

Abstract
AIM: Prostaglandin G/H synthase 2 (PTGS2 or COX2) is one of the key factors in the cellular response to inflammation. PTGS2 is expressed in the affected intestinal segments of patients with inflammatory bowel diseases (IBD). In IBD patients, non-steroidal anti-inflammatory drugs, which have been shown to reduce both the production and activity of PTGS2, may activate IBD and aggravate the symptoms. We aimed at examining genetic variants of PTGS2 that may be risk factors for IBD.
METHODS: We genotyped 291 individuals diagnosed with IBD and 367 controls from the Dutch population for the five most frequent polymorphisms of the PTGS2 gene. Clinical data were collected on all patients. DNA was extracted via normal laboratory methods. Genotyping was carried out using multiplex PCR followed by the Invader Assay and the 5 exonuclease assay (TaqMan). New polymorphism screening was performed by pre-screening with denaturing high-performance liquid chromatography, followed by fluorescent sequencing.
RESULTS: Allele 5209G was weakly associated with Crohn's disease (odds ratio [OR] 1.63, 95% confidence interval [CI] 1.03-2.57), and allele 8473T with ulcerative colitis (OR 1.50, 95%CI 1.00-2.27). The haplotype including both alleles showed a strong association with IBD (OR 13.15, 95%CI 3.17-116.15). This haplotype, while rare (-0.3%) in the general population, is found more frequently in patients (3.5%).
CONCLUSION: Our data suggest that this haplotype of PTGS2 contributes to the susceptibility of IBD.
Keywords: Inflammatory bowel disease, Prostaglandin G/H synthase, Cyclooxygenase, SNP, Haplotype
INTRODUCTION
Risk factors for inflammatory bowel diseases (IBD) include several environmental and genetic exposures. Diet, tobacco smoking, and childhood diseases or poor hygiene have been identified as risk factors for IBD, with some important differences between ulcerative colitis (UC) and Crohn’s disease (CD)[1-3]. Caucasians have been shown to be at higher risk than non-Caucasians, with North-South gradients seen in Europe[4,5].
Familial aggregation of IBD suggests a genetic component in the susceptibility to IBD. One major locus of susceptibility to IBD has been mapped by linkage analysis to chromosome 16, at the CARD15/NOD2 gene[6-8] which has a role in inflammatory responses, through activation of nuclear factor NF-kB. Two more susceptibility genes have recently been identified[9,10]. Population-based association studies focused on polymorphisms of inflammatory genes, such as genes of the interleukin-1bpathway. Few showed significant associations with some degree of contradiction between studies[11-15].
Key genes involved in the regulation of the inflammatory processes, such as prostaglandin G/H synthase/cyclooxygenase (PTGS/COX), are obvious candidates to look for variants predisposing to IBD. One of the two PTGS isoforms, PTGS2/COX2, is expressed in epithelial cells and mononuclear cells in IBD[4] and it is induced in response to pro-inflammatory cytokines, including interleukin-1b[16-18]. PTGS2 is the rate-limiting enzyme in the production of prostaglandins. Prostaglandins are thought to be essential in the process of wound healing in the gastrointestinal tract[19]. The use of non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit both the transcription and activity of PTGS2, exacerbates the symptoms in UC[20] and may even activate quiescent IBD[21]. Thus, the expression of PTGS2 in the inflamed intestine might be a protective response within the wound-healing process[22]. Consequently, polymorphisms that change the amount of prostaglandins produced in inflamed cells could cause susceptibility to IBD.
To address this hypothesis, we have studied 145 UC patients, 146 CD patients, and 367 controls from the Dutch Caucasian population, which has a high incidence of IBD[23]. The present study was to search for associations between IBD and common single nucleotide polymorphisms (SNPs) in the PTGS2 gene.
MATERIALS AND METHODS
Samples
The recruitment of cases took place at the Department of Gastroenterology of the VU University Medical Center (VUmc), a referral center for IBD, between April 1992 and September 2000. Patients with indeterminate colitis were excluded from this study. Seventy-five UC patients were females and seventy males, with a median age of 46 years (range 19-89 years). The gender repartition of CD patients was 101 females and 45 males, with a median age of 40 years (range 15-80 years). The control population consisted of 175 healthy individuals who were students or staff at the VUmc, with a median age of 51 years (range 24-88 years). All subjects were unrelated Dutch Caucasians. Genomic DNA was extracted using the DNAzol procedure (Invitrogen).
