

Hsp70 facilitates maturation of newly synthesized kinases and assists degradation of kinases under normal and stressed conditions. Hsp70 degrades misfolded kinases that partition into different quality-control compartments by promoting their ubiquitination, thus protecting cells from proteotoxic stress.
Abstract
Hsp70 aids in protein folding and directs misfolded proteins to the cellular degradation machinery. We describe discrete roles of Hsp70,SSA1 as an important quality-control machinery that switches functions to ameliorate the cellular environment. SSA1 facilitates folding/maturation of newly synthesized protein kinases by aiding their phosphorylation process and also stimulates ubiquitylation and degradation of kinases in regular protein turnover or during stress when kinases are denatured or improperly folded. Significantly, while kinases accumulate as insoluble inclusions upon SSA1 inhibition, they form soluble inclusions upon Hsp90 inhibition or stress foci during heat stress. This suggests formation of inclusion-specific quality-control compartments under various stress conditions. Up-regulation of SSA1 results in complete removal of these inclusions by the proteasome. Elevation of the cellular SSA1 level accelerates kinase turnover and protects cells from proteotoxic stress. Upon overexpression, SSA1 targets heat-denatured kinases toward degradation, which could enable them to recover their functional state under physiological conditions. Thus active participation of SSA1 in the degradation of misfolded proteins establishes an essential role of Hsp70 in deciding client fate during stress.
INTRODUCTION
The protein quality-control system plays a ubiquitous role in determining the life of cells and ensuring proteins are correctly folded (Chen et al., 2011; Hartl et al., 2013). It is essential in abating the detrimental effects of protein misfolding and aggregation (Zhang and Qian, 2011; Houck et al., 2012). Chaperone families of proteins assist various cellular processes, including protein folding and unfolding and homeostasis of cellular proteins (McClellan et al., 2005). Found in all cellular compartments, they carry out a swarm of functions, all striving toward assuring positive equilibrium within cells (Mayer, 2010). The chaperone of molecular mass of 70 kDa (Hsp70) is the most commonly occurring chaperone and exists as many orthologues in different cellular compartments (Genevaux et al., 2007). In association with its different cochaperones, it carries out diverse functions, including protein folding, translocation across organelle membranes, disaggregation, and protein degradation (Shorter and Lindquist, 2008; Mandal et al., 2011). Hsp70 binds to the hydrophobic peptide stretches of substrates, and this binding is dependent on its nucleotide-binding state (Mayer and Bukau, 2005). The ATP-bound form of Hsp70 has low affinity for its substrate, whereas the ADP-bound form binds the substrate with high affinity. ATP hydrolysis is accelerated by the cochaperone Hsp40, which directly associates with and conveys substrates to Hsp70 (Kampinga and Craig, 2010). Exchange of ADP to ATP is triggered by the nucleotide exchange factor (NEF), and, as a result, the substrate is released from Hsp70. This conformational cycle of Hsp70 is used for stabilizing the unfolded proteins until they fold or for sending them toward degradation (Sharma and Masison, 2009; Priya et al., 2013). Therefore how Hsp70 deals with different states (unfolded, misfolded, aggregated) of substrates and channels them toward their actual fate (refolding/degradation) is highly intriguing.
The functional activity of Hsp70 relies on the structural concord between two domains: the N-terminal nucleotide-binding domain (NBD) and the C-terminal substrate-binding domain (SBD). The NBD binds ATP and is responsible for ATPase activity. A flexible linker that connects the two domains upholds the prime allosteric function of the chaperone (Goloubinoff and De Los Rios, 2007; Stricher et al., 2013). Thus the ATPase activity of Hsp70 serves as the pivot of activation for cochaperones to act upon either to promote recycling or for clearance of substrates (Meimaridou et al., 2009; Kityk et al., 2012). In addition, Hsp70 also coordinates with Hsp100 ATPases in disaggregating large aggregates (Zietkiewicz et al., 2004).
Substantial but diverse members of eukaryotic regulatory proteins are found to be supervised by the Hsp70 chaperone machinery. Proteins, including transcription factors (HSF, c-Myc, pRb), steroid hormone receptors (glucocorticoid receptor), and protein kinases (Raf, eIF2α kinase, cyclinB1/Cdk1), are chaperoned by Hsp70 (Kanelakis et al., 2002; Huang et al., 2004; Arndt et al., 2005; Kriegenburg et al., 2014). Although divergent, it appears that these chaperone–substrate complexes share considerable parity. Essentially, Hsp70 association during de novo synthesis of the substrate is indispensable for formation of an active complex (Caplan et al., 2007). Kinases in general form a majority of client proteins, which are dependent on chaperones. Hsp70 binds with a newly synthesized or unfolded kinase and Hsp40 to form a ternary complex (Mandal et al., 2008). Hsp90 is then recruited by Cdc37 and Hop to form an intermediate complex, which gradually moves toward the late complex, with which Hsp70 is not associated (Bukau and Horwich, 1998; Kampinga and Craig, 2010). Thus Hsp70 plays an imperative role in the initial decision making of substrate fate. Here, cochaperones of Hsp70 have decisive and opposite effects (Mandal et al., 2008, 2010). In the past decade, several genetic and biochemical approaches have been undertaken to manipulate the function of Hsp70. Chemical manipulation of Hsp70 mostly affects the inhibition of the ATPase cycle and blocks the participation of cochaperones that stimulate the activity of Hsp70 (Jinwal et al., 2009, 2010a). Inhibition of Hsp70 showed varied results that differ among various substrates, such as yeast prions, amyloid proteins, and protein kinases (Masison et al., 2009; Jinwal et al., 2010b). Thus understanding the intricate cellular functioning of Hsp70 and how it modulates its decision when to fold, hold, or degrade is of interest.
In Saccharomyces cerevisiae, the SSA subfamily of cytosolic Hsp70 is composed of functionally redundant genes (SSA1–4; Werner-Washburne et al., 1987). SSA1 and SSA2 are 98% identical and constitutively expressed, whereas SSA3 and SSA4 are expressed under stress conditions. Among the four SSA isoforms, the role of SSA1 is the most characterized and is essential for protein folding (Melville et al., 2003). SSA1 is also known for degradation of several cytosolic proteins, which it achieves by maintaining substrate solubility and recruiting the ubiquitin proteasomal machinery (Metzger and Michaelis, 2009; Prasad et al., 2010), facilitating ubiquitylation of cytoplasmic proteins (Han et al., 2007; Park et al., 2007; Guerriero et al., 2013), and actively targeting the misfolded substrates to the ubiquitin ligases San1 and Ubr1 (Guerriero et al., 2013; Summers et al., 2013), which are responsible for clearing misfolded proteins, including protein kinases (Heck et al., 2010; Nillegoda et al., 2010). The cochaperones Hsp40 and Hsp110 cooperate with Hsp70 in these functions. The cytosolic Hsp40 (Ydj1 and Sis1) assists in folding of nascent peptides, prevents protein aggregation, and facilitates degradation of aberrant and terminally misfolded proteins (Cyr, 1995; Lee et al., 1996; Mandal et al., 2008; Summers et al., 2013); whereas the NEFs Sse1 and Fes1 promote ubiquitylation of protein kinases and misfolded cytosolic proteins, respectively, for their degradation by the proteasome (Mandal et al., 2010; Gowda et al., 2013). SSA1 also facilitates degradation of ubiquitylated proteins and prevents their sequestration into inclusions (Shiber et al., 2013). Misfolded proteins that undergo ubiquitylation are sequestered into the juxtanuclear compartment (JUNQ), whereas nonubiquitylated, terminally misfolded proteins are accumulated in the perivacuolar inclusions (insoluble protein deposits [IPODs]; Kaganovich et al., 2008). However, protein aggregation and the factors governing it seem to be more complicated, as noticeable foci appear from misfolded proteins in several cases (Specht et al., 2011; Spokoini et al., 2012; Escusa-Toret et al., 2013). These distinct quality-control foci largely differ in chaperone association. IPOD inclusions associate with Hsp104 but are devoid of Hsp42; whereas stress-induced foci associate with both Hsp42 and Hsp104 (Kaganovich et al., 2008; Specht et al., 2011; Spokoini et al., 2012; Escusa-Toret et al., 2013). Association of Hsp104 with the inclusions suggests its role in disaggregation, and both Hsp70 and Sis1 help to recruit substrate to Hsp104 (Tipton et al., 2008; Winkler et al., 2012b). JUNQ inclusion colocalizes with Hsp70, and sequestration of Hsp70 with toxic inclusions enhances cytotoxicity by slowing down the clearance of misfolded proteins (Weisberg et al., 2012; Ogrodnik et al., 2014).
In this report, we provide a detailed mechanism of Hsp70's function in determining substrate fate from its nascent state to its misfolded and aggregated state in S. cerevisiae. Our studies showed that Hsp70,SSA1 aided in the maturation of newly synthesized kinases; however, it also assisted in degradation of client kinases for their removal by the proteasome under physiological conditions and also under stress conditions. Interestingly, SSA1 inhibition resulted in the formation of insoluble kinase inclusions (IPODs) deposited in the perivacuolar locale. Overexpression of SSA1 removed the inclusions and rescued cells from proteotoxic stresses. Up-regulation of SSA1 accelerated degradation and clearance of heat-denatured kinases, as opposed to them refolding to their native state under physiological conditions. Thus our study suggests an integral function of SSA1 in client fate determination.
