2005). LOX-1 depletion reduces atherogenesis in mice subject to high cholesterol diets (Mehta et al. 2007) and protects against plaque instability in conditions of hypercholesterolemia (Ishino et al. 2007). In addition, OLR1 is up-regulated in cancer tissues and has been proposed to play an active role in cell transformation (Hirsch et al. 2010; Khaidakov et al. 2011), suggesting molecular links between atherosclerosis and cancer.
OLR1 exon 5 alternative splicing leads to the generation of mRNAs encoding two protein isoforms with antagonistic functions in programed cell death. Exon 5 inclusion generates full-length LOX-1 protein harboring an Ox-LDL binding domain that promotes Ox-LDL uptake (Chen et al. 2001; Ohki et al. 2005) and activation of inflammatory and apoptotic responses (Chen et al. 2007). Skipping of exon 5 generates a truncated protein isoform (LOXIN) that lacks the Ox-LDL binding domain and protects against Ox-LDL uptake (Mango et al. 2005; Ohki et al. 2005).
Six single nucleotide polymorphisms (SNPs) in linkage disequilibrium have been identified within introns 4, 5 and the 3′ UTR of OLR1, which correlate with the risk of cardiovascular disease (Chen et al. 2003; Mango et al. 2003; Tatsuguchi et al. 2003). Carriers of the Low-Risk allele series show equilibrated levels of both AS isoforms, while carriers of the High-Risk allele series display higher exon 5 inclusion (Mango et al. 2005). Understanding the mechanisms behind AS regulation and the role of genetic variation in OLR1 exon 5 AS is therefore of significant biomedical interest.
Here we have characterized in detail the contributions of individual SNP sequence variations to OLR1 exon 5 AS and applied computational, biochemical, mutational and high-throughput screening technologies to identify relevant regulatory sequence motifs and factors. Our results reveal antagonistic effects between SR proteins and other regulators as well as long-range functional interactions between SNPs that modulate the ratio between alternatively spliced isoforms and consequently the efficiency of Ox-LDL uptake.
RESULTS
OLR1 minigenes reproduce the splicing phenotype of the SNP allelic series
To study the role of the different OLR1 alleles in AS regulation, we generated minigenes corresponding to the Low-Risk or High-Risk allelic series in which the corresponding full genomic sequences from exon 4 to exon 6 (including the 3′ UTR), obtained from cell lines of the HapMap project (Table 1; Supplemental Table S1; Supplemental Fig. S1; International HapMap Consortium 2005), were cloned into a pCDNA3.1 backbone under the control of Cytomegalovirus (CMV) promoter and Simian Virus 40 (SV40) polyadenylation signal (Fig. 1A). The minigenes were transfected in HeLa (Human Cervix Adenocarcinoma), HEK293-T (Human Embryonic Kidney) and SK-N-SH (Human Neuroblastoma) cells. Twenty-four hours post-transfection, total cellular RNAs were isolated and the ratio between exon 5 inclusion and skipping isoforms analyzed by reverse transcription and quantitative real-time PCR (RT-qPCR) using splice junction oligonucleotides and a minigene-specific primer (Fig. 1A). Higher inclusion/skipping ratios were consistently detected in transcripts from the High-Risk construct in the three cell lines (Fig. 1B), qualitatively and quantitatively consistent with previous analysis of endogenous OLR1 transcripts in patients bearing Low-Risk or High-Risk allele series (Mango et al. 2005). Thus, our minigenes reproduce the endogenous splicing phenotype of the SNP allelic series and thus provide useful tools to study OLR1 AS (see also Supplemental Fig. S2A).
TABLE 1.
Location of exons, introns, and SNPs in OLR1 genomic sequences analyzed in this study

FIGURE 1.

SNP rs3736234 affects OLR1 AS pattern in both allelic series. (A) Schematic representation of the Low- and High-Risk minigenes used in this study, including the nucleotide identities and the relative position to the closest 3′ss (in base pairs) of the 6 SNPs in linkage disequilibrium in the two allelic series. pCMV indicates Cytomegalovirus promoter, SV40 refers to Simian Virus 40 polyadenylation signal, AMPr indicates ampicillin resistance cassette included in the vector backbone, coding exons are represented as rectangles, intronic sequences as thin lines, and 3′ UTR as thick line. Length in base pairs (bp) of exons, introns, and 3′ UTR is indicated by the numbers below. Arrows represent forward (splice junction) and reverse (vector-specific PT1 sequence) primers used for real-time PCR quantification of the exon 5 inclusion (INC) or skipping (SKIP) isoforms. (B) Exon 5 inclusion/skipping ratios of transcripts generated upon transfection of Low- and High-Risk minigenes in HeLa, HEK293-T, or SK-N-SK cells, and measured 24 h post-transfection. RT-qPCR values represent mean and standard deviations of 10 independent biological replicas. (C) Schematic representation of single point mutations introduced by site-directed mutagenesis in the OLR1 Low- and High-Risk minigenes to dissect the effect of each SNP in AS regulation. Nucleotide identities in the High-Risk series are indicated in bold, those in the Low-Risk series in normal lettering. (D,E) Quantification of inclusion/skipping ratios for the different mutants in C. Data represent mean and standard deviation of exon 5 inclusion/skipping ratios after normalization against Low-Risk (D) or High-Risk (E) minigenes for eight independent biological replicas. P-values obtained from Student's two-tailed heteroscedastic t-test comparing to Low- or High-Risk minigenes are indicated: (*) <0.05, (**) <0.01, (***) <0.001.
Intronic SNPs rs3736234 and rs13306593 modulate OLR1 AS in both allelic series
To dissect the contribution of each intronic SNP in the regulation of OLR1 AS, we generated two series of mutant minigenes constructs containing single point substitutions in each of the six SNPs previously described to be part of the linkage disequilibrium block (Table 1). In each of the series, we individually replaced each SNP in the Low- or High-Risk minigenes by the nucleotide present in the other allelic series (Fig. 1C). Each construct was transfected in HeLa cells and 24 h post-transfection, cellular RNAs were isolated and analyzed as described above. Mutation of SNPs rs3736234 (constructs 2 and 8) and rs13306593 (construct 5) displayed the most significant effects on OLR1 AS regulation (Fig. 1D–E). Mutation of rs3736234 in the Low-Risk series to the nucleotide present in the High-Risk series (C- to T-rs3736234) caused a very significant increase in exon 5 inclusion (Fig. 1D, construct 2), while the reciprocal change in the High-Risk series caused the strongest decrease in exon 5 inclusion (Fig. 1E, construct 8). In contrast, mutation of rs13306593 in the Low-Risk series to the nucleotide present in the High-Risk series (G to T) reduced exon 5 inclusion (Fig. 1D, construct 5), while the reciprocal change did not affect significantly the splicing pattern of the High-Risk series. These results reveal the prominent effects on AS regulation of specific SNPs (particularly rs3736234) within the linkage disequilibrium block.
To evaluate the specificity of these effects, we generated another series of mutants in which each SNP in the Low-Risk allelic series was mutated to a nucleotide different from the one present in the High-Risk series (Fig. 2A, upper panel). Interestingly, the prominent effects of substitutions C→T in SNP rs3736234 and of G→T in SNP rs13306593 were not observed with the different nucleotide substitutions (C→G and G→A, respectively) arguing for specific effects of the natural sequence variants on OLR1 AS.
FIGURE 2.

Additional nucleotide identities in the allelic series attenuate the effect of SNP rs3736234 on OLR1 AS. (A) Nucleotide-specific effect of rs3736234 and rs13306593 SNP substitutions. The scheme indicates the individual substitutions from Low- to High-Risk allelic series and from Low to a different nucleotide. Data represent mean and standard deviation of exon 5 inclusion/skipping ratios of the different constructs after normalization to Low-Risk minigenes for six independent biological replicas. Significant P-values obtained from Student's two-tailed heteroscedastic t-test are indicated: (*) <0.05, (**) <0.01. (B) The table indicates the accumulative series mutations introduced in the Low-Risk minigene to reconstruct the genotype of the High-Risk allele series (in bold). (C) Ratio of exon 5 inclusion/skipping for the constructs indicated in B, normalized to the values of the Low-Risk minigene. Values represent mean and standard deviation for eight independent biological replicas. P-values for each of the conditions are indicated.