In order to ensure that the control group was representative of the general population, a second control group was also used, consisting of 192 Dutch subjects (96 males and 96 females). These samples were selected from the European Prospective Investigation on Cancer (EPIC) cohort[24] to provide information on the genetic background in the overall Dutch population. These samples were selected to provide a similar age distribution and sex ratio as the original control group. DNA was extracted from buffy coat using Puregene chemistry (Gentra Systems, Minneapolis, MN, USA). This population-based sample was indistinguishable from the VUmc controls from the point of view of frequencies of PTGS2 alleles and genotypes (data not shown). We take this as confirmation that both groups were drawn from the general Dutch population, at least from the genetic point of view.
Patient classification
Diagnosis and assessment of maximal extent of IBD were based on endoscopic, histopathological, and radiological criteria[25]. Patients with indeterminate colitis were not included in this study, as they can be categorized as UC or CD only at a later stage. UC patients were subdivided into three groups: proctitis, limited to the rectum (12 cases), left-sided colitis with disease activity up to the splenic flexure (72 cases), and pancolitis (61 cases), extending beyond the splenic flexure. In addition, the patients with UC were subdivided into one group with their colon in situ (116 cases), and a second group in which (procto)colectomy had to be performed during the course of disease (29 cases), indicative of severe or intractable disease (i.e., unresponsive to medical therapy). No patient was operated on prophylactic indication. In CD, patients were subdivided according to the Vienna classification[26]. This classification subdivides patients with CD according to age at onset (<40 years [125 cases] or ≥40 years [21 cases]), disease behavior (non-stricturing, non-penetrating [51 cases], stricturing [63 cases], penetrating [32 cases]) and location of disease (terminal ileum [50 cases], colon [32 cases], ileocolonic [62 cases], or upper gastrointestinal tract [2 cases]). Anatomical classification is defined as the maximal extent of the disease prior to the first surgical procedure, while behavior is assessed at any time during the course of the disease.
SNP selection and genotyping
SNPs were selected from our previously collected data[27] to include those SNPs with a prevalence of greater than 5% in Caucasians. Among the SNPs we studied, the only one with a proven functional role is a SNP located in the promoter of PTGS2, at position 926 of GenBank entry D28235 (dbSNP rs20417), which has been shown to affect the expression of PTGS2mRNA[28].
The order of samples of patients and controls was randomized in the PCR plates, so that a uniform number of patients and controls could be analyzed simultaneously in each run. EPIC samples were genotyped separately.
Three SNPs were genotyped with the Invader Assay[29]. PCR for the Invader assay (Third Wave Technologies, Madison, WI, USA) was carried out in 25 mL reactions, using the following concentrations: 20 ng DNA, 20 mmol/L dNTPs, 1×Taq Platinum buffer, 1.5 mmol/L MgCl2, 1.25 U Taq Platinum polymerase (Life Technologies, Inc., Gaithersburg, MD, USA), 0.5mmol/L of each primer. The primers used were as follows (nomenclature of SNPs refers to base numbers in GenBank entry D28235):
PTGS2.401 (rs689465)
401F: AAG GAC TTA GGA CAT AAC TGA ATT TTC
401R: ATG GGT AGT GCT CAG GGA GGA G
PTGS2.3050 (rs5277)
3050F: CGT TGT GAA TAA CAT TCC CTT
3050R: ATT TTT CTT TGA GAA GGC TAA AA
PTGS2.5209 (rs20432)
5209F: ATG ATG TAT GCC ACA ATC TGG CTG
5209R: TTG TCT GGA ACA ACT GCT CAT CAC.
This multiplex reaction was then carried out in a Tetrad DNA Engine PCR machine (MJ Research, Waltham, MA, USA), with the following cycling conditions: 96 °C for 5 min, then 30 cycles of 96 °C for 30 s, 50 °C for 60 s, and 72 °C for 10 s, with a final extension of 72 °C for 5 min. PCR volumes were then brought up to 150 mL, and Invader reactions were carried out as per manufacturer’s instructions. Plates were read in a fluorometer with excitation and emission spectra as recommended by Third Wave.