RESULTS
Hsp70 facilitates maturation of protein kinases
Hsp70 interacts with the newly synthesized proteins via its cochaperone Hsp40 and facilitates their folding. Absolute requirement of Hsp70 has been shown in the reconstitution of substrates like Chk1 kinase and hormone receptors (Cintron and Toft, 2006; Felts et al., 2007). But how Hsp70 specifically aids in the folding or maturation of newly synthesized kinases is still elusive. We have taken an in vivo approach to clarify the role of Hsp70 in folding of protein kinases, mainly Tpk2 (homologue of cAMP-dependent protein kinase) in yeast (S. cerevisiae). Tpk2 is autophosphorylated and switches from the immature, nonphosphorylated form to the mature, phosphorylated form to attain its native state (Mandal et al., 2008). This characteristic of Tpk2 led us to identify the role of Hsp70 on newly synthesized kinase after pulse labeling yeast cells with [35S]methionine. The Hsp70 function was blocked by the inhibitors myricetin or 2-phenylethynesulfonamide (PES). These two compounds affect the function of Hsp70 by inhibiting Hsp70's ATPase activity or substrate-binding capacity as shown previously (Jinwal et al., 2009; Leu et al., 2009, 2011; Chang et al., 2011). In the presence of myricetin (100 μM) or PES (150 μM), Tpk2 shifted from an upper slower migratory band to a lower faster migratory band after a 10-min pulse (Figure 1A). This difference in Tpk2 mobility could happen due to the inefficient autophosphorylation we have seen previously for this kinase in the case of Ydj1 deletion in yeast Hsp40 (Mandal et al., 2008). To investigate this possibility, we targeted two autophosphorylation sites of Tpk2, the Ser-80 and Thr-224 positions identified by the Phosphogrid database. We converted both the autophosphorylation residues to alanine (S80A, T224A) and then labeled the yeast cells with [35S]methionine at different time points. Pulse-labeling reaction showed that wild-type (WT) Tpk2 migrates at a lower band (immature) at 2 min of pulse and then moves to the upper band (mature) at 5 min of pulse; however, the autophosphorylation-defective Tpk2 mutant (S80A, T224A) was unable to mature even after 10 min of pulse. This result indicated that the lower band seen after Hsp70 inhibition was most likely the immature nonphosphorylated form of Tpk2 (Figure 1B). If that was the case, then Tpk2 kinase should have accumulated in an inactive form due to Hsp70 inhibition. To examine that, we measured the Tpk2 kinase activity after expressing it from galactose-inducible promoter in the presence of Hsp70 inhibitors. Tpk2 kinase activity was reduced in the presence of myricetin or PES, similar to the activity of a kinase-dead version of Tpk2 (K99R). Notably, the autophosphorylation-deficient mutant was devoid of kinase activity (Figure 1C). Prolonged Hsp70 inhibition until 6 h did not show any achievable Tpk2 kinase activity (Supplemental Figure S1), which negates the possibility of delayed Tpk2 maturation as observed for Ydj1 deletion (Mandal et al., 2008).
FIGURE 1:
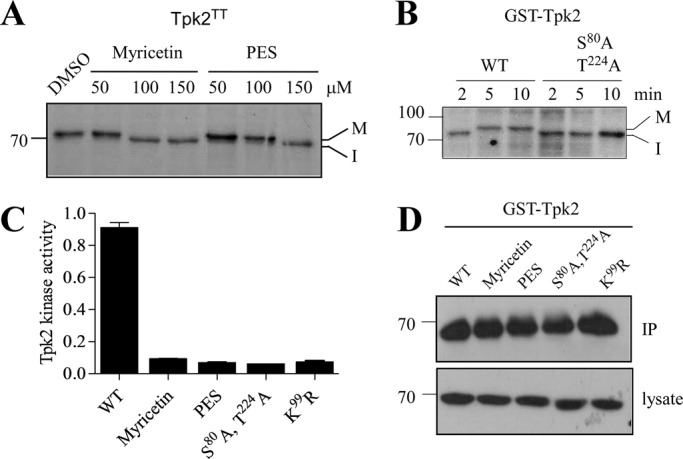
Hsp70 facilitates protein kinase maturation by assisting its autophosphorylation. (A) Tap-tagged Tpk2 kinase (Open Biosystem) was pulse labeled with [35S]methionine for 10 min in the presence of different concentrations of the Hsp70 inhibitors myricetin and PES as indicated. DMSO was used as the solvent. Mature (M, phosphorylated) and immature (I, nonphosphorylated) forms of kinase are denoted. (B) Pulse labeling of GST-tagged WT Tpk2 and autophosphorylation-defective S80A, T224A kinase mutant. Pulse time was as indicated in the figure. (C) Tpk2 kinase activity was measured after treating the cells with the Hsp70 inhibitors myricetin (100 μM) and PES (150 μM) for 2 h. The GST-Tpk2 kinase was purified by glutathione resin, and the activity was assayed by the nonradioactive PKA assay kit as described in Materials and Methods. Kinase-dead K99R Tpk2 mutant was used as control. The kinase activity of autophosphorylation-defective S80A, T224A Tpk2 double mutant was also measured. (D) Western blot with anti-GST antibody showed the amount of GST-Tpk2 kinase used for the kinase assay in C. The bottom panel represents Tpk2 expression in the respective lysate.
The role of Hsp70 in maturation of kinase was also tested for another kinase, Ste11, a MAP3K in yeast. Pulse labeling of Ste11 kinase in the presence of myricetin showed increased kinase mobility (Supplemental Figure S2A), probably hampering the maturation (phosphorylation) of Ste11 kinase, which was not observed in the case of Ste11ΔNK444R (N-terminal regulatory domain–deleted and kinase-dead version of Ste11 kinase; Supplemental Figure S2B; Flom et al., 2008). This defect in Ste11 kinase maturation in the presence of Hsp70 inhibitor was reflected by its kinase activity. The pheromone-induced Ste11 signaling was completely blocked in myricetin-treated yeast cells (Supplemental Figure S2C). Thus we demonstrated that Hsp70 helps in chaperoning newly synthesized kinase to gain its functional state by acting as a mediator of its phosphorylation.
Hsp70 mediates degradation of kinases during normal and stressed conditions
Hsp70 acts as a mediator to facilitate degradation of proteins by the proteasome (Park et al., 2007; Nillegoda et al., 2010). Previously we have seen that the NEF of Hsp70, Sse1 (Hsp110 in yeast), promotes ubiquitylation and degradation of Ste11ΔNK444R kinase by the proteasome. But a mutant of Sse1 (G233D) that is unable to interact with Hsp70,SSA1 (Shaner et al., 2005) cannot degrade Ste11ΔNK444R kinase, suggesting the involvement of SSA1 in Ste11ΔNK444R kinase degradation (Mandal et al., 2010). To examine this, we have taken two approaches by using an Hsp70 temperature-sensitive (ts) SSA1 isoform, ssa1-45 (ssa1tsssa2Δssa3Δssa4Δ), which lacks the other isoform (Becker et al., 1996), and chemically blocking Hsp70 function with myricetin. Ste11ΔNK444R kinase was expressed in ssa1-45 cells and the corresponding isogenic strain, SSA1, at permissive (30°C) and nonpermissive (37°C) temperatures, respectively. The steady-state level of Ste11ΔNK444R kinase was checked by Western blot analysis. We found an increased kinase level in the ssa1-45 strain when incubated for 2 h at 37°C in comparison with its isogenic strain, SSA1 (Figure 2A). This increment in kinase level could be due to the reduced rate of kinase degradation in ssa1-45 cells at nonpermissive temperature. To track this, we analyzed the fate of newly synthesized Ste11ΔNK444R kinase by pulse–chase with [35S]methionine. We saw that the half-life of Ste11ΔNK444R kinase is ∼2 h in SSA1 cells at 37°C, but the kinase was stable until 6 h of chase in ssa1-45 cells (Figure 2B). A similar reduction in kinase degradation was noticed for Tpk2 kinase in ssa1-45 cells at 37°C (Figure 2C). Chemical inhibition of Hsp70 with myricetin also stabilized Ste11ΔNK444R kinase (Figure 2D). Thus identical results were obtained from both genetic and chemical inhibition of SSA1.
FIGURE 2:

Hsp70 facilitates degradation of protein kinases. (A) His-tagged p416-Ste11ΔNK444R plasmid was transformed into the Hsp70 temperature-sensitive mutant ssa1-45 and isogenic SSA1 yeast cells. Steady-state levels of Ste11ΔNK444R kinase from these strains were measured by Western blotting with anti-His antibody after the cells were grown at 30°C and subsequently incubated at 37°C for 2 h. The band intensity was then quantified, normalized with Pgk1, and plotted in the accompanying graph. Bar represents SE of three independent experiments. (B and C) Pulse–chase analysis of His-Ste11ΔNK444R and HA-Tpk2 kinase in SSA1 and ssa1-45 yeast cells at 37°C. Chase times as indicated. The band intensity was quantified by the Typhoon Phosphorimager. The rate of kinase degradation normalized to t = 0 is shown in the accompanying graph. (D) Pulse–chase analysis of Ste11ΔNK444R in the absence and presence of 100 μM myricetin. The turnover of Ste11ΔNK444R kinase was shown in the graph. (E) Pulse–chase of Ste11ΔNK444R was performed in SSA1 and ssa1-45 yeast cells at nonpermissive temperature with protein folding inhibited by the addition of GA (50 μM).
The state of the protein differs extensively from physiological to stress conditions, and the cell makes necessary adjustments to prevent the accumulation of misfolded proteins. So we tested whether Hsp70 can efficiently degrade misfolded proteins generated by stress conditions. We used geldanamycin (GA), the Hsp90 inhibitor that binds competitively to ATP and inhibits Hsp90's ATPase activity. Treatment of cells with GA generates misfolded proteins that create an overall cellular imbalance and induce degradation of kinases by the ubiquitin proteasome system. In the presence of GA, Ste11ΔNK444R kinase was degraded rapidly, with a half-life of ∼30 min as observed previously (Mandal et al., 2010), but the kinase was stable for more than 2 h in ssa1-45 cells (Figure 2E). Altogether our data show that SSA1 is required for degradation both during regular turnover and during cellular stress, when proteins are perpetually misfolded.
