

Abstract
Calcium plays a role in long term plasticity by triggering post-synaptic signaling pathways for both the strengthening (LTP) and weakening (LTD) of synapses. Since these are opposing processes, several hypotheses have been developed to explain how calcium can trigger LTP in some situations and LTD in others. These hypotheses fall broadly into three categories, based on the amplitude of calcium concentration, the duration of the calcium elevation, and the location of the calcium influx. Here we review the experimental evidence for and against each of these hypotheses and the recent computational models utilizing each. We argue that with new experimental techniques for the precise visualization of calcium and new computational techniques for the modeling of calcium diffusion, it is time to take a new look at the location hypothesis.
Keywords: calcium, plasticity, LTP, LTD, STDP, dendrite, spine
Introduction
The relationship between calcium and plasticity is a complicated one. In most cell types, the strengthening of neuronal connections (long term potentiation, LTP) and the weakening of neuronal connections (long term depression, LTD) both require an elevation of intracellular calcium (Artola and Singer, 1993; Cummings et al., 1996; Fino et al., 2010). But LTP and LTD are opposing processes with opposing results. How can calcium cause synaptic strengthening in one situation but synaptic weakening in another?
Calcium acts as a second messenger, triggering complex signaling cascades. In other words, the molecules that bind calcium determine calcium’s function. For example if calcium binds to CaMKII, it will initiate phosphorylation events that often lead to LTP. However if calcium binds to calcineurin, it will initiate dephosphorylation events which often lead to LTD. CaMKII and calcineurin are just two examples of the many calcium binding proteins. Different neuron types express different calcium binding proteins, which makes finding one universal rule governing calcium activity difficult if not impossible. In this review we investigate three factors that could determine which molecules preferentially bind calcium, even within a single cell type.
Three factors describe characteristics of the calcium signal that influence activation of downstream signaling pathways: Amplitude, Duration, and Location. They can be formulated as hypotheses thus: “the X (amplitude, duration, or location) of the calcium signal determines the direction of plasticity.” and therefore can be tested experimentally and evaluated with computational models. A hypothesis positing that calcium amplitude determines the direction of plasticity was the first to emerge (Lisman, 1989; Artola et al., 1990; Artola and Singer, 1993), and is usually articulated as a two-threshold hypothesis where a moderate calcium amplitude is necessary for LTD but an even higher calcium amplitude is necessary for LTP. The duration hypothesis adds to the amplitude hypothesis, predicting that the time course of calcium elevation also helps determine the direction of plasticity. The duration hypothesis is often described as a lower, slower calcium signal resulting in LTD, and a higher, faster calcium transient resulting in LTP. Finally, the location hypothesis states that the specific site of calcium entry on a micro or even nano scale determines the direction of plasticity by controlling which calcium binding proteins are in close proximity to the calcium influx.
Although there are many types of synaptic plasticity, this brief review is limited to homosynaptic plasticity that depends on post-synaptic voltage gated calcium channels (VGCCs) and NMDA receptors (NMDARs). Furthermore, we focus on tissue types with numerous reports of bi-directional plasticity, such as hippocampal area CA1, neocortex and striatum. We examine the experiments supporting and refuting these three hypotheses, as well as the computational models utilizing each. We also discuss the possible interactions between these factors and detail important questions which remain unanswered. Finally, we argue that with new experimental techniques and sophisticated computational algorithms, it is time to take a new look at the location hypothesis.
Amplitude
The two threshold hypothesis
The calcium amplitude hypothesis states that the peak calcium determines the direction of plasticity. Specifically, a low but still significant calcium elevation results in LTD, while a higher calcium elevation results in LTP (Figure 1).
Figure 1. Calcium Amplitude Hypothesis.
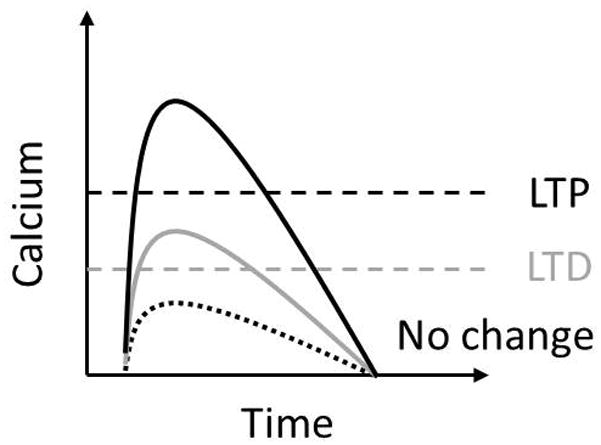
Peak calcium elevation must cross a low threshold to induce LTD, and another, higher threshold to induce LTP.
