

Abstract
Causal genetic changes in oligodendrogliomas (OD) with 1p/19q co-deletion include mutations in IDH1, IDH2, CIC, FUBP1, TERT promoter and NOTCH1. However, it is generally assumed that more somatic mutations are required for tumorigenesis. This study aimed to establish whether genes mutated at low frequency can be involved in OD initiation and/or progression. We performed whole-genome sequencing on three anaplastic ODs with 1p/19q co-deletion. To estimate mutation frequency, we performed targeted resequencing on an additional 39 ODs. Whole-genome sequencing identified a total of 55 coding mutations (range 8–32 mutations per tumor), including known abnormalities in IDH1, IDH2, CIC and FUBP1. We also identified mutations in genes, most of which were previously not implicated in ODs. Targeted resequencing on 39 additional ODs confirmed that these genes are mutated at low frequency. Most of the mutations identified were predicted to have a deleterious functional effect. Functional analysis on a subset of these genes (e.g. NTN4 and MAGEH1) showed that the mutation affects the subcellular localization of the protein (n = 2/12). In addition, HOG cells stably expressing mutant GDI1 or XPO7 showed altered cell proliferation compared to those expressing wildtype constructs. Similarly, HOG cells expressing mutant SASH3 or GDI1 showed altered migration. The significantly higher rate of predicted deleterious mutations, the changes in subcellular localization and the effects on proliferation and/or migration indicate that many of these genes functionally may contribute to gliomagenesis and/or progression. These low-frequency genes and their affected pathways may provide new treatment targets for this tumor type.
Electronic supplementary material
The online version of this article (doi:10.1007/s11060-015-1741-1) contains supplementary material, which is available to authorized users.
Keywords: Oligodendroglioma, Infrequent mutations, Sequencing
Introduction
Oligodendrogliomas (ODs) account for 20 % of all glial tumors and are thought to arise from oligodendroglial precursor cells (OPCs). They are classified as either grade II or grade III and have a more favorable clinical and prognostic outcome with respect to other gliomas [1].
Frequently occurring driver mutations in oligodendrogliomas include mutations in IDH1, CIC, FUBP1, TERT promoter and NOTCH [2–6]. However, tumor formation is assumed to require more somatic mutations [7]. Interestingly, many other somatic mutations within protein-coding genes have also been identified in oligodendrogliomas, albeit at a low frequency [8]. The role of most of these infrequent mutations in tumorigenesis and/or progression is unclear. To date, only few studies have suggested a functional impact of genes mutated at low frequency in gliomas [9–13]. Therefore, low-frequency genes that play a role in the initiation and/or progression of ODs need to be identified to better understand OD pathogenesis and for development of targeted therapies.
Here, we have performed whole-genome sequencing on three ODs to identify all genetic changes in these tumors. We then performed targeted resequencing on an additional 39 tumors to demonstrate that many of these mutations occur at low frequency. Functional analysis of a subset of these low-frequency genes (NTN4, GDI1, MAGEH1, SASH3, ZNF238, OR5D14, ZNF57, DCUN1D2, ARSE, XPO7, GABRE and PGLYRP4) suggest that they can contribute to tumor pathogenesis and therefore are unlikely to be passengers. These genes and their affected pathways open up entirely novel treatment targets for this tumor type.
Materials and methods
Patient material
OD samples were collected from the Erasmus MC tumor archive. Use of patient material was approved by the Institutional Review Board and patients provided written informed consent according to national and local regulations for the clinical study and correlative tissue studies. After surgical resection, all samples were snap-frozen and stored at −80 °C. Non-neoplastic DNA was isolated from blood and stored at −80. Assessment of 1p19q LOH was performed previously [14, 15]. Patient characteristics are listed in supplementary Table 1.
DNA extraction and sequencing
Tumor DNA was extracted from fresh frozen (FF) tumor samples using the AllPrep DNA/RNA mini kit (Qiagen, Venlo, the Netherlands). Matched normal DNA was isolated from 1 ml blood as part of routine diagnostic procedures. Whole-genome sequencing was performed by Complete Genomics (Mountain View, US) using 5 µg DNA. Whole-genome sequencing data analysis was performed using cgatools version 1.4.0 and described in supplementary methods. All mutations were validated by Sanger sequencing. Targeted resequencing on an additional 39 ODs was performed by Baseclear (Leiden, the Netherlands). Library construction and resequencing data analysis were described in supplementary methods.
Cell lines and sorting
Constructs of wildtype and mutated genes were generated by site directed mutagenesis (n = 12), and C-terminally fused to GFP for visualization. Subsequently, Human oligodendroglial (HOG) cells [16] and HEK 293 cells were transiently transfected with wildtype or mutant constructs using lipofectamine according to the manufacturer’s instructions. Stably transfected HOG cells were created as described in supplementary methods.