The SNPs PTGS2.926 and PTGS2.8473 were tested using the TaqMan assay with MGB chemistry (Applied Biosystems, Foster City, CA, USA). The assay was carried out in a 10 mL reaction, with 20 ng DNA, 1×TaqMan master mix (Applied Biosystems), 0.1 mmol/L of each primer, and 0.2 mmol/L of each probe. Primer and probe sequences are as follows:
PTGS2.926 (rs20417)
926F: TTA ACT ATT TAC AGG GTA ACT GCT TAG G
926R: CTT CAC CCC CTC CTT GTT TC
926VIC: CCT TTC CCG CCT CT
926FAM: CTT TCC CCC CTC TC
PTGS2.8473 (rs5275)
8473F: ATG CAC TGA TAC CTG TTT TTG TTT G
8473R: GTT TCC AAT GCA TCT TCC ATG A
8473VIC: TGA CAG AAA AAT AAC CAA AA
8473FAM: TGA CAG AAA AAT GAC CAA A
The cycling conditions for these PCR reactions were as follows: 95 °C for 10 min, then 35 cycles of 95 °C for 15 s, and 60 °C(58 °C for PTGS2.926) for 60 s. Plates were then read in the ABI 7900HT sequence detection system.
Data analysis
For genotype data from the Invader assay, raw intensity counts were corrected with a negative control, and the ratio between the two signals (one color for each allele) was used to call each genotype. For the TaqMan assay, groups of genotypes (homozygote common, heterozygote, and homozygote rare) were determined manually within the SDS software (Applied Biosystems). Each SNP was tested in the control group to ensure that it does not deviate from Hardy-Weinberg equilibrium. Linkage disequilibrium tests were performed using macros developed by the authors in conjunction with the PHASE program[30] to reconstruct haplotype frequencies. Odds ratios (OR) and 95% confidence interval (CI) were calculated in Stata 7.0, using logistic regressions correcting for sex and age in analyzing genotypes at single SNPs. Simple OR were calculated when data consisted of frequencies of alleles or haplotypes in the population. All P values reported are two sided. SNPs were tested for effects on splicing using the Delila Server (http://www.lecb.ncifcrf.gov/-toms/delilaserver.html) at the Laboratory of Experimental and Computational Biology of the National Cancer Institute[31].
New polymorphism screening
Individuals termed “at risk” based on association testing (i.e., the 19 cases carrying the haplotype, which includes the two alleles showing increase in risk at SNPs 5 209 and 8 473) were screened for novel polymorphisms using denaturing high-performance liquid chromatography (DHPLC) via the WAVE system (Transgenomic, Omaha, NE, USA). The areas studied were the promoter region, exons 6 and 10, and the 3 UTR. The protocols followed were as published previously[27].
RESULTS
Figure 1 shows a graphical representation of the structure of the PTGS2 gene. The five major SNPs as well as the polymorphisms discovered as part of this study are depicted. The allele frequencies of the five major SNPs in the control group were 11.4% (PTGS2.401), 13.4% (PTGS2.926), 14.6% (PTGS2.3050), 13.5% (PTGS2.5209), and 33.9% (PTGS2.8473). All SNPs, except PTGS2.3050, were in Hardy-Weinberg equilibrium in the control group. Table 1 shows the genotype counts of the SNPs studied, among cases and controls. The SNP at position 5 209 shows an increase in the carriers of the G allele in cases as compared to controls, with a weak but significant association was found only in CD (OR = 1.63, 95%CI 1.03-2.57, P = 0.04). The SNP at position 8 473 shows an increase in the frequency of the T/T homozygote genotype in cases as compared to controls, with an association of borderline significance found only in UC (OR = 1.50, 95%CI 1.00-2.27, P = 0.05). No association was seen with the SNPs at position 401, 926, or 3 050, although it should be noted that there is a lack of homozygous rare genotypes at bp 3 050 in the control group, which is the reason for its departure from Hardy-Weinberg equilibrium.
Figure 1.
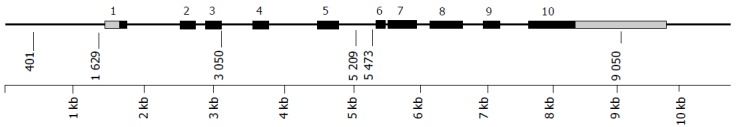
Structure of PTGS2. Positions in base pairs as per GenBank sequence D28235. Exons are shown as dark boxes, untranslated regions as light boxes. Vertical lines represent SNPs, with their positions in the sequence indicated.
Table 1.