Attenuating effects of other nucleotides in the linkage disequilibrium block
The AS changes caused by swapping rs3736234 SNP variants were more dramatic than the differences in inclusion/skipping ratio observed between the two allelic series (either in patient cells or in our minigenes system) (Fig. 1D–E, compare lanes 2 or 8 with Low- and High-Risk). To test whether the presence of other SNPs could modulate the effect exerted by SNP rs3736234, we progressively introduced nucleotide changes in the Low-Risk construct until we reconstructed the SNP composition of the High-Risk allele (Fig. 2B). Remarkably, the strong increase in exon 5 inclusion caused by the natural variant of rs3736234 (Low-Risk mut 2) was progressively attenuated by the presence of additional nucleotide variants present in the linkage disequilibrium block, until the ratio of inclusion/skipping resembled that observed in the High-Risk minigene (Fig. 2C). Reciprocal effects were observed when progressive substitutions were introduced in the High-Risk minigene to reassemble the SNP configuration of the Low-Risk allelic series (data not shown). It is important to point out that additional nucleotide differences (not in linkage disequilibrium) are present in our Low- and High-Risk minigene constructs. These results suggest that the difference in exon 5 inclusion/skipping ratio between the Low- and High-Risk allelic series observed in human populations (Mango et al. 2005) is the result of the balance between the strong effects of variations in SNP rs3736234 and compensatory effects of (at least some of) the other sequence variants present in the linkage disequilibrium block.
Bioinformatic identification of putative regulators of OLR1 AS regulation
To identify sequence motifs relevant for OLR1 AS regulation, we developed a software that compiles potential binding sites for splicing regulatory factors derived from in vitro SELEX and other high-throughput RNA binding techniques, as well as potential splicing regulatory motifs identified by various in silico approaches (see Materials and Methods section). We focused our attention in the region of intron 4 containing the key rs3736234 SNP. A variety of potential regulatory elements were identified in this region (Fig. 3A) and the identity of rs3736234 (C or T) was predicted to significantly influence the presence or not of various motifs. In particular, several elements with predicted splicing enhancer function, including a potential binding site for the SR protein SRSF2 (SC35), partially overlapping with the SRSF1-binding site, were no longer predicted when C-rs3736234 (Low-Risk allele) was replaced by T-rs3736234 (High-Risk allele). Of potential interest, a predicted SRSF1 (ASF/SF2) high score motif (AACACTA) was maintained in both alleles. Therefore, a possible scenario emerging from these in silico analysis is that an intronic enhancer element (such as a SRSF1-binding site) promotes exon 5 inclusion in the High-Risk allele, while regulatory motifs in adjacent sequences act as dominant silencer motifs. Such motifs would be sensitive to the presence of T or C at rs3736234 and somehow prevent, in the case of the Low-Risk C-rs3736234 allele, the function of the SRSF1 enhancer.
FIGURE 3.

Bioinformatic identification of putative sequence regulatory elements in the vicinity of SNP rs3736234. (A) Potential sequence regulatory elements identified in silico in the sequences of Low-Risk (upper) or High-Risk (lower) RNAs around rs3736234. The identity of the nucleotide in each allelic series is indicated by bold bigger case. Light gray indicates sequence motifs common in both alleles, bold indicates motifs identified in the Low-Risk but not in the High-Risk allele, shadowed motif corresponds to a high score putative binding site for the SR protein SRSF1, the italicized motif indicates putative binding site for the SR protein SRSF2. The tables on the right indicate the putative regulatory motifs identified: their source, original classification ([ESS] exonic splicing silencer, [ESE] exonic splicing enhancer, [ISRE] intronic splicing regulatory element), location of the first nucleotide of the motif in the sequence analyzed (left), and description of the regulatory element. (B) Sequence alignment of OLR1 intron 4 3′ region across different vertebrate species. Potential overlapping SRSF2- and SRSF1-binding sites around SNP rs3736234 are indicated by light and dark shadowed boxes, respectively.
A comparative analysis of the sequences around SNP rs3736234 across various vertebrate species revealed conservation of the overlapping human SRSF1 and SRSF2 regulatory elements in different primate species, but not in other vertebrates, including mouse or rat (Fig. 3B). Interestingly, this comparative study identified the High-Risk allele as the ancestor sequence, suggesting that the emergence of the Low-Risk allele is restricted to humans, in line with recent studies reporting frequent gain of novel regulatory elements and increased complexity in AS in particular evolutionary lineages (Barbosa-Morais et al. 2012; Merkin et al. 2012).
Spliceosome-wide screen identifies regulators of OLR1 AS
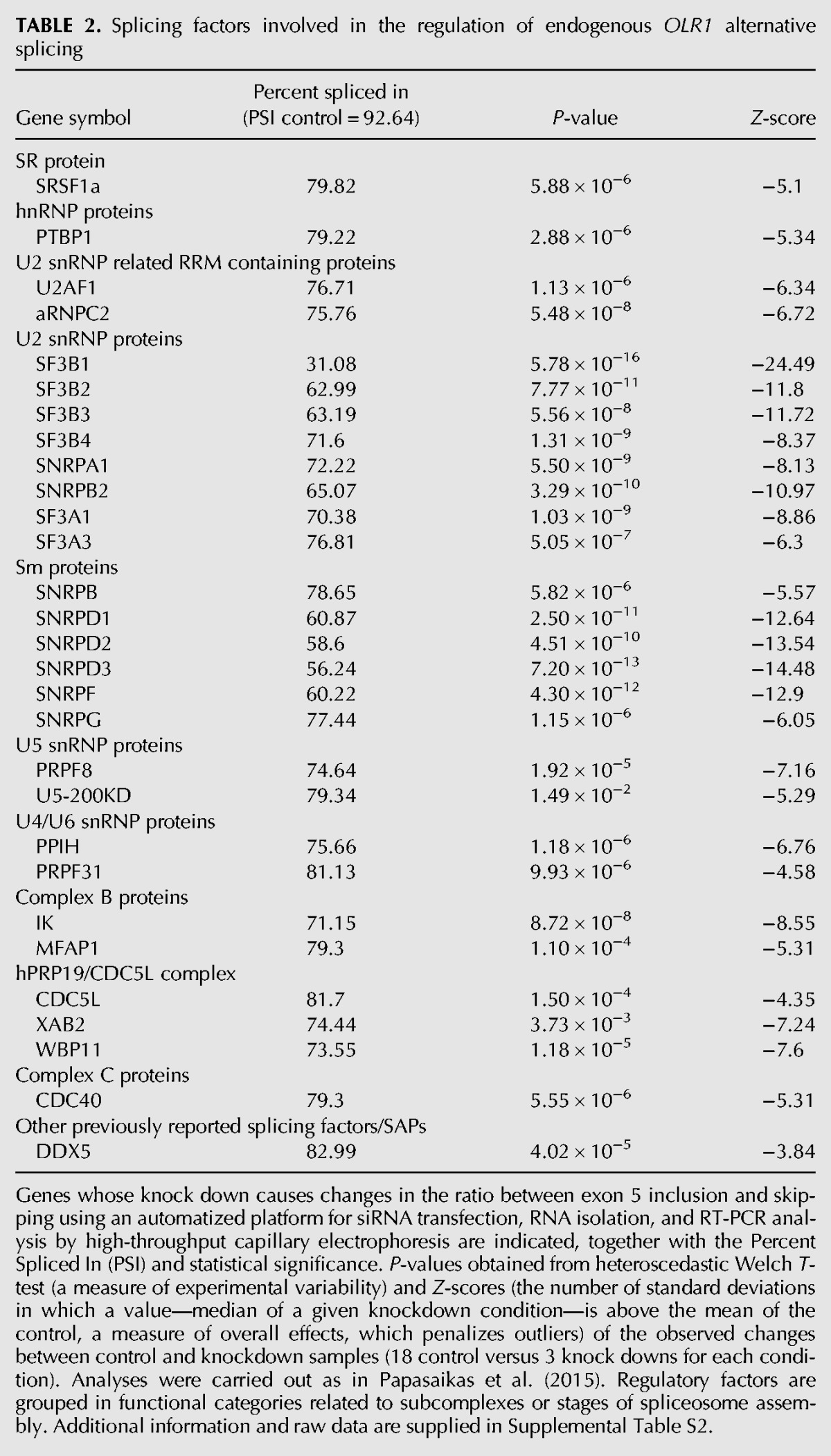
A second, unbiased approach to study OLR1 AS was undertaken, using automatized screening methods recently developed by our laboratory to identify AS regulators by high-throughput RNAi (Papasaikas et al. 2015; Tejedor et al. 2015). Briefly, HeLa cells were transfected with a customized siRNA library individually targeting ∼250 spliceosome components, splicing regulators, and chromatin remodeling factors and 72 h post-transfection mRNAs were isolated using oligo-dT-coated plates. PCR amplification with OLR1 specific primers located in exons 4 and 6, coupled with high-throughput capillary electrophoresis, allowed us to detect changes in the relative levels of endogenous OLR1 splicing isoforms upon each individual knockdown condition, quantified using a robust Z-score (Table 2; Supplemental Table S2; Supplemental Fig. S2B). The difference in the levels of exon inclusion between endogenous and minigene-derived OLR1 transcripts is likely due to differences in expression levels, chromatin organization, and topology of RNAs expressed from episomal DNA (see also Fig. 2A). Table 2 lists the most significant regulators identified, including U2 snRNP and U2 snRNP-associated factors (possibly indicating that intron 4 3′ splice site recognition by U2 is rate-limiting for exon 5 inclusion). Significantly, SRSF1 was also found to regulate OLR1 exon 5 inclusion, consistent with the bioinformatic prediction of a binding site for this SR protein near rs3736234. Other regulators identified include PTBP1 as well as a number of factors acting late in spliceosome assembly, in conformational rearrangements of spliceosome complexes or even in catalysis, consistent with extensive potential of core spliceosome components in the regulation of AS (Park et al. 2004; Saltzman et al. 2011; Papasaikas et al. 2015; Tejedor et al. 2015).