SSA1 promotes ubiquitylation of kinases for degradation via the proteasome
We have seen that Hsp70 mediates degradation of Ste11ΔNK444R kinase by the proteasome when its folding is interrupted by the Hsp90 inhibitor GA (Mandal et al., 2010). Proteasome-mediated degradation requires ubiquitylation and Hsp70,SSA1 plays an active role in facilitating ubiquitylation of several misfolded proteins (Nillegoda et al., 2010; Guerriero et al., 2013). So we tested whether SSA1 promotes ubiquitylation of Ste11ΔNK444R for its degradation. To do this, we expressed His-tagged Ste11ΔNK444R kinase in SSA1 and ssa1-45 yeast cells. The Ste11ΔNK444R kinase was then immunoprecipitated with anti-His antibody, and the ubiquitination was checked by Western blotting followed by immunostaining with antiubiquitin antibody. Ubiquitylated Ste11ΔNK444R was considerably less in ssa1-45 cells at 30°C and also at 37°C in comparison with SSA1 cells, even in the presence of the proteasome inhibitor MG132 (Figure 3A). This result suggests that SSA1 acts as an adapter for delivering substrate to the ubiquitin ligase, as observed in the case of NBD2* (mutated second NBD of Ste6; Guerriero et al., 2013).
FIGURE 3:

Hsp70 promotes ubiquitylation of kinases for their degradation. (A) Ubiquitylation of Ste11ΔNK444R was assayed in SSA1 and ssa1-45 cells at permissive (30°C) and nonpermissive (37°C) temperatures. MG-132 (100 μM) was added for 2 h to block the proteasome. Ste11ΔNK444R was immunoprecipitated from these cells with anti-His antibody. Ubiquitylated Ste11ΔNK444R was visualized by Western blotting followed by immunostaining with antiubiquitin antibody. Five microliters of immunoprecipitated Ste11ΔNK444R from each sample was loaded onto an SDS–PAGE, and the kinase level was identified by Western blotting probed with anti-His antibody. Vector control (NT) was used to visualize any nonspecific ubiquitination. Ubiquitinated Ste11ΔNK444R was quantified and represented in the graph. (n = 3, bar = ±SE). (B) Ubiquitination of Ste11ΔNK444R kinase was checked after WT yeast cells were treated with the proline analogue AZC (50 mM) alone or in combination with myricetin (100 μM) or PES (150 μM) for 2 h. In this case, Myc-tag ubiquitin was expressed from the CUP promoter after induction with 100 μM of copper. The Ste11ΔNK444R was immunoprecipitated, and polyubiquitylation of kinase was identified by the anti-myc antibody. AZC-induced Ste11ΔNK444R ubiquitylation was checked in the ubiquitin ligases of ubr1Δsan1Δ knockout yeast cells. GA (50 μM)-induced Ste11ΔNK444R ubiquitylation in the presence of MG132 (100 μM) was used as a control. The polyubiquitylation of Ste11ΔNK444R was quantified and represented in the associated graph (n = 3, bar = ± SE). * indicates the significance of p ≤ 0.05. (C) Western blotting with anti-myc antibody showed the same level of myc-ub expression in the yeast cell lysates used for Ste11ΔNK444R ubiquination in B. Vector control (NT) was used for nonspecific interaction with the antibody. Pgk1 served as a loading control.
To confirm further Hsp70-mediated ubiquitylation of Ste11ΔNK444R kinase, we analyzed stress-induced ubiquitylation of kinases after treatment with the toxic proline analogue azetidine 2-carboxylic acid (AZC). AZC incorporates competitively into newly synthesized proteins and causes thermal instability resulting in protein misfolding (Grant et al., 1975; Mandal et al., 2010; Bach and Takagi, 2013). Ubiquitylation of Ste11ΔNK444R was checked after expression of myc-tagged ubiquitin in the cells followed by immunoprecipitation and Western blotting with anti-myc antibody. Addition of AZC to the yeast cells enhanced the ubiquitylation of Ste11ΔNK444R kinase at the same level as GA-induced Ste11ΔNK444R ubiquitination. However, treatment with either myricetin or PES decreased AZC-induced Ste11ΔNK444R ubiquitylation (Figure 3B). This result showed that Hsp70 actively participates in the ubiquitylation process of Ste11ΔNK444R kinase. The addition of ubiquitin to the proteins is mediated by the ubiquitin ligases Ubr1 and San1 in yeast, and these two ubiquitin ligases maintain cytoplasmic protein quality control (Eisele and Wolf, 2008; Heck et al., 2010; Nillegoda et al., 2010; Guerriero et al., 2013). So we tested ubiquitination of Ste11ΔNK444R in a ubr1Δsan1Δ double-knockout yeast strain in the presence of AZC, and we found that ubiquitylation of Ste11ΔNK444R is completely abolished in the double-knockout strain.
SSA1 protects kinases from accumulating in insoluble inclusions (IPODs)
We saw that SSA1 favors the degradation of misfolded kinases by the proteasome. As a consequence of this, cellular kinase level was restored when SSA1 function was impaired (Figure 2A). Cells evolve dynamic spatiotemporal measures to counter nonessential accumulation of proteins. Misfolded proteins are degraded by the proteasome or autophagy; if not, they are sequestered temporarily as a dynamic soluble inclusion or as an insoluble aggregate (Chen et al., 2011). To track the fate of the restored Ste11ΔNK444R protein in ssa1-45 cell, we took advantage of a green fluorescent protein (GFP)-labeled kinase that was introduced in the C-terminal of the Ste11ΔNK444R (Theodoraki et al., 2012). Ste11ΔNK444R-GFP was expressed from a galactose-inducible promoter in SSA1 and ssa1-45 yeast cells at nonpermissive temperature. GFP fluorescence showed distinct puncta at the periphery of ssa1-45 cells at 37°C but localized diffusely throughout the SSA1 cells. Counting the number of puncta by taking five individual fields with a minimum of four cells per field showed five times more puncta when SSA1 function was impaired (Figure 4A). We have also seen similar results with the Hsp70 inhibitor myricetin. Myricetin-treated cells generated Ste11ΔNK444R kinase inclusion in a dose-dependent manner. Myricetin (100 μM) increased inclusion formation approximately threefold in comparison with the nontreated cells (Figure 4B), whereas myricetin at a concentration of 50 μM had no appreciable effect; this confirmed our earlier observation with Tpk2 kinase (Supplemental Figures S3A and S1A). Hsp70 impairment by myricetin (100 μM) treatment also formed inclusion of Tpk2-GFP kinase but was less frequent in comparison with Ste11ΔNK444R-GFP (Supplemental Figure S3B).
FIGURE 4:

Hsp70 impairment leads to the formation of insoluble cytosolic aggregates. (A) GFP-Ste11ΔNK444R kinase was expressed under GAL promoter in SSA1 and ssa1-45 yeast cells at nonpermissive temperature. GFP fluorescence was observed under a fluorescence microscope. Kinase aggregates were seen in ssa1-45 cells. Bright-field images of the same cells are also shown. (B) WT cells were tested for aggregate formation after 2 h of treatment with 100 μM myricetin or DMSO. Aggregates were counted in a minimum of three different fields with an average of five cells per field. The number of aggregate formations was plotted as a bar diagram. Bar represents SE of three independent experiments. (C) Ste11ΔNK444R-GFP–expressing yeast cells were treated for 2 h with myricetin (100 μM) and then stained with FM4-64 and Hoechst 33242 for vacuoles and nuclei, respectively. GFP (green), FM4-64 (red), and Hoechst 33242 (blue) fluorescences were overlaid. The merged picture shows the predominant deposition of puncta in the perivacuolar region upon Hsp70 impairment with myricetin. Yeast cells without myricetin treatment are shown as a control. (D) Ste11ΔNK444R-expressing SSA1 and ssa1-45 cells were grown at 30 and 37°C, respectively. The lysates were prepared using 0.1% NP-40 buffer. The soluble and insoluble fractions were separated at 15,000 × g for 15 min. The samples were resolved in SDS–PAGE and immunoblotted with anti-His antibody. The same blot was analyzed with anti-Pgk1. (E) WT and hsp42Δ yeast cells expressing GFP-Ste11ΔNK444R were treated with myricetin (100 μM) or DMSO for 2 h and viewed under a fluorescence microscope. The nuclei were stained with Hoechst dye. Kinases formed puncta distant from the nuclei in both the cases. Aggregates were counted in a minimum of three different fields with an average of five cells per field. The number of aggregate formations was plotted as a bar diagram. Bar represents SE of three independent experiments. (F) RFP-Hsp42 does not colocalize with Ste11ΔNK444R-GFP when Hsp70 is impaired with myricetin. (G) RFP-Hsp104 colocalizes with insoluble Ste11ΔNK444R-GFP inclusion when yeast cells are treated with myricetin. (H) RFP-Ste11ΔNK444R inclusion colocalizes with polyglutamin HttQ97-GFP, which forms an IPOD inclusion. (I) Ste11ΔNK444R-GFP forms an inclusion in ubiquitin conjugating enzyme–deleted yeast strain, ubc4/5Δ.
Thus both chemical inhibition and genetic impairment of Hsp70,SSA1 resulted in inclusion formation. Inclusions are sequestered into specialized and functionally distinct compartments in yeast: the JUNQ compartment, the IPOD compartment, and a peripheral compartment enriched with the small heat shock protein Hsp42 (Kaganovich et al., 2008; Specht et al., 2011; Spokoini et al., 2012; Escusa-Toret et al., 2013). JUNQ inclusions are localized adjacent to the nucleus and are sequestered in a detergent-soluble state, whereas IPOD inclusions are nonubiquitylated aggregated proteins and reside in the cell periphery near to the perivacuolar region. But peripheral inclusions are characteristically different from IPODs, because they colocalize with Hsp42 and, unlike IPOD inclusions, their formation is severely affected in hsp42Δ cells (Specht et al., 2011; Escusa-Toret et al., 2013). Although protein kinase inclusions are not characterized as congruent to their amyloid counterparts, we delineated the localization of inclusions further by staining nuclei with Hoecsht 33242 (Molecular Probes, Invitrogen, Carlsbad, CA) and vacuoles with FM4-64 (Invitrogen, Carlsbad, CA). Interestingly, kinase puncta were observed at the perivacuolar region, identical to IPOD, but were seen far from the nucleus when Hsp70 was inhibited by myricetin (Figure 4C). Separation of soluble and insoluble parts by cell fractionation supported the previously obtained results. Ste11ΔNK444R-GFP kinase significantly accumulated in the insoluble fraction in ssa1-45 cells at 37°C (Figure 4D). Thus Hsp70,SSA1 impairment is hindering degradation of the client kinase, and the restored protein accumulates in the form of insoluble inclusions.