Functional analysis
For proliferation experiments, cells (50.000 cells/well) were plated in a 24-well Greiner plate (Greiner Bio-One, Alphen a/d Rijn, the Netherlands) and followed using an Incucyte (Essenbioscience, Hertfordshire, United Kingdom). Growth curves were constructed using the Confluence v1.5 metric of the Incucyte software.
For migration experiments, cells were grown to confluence in a 24-well Essen ImageLockplate after which a cell-free zone (scratch) was created using a WoundMaker. Wells were then washed in PBS after which serum-free media was added to the plates. All plates were followed for 5 days and images were automatically captured at 3-h intervals from 3 to 4 separate regions within a well. Relative wound density, wound width and wound confluency curves were constructed using data points of every capture. All proliferation and migration experiments were performed at least in triplicate.
Flow cytometric cell cycle analysis using propidium iodide was performed as described in supplementary methods.
Results
Whole-genome sequencing
To identify somatic alterations in oligodendrogliomas, we performed whole-genome sequencing on DNA of three ODs and their matched germline DNA. All tumors were WHO grade III and had 1p19q co-deletion. The mapped sequence of the six samples varied between 237 and 249 Gb, resulting in a coverage between respectively 83 and 90-fold per genome. Confident diploid calls could be made for 94–95 % of the reference genome. On average 3,5 million genetic variants were identified per sample. Of these, 55 (range of 8–32 variants per sample), were localized to coding exons, were neither synonymous nor present in dbSNP130 and had a somatic score >−20 (see supplementary methods) (Fig. 1). Identified variants consisted of missense (84 %), nonsense (9 %) and frameshifts (7 %). All 55 mutations were validated by Sanger sequencing (supplementary Table 2).
Fig. 1.

Somatic genetic alterations in three oligodendrogliomas identified by whole-genome sequencing. Shown are all mutations identified in three tumors that (i) were localized to the coding regions of exons,( ii) were nonsynonymous, (iii) were absent from dbSNP130 and (iv) had a somatic score >−20. Alterations are coded in greyscale by the type of mutation (deletion, insertion, snp, substitution). The number of alterations range from 8 to 32 variants per sample. All 55 mutations were validated by Sanger sequencing
We performed an in silico analysis on all 55 genes to estimate the effect of mutation on protein. Polyphen-2 predictions were available for 43 genes of which 22 were probably damaging, 9 possible damaging, and 12 were benign. In randomly picked mutations identified with a lower confidence score (i.e. somatic scores ≤−20) the rate of damaging mutations was significantly lower: 10 probably damaging, 8 possible damaging and 21 benign changes, (p = 0.033, Chi square test). The identified mutations also had a slight tendency towards a higher conservation compared to mutations with somatic scores ≤−20: GERP-score 2.71 ± 2.96 v 1.75 ± 3.61 p = 0.156). The 55 mutations identified by whole-genome sequencing therefore were often predicted to have deleterious effects on the protein.
Targeted resequencing
Because we aimed to determine functional effects of genes mutated at low frequency in gliomas, we performed targeted resequencing of 44 (of the 55 identified by whole-genome sequencing) genes that were thusfar not implicated in gliomagenesis (supplementary Table 2). Resequencing was performed on the entire coding region of the 44 genes on 39 grade II/III ODs of which 28/39 had 1p/19q LOH, 5/39 had loss of 1p or 19q and 6/39 had no 1p19q loss. We chose this dataset because it represents a typical cohort of histologically diagnosed oligodendrogliomas where most will have 1p19q co-deletion, but some have other, more aggressive genetic changes.
No mutations were found in 39 of the 44 genes in any of the additional 39 tumors. Of the remaining five genes, mutations were identified in only one additional sample (Fig. 2, supplementary Table 4). In our samples, the mutations were all present at a high allele frequency (range 39–84 %) suggesting that the mutation is present in a large proportion of the resected tumor, at least within the tissue investigated. Among the 28 tumors with 1p19q co-deletion, mutations in only three of the 44 genes were identified (supplementary Table 5). Although in individual tumors the allele frequency was high, the overall mutation frequency of all 44 genes was low. The mutations in the 44 genes identified by our screen therefore are ‘low-frequency genes’: genes mutated at low frequency in oligodendrogliomas.
Fig. 2.