TABLE 2.
Splicing factors involved in the regulation of endogenous OLR1 alternative splicing
Biochemical identification of regulatory factors associated with rs3736234 polymorphism and adjacent motifs
To directly identify factors interacting with the region of the RNA comprising SNP rs3736234 and adjacent sequences, we used an efficient RNAse-assisted RNA chromatography protocol (Michlewski and Cáceres 2010) followed by identification of associated proteins by mass spectrometry. RNA pull-downs were carried out using RNA oligonucleotides corresponding to 31 nucleotides of intron 4 centered around rs3736234, in the Low- and High-Risk versions (Fig. 4A), incubated with nuclear extracts from HeLa (three different preparations) or Human Microvascular Endothelial (HMVEC) cells. Proteins retained after extensive washing were analyzed (in duplicates for each condition) by SDS electrophoresis and mass spectrometry (Fig. 4B,C; Supplemental Table S3). After lane-based normalization by overall protein levels, the relative abundance and enrichment score of proteins differentially present in the precipitates of Low-Risk (C-rs3736234) or High-Risk (T-rs3736234) RNAs was assessed (Table 3; Fig. 4D). The splicing factor SRSF1 was clearly enriched in association with the High-Risk RNA oligonucleotide, while the RNA helicase DDX17 and factors corresponding to the family of High-Mobility Group proteins (HMGA1 and HMGN1), previously involved in chromatin remodeling and DNA compaction (Cleynen and Van de Ven 2008), were enriched in association with the Low-Risk RNA oligonucleotide.
FIGURE 4.

RNA affinity selection of potential OLR1 AS regulators. (A) RNA oligonucleotide sequences corresponding to the Low-Risk or High-Risk alleles used for RNA pull down experiments. (B) Proteins isolated after RNase-assisted RNA pull down using Low- or High-Risk RNA oligonucleotides or beads alone, incubated with nuclear extracts from different cell lines ([HeLa com] commercial nuclear extracts, [HeLa#1 and HeLa#2] HeLa nuclear extracts prepared in our laboratory, [HMVEC] human microvascular vein endothelial cells nuclear extracts, [NE] nuclear extracts) were fractionated by electrophoresis in SDS-polyacrylamide gels detected by silver staining. The experiment was performed in duplicate for each of the conditions. (C) Venn diagram indicating the number of overlapping mass spectrometry-determined proteins between the different nuclear extract conditions and replicates. Proteins bound to both alleles as well as proteins present across the different conditions were considered for further analysis. (D) Mass spectrometry analysis of protein abundance and ratio between Low- and High-Risk affinity chromatography results. Enrichment P-value is indicated, together with the ratio between Low and High binding enrichment. (E) Functional hit validation. HeLa cells were co-transfected for 48 h with Low- or High-Risk minigenes and siRNAs against the indicated factors ([0] scramble siRNA 100 nM, [+] siRNA 50 nM, [++] siRNA 100 nM). Values represent mean and standard deviations of exon 5 inclusion/skipping ratios for four independent biological replicas. Significant P-values obtained from Student's two-tailed heteroscedastic t-test are indicated: (*) <0.05, (**) <0.01.
TABLE 3.
Proteins enriched by RNase-assisted RNA affinity chromatography from nuclear extracts, using RNA oligonucleotides corresponding to Low-Risk (a) or High-Risk (b) alleles followed by mass spectrometry

The association of SRSF1 with RNA sequences involved in OLR1 AS regulation is in line with the results of both the bioinformatic predictions (Fig. 3) and the functional knockdown screen (Table 2). Furthermore, the enrichment of SRSF1 in the High-Risk RNA is consistent with a function for this factor in promoting exon 5 inclusion. Also consistent with the enrichment of HMG proteins in the Low-Risk RNA, knock down of HMGA1 increased exon 5 inclusion of the Low-Risk allele, but did not affect the inclusion/skipping ratio of the High-Risk allele (Fig. 4E). Knock down of other factors that did not reach the same enrichment significance in the RNA pull-down experiments (PSIP1 and NONO) did not alter OLR1 AS, while knock down of another such factor (SFPQ) reduced exon 5 inclusion in the two alleles (Fig. 4E), suggesting that additional regulatory factors not identified by our combined approaches remain to be discovered.
SRSF2 and SRSF1 display opposite roles in the regulation of OLR1 AS
To further investigate the involvement of SRSF1 and SRSF2 in OLR1 exon 5 AS, we generated mutants containing deletions in their putative cognate sites in the vicinity of rs3736234, in both the Low- and High-Risk versions of the minigenes (Fig. 5A). Deletion of the SRSF2-binding site led to a significant increase in exon 5 inclusion in the context of the Low-Risk minigene, but not in the context of the High-Risk minigene (Fig. 5B). These results are consistent with the prediction of a SRSF2-binding site in the Low-Risk allele, which is disrupted by the C→T transition in the High-Risk allele (Fig. 3), and with a hypothetical repressive effect of SRSF2 binding to this site on exon 5 inclusion. Consistent with this possibility, SRSF2 overexpression led to a decrease in exon 5 inclusion, while SRSF2 knock down by RNAi led to an increase in exon 5 inclusion (Fig. 5D,F). These effects, however, were observed in both the Low- and High-Risk minigenes (albeit less significantly for the High-Risk minigene under conditions of SRSF2 knock down), suggesting that the repressive effects of SRSF2 on exon 5 inclusion are not limited to its function through the putative binding site at rs3736234 and may rather be mediated by multiple cognate sites. Consistent with this notion, SRSF2 overexpression led to decreased exon 5 inclusion when its putative binding site was deleted from either the Low- or the High-Risk minigene (Fig. 6B,D).