Next we wanted to clarify the nature of these inclusions formed by the Hsp70 inhibition, whether they were IPODs or accumulated as peripheral inclusions. To distinguish that, we have studied inclusion formation of Ste11ΔNK444R-GFP in hsp42-deleted yeast cells. Hsp42 had no effect on the formation of Ste11ΔNK444R-GFP inclusions in the presence of myricetin (Figure 4E). The propensity for forming the kinase inclusions was unchanged in hsp42 knockout cells. No obvious colocalization of Ste11ΔNK444R-GFP inclusions was observed with Hsp42 (Figure 4F). Instead, Ste11ΔNK444R-GFP inclusions colocalized with the IPOD marker proteins amyloidogenic HttQ97 and Hsp104 (Figure 4, H and G), but we did not see any change of inclusion properties (size and number) in hsp104Δ yeast cells (Supplemental Figure S4). In cells, misfolded protein is sequestered into the IPOD inclusion when ubiquitination is inhibited (Kaganovich et al., 2008; Spokoini et al., 2012). Similarly, Ste11ΔNK444R-GFP formed inclusions (Figure 4I) and accumulated more in the insoluble fraction in ubc4/5 knockout cells (unpublished data). In contrast, a distinct phenomenon was observed when we stressed the cells with heat for 1 h at 37°C. Under this condition, Ste11ΔNK444R-GFP inclusions were bigger, and the intensity of the puncta was higher. Interestingly, the puncta colocalize with Hsp42 (Figure 5A), and only a few dim puncta were observed during heat stress in hsp42Δ (unpublished data). Hsp104 also colocalizes with kinase puncta (Figure 5B). Distinct inclusions were observed for kinase with respect to HttQ97 during heat stress (Figure 5C), but they colocalized with thermally denatured Ubc9ts protein, which form stress foci (Figure 5D; Spokoini et al., 2012). Thus proteostatic stress foci are formed during heat stress as a physiological response to Ste11ΔNK444R-GFP, whereas IPOD inclusions are generated upon Hsp70 inhibition.
FIGURE 5:

Kinases form aggregates of a discrete nature upon exposure to particular stress conditions. (A) Yeast cells expressing Ste11ΔNK444R−GFP under GAL promoter were shifted to 37°C for 1 h and viewed under a fluorescence microscope. The nucleus was stained with Hoechst dye (blue), as in all experiments. Kinases formed proteostatic stress foci distant from the nuclei upon a temperature shift to 37°C. The kinase stress foci colocalize with RFP-Hsp42 (red). (B) Ste11ΔNK444R-GFP inclusion colocalizes with the chaperone RFP-Hsp104 (red) upon temperature stress. (C) Ste11ΔNK444R stress foci (red) are distinct from amyloid IPOD inclusions, HttQ97-GFP (green). (D) Ste11ΔNK444R kinase inclusion colocalizes with the misfolded protein CHFP-Ubc9ts (red) that forms stress foci upon a temperature shift to 37°C. (E) Ste11ΔNK444R-GFP–expressing yeast cells were treated with DMSO or the Hsp90 inhibitor GA for 2 h. The kinase inclusions are found adjacent to the nucleus. (F) Upon GA treatment, Ste11ΔNK444R-GFP forms a JUNQ inclusion that colocalizes with the misfolded JUNQ substrate CHFP-VHL. The nucleus was stained with Hoechst dye. (G) WT cells expressing GFP-Ste11ΔNK444R were treated with 50 μM GA for 2 h. The soluble and insoluble fractions were separated by centrifugation and analyzed by SDS–PAGE followed by immunoblotting. Pgk1 was used as a control for soluble protein.
We next examined the nature of Ste11ΔNK444R-GFP inclusions when these were formed by inhibiting another major folding chaperone, Hsp90, by GA. Nuclear staining showed Ste11ΔNK444R-GFP inclusion at the juxtanuclear segments of the cell (Figure 5E), as observed previously (Theodoraki et al., 2012). These inclusions colocalized with misfolded VHL, a JUNQ substrate, at transient stress (Figure 5F). Notably, kinase inclusions remained in the soluble form (Figure 5G), characteristic of JUNQ, a phenomenon antithetical to Hsp70 inhibition. This phenotypic difference showed a distinct demarcation between the functioning of these two chaperones. Taken together, our data suggest that Hsp70 prevents accumulation of kinases as terminally misfolded protein at the peripheral site and acts as a holdase for rescuing the cell from a pernicious fate.
Overexpression of SSA1 clears the kinase inclusions by enhancing ubiquitination
Inclusions formed in the cellular environment serve as functional sites for protein quality control (Kaganovich et al., 2008; Specht et al., 2011). The presence of ubiquitin conjugates, molecular chaperones, and proteasomal subunits within the inclusions supports this view (Cummings et al., 1998; Wang et al., 2009). Dynamic JUNQ inclusions are more ubiquitinated and degraded by the proteasome, whereas terminally misfolded aggregates accumulate as an inactive peripheral compartment that can be disintegrated by the cooperative action of Hsp70/Hsp104/Hsp40 chaperones (Kaganovich et al., 2008; Shorter and Lindquist, 2008), although disaggregation activities have also been reported for the individual chaperones alone (Winkler et al., 2012a; Mattoo et al., 2013). But how Hsp70 handles the fate of such collectively misfolded proteins and determines their fate remains elusive. To understand this phenomenon, we overexpressed SSA1 under promoters of increasing strength from copper (Cu2+)-inducible promoter (CUP) to constitutive translation elongation factor (TEF) and glyceraldehyde 3-phosphate dehydrogenase (GPD), respectively, to modulate SSA1 levels and decide the fate of aggregated kinase under SSA1-overexpressed conditions (Blazeck et al., 2012). SSA1 expression was found to be highest with GPD promoter, moderate with TEF, and minimal with CUP promoter (Figure 6C). Expression of SSA1 from these promoters resulted in gradual disappearance of Ste11ΔNK444R-GFP inclusions that, interestingly, correlated with increasing SSA1 levels under weak to strong promoters (Figure 6, A and B). Thus SSA1 level played a pivotal role in disaggregation of kinase inclusions.
FIGURE 6:

Hsp70 overexpression solubilizes kinase aggregates. (A) SSA1 plasmids with different promoters (CUP, TEF, and GPD) were transformed into ssa1-45 cells containing GFP-Ste11ΔNK444R kinase. ssa1-45 cells were incubated for 2 h at 37°C, and the fluorescence of GFP-Ste11ΔNK444R kinase was observed in presence of SSA1-overexpressing condition. Overnight treatment with 100 μM of copper was used to induce SSA1 from CUP promoter. Isogenic SSA1 and ssa1-45 cells without SSA1 expression were used as a control. (B) Similar to A, but WT cells were treated with DMSO or 100 μM myricetin in the absence and presence of overexpressed SSA1. Aggregates were counted in a minimum of three different fields with an average of five cells per field. The quantification of aggregates is shown as a bar diagram. The error bar represents the SE of three independent experiments. (C) Western blotting of cell lysates showed SSA1 levels after expression from CUP, TEF, and GPD promoters. Pgk1 served as the loading control. The bands are quantified and plotted as a bar diagram. Error bar represents the SE of three different experiments.
Next we wanted to track the effect of SSA1 expression on this disappearance of kinase inclusions. To address this, we adopted a strategy to express SSA1 from an inducible promoter after generation of the inclusions and observed the fate of inclusions with time. To do that, we expressed SSA1 from inducible CUP promoter and determined the optimal expression in different concentrations of copper. SSA1 expression was found to be maximal at 300 μM of copper (Figure 7B), and under this condition, Ste11ΔNK444R-GFP inclusions disappeared with an efficiency similar to that of SSA1 overexpressed under GPD promoter (Figure 7A). At 300 μM copper concentration, the level of SSA1 was saturated at ∼6 h, which enabled us to use it for subsequent studies (Figure 7C). Under this condition, by separating the soluble and insoluble fractions, we also assessed the ability of SSA1 to solubilize Ste11ΔNK444R inclusions. We found that Ste11ΔNK444R kinase was partitioned from a soluble to an insoluble fraction in the presence of the Hsp70 inhibitor myricetin. However, the kinase level was increased in the soluble fraction upon SSA1 expression for 6 h (Figure 7D).
FIGURE 7:

Hsp70 expression from inducible CUP promoter clears kinase inclusions. (A) p426CUP-SSA1 plasmid was transformed into GFP-Ste11ΔNK444R–containing yeast cells. GFP-Ste11ΔNK444R was expressed from the GAL promoter by the addition of galactose. The cells were treated with 100 μM myricetin for 2 h to form the kinase inclusions. SSA1 was then expressed from the CUP promoter for an additional 6 h with varying concentrations of copper ion, as indicated. The GFP fluorescence was observed, and the inclusions were counted in a minimum of three different fields with an average of five cells per field. The quantification of inclusions is shown as a bar diagram. (B) Western blots of cell lysates expressing SSA1 with different concentrations of copper ion. Pgk1 served as a loading control. The bands were quantified by ImageJ software and plotted as a bar diagram. (C) p426CUP-SSA1–containing yeast cells were induced with 300 μM of copper ion for different times, as indicated. The expression of SSA1 was measured by Western blot analysis with anti-His antibody. The bands were quantified, normalized with Pgk1, and plotted as a bar diagram. (D) WT cells were cotransformed with p416GPD-Ste11ΔNK444R (marker-swapped Leu version) and p426CUP-SSA1 plasmids. The cells were treated with 100 μM myricetin for 2 h. The SSA1 was expressed by the addition of 300 μm of copper ion for 6 h. The soluble and insoluble fractions were separated and analyzed by Western blotting. The bands were quantified and plotted as a bar diagram. Bar represents SE of three independent experiments.