Targeted resequencing on 39 ODs confirms that many genes are mutated at low frequency. Of the known cancer genes (IDH1/2, TP53, NOTCH1, EGFR, CDKN2A, NF1, PIK3R1 and PTEN), a total of 48 mutations (range 1–4 mutations per tumor) were identified in 39 tumors (known causal cancer genes). Of the genes identified by whole-genome sequencing that thus far are not implicated in oligodendrogliomas, no mutations were found in 39 of the 44 genes in any of the additional 39 tumors (not shown). In the remaining five genes, we identified mutations in only one additional sample (indicated in the figure as low-frequency genes). Distribution of 1p19q status is reported for each identified mutation. Mutations in EGFR, CDKN2A and PTEN were only found in patients with intact 1p19q status
Apart from these low-frequency genes, we included a set of known cancer genes (IDH1, IDH2, PTEN, TP53, NOTCH1, EGFR, CDKN2A, CDKN2B, NF1 and PIK3R1) for reference on our targeted resequencing effort. Analysis of these known cancer-relevant genes revealed a total of 48 mutations (range of 1–4 mutations per tumor), which consisted of missense (44) and nonsense (4) variants. These include mutations in IDH1 (34 mutations in 39 tumors), TP53 (4/39), NOTCH1 (3/39), EGFR (2/39), IDH2 (1/39), CDKN2A (1/39), NF1 (1/39), PIK3R1 (1/39) and PTEN (1/39) (Fig. 2, supplementary Table 4). Mutations in EGFR, CDKN2A and PTEN were only found in patients with intact 1p19q chromosomes. Targeted resequencing therefore confirmed a high frequency of mutations in genes that drive oligodendrogliomas.
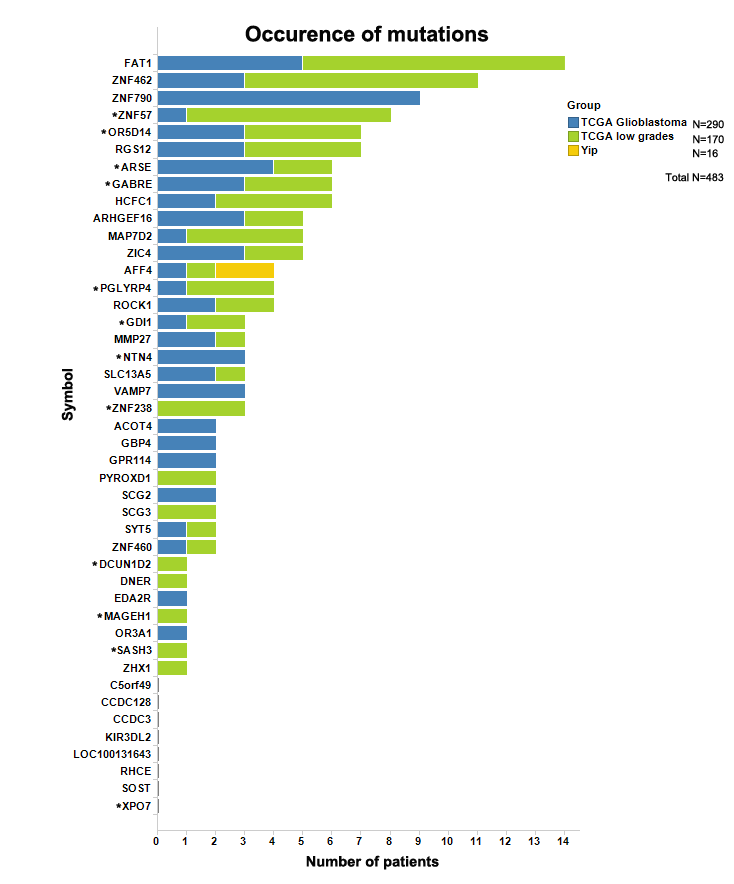
To better determine the incidence of the identified mutations, we analyzed exome sequencing data of 7 oligodendrogliomas from Bettegowda et al. [2], 16 oligodendrogliomas from Yip et al. [5] and 170 low-grade gliomas (LGG) and 290 glioblastomas (GBM) [8] from the TCGA dataset. In this large dataset, most [36/44] mutations identified by whole-genome sequencing that thus far are not implicated in oligodendrogliomas, were also identified in one of these datasets; eight were uniquely identified by us. The frequency was however low: often only one sample was identified in the external datasets with a mutation in that gene (supplementary Fig. 1).
Mutations in low-frequency genes can affect the proteins’ subcellular localization
In order to determine whether these low-frequency genes may be involved in glioma pathogenesis, we performed a more detailed molecular analysis on 12/44 of those genes (Figs. 3, 4, 5). These 12 genes were randomly picked from the 44 low-frequency genes. Transient transfection revealed that in 2/12 constructs, the mutation affects the proteins’ subcellular localization. In the gene NTN4, the mutation resulted in a strong nuclear localization, which was absent in the wildtype (Fig. 3). For MAGEH1, the mutation resulted in a reduced or even absent nuclear localization. No differences in the subcellular localization between wildtype and mutant constructs were found in cells expressing GDI1, ZNF238, SASH3, XPO7, ZNF57, GABRE, OR5D14, PGLYRP4, ARSE and DCUN1D2 (not shown).
Fig. 3.

Mutations in low-frequency genes can affect the subcellular localization of proteins. HOG and HEK 293 cells were transiently transfected with either the wildtype or mutant construct and stained for GFP (green), DAPI (blue) and Alexa fluor phalloidin (red). A total of twelve wildtype and mutant construct pairs were made. In both cell lines, the mutated NTN4 construct showed a stronger nuclear staining pattern compared to the wildtype construct. For MAGEH1, the mutation resulted in a reduced or even absent nuclear staining. No differences in the subcellular localization between wildtype and mutant constructs were found in HOG and HEK 293 cells expressing GDI1, ZNF238, SASH3, XPO7, ZNF57, GABRE, OR5D14, PGLYRP4, ARSE and DCUN1D2 (not shown)
Fig. 4.