FIGURE 5.

Antagonistic effects of SRSF2 and SRSF1 in the regulation of OLR1 exon 5 AS. (A) Schematic representation of the deletion mutants of predicted binding sites for SRSF2 and SRSF1. Predicted binding sites for SRSF2 and SRSF1 in the region of rs3736234 are indicated as gray and black boxes, respectively. The different nucleotide variants in rs3736234, as well as the sequence deletions introduced in the constructs, are also indicated. (B,C) Effects of deletion of putative SRSF2/SRSF1-binding sites on OLR1 exon 5 AS. Ratio of exon 5 inclusion/skipping was determined for the indicated constructs as in previous figures. Data represent mean and standard deviations after normalization by the results of the Low-Risk minigene for four independent experiments. (D,E) Quantification of splicing changes for the Low- and High-Risk minigenes upon SRSF2 (D) or SRSF1 (E) overexpression. Cells were co-transfected for 48 h with the indicated minigenes and the corresponding protein expression vector (numbers indicate ng of expression vector utilized). Total amounts of transfected plasmids were raised to 1000 ng by using pCMV vector. Data represent mean and standard deviation for eight independent biological experiments. Insets show protein levels for the control and the maximum overexpression condition detected with anti-T7 tag antibodies. (F,G) Quantification of splicing changes for the Low- and High-Risk minigenes upon SRSF2 (F) or SRSF1 (G) knock down by RNAi. Numbers below indicate the final concentration of siRNAs used for each of the conditions. Insets show the RNA (RT-PCR, SRSF2) or protein (western blot, SRSF1) levels in scramble siRNA and SRSF2/SRSF1 siRNA knockdown conditions. (+) 100 µM. Values represent mean and standard deviations for six independent biological replicas. P-values obtained from Student's two-tailed heteroscedastic t-test used in this figure are indicated: (*) <0.05, (**) <0.01, (***) <0.001.
FIGURE 6.

Effects of SRSF1/SRSF2 overexpression on OLR1 AS regulation upon deletion of their putative binding sites. (A) Schematic representation of wild type and mutant minigenes used in this experiment. Nomenclature and symbols as in Figure 5A. (B–E) Splicing changes induced in the indicated minigenes by overexpression of the indicated proteins, analyzed as in previous figures. Numbers below indicate nanogram of protein expression vector co-transfected in each condition, with the total amount of transfected plasmid adjusted to 1000 ng using pCMV empty vector. For all the panels, data represent mean and standard deviations of inclusion/skipping ratio for eight independent experiments. P-values obtained from Student's two-tailed heteroscedastic t-test are indicated: (**) <0.01, (***) <0.001.
Surprisingly, deletion of the SRSF1-binding site had no significant effect on exon 5 inclusion in transcripts from either the High- or the Low-Risk minigenes (Fig. 5C), although SRSF1 overexpression led, as expected, to increased exon 5 inclusion and SRSF1 knock down led to reduced exon 5 inclusion—confirming the results of the high-throughput RNAi screen—in both minigene alleles (Fig. 5E,G). Significantly, deletion of the SRSF1-binding site adjacent to rs3736234 eliminated the effects of SRSF1 overexpression on both the Low- and High-Risk minigenes (Fig. 6C,E), consistent with the idea that this putative SRSF1-binding site, predicted bioinformatically and confirmed by RNA pull-down experiments, functionally mediates the effects of SRSF1. In contrast, deletion of the SRSF2-binding site did not affect the response to SRSF1 (Fig. 6C,E).
Although we cannot rule out confounding effects of generating regulatory motifs upon deletion of predicted binding sites, the results of Figures 5 and 6 collectively reveal antagonistic roles of SRSF1 and SRSF2 on OLR1 exon 5 AS, with SRSF1 promoting exon 5 inclusion at least partly through its binding site adjacent to rs3736234, and SRSF2 promoting exon 5 skipping most likely through multiple binding sites.
Both RNA recognition motifs of SRSF1 are necessary for OLR1 AS regulation
SRSF1 is a classical member of the family of the SR proteins harboring a modular structure composed of two RNA recognition motifs (RRMs) and one carboxy-terminal arginine/serine-rich domain (RS) involved in protein–protein and in mediating RNA–RNA interactions during spliceosome assembly (Wu and Maniatis 1993; Shen et al. 2004). To dissect the domain requirements for SRSF1-mediated OLR1 AS regulation, a panel of SRSF1 domain mutants (Fig. 7A) were co-transfected with the Low- or High-Risk OLR1 minigenes and their effects on exon 5 inclusion analyzed as before (Fig. 5E). The results indicated that deletion of RRM1 or RRM2 compromises the function of SRSF1 on OLR1 AS (Fig. 7B,D), while the RS domain was dispensable (Fig. 7F), consistent with previous dissection of domain requirements for SRSF1 function in AS (Cáceres and Krainer 1993). Interestingly, while expression of RRM1 or RRM2 alone was not sufficient to induce exon 5 inclusion, expression of the RS domain alone caused significant increase in exon 5 skipping in both Low- and High-Risk minigenes (Fig. 7C,E,G), suggesting some form of squelching or dominant negative effect of the expression of the RS in isolation.
FIGURE 7.

SRSF1 RRM1 and RRM2 are required for OLR1 AS regulation. (A) Schematic representation of the different SRSF1 protein expression mutants used in this experiment. OE indicates overexpression, WB stands for Western blot. (B–G) Real-time PCR quantification of the ratio of inclusion/skipping upon co-transfection of Low- or High-Risk minigenes with the indicated SRSF1 constructs in HeLa cells (+, empty vector; ++, 250 ng of protein expression vector; +++, 1000 ng of protein expression vector). In all cases, the total amount of transfected DNA was adjusted to 1000 ng with empty vector. Data represent mean and standard deviations of the ratio inclusion versus skipping after normalization by the ratio of the Low-Risk minigene for eight independent experiments. Protein overexpression was validated by Western blot using anti-T7 tag antibody (+, maximal amount of expression plasmid) and tubulin was used as loading control. Endogenous OLR1 isoforms were detected by semi-quantitative PCR. P-values obtained from Student's two-tailed heteroscedastic t-test are indicated: (**) <0.01, (***) <0.001.
Ox-LDL uptake is modulated by changes in OLR1 AS
To further validate the physiological relevance of the proposed mechanisms of OLR1 AS regulation, we combined SRSF1/2 overexpression or Antisense RNA Oligonucleotides (AONs)-mediated splice site switching strategies (Fig. 8A) with measurements of Ox-LDL uptake in HeLa cells. 2′-O-methyl phosphorothioate-modified AONs have been shown to be effective tools to modulate AS of endogenous transcripts, even with therapeutic potential (Hua et al. 2011; Kole et al. 2012; Bechara et al. 2013; Maimon et al. 2014). AONs complementary to the 5′ and 3′ splice sites of OLR1 exon 5 promoted exon skipping, particularly when used in combination (Fig. 8B,C). In contrast, an AON complementary to the region of the rs3736234 polymorphism led to a mild increase in exon 5 inclusion, suggesting an overall repressive effect of this region on exon 5 inclusion of HeLa endogenous (Low-Risk) transcripts (Fig. 8B,C). As observed with minigenes, overexpression of SRSF1 and SRSF2 led to decreased and increased levels of exon skipping, respectively, in the endogenous OLR1 transcripts (Fig. 8C).
FIGURE 8.