We then sought to examine the inclusion clearance capacity of SSA1, in real time, by chasing the fluorescence of Ste11ΔNK444R-GFP inclusions. Ste11ΔNK444R-GFP inclusions were generated by treating the cells with myricetin for 2 h. The inclusions were then followed at 3-h intervals up to 9 h after overexpression of SSA1 from the CUP promoter. Myricetin-treated cells displayed Ste11ΔNK444R-GFP inclusions at the cell periphery at 0 h of SSA1 induction. But, at 3–6 h of SSA1 induction, kinase inclusions were found close to the nucleus. Complete disappearance of inclusions was observed at 9 h (Figure 8A). The disappearance of the inclusions could happen in two possible ways: degradation by the proteasome or refolding by the chaperone machinery. However, degradation by the proteasome was more likely, because inclusions localized at the JUNQ compartment are ubiquitylated and temporarily stored for Hsp70-assisted proteasome-mediated removal (Kaganovich et al., 2008; Weisberg et al., 2012). To test this possibility, we added the proteasome blocker MG-132 after 5.5 h of SSA1 expression. Addition of MG132 restored Ste11ΔNK444R-GFP inclusions, even after 9 h of SSA1 expression. This result proved that Ste11ΔNK444R-GFP inclusions were removed via the proteasomal pathway when SSA1 was overexpressed. Solubility analysis of the same samples used for the chase experiments confirmed the above data. Ste11ΔNK444R-GFP was increased in the soluble fraction and was more ubiquitylated after 6 h of SSA1 induction (Figure 8, B and C). Moreover, single-cell time-lapse study showed that SSA1 overexpression leads to the clearance of IPOD inclusions generated by Hsp70 inhibition (Figure 8D). Altogether these results suggest that SSA1 eliminates kinase inclusions by promoting their ubiquitination.
FIGURE 8:

Hsp70 overexpression compartmentalizes the kinase inclusions from IPOD to JUNQ. (A) SSA1 is expressed from inducible CUP1 promoter in Ste11ΔNK444R-GFP–expressing yeast cells after treatment with 100 μM myricetin for 2 h. Ste11ΔNK444R-GFP aggregation was checked at regular time intervals (0, 3, 6, and 9 h) after SSA1 expression with 300 μM copper. The nucleus was stained with Hoecsht 33242. The proteasome was blocked by adding MG132 (100 μM) at 5.5 h of SSA1 expression with copper. (B) The same cells were analyzed for Ste11ΔNK444R-GFP solubility by separating the soluble and insoluble fractions. Western blotting was performed with the samples; this was followed by immunostaining with anti-GFP antibody. The immunostain was carried out with anti-Pgk1 and anti-Hsp70. The band intensity was quantified and plotted as a bar diagram. The error bar represents the SE (n = 3). (C) Ubiquitination status of Ste11ΔNK444R after the yeast cells were treated with myricetin for 2 h and then SSA1 was overexpressed for 6 h. Ste11ΔNK444R was immunoprecipitated from the soluble fraction with anti-His antibody; this was followed by immunostaining with antiubiquitin antibody. (D) Time-dependent changes in the localization of Ste11ΔNK444R-GFP aggregates in cells treated with myricetin (100 μM) for 2 h followed by SSA1 overexpression. (E) Ste11ΔNK444R-GFP–expressing cells were treated with the Hsp90 inhibitor GA (50 μM) for 2 h. SSA1 was expressed afterward from CUP promoter with 300 μM copper, and the Ste11ΔNK444R-GFP aggregation was measured at 0, 3, 6, and 9 h. MG132 (100 μM) was added to block the proteasome at 5.5 h of SSA1 expression.
To study the capacity of SSA1 in modulation of insoluble protein deposits, we examined the well-characterized amyloid protein RNQ1, which agglomerates in the IPOD compartments in yeast (Specht et al., 2011; Escusa-Toret et al., 2013). RNQ1-mRFP (monomeric red fluorescent protein) was expressed from GAL-promoter cotransformed with SSA1 under inducible CUP promoter. The fate of RNQ1 aggregates were then observed after SSA1 induction (Supplemental Figure S5). Quite distinctly, at 0 h of SSA1 induction, RNQ1 formed peripheral aggregates distant from the nucleus. The aggregates were mostly hefty and coalesced into specific well-defined structures. At 3 h of induction, there was no diacritic variation or transition of aggregate foci, except that a few of the previously formed puncta seemed diffused and dimmer. The initially formed hefty aggregates appeared to disperse all through the cytosol. At 6 h, the puncta were much fewer in number and were much smaller compared with the initial bulky aggregates. The miniscules seemed to locate themselves comparatively near the nucleus. At 9 h, most of the puncta disappeared, but <25% of cells still retained the puncta, although they were much diminished in size and intensity. A phenotypic observation was that the cells lost their essential fitness after 6 h of SSA1 induction. At this stage, most of the cells seemed to get disoriented and lose their cardinal architecture, as was reported previously (Douglas et al., 2008). Thus the overexpression of SSA1 clears RNQ1, a true IPOD substrate, but is unable to suppress its toxicity. Henceforth IPOD substrates of completely varying nature behaved in a comparatively analogous pattern, suggesting that SSA1 handles IPOD deposition in much the same manner, despite the distinct and unique constitution of the substrate.
A distinct phenomenon of cellular surveillance and quality control lies in the functioning of the two major chaperones, Hsp70 and Hsp90. Inhibition of the individual chaperone showed formation of kinase inclusions but at a different location in the cellular compartments. Hsp90 inhibition with GA generated Ste11ΔNK444R-GFP inclusions mostly at the juxtanuclear location (Theodoraki et al., 2012), whereas inclusions were formed at the peripheral sites when Hsp70,SSA1 was inhibited. Because SSA1 overexpression degraded the IPOD inclusions by the proteasome, we anticipated the same operation could eliminate juxtanuclear, soluble inclusions formed upon Hsp90 inhibition. GA (50 μM) treatment for 1 h accelerated formation of considerably hefty and oversized puncta in the juxtanuclear regions (Figure 8E). Overexpression of SSA1 indeed displayed an analogous trend of puncta clearance. Deterring the proteasome by MG-132 restored the inclusions at the juxtanuclear position, implying proteasome-mediated eviction (Figure 8E).
Cellular SSA1 level determines the substrate fate
Cellular stress leads to harmful impact in the cellular milieu, causing heat shock factor–induced induction of heat shock proteins, including Hsp70. To ascertain how Hsp70 determines the fate of protein kinases under physiological and stressed conditions, we integrated folding, degradation, and aggregation—the three major outcomes of triage—and analyzed the phenomenon that occurs upon alteration of Hsp70 level. GST-Tpk2 under the inducible galactose promoter was transformed into yeast cells. Cells were first grown at 30°C and then shifted to 42°C for 2 h to induce protein misfolding that generated inactive kinases (Supplemental Figure S6). Cycloheximide was added to the cells before heat shock to stop further protein synthesis. The cells were then divided into two groups and incubated for another 3 h for recovery at 30°C or under moderately stressed conditions at 37°C. Next we checked the fate of the kinases under these conditions by evaluating the ubiquitylation status and the kinase activity (Schroder et al., 1993). The result showed that Tpk2 ubiquitylation was increased at 42°C, and this ubiquitylation was reduced to the basal level (observed for nonstressed kinases, Figure 9B, lane 4) at the 30°C recovery period. Surprisingly, a comparable level of Tpk2 ubiquitylation was observed when cells were incubated at 37°C after heat shock (compare lanes 2 and 4 in Figure 9B). Therefore kinase degradation was promoted under denaturing conditions (42°C), with the kinase probably refolding to the native state during recovery periods. We have checked the Tpk2 kinase activity under these conditions. The results showed that Tpk2 kinase lost its activity after exposure to the heat stress at 42°C, but the kinase activity was regained after recovery at 30°C. However, the Tpk2 kinase was inactive at 37°C (Figure 9D). This suggests that, after denaturation under stress conditions, kinases can refold to their native state under physiological conditions but not under the stress conditions.
FIGURE 9:

Hsp70 overexpression switches the cellular fate of kinases. (A) Schematic representation of the experiments. (B) Ubiquitylation of GST-Tpk2 kinase after incubation of the yeast cells at 42°C for 2 h followed by recovery at 30°C or 37°C for an additional 3 h in normal or SSA1-overexpressing conditions. SSA1 was expressed from CUP promoter after addition of 300 μM of copper during the heat shock. GST-Tpk2 kinase was purified by glutathione resin. The ubiquitylation of GST-Tpk2 was analyzed by Western blotting with antiubiquitin antibody. The same samples were Western blotted and probed with anti-GST antibody. (C) The same blot (probed with anti-GST antibody in B) was probed with anti-Hsp70 antibody to measure the bound Hsp70 with GST-TPK2. The bands were quantified and are represented as a bar diagram. The error bar is the SE of three individual experiments. (D) The kinase activity of GST-Tkp2 from the same samples was measured with a nonradioactive PKA kinase assay kit, using kemptide as the substrate. The bar represents the average of two independent experiments. (E) pADH-GFP-Tpk2–expressing yeast cells were incubated at 42°C for 2 h and then placed at 30°C or 37°C for 3 h in the absence or presence of SSA1 overexpression. SSA1 was expressed from CUP1 promoter after addition of copper (300 μM) during incubation at 42°C. GFP fluorescence was observed under a fluorescence microscope.