Experimental evidence
There are several key experiments that suggest calcium amplitude determines the direction of synaptic plasticity. These experiments fall into two categories, those testing frequency dependent plasticity and those testing spike timing dependent plasticity (STDP). It is well established that in some brain regions high frequency stimulation (HFS) can cause LTP, while low frequency stimulation (LFS) can cause LTD (reviewed in Stanton, 1996). In contrast to frequency dependent plasticity, STDP depends on the timing of the post-synaptic action potential relative to the pre-synaptic stimulation to determine the direction of plasticity. When the action potential (AP) precedes the pre-synaptic stimulation (post-pre), LTD is induced, but when the AP comes after (pre-post), LTP is induced (reviewed in Bi and Poo, 2001). For both types of induction paradigm, it has been postulated that the direction of plasticity is due to the difference in calcium amplitude induced by either HFS versus LFS or pre-post pairing versus post-pre pairing.
Artolaet al. (1990) showed that post-synaptic depolarization level could alter the direction of plasticity while the frequency of stimulation was kept constant. A stimulation that induced LTD under baseline conditions converted to LTP when given in conjunction with a strong post-synaptic depolarization. However, the same stimulus in conjunction with a post-synaptic hyperpolarization resulted in no change in synaptic strength. This experiment was the first to suggest two ‘excitability’ thresholds for plasticity, a lower one for LTD and a higher one for LTP. Imaging of calcium concentration during application of either HFS or LFS to neocortical cells revealed that the calcium peak was highest during the HFS protocol which induces LTP, and that the calcium signal was lower, but still significant during the LFS protocol which induces LTD (Hansel et al., 1996). While this experiment does not show a causal relationship between calcium amplitude and plasticity, it supports the two threshold hypothesis in that the stimulation pattern that induces LTP correlates with higher calcium amplitudes than the stimulation pattern that induces LTD (Hansel et al., 1996:Figure 1).
More direct evidence is provided by Mulkey and Malenka (1992) who directly manipulate the extracellular calcium concentration in the hippocampus during frequency dependent plasticity induction. They showed that lowering the extracellular calcium concentration turned LTP into LTD when the same stimulation frequency was applied (Mulkey and Malenka, 1992). This supports the amplitude hypothesis because the same channels would be opened in both stimulation conditions, but the calcium through the channels would be decreased due to the lower extracellular concentration. Similar results were shown in the cortex using different concentrations of calcium chelators (Cho et al., 2001).
Similar to the results seen in frequency dependent plasticity, the specific timing of a pre synaptic stimulation and a post-synaptic AP can control both the calcium amplitude and the direction of plasticity. A pairing in which the pre-synaptic stimulation precedes the post-synaptic AP (pre-post) induces LTP; while a pairing in which the post-synaptic AP precedes the pre-synaptic stimulation induces LTD (Bi and Poo, 1998). Koester and Sakmann (1998) demonstrate that a post-pre stimulation (known to induce LTD), triggers a higher calcium elevation than either pre or post stimulation alone. However, during a pre-post stimulation (known to induce LTP), calcium elevation is even higher than during the post-pre stimulation. The spike timing dependent plasticity (STDP) is thought to be a more physiologically realistic approximation of how plasticity may be induced in vivo; however it is not without its limitations, as plasticity can occur without synaptic input (Cummings et al., 1996)and without spikes (Fino et al., 2009). Nonetheless, as in frequency dependent plasticity, LTP correlates with higher calcium and LTD correlates with lower calcium.
The high calcium amplitude that leads to LTP is thought to be generated by calcium influx through the NMDAR. The low calcium leading to LTD has more diverse sources. In particular, most LTD in the hippocampus depends on NMDAR (Malenka and Nicoll, 1993), whereas LTD in the striatum requires L type calcium channels (Fino et al., 2010). However, a recent study by Nabavi et al. (2013) has shown that NMDAR-dependent LTD in the hippocampus is due to a metabotropic action of the NMDAR rather than calcium influx through its pore. They show that this form of LTD requires basal levels of intracellular calcium, but does not require an elevation in calcium.
Computational models
Because the experimental measurement of calcium elevation requires the use of a calcium indicator dye, which binds to and buffers calcium, it is difficult to precisely control and measure the calcium amplitude. Therefore biophysical computational models have been constructed to manipulate calcium amplitude and evaluate frequency-based or STDP-based plasticity rules (Holmes and Levy, 1990; Gold and Bear, 1994; Shouval et al., 2002; Helias et al., 2008; Bush and Jin, 2012; Evans et al., 2012; Cutsuridis, 2013).
An early computational model of calcium influx, diffusion, and buffering in a single cortical dendritic spine compared the calcium amplitudes occurring in response to differing stimulation frequencies (Gamble and Koch, 1987). The model predicts a supralinear relationship between stimulation frequency and calcium amplitude in the spine with high frequency stimulation generating higher calcium peaks than low frequency stimulation. This model also predicts that changes in the calcium amplitude will strongly determine the activation of calmodulin, with a 5-fold increase in calcium amplitude causing an almost 1000-fold increase in active calmodulin (Gamble and Koch, 1987). A subsequent computational model, implementing calcium influx, diffusion, and buffering in the spine head of dentate granule cell, added specific NMDA and non-NMDA receptor components to the stimulation (Holmes and Levy, 1990). The beauty of this model was in attaching the spine to an entire morphologically reconstructed neuron. This model was used to test whether the non-linear relationship between input frequency and calcium amplitude due to the NMDAR voltage dependence was strong enough to explain the associative plasticity seen in the dentate gyrus. The model predicts that calcium influx through the NMDAR is fourfold higher in response to a 200 Hz stimulation than a 10 Hz stimulation, but that this fourfold amplitude increase is not enough to explain the experimentally observed plasticity. The model suggests that subcellular processes such as calcium buffer saturation serve to enhance the non-linear relationship between frequency and calcium amplitude (Holmes and Levy, 1990). This is an example where the disagreement between model and experiments highlights critical properties of the tissue not previously discovered through experiments alone.