Modulation of Ox-LDL uptake by OLR1 AS regulation. (A) Schematic representation of targeting sites for AONs used to modulate OLR1 AS. SNP corresponds to an AON complementary to the region if intron 4 containing rs3736234 in its Low-Risk configuration. (B) Modulation of OLR1 AS by AONs. Exon 5 percent spliced in (PSI) values for endogenous OLR1 transcripts from HeLa cells upon transfection of the indicated AONs, measured by capillary electrophoresis, are indicated. Values represent mean and standard deviation of nine independent biological experiments. (C) RT-qPCR quantification of expression of exon 5 skipping isoform (LOXIN) normalized to HPRT1 mRNA expression upon transfection of the indicated AONs or SRSF expression vectors. Values represent mean and standard deviation of three independent biological experiments. (D) Measurement of Ox-LDL uptake under the same conditions as in C, monitored by FACS analysis quantification of Dil-Ox-LDL. Values correspond to mean and standard deviation for three independent experiments. P-values of results in this figure correspond to Student's two-tailed heteroscedastic t-test: (*) <0.05, (**) <0.01, (***) <0.001.
To evaluate the functional consequences of endogenous OLR1 AS modulation, Ox-LDL uptake was traced by using Ox-LDL labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine fluorophore (Dil-Ox-LDL, Biomedical Technologies Inc.). HeLa cells were transfected with AONs or protein expression vectors for 24 h, subsequently incubated with Dil-Ox-LDL for another period of 24 h and internal Ox-LDL levels monitored by FACS analysis. A significant decrease of Ox-LDL uptake upon transfection with AONs targeting the 5′ss and 3′ss was observed, while the opposite effect was observed upon transfection with AONs targeting SNP rs3736234 (Fig. 8D), consistent with the predicted changes in ratio between OLR1 isoforms. Also consistent with the RNA changes induced by SRSF1/2 overexpression on OLR1 transcripts, higher levels of SRSF1 led to increased Ox-LDL uptake, while the opposite was observed upon overexpression of SRSF2 (Fig. 8D). Taken together, these results recapitulate the antagonistic roles of SRSF1 and SRSF2 in the modulation of OLR1 AS and argue for the physiological relevance of the splicing regulatory mechanisms described in this manuscript (Fig. 9).
FIGURE 9.

Model of OLR1 AS regulation. (A) The identity of the nucleotide in SNP rs3736234 has strong effects on exon 5 inclusion (increased inclusion upon C→T), while the rest of the SNPs in the linkage disequilibrium block attenuate the strong effects of this substitution. (B) Association of SRSF1 to a potential binding site adjacent to rs3736234 promotes exon 5 inclusion. The presence of a C at rs3736234 appears to repress the association of SRSF1 with the adjacent intronic enhancer, leading to higher levels of exon skipping. SRSF2 and HMGA1 may mediate these repressive effects, and while HMGA1 associates with the C-version of the rs3736234 region, SRSF2 likely acts through additional binding sites.
DISCUSSION
Cardiovascular disease is a multifactorial disorder in which both external factors and genetic predisposition influence the risk of developing coronary failure. Previous association studies reported a correlation between intragenic OLR1 single nucleotide polymorphisms and genetic predisposition to cardiovascular disorders (Chen et al. 2003; Mango et al. 2003; Tatsuguchi et al. 2003). In this study, we have dissected the effect on OLR1 AS of the different SNPs present in a linkage disequilibrium block and investigated molecular mechanisms behind the prominent role of SNP rs3736234 on the regulation of exon 5 inclusion.
Bioinformatic, biochemical, functional, and mutational data revealed a role for SRSF1 in promoting OLR1 exon 5 inclusion (Fig. 9). This SR protein has been involved in the regulation of numerous AS events, frequently working as a key component of exonic splicing enhancers, and can play a decisive role in cell transformation and cancer progression (Karni et al. 2007; Das and Krainer 2014). Our pull-down experiments revealed that SRSF1 binds to an RNA containing T-rs3736234 (associated with higher exon inclusion and higher risk of coronary disease), while the protein did not associate with the same RNA containing C-rs3736234 (associated with exon skipping and lower risk of coronary disease). This suggests that the levels of SRSF1 could represent an additional risk factor in cardiovascular disease for individuals harboring the High-Risk allele. Deletion of the predicted SRSF1-binding site adjacent to rs3736234 made a minigene refractory to the exon inclusion-promoting effects of SRSF1 overexpression, arguing that SRSF1 indeed functions through this sequence element. Surprisingly, however, deletion of this motif did not by itself increase exon skipping significantly, suggesting that exon inclusion is maintained by the function of other factors and regulatory sequences under these conditions. Kanopka et al. (1996) previously reported that relocation of SRSF1 binding from an exonic to an intronic location in adenovirus pre-mRNA results in splicing inhibition, which is in contrast with the enhancing effect of SRSF1 proposed for OLR1. This illustrates the context-dependent effects of classical splicing regulators, which facilitate combinatorial regulation of splice site selection (Pandit et al. 2013).
rs3736234 is not within the boundaries of the predicted SRSF1-binding site, but C-rs3736234 is predicted to harbor a binding site for SRSF2 (Liu et al. 2000), which is disrupted by the T-rs3736234 polymorphism. Antagonism between SRSF1 and SRSF2 through intronic enhancer sequences has been reported before (Gallego et al. 1997). Both the results of SRSF2 overexpression/knock down and the effect of deletion of its predicted binding site argue that C-rs3736234 creates a SRSF2 silencer element, although additional sequence motifs allow SRSF2 to exert its inhibitory effects on exon 5 inclusion. Our RNA pull-down assays did not detect a significant enrichment of SRSF2 on C-rs3736234, possibly due to limited sensitivity of the assay or to transient association of SRSF2 with the RNA in this region, e.g., during co-transcriptional splicing. As a matter of fact, SRSF2 is known to play an active role in transcriptional elongation by facilitating RNA polymerase II pause release (Lin et al. 2008; Ji et al. 2013) and SRSF2-mediated enhanced elongation rates could indeed promote exon skipping by facilitating kinetic competition between alternative sites (Dujardin et al. 2013). Nucleosome positioning was also proposed to influence RNA polymerase II elongation rates and AS (Schwartz and Ast 2010; Gómez et al. 2013; Iannone and Valcárcel 2013; Iannone et al. 2015) and it is interesting, in this regard, that several members of the family of chromatin-associated High-Mobility Group (HMG) proteins were identified in our proteomic analysis. HMGA1 was enriched in C-rs3736234 RNA and knock down of HMGA1 increased OLR1 exon 5 inclusion specifically in the C-rs3736234 minigene. It is therefore conceivable that SRSF2 and HMGA1 (and perhaps also the DEAD box helicase DDX17, also enriched in T-rs3736234) jointly influence co-transcriptional regulation of OLR1 AS via chromatin/kinetic effects. In addition, the association of these proteins (and/or additional interacting partners) with C-rs3736234 could more directly influence AS by preventing SRSF1 binding to its adjacent cognate site. Indeed, previous reports identified a direct role for HMGA1a in splicing regulation in presenilin-2 transcripts, based upon sequestration of U1 snRNP on a decoy sequence through interaction of HMGA1a with the U1 snRNP protein U1-70K (Manabe et al. 2003).
However, the most significant splicing factor category identified by our screening approach corresponds to components of the core splicing machinery. Our results indicate a clear role not only for components of U2 snRNP and Sm proteins in the modulation of OLR1 AS but also for core components identified in different stages of spliceosome assembly, including U5 snRNP or PRP19/CDC5L complexes. Similar results were reported from a genome-wide screen of FAS cassette exon 6 regulators (Tejedor et al. 2015) and for 35 other events in a focused screen for spliceosomal components (Papasikas et al. 2015). These core splicing components could be rate-limiting for proper recognition and processing of weak splice sites, or reveal extensive functional flexibility and reversibility in the assembly of these complexes (Hoskins and Moore 2012), reinforcing the idea of extensive potential of the core splicing machinery to regulate splice site choice (Park et al. 2004; Saltzman et al. 2011; Tejedor et al. 2015; Papasaikas et al. 2015).