Our results showed that Hsp70,SSA1 plays an essential role in clearing the misfolded kinase inclusions. Therefore we wanted to test whether the level of SSA1 can alter the triage decision of Tpk2 kinase. For this, we overexpressed SSA1 from CUP promoter before heat shocking the cells at 42°C and then determined the ubiquitylation level and the kinase activity of Tpk2. Indeed, we have seen overall enhancement of Tpk2 ubiquitylation at 42°C and also during its recovery at 30 and 37°C (Figure 9B). Interestingly, the kinase activity measured at these conditions showed a notable difference. No Tpk2 activity was found after recovery from heat shock, even at 30°C (Figure 9D). But an increased binding of Hsp70 with the immunoprecipitated Tpk2 kinase was found with Western blot analysis (Figure 9C). Therefore, under stress conditions, Hsp70 interacted with the misfolded kinases, and the aberrant proteins were targeted toward degradation. Up-regulating the level of SSA1 led to increased binding and degradation of misfolded protein that could otherwise have refolded under physiological conditions. This observation was further supported when we treated the cells with proteotoxic agent, hydrogen peroxide, or AZC. Treatment with either of the toxic agents retarded growth of WT cells, whereas overexpression of SSA1 rescued the growth phenotype (Supplemental Figure S7). Thus, as part of cellular quality control, degradation of misfolded proteins is fostered by SSA1 to protect the cells from toxic accumulations.
We found an apparent paradox in which Tpk2 kinase was inactive but less ubiquitylated in the 37°C recovery period after heat shock. To resolve this, we used GFP-TPK2 to visualize the kinase by fluorescence microscopy. Under heat shock conditions (42°C) the GFP-Tpk2 distribution throughout the cell was similar to that observed for recovery at 30°C. However, kinase inclusions were formed near the cell periphery during recovery at the moderate stress condition of 37°C (Figure 9E). These inclusions were cleared by the proteasome when SSA1 was overexpressed (Figure 9E). Thus, under stress conditions, the kinase tends to form inclusions that are actively degraded by the SSA1 when it is overexpressed. We propose that SSA1 acts in deciding the fate of the protein and triaging it toward folding or degradation or holding it until Hsp70 can assess the situation. This partitioning of misfolded proteins under physiological and stressed conditions by Hsp70 is quite distinct from other chaperones that essentially maintain the cellular protein quality control.
Hsp70 has a bipartite structure in which the participation of the amino terminal domain, which binds ATP, and that of SBD, which binds to hydrophobic molecules, are equally important (Mayer and Bukau, 2005; Kityk et al., 2012). To assess whether both the domains of Hsp70,SSA1 are necessary for clearing kinase inclusions, we used two mutant forms of SSA1, K69Q and G199D, based on SSA1 structure. K69Q mutant inhibits ATP binding at the NBD and is unable to hydrolyze ATP, whereas the G199D mutant is in the α-helical lid of SBD that impedes the opening and closing of the C-terminus that interacts with the substrate (Han and Christen, 2001; Sharma and Masison, 2009; Kityk et al., 2012). Overexpression of K69Q mutant under CUP promoter was unable to clear Ste11ΔNK444R kinase inclusions, whereas G199D behaved in a manner similar to WT SSA1 (Figure 10A). Thus the ATPase activity of SSA1 is crucial in removal of aggregates from the cell. This structural patterning of Hsp70 suggests discrete functioning of SSA1 based on its structure and conformation. We then checked whether SSA1 overexpression can alter the turnover of the protein. We analyzed Ste11ΔNK444R kinase turnover after overexpressing SSA1 by pulse–chase analysis. We found that Ste11ΔNK444R degraded faster when SSA1 was overexpressed (Figure 10B). However, the ATPase-deficient mutant of SSA1, K69Q, had no effect on Ste11ΔNK444R kinase turnover, but G199D showed a moderate effect. Thus the ATPase activity of SSA1 is essential for increased turnover of Ste11ΔNK444R. This suggests that SSA1 plays an essential role in monitoring the substrate fate and shifts the balance toward degradation when its level is increased.
FIGURE 10:

Hsp70's ATPase activity is indispensable for its function. (A) WT cells were transformed with GAL-GFP- Ste11ΔNK444R and variant SSA1 constructs (WT, K69Q, and G199D). GFP-Ste11ΔNK444R–expressing yeast cells were treated with 100 μM myricetin for 2 h. Variants of SSA1 (WT, K69Q, and G199D) were expressed from inducible CUP1 promoter with 300 μM copper after myricetin treatment. The number of inclusions was counted and plotted as a bar diagram. (B) Pulse–chase analysis of Ste11ΔNK444R kinase after overexpressing WT, K69Q, and G199D mutants of SSA1. The band intensity was measured by phosphorimager and plotted in the associated graph. The graph represents the average of two independent experiments.
DISCUSSION
Hsp70 participates in various cellular functions from nascent protein folding to the remodeling of protein complexes. In addition to that, Hsp70 favors degradation of proteins. But how Hsp70 determines the fate of substrates remains unclear. In this report, we describe diverse functions of Hsp70 in maintaining cellular quality control and the triage decisions of Hsp70 based on the substrate status and cellular environment.
We establish here the functions of Hsp70, using protein kinases as substrates. Two functionally distinct kinases, Tpk2 and Ste11, were selected as substrates based on their unique characteristics. Differences in mobility of mature and immature Tpk2 in SDS–PAGE helps to assess the status of the kinase during its folding process. The mutated form of Ste11 kinase, Ste11ΔNK444R, is highly dependent on chaperone machinery (Mandal et al., 2008; Flom et al., 2012), and its conformation is sensitive to the proteostatic condition of the cell, which makes it an ideal substrate for exploring cellular protein quality control (Theodoraki et al., 2012).
We elucidated the function of Hsp70,SSA1 by using chemical inhibitors (myricetin or PES) or genetically challenged temperature-sensitive hsp70 mutant strains (ssa1-45). The chemical inhibition of Hsp70 affects the maturation of Tpk2 kinase (Figure 1A), similarly observed in the case of ydj1Δ (Mandal et al., 2008). Distinctly, Tpk2 maturation was only delayed in ydj1Δ, but no alteration in kinase activity was observed. Possibly, the presence of many J proteins compensates for the loss of function of the Ydj1 chaperone. But Tpk2 did not mature at all and was found to be inactive in the case of Hsp70 inhibition (Figure 1C). Moreover, inefficient maturation of Tpk2 was seen when Hsp90 was inhibited by GA (Nillegoda et al., 2010). Because Tpk2 maturation occurs after recruitment of Hsp90 in the intermediate complex, our data suggest that Hsp70 inhibition hinders the delivery of the kinase to the intermediate complex.
Our results show that Hsp70,SSA1 maintains the homeostasis of kinase by assisting folding and promoting ubiquitylation of kinases for their regular turnover via the proteasome. Importantly, the ability of SSA1 to rescue the cells from stress-induced protein damage was further reflected in its role in protecting cells against proteotoxic stress caused by AZC, hydrogen peroxide, or GA (Figures 2 and 3 and Supplemental Figure S7). The elimination of misfolded proteins was carried out in conjunction with the ubiquitin ligases Ubr1 and San1 (Heck et al., 2010; Nillegoda et al., 2010; Guerriero et al., 2013). Complete absence of Ste11ΔNK444R ubiquitylation in ubr1Δsan1Δ cells justifies that. Facilitation of ubiquitylation of kinases by SSA1 suggests its active participation in transferring misfolded proteins to the ubiquitin ligases (Figure 3). Thus the function of Hsp70,SSA1 in both folding and degradation, as and when required, is quite obvious from our results.
The protective role of Hsp70,SSA1 against aggregate formation was reflected by the formation of kinase puncta at the cell periphery upon Hsp70 inhibition. Similar observations were made with the mildly misfolded protein GFP-DegAB (Shiber et al., 2013). Our findings revealed that kinase accumulated as insoluble inclusions (IPOD) upon Hsp70 inhibition (Figure 4). Formation of these IPOD inclusions is independent of Hsp42 (Figure 4), as observed previously (Escusa-Toret et al., 2013). The presence of Hsp104 in the IPOD inclusion is probably due to its ability to disintegrate the inclusions (Cushman-Nick et al., 2013). Inclusions located in the IPOD are less toxic in comparison with those in the soluble JUNQ compartment, although toxicity not only depends on the location but is also governed by the nature of the substrate (Weisberg et al., 2012). The same precept applies to protein kinases. Misfolded kinases sequestered in IPOD inclusions appear to be less toxic in comparison with those in soluble juxtanuclear inclusions (Supplemental Figure S8). Interestingly, in our study, we found that aggregates were partitioned into different compartments depending on the nature of the stress. Kinases were sequestered into stress foci during heat stress similar to that observed for Ubc9ts protein (Figure 5; Spokoini et al., 2012). In contrast, more soluble juxtanuclear puncta (JUNQ) were formed that are susceptible to degradation when Hsp90 is inhibited (Figure 5). Our result supports the observation by Taipale and colleagues that Hsp90 client kinases undergo repeated rounds of binding and release for their chaperoning and that inhibition of Hsp90 prevents reloading of kinases to the chaperone and, as a consequence, increases targeting of misfolded kinases to the JUNQ compartment (Taipale et al., 2012). Thus a distinct mechanism underlies the operation of the two chaperones, one in which Hsp90 keeps the kinases in functional state, whereas Hsp70 deals with them once they misfold.
Previously it was shown that Hsp70 overexpression clears aggregation-prone proteins (Tau, Ure2, and cytosolic Prp) and increases cell survival (Petrucelli et al., 2004; Weisberg et al., 2012; Zhang et al., 2012; Xu et al., 2013). We identified that Hsp70,SSA1 overexpression clears IPOD inclusions by the proteasome by enhancing their ubiquitination (Figure 8 and Supplemental Figure S5). SSA1 targeted not only IPOD inclusions for degradation but also JUNQ inclusions that were formed by the Hsp90 inhibition (Figure 8). Interestingly, the N-terminal ATPase domain of SSA1 is indispensable for the removal of the aggregates (Figure 10; Basha et al., 2006). Thus Hsp70 primarily acts as a holdase and targets misfolded proteins for degradation by the proteasome.