Mutations in low-frequency genes can affect cell growth. a HOG cells stably expressing wildtype GDI1 show increased proliferation compared to GDI1 R193C expressing cells (p = 0.003, n = 2 independent experiments). b Cells stably expressing XPO7 D237N show a higher rate of proliferation during the first 24 h, compared to wildtype expressing cells (p = 0.02, (n = 2)). After 24 h, similar proliferation rates were observed in wildtype and XPO7 D237N. c HOG cells stably expressing SASH3 wildtype or SASH3 R288* and d wildtype or ZNF238 V121I show similar proliferation rates (n = 3)
Fig. 5.

Mutations in low-frequency genes can affect cell migration. a HOG cells stably expressing GDI1 wildtype have a higher migration rate compared to GDI1 R193C expressing cells (p = 0.003, n = 2 independent experiments). b No differences in migration were observed between cells expressing wildtype and XPO7 D237N or between cells expressing ZNF238 wildtype and mutant (d) (p = 0.5). c Cells expressing wildtype SASH3, show increased migration compared to SASH3 R288*(p = 0.039)
Low-frequency genes can affect proliferation and/or migration
To further examine the functional properties of low-frequency genes, we generated cell lines stably expressing the mutant or wildtype variant of four genes and performed functional analysis on proliferation and migration (Figs. 4, 5).
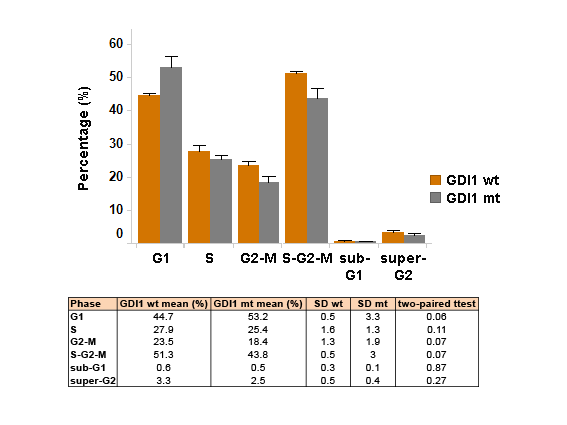
The first gene examined was GDI1, in which the mutation was located in the geranyl–geranyl transferase domain (c.577C > T, p.R193C). HOG cells stably expressing wildtype GDI1 have increased proliferation compared to those expressing GDI1 R193C or eGFP (p = 0.003, p = 0.04, respectively, n = 2). This is also confirmed by flow cytometry, showing that in cells expressing wildtype GDI1, 7.5 % more cells are found in the S-G2-M phase and 8.5 % less in the G1 compared to those expressing GDI1 R193C (supplementary Fig. 2). No differences in proliferation were observed between GDI1 R193C and eGFP cells (p = 0.7). Similarly, HOG cells stably expressing GDI1 wildtype showed increased migration compared to GDI1 R193C or eGFP (p = 0.003, p = 0.0005 respectively, n = 2). No differences in migration were observed between GDI1 R193C and eGFP cells (p = 0.3). The differences in both proliferation and migration between wildtype and mutant constructs indicate that the mutation in GDI1 affects the functional property of the protein.
Our second gene examined was XPO7, in which a mutation was identified in the ARM-type fold domain (c.709G > A, p.D237N). An increase in proliferation was observed during the first 24 h in cells stably expressing XPO7 D237N compared to wildtype and eGFP cells (p = 0.02, p < 0.001 (n = 2), respectively). After 24 h, the proliferation rate was similar in cells stably expressing wildtype, XPO7 D237N and eGFP. Because of the differences in proliferation at the start of the experiment, XPO7 D237N cells reach confluency more rapid than wildtype or eGFP cells. The initial difference was consistently observed in multiple experiments with 3–6 wells per experiment and 4 positions per well. Because cells were plated at 2 different densities (50.000 and 100.000 cells per well) the observed difference in initial proliferation rates appeared independent of plating density. A possible explanation for this difference is that XPO7 D237N cells recover more rapidly after plating. Migration of cells expressing XPO7 D237N or wildtype was higher than those expressing eGFP (p = 0.05, p = 0.03, respectively (n = 2)). No differences in migration were observed between cells expressing XPO7 D237N and wildtype (p = 0.5). The effect of the identified point mutation in XPO7 on proliferation highlights its importance for protein function in these cells.