Our most intriguing observation was the progressive attenuating effect of other SNPs present in the linkage disequilibrium block on the strong exon inclusion caused by T-rs3736234 when combined with the other polymorphisms associated with the C-rs3736234 allele (Fig. 9). It is indeed remarkable that single nucleotide sequence variants, hundreds of bases away from each other in a region spanning two introns and a long 3′ UTR, which have little individual effects on their own, can work in concert to fine-tune the levels of exon 5 inclusion. The existence of composite splicing regulatory motifs whose coordinated effects can involve individual elements that span long genomic distances opens interesting mechanistic questions, perhaps involving a particular 3D organization of the genomic locus. From an evolutionary point of view, C-rs3736234 is present in humans but not in other primate species, suggesting that the Low-Risk block evolved in certain human populations as a defense against detrimental effects of high Ox-LDL uptake on the development of cardiovascular disorders. This is in line with the recent proposal that small changes in cis-acting regulatory elements allow differential rewiring of splicing networks in different species (Barbosa-Morais et al. 2012; Merkin et al. 2012). Furthermore, the attenuating effect of other polymorphisms in the T-rs3736234 linkage disequilibrium block may represent another genetic solution adopted by other populations to limit excessive Ox-LDL uptake.
Many reports have linked genetic variations with altered ratios of alternatively spliced isoforms (Coulombe-Huntington et al. 2009; Dayem Ullah et al. 2012), including associations with disease (Sterne-Weiler and Sanford 2014) and alterations in LDL metabolism (Yu et al. 2014a). It is therefore expected that large-scale computational and experimental tools aimed at revealing the functional effects of genetic variants on RNA processing (Soemedi et al. 2014; Xiong et al. 2015) will have a significant impact on understanding disease etiology and possibly on the design of novel therapies.
Modified antisense oligonucleotides (AONs) have been successfully used for the modulation of endogenous AS, including potential therapeutic effects in Spinal Muscular Atrophy and cancer (Hua et al. 2008, 2011; Kole et al. 2012; Bechara et al. 2013; Maimon et al. 2014). AONs directed against RNA regions containing splicing-altering SNPs can also be used to modulate AS, e.g., with effects on cell differentiation and cytokine expression (Yu et al. 2014b). Our combined use of AONs complementary to the splice sites flanking endogenous OLR1 exon 5 induced some exon skipping and resulted in reduced Ox-LDL uptake in cells in culture. The LOX-1 receptor acts as an oligomer to favor Ox-LDL recognition and uptake (Matsunaga et al. 2007) and changes in the abundance of OLR1 skipping isoform (LOXIN) affect oligomer distribution and disrupt Ox-LDL uptake (Biocca et al. 2008). Modulation of endogenous OLR1 AS by AONs could eventually offer a potential therapy to reduce Ox-LDL levels and atheroma plaque formation in patients with high susceptibility to cardiovascular disease. Further investigation of the mechanisms of OLR1 AS may offer even more efficient and specific therapeutic possibilities.
MATERIALS AND METHODS
siRNA transfection and mRNA isolation
Knockdown experiments were carried out in HeLa cells transfected with siRNA pools against the indicated genes. Cells were reverse transfected in triplicate with a siRNA library comprising 250 spliceosome related siRNAs (SMARTpool ON TARGET PLUS, Thermo-Scientific, listed in Supplemental Table S2) or other factors (PSIP1, NONO, SFPQ, HMGA1, SRSF2, SRSF1, SMARTpools ON TARGET PLUS, Thermo-Scientific) and cells were added to a previously incubated mixture containing SMARTpool siRNAs (25 nM final concentration) and Lipofectamine RNAiMAX (Life Technologies) following the manufacturer's recommendations. Cellular mRNAs were isolated 48 or 72 h post-transfection by an automated procedure using oligo-dT-coated 96-well plates (mRNA catcher PLUS, Life Technologies) or with RNAeasy mRNA extraction columns (Qiagen) following the manufacturer's instructions, as described elsewhere (Papasaikas et al. 2015; Tejedor et al. 2015).
Reverse transcription and semi-quantitative PCR
Purified mRNAs were reverse transcribed with superscript III (Life Technologies) in the presence of oligo-dT (Sigma-Aldrich) and random primers (Life Technologies) following the manufacturer's recommendations. Semi-quantitative PCR primers were designed to amplify either endogenous or minigene OLR1 isoforms corresponding to the inclusion and skipping of OLR1 exon 5 (endogenous forward primer: 5′-GGCATGGAGAAAACTGTTACCTATTTTCCTC; endogenous reverse primer: 5′-CACTGTGCTCTTAGGTTTGCCTTCTTCTG; minigene forward primer: 5′-GTCGACGACACTTGCTCAAC; minigene reverse primer: 5′-CACTGTGCTCTTAGGTTTGCCTTCTTCTG). PCR reactions were performed in a final reaction volume of 30 µL, containing 2 µL of previously synthetized cDNA, forward and reverse OLR1 primers (0.5 µM final concentration), dNTPs (0.2 mM each, Quimigen) and reagents provided in the GoTaq DNA polymerase kit (GoTaq, Promega). PCRs were carried out in Tetrad 2 thermal cyclers (Bio-Rad) with the following parameters: 1 cycle of 95°C (1 min), 35 cycles of 95°C, 60°C, 72°C (30 sec each), and a final cycle of 72°C (1 min).
Primer design
Forward and reverse OLR1 primers (Sigma-Aldrich) used for semi-quantitative PCR, quantitative Real-Time PCR, or site-directed mutagenesis were designed using primer 3 Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) (Koressaar and Remm 2007). A detailed list of primers used in this study is available upon request.
Real-time qPCR
First strand cDNA synthesis was set up with 0.5–1 µg of RNA, 50 pmol of oligo-dT (Sigma-Aldrich), 75 ng of random primers (Life Technologies), and superscript III reverse transcriptase (Life Technologies) in 20 µL final volume, following the manufacturer's instructions. Quantitative PCR amplification was carried out using 1 µL of 1:4 diluted cDNA with 5 µL of 2X SYBR Green Master Mix (Roche) and 4 pmol of specific primer pairs (inclusion isoform minigene 5′-GTCGACGACACTTGCTCAAC–5′-CTTGCTGGATGAAGTCCAGAT; skipping isoform minigene 5′-GTCGACGACACTTGCTCAAC–5′-GCCTCGGACTCTAAATCAGAT; expressed transcripts minigene 5′-GTCGACGACACTTGCTCAAC–5′-GCTGTGCTATTAATTTTCAGCAAC; inclusion isoform endogenous OLR1 5′-GGCATGGAGAAAACTGTTACCTATTTTCCTC–5′-CTTGCTGGATGAAGTCCAGAT; skipping isoform endogenous OLR1 5′-GGCATGGAGAAAACTGTTACCTATTTTCCTC–5′-GCCTCGGACTCTAAATCAGAT; expressed transcripts endogenous OLR1 5′-GTGCTGGGCATGCAATTATCC–5′-GGTGGTGAAGTTCCATTTGC; endogenous HPRT1 5′-CTTTGCTGACCTGCTGGATT–5′-CCCCTGTTGACTGGTCATTACA) in a final volume of 10 µL in 384 well-white microtiter plates, Roche). qPCR mixes were analyzed in triplicates in a Light Cycler 480 system (Roche) and fold change ratios between inclusion and skipping isoforms were calculated according to the Pfaffl method (Pfaffl 2001).
Cell lines
HeLa CCL-2 cells were purchased from the American Type Culture Collection (ATCC). HEK293 and SK-N-SH cells were obtained from the PRBB cell bank repository. For all the cell lines, cell culture was carried out in cell culture dishes or flasks in a humidified incubator at 37°C under 5% CO2 and cells were cultivated in Glutamax Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics (penicillin 500 units/mL; streptomycin 0.5 mg/mL, Life Technologies).