Genotype frequencies and odds ratios for UC and CD of PTGS2 SNPs
| Genotype |
Controls |
UC |
CD |
|||||
| n1 | % | n1 | % | OR(95%CI)2 | n1 | % | OR(95%CI)2 | |
| PTGS2.401 | ||||||||
| A/A | 277 | 79 | 115 | 82 | 1 | 107 | 77 | 1.00 |
| A/G | 66 | 19 | 24 | 17 | 0.82 (0.48 – 1.39)3 | 30 | 22 | 1.17 (0.70 –1.94)3 |
| G/G | 7 | 2 | 1 | 1 | 1 | 1 | ||
| Total | 350 | 100 | 140 | 100 | 138 | 100 | ||
| PTGS2.926 | ||||||||
| G/G | 256 | 75 | 112 | 81 | 1 | 106 | 76 | 1.00 |
| G/C | 75 | 22 | 23 | 17 | 0.77 (0.47 – 1.28)3 | 31 | 22 | 1.06 (0.65 –1.73)3 |
| C/C | 8 | 2 | 4 | 3 | 3 | 2 | ||
| Total | 339 | 100 | 139 | 100 | 140 | 100 | ||
| PTGS2.3050 | ||||||||
| G/G | 250 | 71 | 109 | 75 | 1 | 102 | 71 | 1 |
| G/C | 99 | 28 | 32 | 22 | 0.83 (0.52 – 1.32)3 | 38 | 26 | 0.97 (0.62 –1.53)3 |
| C/C | 1 | 1 | 4 | 3 | 4 | 3 | ||
| Total | 350 | 100 | 145 | 100 | 144 | 100 | ||
| PTGS2.5209 | ||||||||
| T/T | 265 | 75 | 110 | 76 | 1 | 95 | 66 | 1 |
| T/G | 81 | 23 | 30 | 21 | 1.00 (0.62 - 1.61)3 | 46 | 33 | 1.63 (1.03 - 2.57)3 |
| G/G | 7 | 2 | 5 | 3 | 2 | 1 | ||
| Total | 353 | 100 | 145 | 100 | 143 | 100 | ||
| PTGS2.8473 | ||||||||
| C/C | 44 | 13 | 18 | 14 | 1.003 | 21 | 15 | 1.003 |
| T/C | 147 | 42 | 49 | 34 | 54 | 38 | ||
| T/T | 155 | 45 | 78 | 54 | 1.50 (1.00 - 2.27) | 68 | 47 | 1.10 (0.72 - 1.68) |
| Total | 346 | 100 | 145 | 100 | 143 | 100 | ||
1Numbers may not sum up to the totals of controls or cases due to genotyping failure. All samples that did not give a reliable result in the first round of genotyping were resubmitted to up to three additional rounds of genotyping. Data points that were still not filled after this procedure were left blank.
All ORs are adjusted for sex and age.
Heterozygotes and homozygotes for the rare allele have been grouped in order to improve statistical power
We analyzed differences in genotype counts among the different clinical variables for CD and UC. No statistically significant differences between classification levels were observed (data not shown).
Significant levels of linkage disequilibrium were seen among the most common SNPs in the PTGS2 gene (data not shown). Table 2 shows the frequencies of haplotypes of the PTGS2 gene in both IBD patients and controls. The SNP at bp 3 050 was not included in haplotype analysis, as it was not in Hardy-Weinberg equilibrium in the control population, and this has been shown to affect the accuracy of haplotype reconstruction methods[32]. All individuals were genotyped for the SNP at bp 3 050, eliminating the risk that its lack of Hardy-Weinberg equilibrium is caused by bias due to the genotyping technique missing preferentially one genotype. Haplotype frequencies were calculated using all four valid SNPs, and all data are shown. There were no differences in haplotype frequency between the two groups of IBD patients and the controls, except for haplotype AGGT, which shows an OR of 11.9 (95%CI 2.83-105.76, P<0.00005, for UC and CD combined). Interestingly, this haplotype includes the two alleles showing an increase in risk at SNPs 5 209 and 8 473. If we consider the haplotype including the 5209.G and 8473.T alleles (by lumping haplotypes AGGT and GCGT), we observe an increase in its frequency in patients with IBD (0.035, UC and CD combined) as compared to controls (0.003). This increase in frequency yields an OR of 13.15 (95%CI 3.17-116.15, P<0.00005, for UC and CD combined).
Table 2.