We wanted to find out how Hsp70 protects the cells from adverse cellular conditions. In vivo experiments indicated that heat-denatured kinases were susceptible to degradation but refolded to the active state during recovery at physiological temperature (Figure 9). In contrast, at increased Hsp70,SSA1 levels, kinases were destined for degradation both at refolding and stressed conditions. Hsp70 actively binds to the misfolded kinases and sends them for degradation by the proteasome. It also facilitates clearing the kinase inclusions generated due to heat stress at 37°C. SSA1 level plays a deciding factor here for fate of client proteins, and this substrate fate determination of SSA1 requires a functional ATPase cycle. Moreover, cochaperones of Hsp70 and other cofactors participate in the triage decision (Shiber and Ravid, 2014) and aggregate clearance (Glover and Lindquist, 1998; Shorter and Lindquist, 2008; Shorter, 2011; Cushman-Nick et al., 2013).
Surprisingly, aggregates were not observed during the initial heat shock but appeared after the cells were incubated at moderate stress conditions (37°C). This phenomenon supports the hypothesis that protein deposition sites are mainly quality-control compartments by which the cell maneuvers quality-control affairs to keep itself functioning. Extreme heat shock hinders the formation of quality-control compartments and denatures cellular protein irreversibly. On the other hand, under moderate stress conditions, the cell tries to protect itself by establishing quality-control compartments, resulting in the formation of functional quality-control foci.
In summary, our findings demonstrate the overall functions of Hsp70,SSA1 in maintaining cellular protein quality control, wherein it facilitates folding of newly synthesized proteins, degradation of misfolded proteins, clearing of inclusions, and protecting the cells from accumulation of misfolded proteins. SSA1 targets misfolded proteins for degradation, and its level acts as a rate-limiting factor for the manipulation of misfolded proteins inside the cell. Thus the role of Hsp70 in eliminating misfolded proteins raises the possibility of controlling diseases associated with protein misfolding. Finally, we have shown a broader role for Hsp70 in client fate determination that ameliorates the cellular environment in favor of quality assurance.
MATERIALS AND METHODS
Yeast strains, media and chemicals
The BY4741 (MATa his3∆0 leu2∆0 met15∆0 ura3∆0) strain was used for most of the studies. Hsp70 temperature-sensitive mutants JB67 (MATá his3-11,15 leu2-3112 ura3-52, trp1-Δ1 lys2 ssa1::ssa1-45 ssa2::LEU2 ssa3::TRP1 ssa4::LYS2), termed as ssa1-45, and its corresponding isogenic strain, JN516 (MATa his3-11,15 leu2-3112 ura3-52, trp1-Δ1 lys2 SSA1 ssa2::LEU2 ssa3::TRP1 ssa4::LYS2), termed as SSA1, were obtained from E. Craig (University of Wisconsin, Madison). ubc4/5Δ (MHY508) and its isogenic strain, MHY501, were used. Yeast cells were cultured in YPD (1% Bacto-yeast extract, 2% Bacto-peptone, 2% dextrose) media or in selective dropout media according to the standard protocols. For treatment with inhibitors, erg6::KanMx4 or erg6::nat yeast strain was used. G418 sulfate from Sigma (St. Louis, MO) and nourseothricin from Werner BioAgents (Jena, Germany) were added to a final concentration of 400 μg/ml and 200 μg/ml, where necessary. GA was obtained from Invivogen (San Diego, CA); myricetin from SRL (Mumbai, India), PES, and l-azetidine-2-carboxylic acid (AZC) were obtained from Sigma. Antibodies anti-His, antihemagglutinin (anti-HA), anti–GST antibodies, and concanavalin A were obtained from Sigma; anti-GFP from Roche (Mannheim, Germany); anti-TAP and anti-Myc from Thermo Scientific; and antiubiquitin from Cell Signaling (Danvers, MA). GST-resin was purchased from Qiagen (Hilden, Germany).
Generation of plasmids
Plasmids used in this study are listed in the Supplemental Material.
Pulse–chase assays
Pulse labeling of yeast cells was done following Mandal et al. (2011). Briefly, yeast cells were grown in selective media to mid–log phase (A600 = 0.4–0.6), washed twice with water and once with SD-methionine, and then resuspended in SD-methionine at a concentration of 6 OD per ml. Cells were incubated either at 30 or 37°C (for temperature-sensitive mutant yeast cells) for 45 min with constant shaking. GA and myricetin were added 20 min before pulse labeling. Proteins were labeled with [35S]methionine (100 μCi/ml; BRIT, Hyderabad, India) for 10 min. The pulse was quenched with cycloheximide (200 μg/ml) and cold methionine (1 mM) for chase reactions only. Samples were taken at different time points of chase reactions and added to an equal volume of ice-cold trichloroacetic acid. The cells were pelleted and washed twice with chilled acetone (−20°C) before vacuum drying. The cell pellets were resuspended in ice-cold extraction buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 1% SDS, and 1× Halt protease inhibitor cocktail [Thermo Scientific, Rockford, IL]); this was followed by lysing in a bead beater at 4°C four times for 1min after an equal volume of glass beads was added. Extracts were clarified at 13,000 × g for 15 min. 35S incorporation was measured in a scintillation counter, and an equal amount of counts was used for the subsequent immunoprecipitation. Extracts were diluted at least 10-fold with IP-dilution buffer (60 mM Tris-HCl, pH 7.5, 190 mM NaCl, 1.25% Triton-X-100, and 6 mM EDTA). The respective antibody was added, and the samples were incubated overnight at 4°C with rotation. Immunoprecipitates were adsorbed onto protein A/G-agarose resin (Pierce, Rockford, IL) for 1 h and washed four times with IP-dilution buffer. The samples were boiled in 1× SDS-sample buffer and resolved by denaturing gel electrophoresis. The gels were fixed (10% acetic acid, 30% methanol) for 30 min, washed twice in water for 15 min, and incubated in 1 M sodium salicylate for 30 min before being dried and exposed to x-ray film or a phosphorimager screen.
TPK2 kinase assay
The Tpk2 kinase assay was performed according to Mandal et al. (2008) with some modifications. Yeast cells containing pGAL1-GST-TPK2 in YEplac195 were grown overnight at 30°C in synthetic dropout medium without uracil to log phase in the presence of 2% raffinose. Tpk2 kinase was expressed from GAL1 promoter by the addition of 2% galactose for 4–6 h. The Hsp70 inhibitor myricetin or PES was added at final concentrations of 100 and 150 μM, respectively, in the last 2 h of induction. Cells were harvested after 6 h of galactose induction. Cell extracts were prepared in IPP150 buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% NP-40), and the amount of protein was quantified using Bradford reagent. Cell extracts (0.3 mg) from each set were diluted to 0.5 ml of GST pull-down binding buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.1%NP-40, and 10 mM MgCl2) and incubated with 50% (vol/vol) glutathione resin (Qiagen) for 2 h at 4°C. The resin was washed three times and divided into two aliquots. One aliquot was used for the kinase assay, and the other was used for Western blot analysis with GST antibody. The Western blots were quantified by using a chemiluminescence imaging detection system. Tpk2 kinase was assayed according to the manufacturer's protocol by using a Pep-Tag nonradioactive protein kinase assay kit (Promega, Madison, WI).
Generation of the san1::kanMx ubr1::his5 double-deletion yeast strain
Deletion of ubiquitin ligase Ubr1 was done according to Longtine et al. (1998) by a single-step PCR-mediated gene-deletion strategy with HIS5 of Saccharomyces pombe. HIS5 of S. pombe was amplified from a pFA6a plasmid with the following forward 5′AATCTTTACAGCTCACACAAATTACATAGAACATTCCAATCGGATCCCCGGGTTAATTAA3′ and reverse 5′ACAAATATGTCAACTATAAAACATAGTAGAGGGCTTGAATGAATTCGAGCTCGTTTAAAC3′ primers. The amplified fragment was purified and transformed into san1Δ BY4741 yeast cells and selected in SD medium without histidine to create the double knockout strains. The site-specific deletion was further confirmed by PCR.
In vivo ubiquitylation assay
In vivo ubiquitylation of the kinase was assayed following Mandal et al. (2010) with minor modifications. Yeast cells were grown to mid–log phase (OD ∼ 0.8) in selective media. Equal amounts of cells were taken and treated with dimethyl sulfoxide (DMSO), MG132 (100 μM), GA (50 μM), or AZC (50 mM) alone or in combination with the Hsp70 inhibitors myricetin or PES for 2 h. Copper-inducible myc-ubiquitin was transformed to the yeast cell when necessary, and myc-ub was induced in the presence of 100 μM CuSO4 (Nakatsukasa et al., 2008). Cells were harvested and washed once with ice-cold water. The supernatant was discarded, and the cell pellet was resuspended in 500 μl of ice-cold 100% ethanol freshly dissolved in 50 mM NEM (N-ethyl maleimide). The cell pellets were vortexed for 5 min keeping in ice at regular intervals before the solution was transferred to new tubes. Cells were again harvested, the supernatant was discarded, and the cells were washed three times with ice-cold sterile water containing 4 mM NEM. Cell pellets were resuspended in ice-cold extraction buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 1% SDS, 8 M urea, 1 mM dithiothreitol [DTT], 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM NEM, and 1× protease inhibitor cocktail); this was followed by lysis in a bead beater at 4°C after an equal volume of glass beads preincubated with ice-cold water containing 4 mM NEM was added. Extracts were cleared at 13,000 × g. Supernatants were diluted 10-fold with IP-dilution buffer (60 mM Tris-HCl, pH 7.5, 190 mM NaCl, 1.25% Triton-X-100, 6 mM EDTA, 1 mM DTT, 1 mM PMSF, 10 mM NEM, and protease inhibitor cocktail). Antibody against the kinase was added for immunoprecipitation, and the samples were rotated overnight at 4°C. Immunoprecipitates were adsorbed onto protein A/G-agarose resin at 4°C and washed three times with IP-wash buffer (10 mM Tris, pH 7.5, 50 mM NaCl, and 10 mM NEM, supplemented with 0.05% [wt/vol] SDS). The samples were boiled in 2× urea-sample buffer (75 mM MOPS, pH 6.8, 8 M urea, 4% SDS, 0.2 M β-ME, 10 mM NEM, and 0.2 mg/ml bromophenol blue) and resolved in a 4–20% SDS gradient gel (Bio-Rad, Hercules, CA).