Transient transfections, mammalian protein expression vectors, and OLR1 minigenes
OLR1 Low- and High-Risk minigenes used for transfections were generated by cloning a PCR amplification product obtained from human genomic DNA spanning from exon 4 to the 3′ UTR of the OLR1 gene in the pCMV56 expression vector (Clontech, derived from pCDNA3.1 vector) between EcoRV and XhoI restriction sites. PCR amplification was carried out using genomic DNA from Low- or High-Risk carriers identified by the HapMap project (International HapMap Consortium 2005), using DNA oligos containing sequence tags (PT1, Sakamoto et al. 1992) that allow specific analysis by qRT-PCR of transcripts derived from the expression vector. Other OLR1 constructs were generated using the QuickChange site-directed mutagenesis kit (Stratagene) or by performing PCR with primers oriented in opposite orientations. T7 tag SRSF1 protein expression vectors and related constructs containing the different RRMs or RS domain from SRSF1 were obtained from A. Krainer (Cold Spring Harbor Laboratory). T7 tag SRSF2 protein expression vector was a gift from J. Cáceres (MRC, Edinburgh). Cell transfections were carried out using 200 ng of Low- or High-Risk allele minigenes and mutants, combined with Lipofectamine 2000 (Life Technologies) following the manufacturer's recommendations. For co-transfection experiments, 200 ng of the previous minigenes were combined with the indicated amounts of empty vector or scramble siRNAs and the corresponding mammalian protein expression vector or specific siRNAs as indicated for each experimental condition.
Immunoblotting
Protein extraction was carried out in RIPA buffer (10 mM Tris–HCl pH 8.0; 1 mM EDTA; 1% Triton X-100; 0.1% sodium deoxycholate, 0.1% SDS; 140 mM NaCl, and 1 mM PMSF added immediately before use). Protein abundance was measured by Bradford assay. Forty micrograms of total protein were fractionated in 10%–15% Bis–Tris polyacrylamide gels and transferred to PVDF membranes. Primary antibodies used for this purpose are listed below: B-Tubulin (mouse monoclonal, Sigma-Aldrich T4026), T7 tag (rabbit T7 polyclonal antibody novus biological NB600-372), SRSF1 (mouse monoclonal, gift from Adrian Krainer).
Bioinformatic analysis
Motif prediction software used to scan for putative splicing regulatory sequences can be found at this site (http://genome.imim.es/~htilgner/pubRegSeqs/) (login: pubregseqs; password: r3gulator). To build this tool, we searched the literature for published exonic splicing enhancers (ESE), silencers (ESS), exonic splicing regulators (ESR) as well as intronic motifs. For publications that reported word lists, we downloaded these. For publications providing position-weight-matrices (PWMs) of length n along with a cutoff c, we determined all n-mers that received a score ≥c. All published n-mers (n = 3, …, 10) are loaded into a hash and upon pasting a nucleotide sequence into the window, all found instances of the hashed n-mers are highlighted on the screen using as many different lines as necessary in order to visualize overlapping n-mers separately. Hits can be shown in tabular-html format along with further information (e.g., the publication[s] in which the n-mer was published, as well as its supposed mode of action [ESS/ESE/etc.] for direct visual inspection). To allow follow-up investigation, all hits and extra information can also be downloaded as a gtf-file.
RNAse-assisted RNA chromatography and mass spectrometry analysis
RNA pull-down assay were carried out as described by Michlewski and Cáceres (2010). Sodium-m-periodate and Adipic acid dehydrazide agarose beads were purchased from Sigma-Aldrich, and A/T1 ribonuclease cocktail (Ambion) was used for RNAse-assisted digestion of RNA–protein complexes formed after incubation of the beads with HeLa or HMVEC Nuclear Extracts. Supernatants from duplicated experiments were subject to mass spectrometry analysis by the CRG proteomic facility and data were normalized against total internal protein content for each of the conditions. Additional information and raw data are supplied in Supplemental Table S3.
Ox-LDL uptake and flow cytometry analysis
Ox-LDL uptake was monitored using Dil-labeled Ox-LDL (Biomedical Technologies Inc.). HeLa cells were transfected with AONs or protein expression vectors for 48 h. Twenty-four hours post-transfection, cells were incubated with Dil-Ox-LDL (20 µg/mL) for a period of 24 h. Cells were subsequently collected and washed three times with cold PBS 1×, and samples were subject to FACS analysis (FACSCalibur, BD).
Antisense oligonucleotides and RNA substrates
RNA oligonucleotides used for pull-down experiments and Antisense RNA Oligonucleotides (AONs) used for endogenous modulation of OLR1 exon 5 inclusion were purchased from Sigma-Aldrich. AONs were ordered with full Phosphothioate and 2′-O-Me modifications in the RNA backbone. RNA sequences are listed below: Low-Risk pull-down: 5′-UUAGUUUUGUGGAUCCAACACUAACUGACUU; High-Risk pull-down: 5′-UUAGUUUUGUGGAUCUAACACUAACUGACUU; AON SNP RS3736234 OLR1: 5′-AGUCAGUUAGUGUUGGAUCCACA; AON 3′ss OLR1: 5′-CUGGAUGAAGUCCUGUGGGGAGUA; AON 5′ss OLR1: 5′-AUGGGAAACUUACAAGUGGGGCAU. Transfection experiments involving AONs were performed in HeLa cells using Lipofectamine RNAiMAX (Life Technologies) and a final concentration of 200 nM of AONs RNA oligos following the manufacturer's recommendations. Cellular mRNAs were isolated 48 h post-transfection and isoform abundance was analyzed by either quantitative Real-Time PCR or Capillary electrophoresis.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank many CRG colleagues, laboratory members, and EURASNET members for their advice, encouragement, and critical reading of the manuscript, Xavier Estivill and the Genomics and Disease group of the CRG for their help with accession to HapMap samples, and Adrian Krainer and Javier Cáceres for valuable reagents. We also thank an anonymous referee for detailed reviewing of data presentation. We acknowledge the excellent technical support of the CRG Robotics, Cell Sorting, and Proteomics Facilities. J.R.T. and C.I. were supported by PhD fellowships from Fondo de Investigaciones Sanitarias and La Caixa, respectively. This work was supported by Fundación Botín, by Banco de Santander through its Santander Universities Global Division, and by Consolider RNAREG, Ministerio de Economía e Innovación, and AGAUR.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.049890.115.