PTGS2 haplotype frequencies and odds ratios for IBD
| Haplotype1 | Controls |
UC cases |
CD cases |
UC+CD cases2 |
|||
| n (%) | n (%) | OR(95% CI) | n (%) | OR (95% CI) | n (%) | OR (95% CI)3 | |
| AGTT | 471 (64.7) | 193 (65.6) | 1.00 (Ref.) | 183 (62.7) | 1.00 (Ref.) | 376 (64.2) | 1.00 (Ref.) |
| AGTC | 147 (20.2) | 55 (18.7) | 0.91 (0.64 - 1.30) | 54 (18.5) | 0.95 (0.66 - 1.35) | 109 (18.6) | 0.93 (0.69 - 1.24) |
| GCGC | 64 (8.8) | 22 (7.5) | 0.84 (0.50 - 1.39) | 27 (9.2) | 1.09 (0.67 - 1.75) | 49 (8.4) | 0.96 (0.63 - 1.45) |
| ACGC | 25 (3.4) | 8 (2.7) | 0.78 (0.35 - 1.73) | 7 (2.4) | 0.72 (0.31 - 1.66) | 15 (2.6) | 0.75 (0.36 - 1.51) |
| AGGT | 2 (0.3) | 10 (3.4) | 12.2 (2.97 – inf.) | 9 (3.1) | 11.58 (2.79 -inf.) | 19 (3.2) | 11.9 (2.83 – 105.76) |
| Rare4 | 19 (2.6) | 6 (2.0) | 0.77 (0.31 - 1.90) | 12 (4.1) | 1.63 (0.78 - 3.37) | 18 (3.1) | 1.19 (0.61 – 2.29) |
The order of SNPs in the haplotypes is PTGS2.401, PTGS2.926, PTGS2.5209, PTGS2.8473 .
UC and CD combined.
ORs are not corrected, they are based on population level haplotype frequencies and are relative to the most common haplotype (AGTT).
Rare haplotypes (frequency ≤1% in both cases and controls)
In order to test the possibility that these positive associations are in reality indicative of linkage disequilibrium with yet undiscovered nearby polymorphisms, we studied by DHPLC the 19 cases who could carry the risk haplotype. Regions of PTGS2surrounding the three SNPs, and in addition the promoter region, exon 1 and the UTRs were analyzed. No novel polymorphisms were discovered in the coding sequence of the gene. One SNP was discovered at bp 9 850 (A-G), with a minor allele frequency of lower than 2%. Therefore, we considered it unlikely that polymorphisms other than 5 209 or 8 473 could explain the observed associations.
DISCUSSION
PTGS2 is a critical enzyme involved in the production of prostaglandins, which are essential in the process of healing bowel wounds. The use of NSAIDs, which inhibit both the translation and activity of PTGS2, can induce a flare-up[21] and actually exacerbate the symptoms of IBD[20]. We hypothesized that polymorphisms in PTGS2 might influence prostaglandin production in inflamed cells, thus affecting susceptibility to IBD. In this study, we have found a slightly increased risk of IBD associated toPTGS2.5209 and PTGS2.8473, and a strong association with a haplotype including alleles of these two SNPs.
PTGS2.5209 is in intron 5 of PTGS2. In silico analysis of this SNP for splice site mutations[31] reveals that the T-G substitution creates a new splicing acceptor sequence with nearly the same strength as that found in normal splice sites. While this is most probably not enough of a change to exclude completely the normal splicing of exons 5-6, it could cause some搇eaking, reducing the amount of normal PTGS2 mRNA. PTGS2.8473, being in the 3 untranslated region (UTR), could affect the stability of PTGS2 mRNA[33]. In fact, the unusually long 3 UTR plays an important role in determining the half-life of the mRNA. Various proteins bind to it and cause either acceleration or protection of the degradation of the mRNA[34,35].
The associations we have found between CD and PTGS2.5209 G/G, and UC and PTGS2.8473 T/T are statistically significant, but of moderate importance. We propose that it is the combination of the G allele at PTGS2.5209 with the T allele atPTGS2.8473, on the same chromosome (GT haplotype), that would have the most influence on disease status.
The association between IBD and the GT haplotype could be due to linkage disequilibrium with another polymorphism that lies nearby. To test this hypothesis, we have analyzed subjects who carry the GT haplotype for the presence of novel functional polymorphisms of PTGS2, but this search has not yielded any additional candidates. Linkage disequilibrium with polymorphisms in neighboring genes is not a likely explanation either, as there is no gene mapping of at less than 100 kbp on either side ofPTGS2.