Some computational models attempt to reproduce both STDP and frequency dependent plasticity induction (For review of computational models using calcium amplitude to predict primarily STDP curves see Graupner and Brunel, 2010). Assuming that the main source of calcium in the dendrites is the NMDAR, Shouval et al. (2002) created a plasticity-predictive model based on calcium amplitude. Using this model, the authors were able to match a frequency based (0.5–20 Hz) plasticity curve and partially match a spike timing (STDP) based plasticity curve (Shouval et al., 2002:figure 3). A more recent model of spines on CA1 hippocampal neurons implements stochastic transitions between high and low synaptic states. The transition probabilities are calculated from simplified, calcium based models of kinase and phosphatase activity. The model was tuned to match hippocampal STDP experiments, and then tested using stimulation paradigms other than those used to tune the model. The model was not only able to predict frequency dependent plasticity, but also replicated the observed plasticity due to a variety of other induction protocols such as subthreshold depolarizations and burst pairings (Bush and Jin, 2012).
Outstanding questions
That calcium amplitude would determine the direction of plasticity seems both reasonable and useful at first glance, but further investigation reveals that it is incomplete. The main problem is revealed through the computational models of calcium-based STDP. Both Shouval et al. (2002) and Bush and Jin (2012) show an STDP curve with two windows for LTD. The first window is in the post-pre timing range and matches up with experimental data, but the second window is in the pre-post range between about 70 and 120 ms time difference between the pre-synaptic stimulation and the post-synaptic action potential. This model discrepancy is not due to specific model assumptions, because Rubin et al. (2005) show that any computational model based solely on calcium amplitude will always exhibit a second LTD window at long positive timing intervals. This is a problem because the experimental data does not support this shape for the STDP curve. Most experiments demonstrating STDP show no LTD during pre-post intervals (Markram et al., 1997; Bi and Poo, 1998; Feldman, 2000; Pawlak and Kerr, 2008) (but see Nishiyama et al., 2000 and Wittenberg and Wang, 2006) (Figure).
Direct evidence against the amplitude hypothesis comes from an experimental study (Nevian and Sakmann, 2006) in which calcium amplitude in the spine can predict LTP and LTD at extreme concentrations, but there is a middle range where calcium amplitude does not predict the direction of plasticity. Specifically, while inducing opposing directions of plasticity, both pre-post (LTP) and post-pre (LTD) timing intervals can have the same measured calcium amplitude. In summary, amplitude provides a reasonable explanation of plasticity in certain cases, but is not always a sufficient predictor, and other aspects of calcium dynamics may be critical in determining the direction of plasticity.
Duration
The low and slow, high and fast hypothesis
The calcium duration hypothesis is not an alternative to the amplitude one, but rather it adds additional criteria. It states that the kinetics of the calcium elevation in conjunction with differences in peak calcium strongly influences the direction of plasticity. Specifically it is thought that a high and fast calcium signal will trigger LTP, while a low and slow calcium signal will trigger LTD (Figure).
Experimental evidence
To test whether calcium kinetics determine plasticity, experimenters need to control the duration that calcium is elevated within the cell. One study using photolytic uncaging of intracellular calcium in hippocampal slices showed that the same calcium uncaging protocol could induce either LTP or LTD. The estimated concentration (300–500 nM) released during the photolysis did not predict the direction of plasticity (Neveu and Zucker, 1996). Therefore this study did not support the two-threshold amplitude hypothesis. However, the calcium uncaging method used in this study caused only moderate calcium increases (Yang et al., 1999), possibly similar to the ‘middle range’ described by Nevian and Sakmann (2006) in which the calcium amplitude did not consistently predict the direction of plasticity. Thus, rather than refuting the calcium amplitude hypothesis, these results may only limit its application to all but this middle range of calcium concentration.
In a subsequent study, a newer calcium uncaging method allowed for more precise temporal control of calcium elevation and higher calcium peaks (Yang et al., 1999). Sustaining an estimated concentration of 750 nM calcium for 1 minute induced LTD, while a rise in calcium concentration >10μM for just a few seconds resulted in LTP. This study supports the idea that a high and fast elevation of calcium produces LTP, while a low and slow elevation in calcium produces LTD (Yang et al., 1999: figure 2). These experiments showed that the duration of the calcium elevation along with the peak amplitude can determine the direction of plasticity.
While direct experiments testing the duration hypothesis are scant, indirect support comes from experiments manipulating calcium release from internal stores. Because calcium release from internal stores is secondary to calcium influx or to activation of metabotropic glutamate receptors, the calcium signal issuing from the internal stores is of longer duration when compared with the direct calcium influx through VGCCs and NMDAR. Consistent with the idea that slow calcium signals would trigger LTD, several experimental studies have implicated calcium release from internal stores in the control of LTD (Nishiyama et al., 2000; Jo et al., 2008; Camiré and Topolnik, 2014).