The third gene examined was SASH3, in which the mutation was located in the SAM domain (c.862C > T), partially disrupting this domain in the C-terminal region (p.R288*). HOG cells stably expressing SASH3 wildtype, SASH3 R288* or eGFP have a similar proliferation rate (n = 3). However, cells expressing wildtype SASH3, show increased migration compared to SASH3 R288* or eGFP. The difference in migration between wildtype and SASH3 R288*indicates that the mutation affects the function of the wildtype protein (p = 0.001, n = 2).
Our last gene examined was ZNF238, in which the mutation was located in a BTB/POZ fold domain (c.361G > A, p.V121I). In HOG cells stably expressing ZNF238 wildtype or ZNF238 V121I constructs, we observed a slight decrease in proliferation compared to eGFP expressing cells (p < 0.001, n = 4). A more pronounced effect was observed in migration: HOG cells stably expressing ZNF238 wildtype and ZNF238 V121I show a strong decrease compared to eGFP. These results were consistently observed in multiple experiments (p < 0.001, n = 4). Although no differences were observed between wildtype and mutant constructs, the results do indicate that wildtype or ZNF238 V121I constructs affect cellular proliferation and migration.
Discussion
In this study, we have performed whole-genome and targeted resequencing on 3 and 39 oligodendrogliomas to identify somatic mutations. Apart from the known frequently mutated genes such as IDH1, CIC and FUBP1, our study also identified genes that were infrequently mutated (i.e. in one or two samples only). Most low-frequency genes are predicted to affect the protein’s function by in silico analysis. A significantly higher proportion of genes had a potential deleterious effect compared to mutations in genes identified with a lower confidence score (i.e. somatic scores ≤−20). Functional analysis of these low-frequency genes indicated that the identified mutation can affect protein subcellular localization and/or cell physiology. Our results therefore suggest that (at least some of) these genes may be relevant for gliomagenesis and/or contribute to progression.
The frequency of mutations identified at a higher frequency in our study (IDH1, CIC and FUBP1) are similar to those reported by others for this tumor type. Mutations in the ATRX gene were not identified; this gene is frequently mutated in other glioma subtypes including WHO grade II-III astrocytomas (71 %), oligoastrocytomas (68 %) and secondary glioblastomas (57 %) [2, 5]. Also similar to reported by others, mutations in EGFR, CDKN2A and PTEN were mutually exclusive with 1p19q co-deletion [2–5, 8].
To our knowledge, our study is the first to functionally study low-frequency genes on a larger scale. Several other studies have demonstrated a functional impact of genes mutated at a low frequency [9–13, 17]. Our data therefore are in line with the hypothesis that genes mutated at low frequency in gliomas can functionally contribute to gliomagenesis.
Of the genes examined, the mutations identified in NTN4 and MAGEH1 were predicted as “probably damaging” by Polyphen-2 analysis and affected the protein subcellular localization. Both mutations in NTN4 and MAGEH1 are not located in any of the known signal peptides for protein localization . These mutations may affect protein folding and sorting, however this would be accompanied by an accumulation of proteins in the ER, and we did not observe such accumulation in our transfected cells.
Of the genes examined in more detail, the mutation identified in GDI1 was predicted as “probably damaging” by Polyphen-2 analysis and did not affect the protein subcellular localization. Our data show that HOG cells stably expressing wildtype GDI1 have increased proliferation and migration compared to those expressing GDI1 R193C or eGFP. The proliferation experiments were validated by flow cytometry, showing that wildtype GDI1 expressing cells are more present in the S-G2-M phase and less in the G1 compared to mutant GDI1 expressing cells. GDI1 encodes for GDP dissociation inhibitor 1, which is involved in recycling of Rab proteins and contains a GTPase activation GDI1-β2/GDI1-β and geranyl–geranyl transferase domain (supplementary Fig. 3) [18–20]. The GTPase activating domain of GDI1 interacts with the GDP-bound Rab proteins, while the geranyl–geranyl domain interacts with the prenylated binding motif of Rab protein [21].
The altered migration may be caused by a differential activation of Rab proteins by GDI. Rab proteins belong to the Ras superfamily and are involved in vesicle trafficking between cellular compartments along actin or microtubules [22]. Binding of GDI to prenylated GDP-bound Rab protein in the cytosol mediates the delivery of Rab proteins to membranes during vesicle formation and their return into the cytosol after vesicle fusion [23, 24]. Importantly, Rab proteins seem to direct migration of cancer cells by regulating integrin recycling [25]. This mutation is functionally important as mutated GDI1 does not stimulate cell proliferation and migration whereas wildtype GDI1 does.
The second gene examined was XPO7, in which the mutation was also predicted to be probably damaging by Polyphen-2 analysis. The mutation did not affect the protein subcellular localization. Our data indicate that both wildtype and XPO7 D237N increase the proliferation rate of HOG cells. XPO7 encodes for exportin 7, which is a nuclear transport receptor that exports cargos from the nucleus into the cytoplasm [26]. This protein contains a N-terminal Importin-beta domain that binds to RAN and an ARM-type fold domain [26]. Its C-terminal region is thought to be involved in the recognition of substrates with broad specificity [27]. The fact that both wildtype and XPO7 D237N stimulate cell proliferation and migration, highlights the importance of this gene.