We are inclined to think that this association is not a finding by chance. First of all, we have found a higher level of linkage disequilibrium between alleles at bp 5 209 and 8 473 in controls than in cases. This could mean that natural selection has exerted pressure toward the disappearance of the GT haplotype, i.e. this haplotype might negatively affect the fitness of individuals carrying it. Additionally, we have observed that this haplotype is virtually absent not only in the Dutch population, but in other Northern European populations as well (Cox et al unpublished data).
These results are the first linking of PTGS2 to IBD. While the GT haplotype of PTGS2 is rare, the two alleles that compose it are much more frequent, they both show a modest association with the disease, and could therefore be important for a large proportion of the population. Future steps are to replicate these findings by independent studies, to determine experimentally the extent to which PTGS2 polymorphisms alter the expression or function of the gene, and to discover genetic or environmental factors that may alter the risk of IBD interacting with PTGS2 alleles.
ACKNOWLEDGMENTS
The authors thank Pietro Ferrari (Nutrition and Cancer Group, IARC) for discussion on statistical issues, Stephanie Monnier (Genome Analysis Team, IARC) for bioinformatic support and AA van Bodegraven MD, PhD for facilitating the clinical database of the IBD patients at the VUmc.
Footnotes
Science Editor Guo SY Language Editor Elsevier HK
Supported by The Grants from the International Agency for Research on Cancer (Special Training Award to DGC), the French Association for Research on Cancer (grant #7478), and the Crohn's and Colitis Foundation of America (to ASP)
References
- 1.Van Kruiningen HJ, Freda BJ. A clustering of Crohn's disease in Mankato, Minnesota. Inflamm Bowel Dis. 2001;7:27–33. doi: 10.1097/00054725-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Mahmud N, Weir DG. The urban diet and Crohn's disease: is there a relationship? Eur J Gastroenterol Hepatol. 2001;13:93–95. doi: 10.1097/00042737-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Koutroubakis I, Manousos ON, Meuwissen SG, Pena AS. Environmental risk factors in inflammatory bowel disease. Hepatogastroenterology. 1996;43:381–393. [PubMed] [Google Scholar]
- 4.Farrokhyar F, Swarbrick ET, Irvine EJ. A critical review of epidemiological studies in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:2–15. doi: 10.1080/00365520150218002. [DOI] [PubMed] [Google Scholar]
- 5.Karlinger K, Györke T, Makö E, Mester A, Tarján Z. The epidemiology and the pathogenesis of inflammatory bowel disease. Eur J Radiol. 2000;35:154–167. doi: 10.1016/s0720-048x(00)00238-2. [DOI] [PubMed] [Google Scholar]
- 6.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 7.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 8.Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, Frenzel H, King K, Hasselmeyer A, MacPherson AJ, et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. 2001;357:1925–1928. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 9.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 10.Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, Rosenstiel P, Albrecht M, Croucher PJ, Seegert D, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476–480. doi: 10.1038/ng1345. [DOI] [PubMed] [Google Scholar]
- 11.Watts DA, Satsangi J. The genetic jigsaw of inflammatory bowel disease. Gut. 2002;50 Suppl 3:III31–III36. doi: 10.1136/gut.50.suppl_3.iii31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemetz A, Nosti-Escanilla MP, Molnár T, Köpe A, Kovács A, Fehér J, Tulassay Z, Nagy F, García-González MA, Peña AS. IL1B gene polymorphisms influence the course and severity of inflammatory bowel disease. Immunogenetics. 1999;49:527–531. doi: 10.1007/s002510050530. [DOI] [PubMed] [Google Scholar]
- 13.Tagore A, Gonsalkorale WM, Pravica V, Hajeer AH, McMahon R, Whorwell PJ, Sinnott PJ, Hutchinson IV. Interleukin-10 (IL-10) genotypes in inflammatory bowel disease. Tissue Antigens. 1999;54:386–390. doi: 10.1034/j.1399-0039.1999.540408.x. [DOI] [PubMed] [Google Scholar]
- 14.Heresbach D, Alizadeh M, Dabadie A, Le Berre N, Colombel JF, Yaouanq J, Bretagne JF, Semana G. Significance of interleukin-1beta and interleukin-1 receptor antagonist genetic polymorphism in inflammatory bowel diseases. Am J Gastroenterol. 1997;92:1164–1169. [PubMed] [Google Scholar]