Computational models
It is extremely difficult to experimentally manipulate the duration of calcium elevation. Furthermore, measurements of the duration of the calcium elevation are altered by the binding kinetics of the calcium reporters used for imaging calcium concentration. Because of these difficulties, experimental evidence for the duration hypothesis is scarce. However, computational models are particularly useful in investigating the effects of calcium duration because they provide the ability to precisely control the total quantity of calcium influx. Thus, they can demonstrate the plausibility of the hypothesis and help direct experiments toward testing potential mechanisms. Therefore several computational models evaluate the consequences of different durations of calcium influx (Gamble and Koch, 1987; Bi and Rubin, 2005; Goldberg and Wilson, 2005; Rubin et al., 2005; Carlson and Giordano, 2011; Kumar and Mehta, 2011; Graupner and Brunel, 2012; Li et al., 2012).
Some models use calcium amplitude thresholds, and explicitly take signal duration into account (Rubin et al., 2005; Kumar and Mehta, 2011; Graupner and Brunel, 2012). Graupner and Brunel (2012) use a simplified model of calcium dynamics in a dendritic spine to produce a range of STDP curves as seen in different cell types. They make use of two calcium amplitude thresholds (Graupner and Brunel, 2012: Figure 1), but track how much time the calcium elevation is above each threshold to determine the strength and direction of plasticity. Rubin et al. (2005) accurately replicate experimental STDP curves in a simplified hippocampal pyramidal neuron model by using calcium-dependent equations for kinase and phosphatase activity. The phosphatase equation with a low calcium threshold requires a minimum duration to trigger LTD induction. The kinase equation has a high calcium threshold, which must be exceeded for LTP to occur. An additional equation detects a middle level of calcium, and can trigger a ‘veto mechanism’ to prevent LTD induction. This manipulation permits the post-pre (-Δt) LTD window because the calcium signals are long enough to cause LTD during those post-pre intervals. However it eliminates the problematic second LTD window because the calcium signals during the pre-post (+Δt) timing intervals trigger the LTD veto mechanism (Figure 2).
Figure 2.
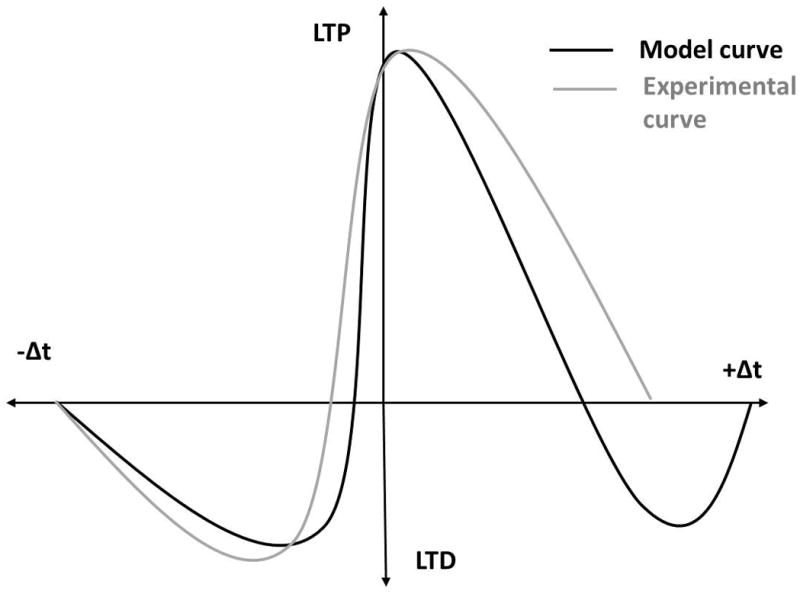
Schematic graph showing the difference between a computational model STDP curve (similar to Shouval et al., 2002) and an experimental STDP curve (similar to Bi and Poo, 1998). When using a calcium amplitude hypothesis, model STDP curves do not match the experimental STDP. The models show a second LTD window during the positive Δt (black trace) that is not reflected in experimental data (gray trace).
Computational models using calcium pools with single time constants of decay cannot isolate calcium duration from calcium amplitude because as the signal increases in amplitude, the duration that it exceeds a fixed threshold also increases. To more precisely control the time course and amount of calcium, a second group of models simulate calcium binding to different downstream molecules (kinases and phosphatases) which are known to produce either LTP or LTD. The proportion of calcium binding to ‘LTP’ molecules compared to the calcium binding to ‘LTD’ molecules can predict which stimulation patterns will produce which direction of plasticity. Calcium amplitude differentially controls these processes because LTP molecules, e.g. CaMKII, may have a different affinity for calcium than LTD molecules, e.g. calcineurin. Calcium duration can determine which molecules bind calcium because some molecules are fast binding (e.g. calmodulin N site) and some slow binding (e.g. calmodulin C site).