The third gene examined was SASH3, in which the mutation was predicted as probably damaging by Polyphen-2 analysis and did not affect the protein subcellular localization. The current study found that cells expressing wildtype SASH3, but not SASH3 R288*, show increased migration, indicating that the mutation affects the function of the wildtype protein. SASH3 encodes a signaling adapter protein, containing a SLY motif in the N-terminal region, a SH3 motif and a SAM motif in the C-terminal region [28]. SAM families of receptors are known to play a role in many developmental processes including cell migration, neuronal formation and angiogenesis [28]. They seem to mediate signal transduction by connecting downstream effector proteins to cell surface receptors [29, 30]. The altered migration pattern in SASH3 R288* expressing cells may therefore be caused by disturbed signal transduction pathways that lead to cell migration. This mutation is functionally important as SASH3 R288* does not stimulate cell migration where wildtype SASH3 does.
The last gene examined was ZNF238, in which the mutation was predicted as “probably damaging” by Polyphen-2 analysis and did not affect the protein subcellular localization. Our data indicate that cells stably expressing wildtype or ZNF238 V121I decrease proliferation and migration compared to control. The present finding is consistent with other studies in which ZNF238 was found to decrease proliferation [31, 32] and that ZNF238 is essential for neuronal migration in experimental mouse models [32–34]. ZNF238 encodes a transcriptional repressor that contains a BTB/POZ fold domain in the N-terminal region and four zinc fingers in the C-terminal region [35, 36]. This protein family plays a role in many processes, including DNA damage response, cell cycle and developmental processes [31–34, 37]. Our observation that wildtype and mutant constructs affect cellular proliferation and migration, highlights the important role of this gene.
Although our data argue that many genes are functionally relevant for gliomas, there are some limitations to our study. For all functional experiments, we have only utilized the HOG cell line, as these cells have been well characterized and resemble immature oligodendrocytes [16]. Whether these effects are retained in other cell lines remains to be determined. For example, to determine whether these genes indeed contribute to gliomagenesis, similar experiments need to be performed in stem cells, patient derived xenografts and various in vivo experiments. However because mutations occur at such low frequency, patient-derived primary cultured tumor cell lines containing these mutations are hard to obtain. Moreover, oligodendrogliomas with 1p19qLOH and/or IDH1 mutated tumors virtually cannot be propagated in vitro. Nevertheless, our data shows that even in a single cell line, a substantial proportion of low-frequency genes functionally affects cell physiology.
Another limitation of this study is that we have only performed functional analysis of a few genes. However, in a substantial proportion of the examined genes, the mutation affected the function of the wildtype protein. Our data therefore suggest that a substantial proportion of low-frequency genes are functionally relevant for glioma initiation and/or progression.
In contrast to may be expected for oncogenic mutations, we found that the proliferation and/or migration rate was reduced in cells expressing GDI1 R1193C, ZNF238 V121I and SASH3 R288*. It is not uncommon for ectopic expression of oncogenes in various cell lines to result in reduced proliferation and altered migration. In fact, Sun et al. have shown that expression of EGFR in mutant melanoma cells confers a growth disadvantage that is further strengthened by the addition of EGFR ligand [38]. A similar growth disadvantage (and altered migration pattern) was observed when expressing IDH1 R132H [10, 39]. Perhaps, this is a relatively common physiological response of cells that have never been dependent on a specific oncogene. It is however also possible that the mutation does not contribute to tumor formation/progression but simply has deleterious effects on the functioning of the wildtype protein.