REFERENCES
- Aoyama T, Sawamura T, Furutani Y, Matsuoka R, Yoshida MC, Fujiwara H, Masaki T 1999. Structure and chromosomal assignment of the human lectin-like oxidized low-density-lipoprotein receptor-1 (LOX-1) gene. Biochem J 339(Pt 1):177–184. [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. [DOI] [PubMed] [Google Scholar]
- Bechara EG, Sebestyén E, Bernardis I, Eyras E, Valcárcel J 2013. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 52: 720–733. [DOI] [PubMed] [Google Scholar]
- Biocca S, Filesi I, Mango R, Maggiore L, Baldini F, Vecchione L, Viola A, Citro G, Federici G, Romeo F, Novelli G 2008. The splice variant LOXIN inhibits LOX-1 receptor function through hetero-oligomerization. J Mol Cell Cardiol 44: 561–570. [DOI] [PubMed] [Google Scholar]
- Cáceres JF, Krainer AR 1993. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. EMBO J 12: 4715–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Narumiya S, Masaki T, Sawamura T 2001. Conserved C-terminal residues within the lectin-like domain of LOX-1 are essential for oxidized low-density-lipoprotein binding. Biochem J 355(Pt 2):289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Reis SE, Kammerer C, Craig WY, LaPierre SE, Zimmer EL, McNamara DM, Pauly DF, Sharaf B, Holubkov R, et al. 2003. Genetic variation in lectin-like oxidized low-density lipoprotein receptor 1 (LOX1) gene and the risk of coronary artery disease. Circulation 107: 3146–3151. [DOI] [PubMed] [Google Scholar]
- Chen XP, Zhang TT, Du GH 2007. Lectin-like oxidized low-density lipoprotein receptor-1, a new promising target for the therapy of atherosclerosis? Cardiovasc Drug Rev 25: 146–161. [DOI] [PubMed] [Google Scholar]
- Cleynen I, Van de Ven WJ 2008. The HMGA proteins: a myriad of functions. Int J Oncol 32: 289–305. [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G 2009. RNA and disease. Cell 136: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe-Huntington J, Lam KC, Dias C, Majewski J 2009. Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet 5: e1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Krainer AR 2014. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 12: 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayem Ullah AZ, Lemoine NR, Chelala C 2012. SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update). Nucleic Acids Res 40(Web Server issue): W65–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin G, Lafaille C, Petrillo E, Buggiano V, Gómez Acuña LI, Fiszbein A, Godoy Herz MA, Nieto Moreno N, Muñoz MJ, Alló M, et al. 2013. Transcriptional elongation and alternative splicing. Biochim Biophys Acta 1829: 134–140. [DOI] [PubMed] [Google Scholar]
- Gallego ME, Gattoni R, Stévenin J, Marie J, Expert-Bezançon A 1997. The SR splicing factors ASF/SF2 and SC35 have antagonistic effects on intronic enhancer-dependent splicing of the β-tropomyosin alternative exon 6A. EMBO J 16: 1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez Acuña LI, Fiszbein A, Alló M, Schor IE, Kornblihtt AR 2013. Connections between chromatin signatures and splicing. Wiley Interdiscip Rev RNA 4: 77–91. [DOI] [PubMed] [Google Scholar]
- Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, Tsichlis PN, Shirley Liu X, Struhl K 2010. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell 17: 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Moore MJ 2012. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR 2008. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82: 834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR 2011. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478: 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannone C, Valcárcel J 2013. Chromatin's thread to alternative splicing regulation. Chromosoma 122: 465–474. [DOI] [PubMed] [Google Scholar]
- Iannone C, Pohl A, Papasaikas P, Soronellas D, Vicent GP, Beato M, Valcárcel J 2015. Relationship between nucleosome positioning and progesterone-induced alternative splicing in breast cancer cells. RNA 21: 360–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. 2005. A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino S, Mukai T, Kume N, Asano D, Ogawa M, Kuge Y, Minami M, Kita T, Shiomi M, Saji H 2007. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability—analysis in hypercholesterolemic rabbits. Atherosclerosis 195: 48–56. [DOI] [PubMed] [Google Scholar]
- Jangi M, Sharp PA 2014. Building robust transcriptomes with master splicing factors. Cell 159: 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD 2013. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153: 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanopka A, Mühlemann O, Akusjärvi G 1996. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 381: 535–538. [DOI] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR 2007. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaidakov M, Mitra S, Kang BY, Wang X, Kadlubar S, Novelli G, Raj V, Winters M, Carter WC, Mehta JL 2011. Oxidized LDL receptor 1 (OLR1) as a possible link between obesity, dyslipidemia and cancer. PLoS One 6: e20277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S 2012. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11: 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar T, Remm M 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23: 1289–1291. [DOI] [PubMed] [Google Scholar]
- Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD 2008. The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 15: 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR 2000. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol 20: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimon A, Mogilevsky M, Shilo A, Golan-Gerstl R, Obiedat A, Ben-Hur V, Lebenthal-Loinger I, Stein I, Reich R, Beenstock J, et al. 2014. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep 7: 501–513. [DOI] [PubMed] [Google Scholar]
- Manabe T, Katayama T, Sato N, Gomi F, Hitomi J, Yanagita T, Kudo T, Honda A, Mori Y, Matsuzaki S, et al. 2003. Induced HMGA1a expression causes aberrant splicing of Presenilin-2 pre-mRNA in sporadic Alzheimer's disease. Cell Death Differ 10: 698–708. [DOI] [PubMed] [Google Scholar]
- Mango R, Clementi F, Borgiani P, Forleo GB, Federici M, Contino G, Giardina E, Garza L, Fahdi IE, Lauro R, et al. 2003. Association of single nucleotide polymorphisms in the oxidised LDL receptor 1 (OLR1) gene in patients with acute myocardial infarction. J Med Genet 40: 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mango R, Biocca S, del Vecchio F, Clementi F, Sangiuolo F, Amati F, Filareto A, Grelli S, Spitalieri P, Filesi I, et al. 2005. In vivo and in vitro studies support that a new splicing isoform of OLR1 gene is protective against acute myocardial infarction. Circ Res 97: 152–158. [DOI] [PubMed] [Google Scholar]
- Matsunaga S, Xie Q, Kumano M, Niimi S, Sekizawa K, Sakakibara Y, Komba S, Machida S 2007. Lectin-like oxidized low-density lipoprotein receptor (LOX-1) functions as an oligomer and oligomerization is dependent on receptor density. Exp Cell Res 313: 1203–1214. [DOI] [PubMed] [Google Scholar]
- Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G 2006. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res 69: 36–45. [DOI] [PubMed] [Google Scholar]
- Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, Satoh H, Inoue K, Kawase Y, Jishage K, et al. 2007. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res 100: 1634–1642. [DOI] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlewski G, Cáceres JF 2010. RNase-assisted RNA chromatography. RNA 16: 1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki I, Ishigaki T, Oyama T, Matsunaga S, Xie Q, Ohnishi-Kameyama M, Murata T, Tsuchiya D, Machida S, Morikawa K, et al. 2005. Crystal structure of human lectin-like, oxidized low-density lipoprotein receptor 1 ligand binding domain and its ligand recognition mode to OxLDL. Structure 13: 905–917. [DOI] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr, Fu XD 2013. Genome wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 50: 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcarcel J 2015. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell 57: 7–22. [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci 101: 15974–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Inoue K, Higuchi I, Ono Y, Shimura Y 1992. Control of Drosophila Sex-lethal pre-mRNA splicing by its own female-specific product. Nucleic Acids Res 20: 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ 2011. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev 25: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Ast G 2010. Chromatin density and splicing destiny: on the cross-talk between chromatin structure and splicing. EMBO J 29: 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Kan JL, Green MR 2004. Arginine-serine-rich domains bound at splicing enhancers contact the branchpoint to promote prespliceosome assembly. Mol Cell 13: 367–376. [DOI] [PubMed] [Google Scholar]
- Soemedi R, Vega H, Belmont JM, Ramachandran S, Fairbrother WG 2014. Genetic variation and RNA binding proteins: tools and techniques to detect functional polymorphisms. Adv Exp Med Biol 825: 227–266. [DOI] [PubMed] [Google Scholar]
- Sterne-Weiler T, Sanford JR 2014. Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol 15: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuguchi M, Furutani M, Hinagata J, Tanaka T, Furutani Y, Imamura S, Kawana M, Masaki T, Kasanuki H, Sawamura T, et al. 2003. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem Biophys Res Commun 303: 247–250. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Stamm S 2009. Alternative splicing and disease. Biochem Biophys Acta 1792: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor JR, Papasaikas P, Valcárcel J 2015. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol Cell 57: 23–38. [DOI] [PubMed] [Google Scholar]
- Vohra RS, Murphy JE, Walker JH, Ponnambalam S, Homer-Vanniasinkam S 2006. Atherosclerosis and the Lectin-like OXidized low-density lipoprotein scavenger receptor. Trends Cardiovasc Med 16: 60–64. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wang GS, Cooper TA 2007. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8: 749–61. [DOI] [PubMed] [Google Scholar]
- Wu JY, Maniatis T 1993. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 75: 1061–1070. [DOI] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CY, Theusch E, Lo K, Mangravite LM, Naidoo D, Kutilova M, Medina MW 2014a. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum Mol Genet 23: 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RY, Brazaitis J, Gallagher G 2014b. The human IL-23 receptor rs11209026 A allele promotes the expression of a soluble IL-23R–encoding mRNA species. J Immunol 194: 1062–1068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.