- 15.Klein W, Tromm A, Griga T, Fricke H, Folwaczny C, Hocke M, Eitner K, Marx M, Runte M, Epplen JT. The IL-10 gene is not involved in the predisposition to inflammatory bowel disease. Electrophoresis. 2000;21:3578–3582. doi: 10.1002/1522-2683(200011)21:17<3578::AID-ELPS3578>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 16.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 17.Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem. 1990;265:10805–10808. [PubMed] [Google Scholar]
- 18.Masferrer JL, Seibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci USA. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace JL. Prostaglandin biology in inflammatory bowel disease. Gastroenterol Clin North Am. 2001;30:971–980. doi: 10.1016/s0889-8553(05)70223-5. [DOI] [PubMed] [Google Scholar]
- 20.Eberhart CE, Dubois RN. Eicosanoids and the gastrointestinal tract. Gastroenterology. 1995;109:285–301. doi: 10.1016/0016-5085(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann HJ, Taubin HL. Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann Intern Med. 1987;107:513–516. doi: 10.7326/0003-4819-107-4-513. [DOI] [PubMed] [Google Scholar]
- 22.Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology. 1998;115:297–306. doi: 10.1016/s0016-5085(98)70196-9. [DOI] [PubMed] [Google Scholar]
- 23.Russel MG, Dorant E, Volovics A, Brummer RJ, Pop P, Muris JW, Bos LP, Limonard CB, Stockbrügger RW. High incidence of inflammatory bowel disease in The Netherlands: results of a prospective study. The South Limburg IBD Study Group. Dis Colon Rectum. 1998;41:33–40. doi: 10.1007/BF02236893. [DOI] [PubMed] [Google Scholar]
- 24.Bingham S, Riboli E. Diet and cancer--the European Prospective Investigation into Cancer and Nutrition. Nat Rev Cancer. 2004;4:206–215. doi: 10.1038/nrc1298. [DOI] [PubMed] [Google Scholar]
- 25.Lennard-Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol Suppl. 1989;170:2–6; discussion 16-9. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]
- 26.Gasche C, Scholmerich J, Brynskov J, D'Haens G, Hanauer SB, Irvine EJ, Jewell DP, Rachmilewitz D, Sachar DB, Sandborn WJ, et al. A simple classification of Crohn's disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm Bowel Dis. 2000;6:8–15. doi: 10.1097/00054725-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Cox D, Boillot C, Canzian F. Data mining: Efficiency of using sequence databases for polymorphism discovery. Hum Mutat. 2001;17:141–150. doi: 10.1002/1098-1004(200102)17:2<141::AID-HUMU6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Papafili A, Hill MR, Brull DJ, McAnulty RJ, Marshall RP, Humphries SE, Laurent GJ. Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acute-phase inflammatory response. Arterioscler Thromb Vasc Biol. 2002;22:1631–1636. doi: 10.1161/01.atv.0000030340.80207.c5. [DOI] [PubMed] [Google Scholar]
- 29.Mein CA, Barratt BJ, Dunn MG, Siegmund T, Smith AN, Esposito L, Nutland S, Stevens HE, Wilson AJ, Phillips MS, et al. Evaluation of single nucleotide polymorphism typing with invader on PCR amplicons and its automation. Genome Res. 2000;10:330–343. doi: 10.1101/gr.10.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12:153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 32.Single RM, Meyer D, Hollenbach JA, Nelson MP, Noble JA, Erlich HA, Thomson G. Haplotype frequency estimation in patient populations: the effect of departures from Hardy-Weinberg proportions and collapsing over a locus in the HLA region. Genet Epidemiol. 2002;22:186–195. doi: 10.1002/gepi.0163. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Pascual F, Hausding M, Ihrig-Biedert I, Furneaux H, Levy AP, Förstermann U, Kleinert H. Complex contribution of the 3'-untranslated region to the expressional regulation of the human inducible nitric-oxide synthase gene. Involvement of the RNA-binding protein HuR. J Biol Chem. 2000;275:26040–26049. doi: 10.1074/jbc.M910460199. [DOI] [PubMed] [Google Scholar]
- 34.Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3' untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- 35.Sheng H, Shao J, Dixon DA, Williams CS, Prescott SM, DuBois RN, Beauchamp RD. Transforming growth factor-beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in intestinal epithelial cells via stabilization of mRNA. J Biol Chem. 2000;275:6628–6635. doi: 10.1074/jbc.275.9.6628. [DOI] [PubMed] [Google Scholar]