In conclusion, we have demonstrated that low-frequency genes can affect the proteins’ subcellular localization and/or physiology in HOG cells. These findings indicate that low-frequency genes functionally can contribute to gliomagenesis and/or progression and suggest these genes as new therapeutic targets for treatment for this tumor type.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported from grants from the Dutch Foundation for Scientific Research ZonMw (Grants No. 95110051 and 92003560), Programme Translational Research.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Bromberg JE, van den Bent MJ. Oligodendrogliomas: molecular biology and treatment. Oncologist. 2009;14:155–163. doi: 10.1634/theoncologist.2008-0248. [DOI] [PubMed] [Google Scholar]
- 2.Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, Velculescu VE, Oba-Shinjo SM, Marie SK, Vogelstein B, Bigner D, Yan H, Papadopoulos N, Kinzler KW. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahm F, Koelsche C, Meyer J, Pusch S, Lindenberg K, Mueller W, Herold-Mende C, von Deimling A, Hartmann C. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 5.Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, Birol I, Chesnelong C, Chiu R, Chuah E, Corbett R, Docking R, Firme M, Hirst M, Jackman S, Karsan A, Li H, Louis DN, Maslova A, Moore R, Moradian A, Mungall KL, Perizzolo M, Qian J, Roldan G, Smith EE, Tamura-Wells J, Thiessen N, Varhol R, Weiss S, Wu W, Young S, Zhao Y, Mungall AJ, Jones SJ, Morin GB, Chan JA, Cairncross JG, Marra MA. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, Hruban RH, Jallo GI, Mandahl N, Meeker AK, Mertens F, Netto GJ, Rasheed BA, Riggins GJ, Rosenquist TA, Schiffman M, Shih IM, Theodorescu D, Torbenson MS, Velculescu VE, Wang TL, Wentzensen N, Wood LD, Zhang M, McLendon RE, Bigner DD, Kinzler KW, Vogelstein B, Papadopoulos N, Yan H. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bralten LB, Gravendeel AM, Kloosterhof NK, Sacchetti A, Vrijenhoek T, Veltman JA, van den Bent MJ, Kros JM, Hoogenraad CC, Sillevis Smitt PA, French PJ. The CASPR2 cell adhesion molecule functions as a tumor suppressor gene in glioma. Oncogene. 2010;29:6138–6148. doi: 10.1038/onc.2010.342. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Pan L, Che X, Cui D, Li C. Gli1 inhibition induces cell-cycle arrest and enhanced apoptosis in brain glioma cell lines. J Neurooncol. 2010;98:319–327. doi: 10.1007/s11060-009-0082-3. [DOI] [PubMed] [Google Scholar]
- 11.Basto D, Trovisco V, Lopes JM, Martins A, Pardal F, Soares P, Reis RM. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol. 2005;109:207–210. doi: 10.1007/s00401-004-0936-x. [DOI] [PubMed] [Google Scholar]
- 12.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 13.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bralten LB, Kloosterhof NK, Gravendeel LA, Sacchetti A, Duijm EJ, Kros JM, van den Bent MJ, Hoogenraad CC, Sillevis Smitt PA, French PJ. Integrated genomic profiling identifies candidate genes implicated in glioma-genesis and a novel LEO1-SLC12A1 fusion gene. Genes Chromosom Cancer. 2010;49:509–517. doi: 10.1002/gcc.20760. [DOI] [PubMed] [Google Scholar]
- 15.French PJ, Swagemakers SM, Nagel JH, Kouwenhoven MC, Brouwer E, van der Spek P, Luider TM, Kros JM, van den Bent MJ, Sillevis Smitt PA. Gene expression profiles associated with treatment response in oligodendrogliomas. Cancer Res. 2005;65:11335–11344. doi: 10.1158/0008-5472.CAN-05-1886. [DOI] [PubMed] [Google Scholar]
- 16.Buntinx M, Vanderlocht J, Hellings N, Vandenabeele F, Lambrichts I, Raus J, Ameloot M, Stinissen P, Steels P. Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: morphology and oligodendrocyte-specific gene expression. J Neurocytol. 2003;32:25–38. doi: 10.1023/A:1027324230923. [DOI] [PubMed] [Google Scholar]
- 17.Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H, Housman D. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21) Genes Chromosom Cancer. 2003;37:58–71. doi: 10.1002/gcc.10207. [DOI] [PubMed] [Google Scholar]
- 18.D’Adamo P, Menegon A, Lo Nigro C, Grasso M, Gulisano M, Tamanini F, Bienvenu T, Gedeon AK, Oostra B, Wu SK, Tandon A, Valtorta F, Balch WE, Chelly J, Toniolo D. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat Genet. 1998;19:134–139. doi: 10.1038/487. [DOI] [PubMed] [Google Scholar]
- 19.Garrett MD, Zahner JE, Cheney CM, Novick PJ. GDI1 encodes a GDP dissociation inhibitor that plays an essential role in the yeast secretory pathway. EMBO J. 1994;13:1718–1728. doi: 10.1002/j.1460-2075.1994.tb06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luan P, Balch WE, Emr SD, Burd CG. Molecular dissection of guanine nucleotide dissociation inhibitor function in vivo. Rab-independent binding to membranes and role of Rab recycling factors. J Biol Chem. 1999;274:14806–14817. doi: 10.1074/jbc.274.21.14806. [DOI] [PubMed] [Google Scholar]