Li et al. (2012) used this biophysical modeling approach to compare the activation of CaMKII (an LTP facilitating molecule) and calcineurin (CaN or PP2B, a LTD facilitating molecule) while holding the total number of calcium ions constant. They found that higher frequencies (higher amplitude, shorter durations) more efficiently activated CaMKII, while lower frequencies (lower amplitude, longer durations) facilitated phosphatase activation. The ratio of kinase to phosphatase activity defines a frequency curve with the ratio favoring LTD between 1 and 3 Hz, and LTP between 10 and 200 Hz (Li et al., 2012: figure 6B). However, this frequency curve is modulated by calcium amplitude. When the total number of calcium ions is increased, CaMKII becomes sensitive to lower frequencies, increasing the range of frequencies that favor LTP. When the number of calcium ions is decreased, the range of frequencies favoring LTP is reduced (Li et al., 2012: figure 7).
Using an existing CaMKII/PP2A biophysical compartment model (Pi and Lisman, 2008), Carlson and Giordano (2011) demonstrate that calcium kinetics can prevent the problematic second LTD window (Carlson and Giordano, 2011: figure 6) (Figure 2). Similar to the results of Rubin et al. (2005) described above, they show that beyond a certain calcium amplitude threshold, LTP is induced, while below that threshold, LTD is induced, but only when the duration of the calcium elevation is greater than 45 ms (Carlson and Giordano, 2011: figure 2).
Outstanding questions
The duration component added to the amplitude hypothesis solves the problem of the second LTD window; however, neither the amplitude nor the duration of a calcium signal is constant throughout a dendrite or even within a single spine. Calcium binding molecules micrometers or even nanometers away from the calcium source will experience different calcium peaks and kinetics from calcium binding proteins adjacent to a calcium channel. Therefore the functional amplitude and duration of a calcium signal is governed by the location of both the calcium source and the calcium target.
Location
The microdomain/nanodomain hypothesis
The calcium location hypothesis states that the specific location of the calcium influx is what predicts which molecules calcium will bind to and therefore the direction of plasticity that calcium will trigger. The location for calcium influx required for LTP and LTD can be as far apart as spine and dendrite (microdomains, Figure 4A&C), or as close together as the PSD and adjacent membrane (nanodomains, Figure 4B&D). The location of calcium influx can be determined by different channel types, for example if VGCCs and NMDARs are located in different areas (Figure 4A&B). The location can determine which molecules bind calcium because different calcium sources (NMDAR, VGCC) may be anchored close to different calcium targets (CaM, Calcineurin). In contrast, the same calcium channel type can be located in different places, such as synaptic NMDARs (S-NMDAR) compared to extra synaptic NMDARs (ES-NMDAR) (Figure 4C&D).
Figure 4. Calcium Location Hypothesis.

A. Different calcium sources may provide calcium influx in different locations creating dendrite or spine specific microdomains. VGCC=voltage gated calcium channel B. Different calcium sources can cause nanodomain specificity within the spine. C. The same calcium channel type can be in different locations, creating specific calcium microdomains. S-NMDA=synaptic NMDA receptor, ES-NMDA=extrasynaptic NMDA receptor. D. The same channel type can cause nanodomain specificity within the spine.
Experimental evidence
Experiments demonstrating that a specific calcium channel is necessary for LTP or LTD suggest that calcium is required in a specific location. In the striatum, several studies have shown that NMDARs are specifically necessary for LTP, while other calcium sources such as L-type VGCCs are necessary for LTD (Fino et al., 2010; Shindou et al., 2011). Similarly in the hippocampus, NMDARs are necessary for both LTP and LTD, while L-type VGCCs are necessary for LTD only (Bi and Poo, 1998). Channel specificity can take the form of a microdomain hypothesis if one channel is on the dendrite and one is on the spine (Figure 4A), or a nanodomain hypothesis if one channel is synaptic (anchored to the PSD) while the other is on the side of a spine (Figure 4B).
Diverse effects of calcium influx through the same channel type in different locations (Figure 4C&D) also support the location hypothesis. While post-synaptic NMDARs contribute to LTP, selectively activating the extrasynaptic NMDARs causes LTD (Liu et al., 2013). In striatal cultures, stimulating synaptic NMDARs increases phosphorylated CREB, while activating extrasynaptic NMDARs decreases it. It is not clear how calcium through extrasynaptic NMDARs triggers one set of mechanisms while calcium through synaptic NMDARs triggers an opposing set. One possibility is that different calcium binding proteins are anchored to synaptic versus extra-synaptic NMDARs. Another possibility is that synaptic and extra-synaptic NMDARs different in their kinetics or calcium permeability. However, more experiments are needed to investigate how the exact location of an NMDAR can alters its function.
The above evidence suggests that calcium location is important for plasticity on the microdomain level. However, calcium location at the nanodomain level, within a single spine, is more difficult to study experimentally. Nonetheless, the existence and importance of calcium nanodomains have been demonstrated in phenomena other than synaptic plasticity. In dendritic spines, NMDARs represent the dominant calcium source, while the L-type calcium channels do not noticeably contribute to the total calcium elevation in the spine (Yasuda et al., 2003). Nonetheless, opening of L-type calcium channels is essential for the depression of R-type calcium channels due to back propagating action potentials (Yasuda et al., 2003). Similarly, local calcium through R-type calcium channels selectively activates the small conductance calcium-activated potassium channel (SK) in the spines of hippocampal neurons. When R-type channels are blocked, the effect of SK blockade was occluded, even though calcium in the spine head was increased. This result demonstrates that the calcium through R-type channels selectively activated the SK channel (Bloodgood and Sabatini, 2007). These observations show that distinct calcium pools within a single spine can activate distinct downstream molecules.
The different effects of fast versus slow calcium buffers also suggest that calcium nanodomains are important. BAPTA binds calcium quickly before it can diffuse from its entry point, while EGTA binds calcium more slowly allowing it to diffuse a small distance away. Therefore BAPTA will constrain the spatial specificity of calcium much more than EGTA (Naraghi and Neher, 1997). Consequently, the observation that BAPTA prevents the activation of BK channels by NMDA receptors, while EGTA does not, suggests a nano-scale calcium domain in olfactory granule cells (Isaacson and Murphy, 2001). Similarly, the calcium dependent inactivation of L type calcium channels is not sensitive to either BAPTA or EGTA, while the calcium dependent inactivation of N and R type calcium channels is eliminated by EGTA. This result suggests that N and R type calcium channels are sensitive to global changes in calcium, while L type calcium channels respond to local calcium elevation (Liang et al., 2003). Although calcium nanodomains have not yet been shown to play a role in plasticity, their importance in these other calcium dependent processes suggests that future research should investigate the importance of calcium nanodomains in synaptic plasticity.
Computational models
Computational models are an excellent tool for exploring the role of calcium microdomains and nanodomans in plasticity because they allow for complete control of calcium channel location. Nonetheless, evaluating the function of calcium microdomains requires detailed, spatial models of the myriad mechanisms controlling calcium dynamics. Therefore only a few such models have been developed.
A minimalistic model ‘co-localized’ specific calcium binding molecules with specific calcium sources by allowing activation of a downstream molecule by only one calcium source (Mihalas, 2011). CaMKII was activated by NMDARs, PP2B (calcineurin) was activated by VGCCs, and phosphodiesterases (PDEs) were activated by either NMDARs (to mimic spines in cortical slices) or IP3 receptors (to mimic spines in hippocampal cultures). Simulations from this model replicated the standard STDP curve and accounted for the difference seen in STDP-like triplets in cortical slices compared to hippocampal cultures. Although calcium diffusion and buffering were not specifically modeled in this study, it demonstrated that selective activation of calcium binding molecules by specific calcium sources might determine plasticity in place of calcium amplitude or duration.
A spine model with diffusion, buffering and pumps was created using MCell in order to investigate the spatial calcium gradients resulting from STDP induction protocols (Keller et al., 2008). Voltage gated calcium channels (VGCCs) were added uniformly to the spine, while the NMDARs were concentrated at the post-synaptic density (PSD). In this model, a pre-post stimulation caused a steep spatial calcium gradient with the densest calcium in the PSD and the lowest calcium at the base of the spine, while a post-pre stimulation resulted in a weaker gradient (Keller et al., 2008: figure 6). These results indicate that STDP stimulation patterns would cause different spatial profiles of calcium concentration in the spine. These differing calcium gradients could determine which downstream molecules calcium binds to, and consequently which plasticity mechanisms are set into motion.
Another spine model with spatial detail was created using the cellular dynamics simulator to evaluate whether the two lobes of calmodulin could be differentially sensitive to calcium nanodomains within the spine (Kubota and Waxham, 2010). Simulations showed that the fast binding N lobe of calmodulin is much more sensitive to calcium channel location than the slow binding C lobe, and that saturated calmodulin N lobe itself forms a distinct gradient within the spine. Because calcium binding to N or C lobes of calmodulin differentially activates CaMKII (Forest et al., 2008), this may be a mechanism by which calcium location can affect plasticity.
Outstanding questions
While it is tempting to conclude that channel specificity means location specificity, each channel has its own kinetics and therefore affects the duration of calcium influx as well as the location. For example, though certain calcium channels selectively activate certain potassium channels, the selective activation may be due to the specific time course of calcium influx, not the co-localization of calcium and potassium channels (Goldberg et al., 2009). Similarly, channel specificity could support the amplitude hypothesis because certain channels are more densely expressed and conduct more calcium. For example, the calcium that permeates NMDARs is likely much greater than the calcium permeating all other calcium channels in the spine taken together. NMDARs may be necessary simply because they facilitate high calcium amplitude in the spine. Supporting this, a very strong activation of L-type calcium channels can allow in a high enough amplitude of calcium to facilitate LTP even when NMDARs are blocked (Grover and Teyler, 1990). Due to these confounds, the experiments showing that VGCCs are essential for LTD and NMDARs are essential for LTP do not necessarily mean that the location of these specific calcium sources is important. Directly testing the role of calcium location in plasticity requires the most sophisticated calcium uncaging and imaging techniques.