- 21.Gavriljuk K, Itzen A, Goody RS, Gerwert K, Kotting C. Membrane extraction of Rab proteins by GDP dissociation inhibitor characterized using attenuated total reflection infrared spectroscopy. Proc Natl Acad Sci USA. 2013;110:13380–13385. doi: 10.1073/pnas.1307655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 23.Sivars U, Aivazian D, Pfeffer SR. Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature. 2003;425:856–859. doi: 10.1038/nature02057. [DOI] [PubMed] [Google Scholar]
- 24.Ullrich O, Stenmark H, Alexandrov K, Huber LA, Kaibuchi K, Sasaki T, Takai Y, Zerial M. Rab GDP dissociation inhibitor as a general regulator for the membrane association of rab proteins. J Biol Chem. 1993;268:18143–18150. [PubMed] [Google Scholar]
- 25.Recchi C, Seabra MC. Novel functions for Rab GTPases in multiple aspects of tumour progression. Biochem Soc Trans. 2012;40:1398–1403. doi: 10.1042/BST20120199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koch P, Bohlmann I, Schafer M, Hansen-Hagge TE, Kiyoi H, Wilda M, Hameister H, Bartram CR, Janssen JW. Identification of a novel putative Ran-binding protein and its close homologue. Biochem Biophys Res Commun. 2000;278:241–249. doi: 10.1006/bbrc.2000.3788. [DOI] [PubMed] [Google Scholar]
- 27.Kutay U, Hartmann E, Treichel N, Calado A, Carmo-Fonseca M, Prehn S, Kraft R, Gorlich D, Bischoff FR. Identification of two novel RanGTP-binding proteins belonging to the importin beta superfamily. J Biol Chem. 2000;275:40163–40168. doi: 10.1074/jbc.M006242200. [DOI] [PubMed] [Google Scholar]
- 28.Beer S, Simins AB, Schuster A, Holzmann B. Molecular cloning and characterization of a novel SH3 protein (SLY) preferentially expressed in lymphoid cells. Biochim Biophys Acta. 2001;1520:89–93. doi: 10.1016/S0167-4781(01)00242-1. [DOI] [PubMed] [Google Scholar]
- 29.Nakamoto M. Eph receptors and ephrins. Int J Biochem Cell Biol. 2000;32:7–12. doi: 10.1016/S1357-2725(99)00096-5. [DOI] [PubMed] [Google Scholar]
- 30.Pasquale EB. The Eph family of receptors. Curr Opin Cell Biol. 1997;9:608–615. doi: 10.1016/S0955-0674(97)80113-5. [DOI] [PubMed] [Google Scholar]
- 31.Okado H, Ohtaka-Maruyama C, Sugitani Y, Fukuda Y, Ishida R, Hirai S, Miwa A, Takahashi A, Aoki K, Mochida K, Suzuki O, Honda T, Nakajima K, Ogawa M, Terashima T, Matsuda J, Kawano H, Kasai M. The transcriptional repressor RP58 is crucial for cell-division patterning and neuronal survival in the developing cortex. Dev Biol. 2009;331:140–151. doi: 10.1016/j.ydbio.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Tatard VM, Xiang C, Biegel JA, Dahmane N. ZNF238 is expressed in postmitotic brain cells and inhibits brain tumor growth. Cancer Res. 2010;70:1236–1246. doi: 10.1158/0008-5472.CAN-09-2249. [DOI] [PubMed] [Google Scholar]
- 33.Ohtaka-Maruyama C, Hirai S, Miwa A, Heng JI, Shitara H, Ishii R, Taya C, Kawano H, Kasai M, Nakajima K, Okado H. RP58 regulates the multipolar-bipolar transition of newborn neurons in the developing cerebral cortex. Cell Rep. 2013;3:458–471. doi: 10.1016/j.celrep.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Xiang C, Baubet V, Pal S, Holderbaum L, Tatard V, Jiang P, Davuluri RV, Dahmane N. RP58/ZNF238 directly modulates proneurogenic gene levels and is required for neuronal differentiation and brain expansion. Cell Death Differ. 2012;19:692–702. doi: 10.1038/cdd.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoki K, Meng G, Suzuki K, Takashi T, Kameoka Y, Nakahara K, Ishida R, Kasai M. RP58 associates with condensed chromatin and mediates a sequence-specific transcriptional repression. J Biol Chem. 1998;273:26698–26704. doi: 10.1074/jbc.273.41.26698. [DOI] [PubMed] [Google Scholar]
- 36.Meng G, Inazawa J, Ishida R, Tokura K, Nakahara K, Aoki K, Kasai M. Structural analysis of the gene encoding RP58, a sequence-specific transrepressor associated with heterochromatin. Gene. 2000;242:59–64. doi: 10.1016/S0378-1119(99)00477-1. [DOI] [PubMed] [Google Scholar]
- 37.Heng JI, Qu Z, Ohtaka-Maruyama C, Okado H, Kasai M, Castro D, Guillemot F, Tan SS. The zinc finger transcription factor RP58 negatively regulates Rnd2 for the control of neuronal migration during cerebral cortical development. Cereb Cortex. 2013 doi: 10.1093/cercor/bht277. [DOI] [PubMed] [Google Scholar]
- 38.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, Zecchin D, Hobor S, Bajpe PK, Lieftink C, Mateus C, Vagner S, Grernrum W, Hofland I, Schlicker A, Wessels LF, Beijersbergen RL, Bardelli A, Di Nicolantonio F, Eggermont AM, Bernards R. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 39.Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM, Jansen EE, Struys EA, Jakobs C, Salomons GS, Diks SH, Peppelenbosch M, Kremer A, Hoogenraad CC, Smitt PA, French PJ. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69:455–463. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.