In the past five years, great leaps have been made in our ability to precisely visualize and computationally model calcium location. New techniques like array tomography (Micheva et al., 2010), SBEM (Briggman et al., 2011), and STORM (Xu et al., 2013) now permit unprecedented levels of specificity in defining location of calcium channels on spine heads, necks and dendritic shafts. New advances in calcium imaging such as LOTOS (Chen et al., 2012) and GCaMP6 (Chen et al., 2013) create the ability to image single spines in vivo. Similarly, advances in sophisticated computational modeling software such as NeuroRD (Oliveira et al., 2010), Smoldyn (Andrews et al., 2010), and Vcell (Cowan et al., 2012) allow for the simulation of realistic calcium gradients in computational models (reviewed in Blackwell, 2013). Now is the time to re-examine the location hypothesis of calcium-dependent plasticity in light of these new tools.
Figure 3. Calcium Duration Hypothesis.
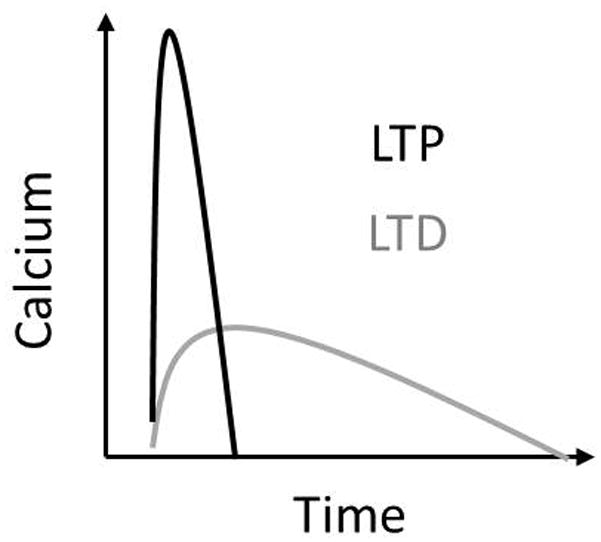
A low, slow calcium elevation results in LTD, while a high and fast calcium elevation results in LTP.
Acknowledgments
We would like to thank all of CENlab for helpful comments on earlier drafts of this manuscript. RCE was supported by NRSA1F31NS066645 from NINDS and a Dissertation Completion Grant from George Mason University. KTB was supported by the NSF NIH CRCNS program (R01AA016022 & R01AA18066) and by ONR (MURI N00014-10-1-0198).
Footnotes
Conflict of Interest: The authors have no conflicts of interest
References
- Andrews SS, Addy NJ, Brent R, Arkin AP. Detailed simulations of cell biology with Smoldyn 2.1. PLoSComput Biol. 2010;6:e1000705. doi: 10.1371/journal.pcbi.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola A, Bröcher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb’s postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci Off J Soc Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G-Q, Rubin J. Timing in synaptic plasticity: from detection to integration. Trends Neurosci. 2005;28:222–228. doi: 10.1016/j.tins.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Blackwell KT. Approaches and tools for modeling signaling pathways and calcium dynamics in neurons. J Neurosci Methods. 2013 doi: 10.1016/j.jneumeth.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471:183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Bush D, Jin Y. Calcium control of triphasic hippocampal STDP. J Comput Neurosci. 2012 doi: 10.1007/s10827-012-0397-5. [DOI] [PubMed] [Google Scholar]
- Camiré O, Topolnik L. Dendritic calcium nonlinearities switch the direction of synaptic plasticity in fast-spiking interneurons. J Neurosci Off J Soc Neurosci. 2014;34:3864–3877. doi: 10.1523/JNEUROSCI.2253-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson KD, Giordano N. Interplay of the magnitude and time-course of postsynaptic Ca2+ concentration in producing spike timing-dependent plasticity. J Comput Neurosci. 2011;30:747–758. doi: 10.1007/s10827-010-0290-z. [DOI] [PubMed] [Google Scholar]
- Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Varga Z, Jia H, Deca D, Rochefort NL, Konnerth A. LOTOS-based two-photon calcium imaging of dendritic spines in vivo. Nat Protoc. 2012;7:1818–1829. doi: 10.1038/nprot.2012.106. [DOI] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI. An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol. 2001;532:459–466. doi: 10.1111/j.1469-7793.2001.0459f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AE, Moraru II, Schaff JC, Slepchenko BM, Loew LM. Spatial modeling of cell signaling networks. Methods Cell Biol. 2012;110:195–221. doi: 10.1016/B978-0-12-388403-9.00008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Cutsuridis V. Interaction of inhibition and triplets of excitatory spikes modulates the NMDA-R-mediated synaptic plasticity in a computational model of spike timing-dependent plasticity. Hippocampus. 2013;23:75–86. doi: 10.1002/hipo.22057. [DOI] [PubMed] [Google Scholar]
- Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH, Venance L, Blackwell KT. The Effects of NMDA Subunit Composition on Calcium Influx and Spike Timing-Dependent Plasticity in Striatal Medium Spiny Neurons. PLoS Comput Biol. 2012;8:e1002493. doi: 10.1371/journal.pcbi.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L. Brief subthreshold events can act as Hebbian signals for long-term plasticity. PloS One. 2009;4:e6557. doi: 10.1371/journal.pone.0006557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forest A, Swulius MT, Tse JKY, Bradshaw JM, Gaertner T, Waxham MN. Role of the N- and C-lobes of calmodulin in the activation of Ca(2+)/calmodulin-dependent protein kinase II. Biochemistry (Mosc) 2008;47:10587–10599. doi: 10.1021/bi8007033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble E, Koch C. The dynamics of free calcium in dendritic spines in response to repetitive synaptic input. Science. 1987;236:1311–1315. doi: 10.1126/science.3495885. [DOI] [PubMed] [Google Scholar]
- Gold JI, Bear MF. A model of dendritic spine Ca2+ concentration exploring possible bases for a sliding synaptic modification threshold. Proc Natl Acad Sci U S A. 1994;91:3941–3945. doi: 10.1073/pnas.91.9.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Teagarden MA, Foehring RC, Wilson CJ. Nonequilibrium calcium dynamics regulate the autonomous firing pattern of rat striatal cholinergic interneurons. J Neurosci Off J Soc Neurosci. 2009;29:8396–8407. doi: 10.1523/JNEUROSCI.5582-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci Off J Soc Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Brunel N. Mechanisms of induction and maintenance of spike-timing dependent plasticity in biophysical synapse models. Front Comput Neurosci. 2010;4 doi: 10.3389/fncom.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Brunel N. Calcium-based plasticity model explains sensitivity of synaptic changes to spike pattern, rate, and dendritic location. Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21551. doi: 10.1073/pnas.1109359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Different threshold levels of postsynaptic [Ca2+]i have to be reached to induce LTP and LTD in neocortical pyramidal cells. J Physiol Paris. 1996;90:317–319. doi: 10.1016/s0928-4257(97)87906-5. [DOI] [PubMed] [Google Scholar]
- Helias M, Rotter S, Gewaltig MO, Diesmann M. Structural plasticity controlled by calcium based correlation detection. Front Comput Neurosci. 2008;2:7. doi: 10.3389/neuro.10.007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes WR, Levy WB. Insights into associative long-term potentiation from computational models of NMDA receptor-mediated calcium influx and intracellular calcium concentration changes. J Neurophysiol. 1990;63:1148–1168. doi: 10.1152/jn.1990.63.5.1148. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–1034. doi: 10.1016/s0896-6273(01)00428-7. [DOI] [PubMed] [Google Scholar]
- Jo J, Heon S, Kim MJ, Son GH, Park Y, Henley JM, Weiss JL, Sheng M, Collingridge GL, Cho K. Metabotropic glutamate receptor-mediated LTD involves two interacting Ca(2+) sensors, NCS-1 and PICK1. Neuron. 2008;60:1095–1111. doi: 10.1016/j.neuron.2008.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DX, Franks KM, Bartol TM, Sejnowski TJ. Calmodulin activation by calcium transients in the postsynaptic density of dendritic spines. PloS One. 2008;3:e2045. doi: 10.1371/journal.pone.0002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci U S A. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Waxham MN. Lobe specific Ca2+-calmodulinnano-domain in neuronal spines: a single molecule level analysis. PLoS Comput Biol. 2010;6:e1000987. doi: 10.1371/journal.pcbi.1000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Mehta MR. Frequency-Dependent Changes in NMDAR-Dependent Synaptic Plasticity. Front Comput Neurosci. 2011;5:38. doi: 10.3389/fncom.2011.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stefan MI, Le Novère N. Calcium input frequency, duration and amplitude differentially modulate the relative activation of calcineurin and CaMKII. PloS One. 2012;7:e43810. doi: 10.1371/journal.pone.0043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yang Q, Li S. Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res Bull. 2013;93:10–16. doi: 10.1016/j.brainresbull.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O’Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalas S. Calcium messenger heterogeneity: a possible signal for spike timing-dependent plasticity. Front Comput Neurosci. 2011;4:158. doi: 10.3389/fncom.2010.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A. 2013;110:4027–4032. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci Off J Soc Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci Off J Soc Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, Kim M, Zaccolo M, Blackwell KT. The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PloS One. 2010;5:e11725. doi: 10.1371/journal.pone.0011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Kerr JND. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci Off J Soc Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Lisman JE. Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J Neurosci Off J Soc Neurosci. 2008;28:13132–13138. doi: 10.1523/JNEUROSCI.2348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JE, Gerkin RC, Bi GQ, Chow CC. Calcium time course as a signal for spike-timing-dependent plasticity. J Neurophysiol. 2005;93:2600–2613. doi: 10.1152/jn.00803.2004. [DOI] [PubMed] [Google Scholar]
- Shindou T, Ochi-Shindou M, Wickens JR. A Ca(2+) threshold for induction of spike-timing-dependent depression in the mouse striatum. J Neurosci Off J Soc Neurosci. 2011;31:13015–13022. doi: 10.1523/JNEUROSCI.3206-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK. LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus. 1996;6:35–42. doi: 10.1002/(SICI)1098-1063(1996)6:1<35::AID-HIPO7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, Wang SSH. Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J Neurosci Off J Soc Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]