Solubility assay and Western blot analysis
Solubility assays of kinases were performed according to Theodoraki et al. (2012). Yeast cells were grown to log phase and an OD600 of 6 were collected for each sample. Cells were pelleted down by centrifugation for 4 min at 5000 rpm. The cell pellets were resuspended in 250 μl of lysis buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 1% glycerol, 0.1% NP-40, 1 mM PMSF, and 1× protease inhibitor) and were added into prechilled glass beads (0.5 mm) in 0.5-ml microfuge tubes. The cells were lysed in a bead beater at 4°C and centrifuged at 2000 rpm. The supernatant was transferred into new tubes and was centrifuged at 3000 rpm. The supernatant was again collected and centrifuged for 15 min at 15,000 × g. After centrifugation, the soluble fraction (supernatant) was removed and transferred into new tubes. The insoluble fraction (pellet) was washed three times with 400 μl of wash buffer (2% NP-40, 50 mM Tris, pH 7.4, 1 mM EDTA, and 1% glycerol) and centrifuged for 15 min at 15,000 × g. After the washes, the insoluble fraction was resuspended in lysis buffer and 2× SDS sample buffer was added to the same volume in both soluble and insoluble fraction samples. The samples were then boiled for 5 min, and equal volumes were analyzed in 10% SDS–PAGE.
Swapping Ura-3 marker in p316-Ste11ΔNK444R-GFP plasmid to Leu-2
The Leu2-KanR fragment was excised from pUL9 plasmid with Sma1 and transformed to erg6Δ yeast strains containing p316-GFP-Ste11ΔNK444R (Cross, 1997). Ura-3 and Leu-2 were in the same orientation. The colonies were selected in SD medium without leucine. No colonies were observed in SD medium lacking uracil, which confirmed the disruption of the Ura-3 fragment by Leu-2.
Microscopy and image analysis
The formation of Ste11ΔNK444R kinase aggregate was checked in vivo using a GFP-tagged kinase construct in a temperature-sensitive strain of Hsp70 or by treatment with the Hsp70 inhibitor myricetin. Yeast strains containing Ste11ΔNK444R-GFP were grown overnight in SD media containing glucose and then transferred into SD media containing 2% raffinose; this was followed by induction using 2% galactose. The cell was supplemented with 2% glucose to shut up kinase expression for the time-lapse study. Tpk2 kinase aggregates were checked from cells transformed with pADH1-GFP-TPK2 in YCplac33. The OD600 of 3–4 was collected for each sample. For temperature-sensitive mutants, yeast cells were grown at 30°C and then shifted to 37°C for 2 h before viewing the fluorescence in live cells; whereas for inhibitor treatment, erg6Δ cells were treated with myricetin for 2 h. Cells were collected by centrifugation, washed twice with 1× phosphate-buffered saline (PBS), and resuspended in fresh SC (synthetic complete) medium. The suspension (2.5 μl) was dropped onto a 76 × 26-mm microscopy slide coated with poly-l-lysine, covered with a clean coverslip, air dried, and subjected to immediate viewing.
For FM4-64 staining, cells were pelleted down from an OD600 of 2.5, spun at 2500 rpm for 2 min, washed twice with chilled 1× PBS, and then resuspended in YPD medium. FM4-64 (Invitrogen, Carlsbad, CA) at a final concentration of 1 μg/ml was added to the cells to stain the cell vacuole. Cells were labeled for 20 min at 30°C under shaking conditions. Ste11ΔNK444R-GFP was observed using the GFP filter, and the FM4-64–stained cell membrane was viewed using the TRIC/CCT filter. The nucleus was stained with Hoechst 33242 dye (2 μg/ml) at 30°C for 15 min (Invitrogen) and viewed under a 4′,6-diamidino-2-phenylindole (DAPI) filter.
General fluorescence microscopy was performed using an Olympus BX61 microscope with a 100× oil-immersion objective (UPlan FLN/1.40 NA oil). The imaging was done with an Olympus DL604 Low-light Imaging EMCCD Camera. The colocalization studies were done using a Nikon Inverted Research Microscope ECLIPSE TI-U (60×/1.40 NA oil-immersion objective) with a digital 1.5× zoom. The images were collected with Nikon DS-Ri1 mounted on a Ti-U fluorescence microscope. The excitation filters used were DAPI (405 nm), GFP (488 nm), RFP (555 nm), and bright field as a differential interference contrast image. All images were deconvolved using standard softWRx deconvolution algorithms (enhanced ratio, high-to-medium noise filtering). Representative cells are shown in all figures, and each experiment was performed independently three times. We performed several control experiments with only the GFP,RFP constructs alone (unpublished data). In these experiments, we did not observe coalescence of GFP,RFP into fluorescent foci or association with PQC components. In addition, GFP,RFP did not colocalize with foci-forming proteins such as Hsp104 and Hsp42.
For live-cell imaging, cells were immobilized using concanavalin A (0.1 mg/ml stock solution made with sterile RO [reverse osmosis] water), spotted onto a clean glass slide (wiped with 80% ethanol), and dried in a 30°C incubator overnight. The slide was rinsed with enough RO water to submerge the slide. Yeast cell suspension (around 30 μl) was spotted on the same area and left on the slide for 10 min (inside incubator). After 10 min, excess cells were rinsed off by gently submerging the slide in RO water in a beaker of appropriate size. This step was repeated several times to prevent cells floating between the slide and the coverslip. Excess water was dried with a Kimwipe, the coverslip was placed on the area of interest, and the slide was subjected to immediate viewing. Images were obtained by confocal microscopy using a Nikon Eclipse TiE confocal microscope with a 100×/NA 1.4 oil-immersion objective). The imaging was done with A1R+ scanner head mounted on Ti-E inverted microscope: blue channel (DAPI) with a 405-nm laser and green channel (GFP) with 488-nm laser. The slide was subsequently placed into a custom-made metal holder that was connected to a Peltier element, which allowed accurate temperature control. The entire setup was controlled by NIS Elements AR software. ImageJ (National Institutes of Health [NIH], Bethesda, MD) was used for image processing, analysis, and assembly.
Kinase refolding assay
Wild-type strains expressing GAL-GST-Tpk2 were grown overnight at 30°C in synthetic dropout medium to log phase (0.4 OD). The culture was then incubated at 42°C for 2 h to denature the kinase and was then shifted to 30 and 37°C for 3 h for the refolding reaction. Cells were harvested, and extracts were prepared in IPP150 buffer as described earlier (see Tpk2 kinase assay). The amount of protein was quantified by Bradford reagent. Cell extracts (0.3 mg) were diluted to 0.5 ml with GST binding buffer as mentioned previously (see Tpk2 kinase assay) and incubated with 50 μl of 50% (vol/vol) glutathione resin (Qiagen) for 2 h at 4°C. The resin was washed three times and divided into three aliquots. One aliquot was used for the kinase assay, and the others were used for Western blot analysis with anti-GST antibody and antiubiquitin antibody. Kinase assays for Tpk2 were performed by using a Pep-Tag nonradioactive protein kinase assay kit as described by the manufacturer (Promega). The bands in the Western blots were quantified by using ImageJ software (NIH).
Supplementary Material
Acknowledgments
We thank Avrom J. Caplan, Jeff Brodsky, Kevin Morano, Elizabeth Craig, Daniel Kaganovich, and Axel Mogk for strains and plasmids. Thanks to Srimonti Sarkar for the generous gift of FM4-64 dye; Anirban Siddhanta, Rajarshi Chakrabarti, and Prabal Chakraborty for their assistance in microscopic analysis; and Maria Theodoraki and Avrom Caplan for reading the manuscript. This work was supported by CSIR (Council of Scientific and Industrial Research) 38(1277)/11/EMR-II (AKM) and the Bose Institute intramural fund.
Abbreviations used:
- AZC
l-azetidine 2-carboxylic acid
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- GA
geldanamycin
- GPD
glyceraldehyde 3-phosphate dehydrogenase
- Hsp
heat shock protein
- IPOD
insoluble protein deposit
- JUNQ
juxtanuclear compartment
- NBD
nucleotide-binding domain
- NEF
nucleotide exchange factor
- PBS
phosphate-buffered saline
- PES
2-phenylethynesulfonamide
- PMSF
phenylmethylsulfonyl fluoride
- SBD
substrate-binding domain
- TEF
translation elongation factor
- ts
temperature-sensitive
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1262) on March 4, 2015.
REFERENCES
- Arndt V, Daniel C, Nastainczyk W, Alberti S, Hohfeld J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol Biol Cell. 2005;16:5891–5900. doi: 10.1091/mbc.E05-07-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach TM, Takagi H. Properties, metabolisms, and applications of (L)-proline analogues. Appl Microbiol Biotechnol. 2013;97:6623–6634. doi: 10.1007/s00253-013-5022-7. [DOI] [PubMed] [Google Scholar]
- Basha E, Friedrich KL, Vierling E. The N-terminal arm of small heat shock proteins is important for both chaperone activity and substrate specificity. J Biol Chem. 2006;281:39943–39952. doi: 10.1074/jbc.M607677200. [DOI] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazeck J, Garg R, Reed B, Alper HS. Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol Bioeng. 2012;109:2884–2895. doi: 10.1002/bit.24552. [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER, Gestwicki JE. Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chem Biol. 2011;18:210–221. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintron NS, Toft D. Defining the requirements for Hsp40 and Hsp70 in the Hsp90 chaperone pathway. J Biol Chem. 2006;281:26235–26244. doi: 10.1074/jbc.M605417200. [DOI] [PubMed] [Google Scholar]
- Cross FR. “Marker swap” plasmids: convenient tools for budding yeast molecular genetics. Yeast. 1997;13:647–653. doi: 10.1002/(SICI)1097-0061(19970615)13:7<647::AID-YEA115>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Cushman-Nick M, Bonini NM, Shorter J. Hsp104 suppresses polyglutamine-induced degeneration post onset in a Drosophila MJD/SCA3 model. PLoS Genet. 2013;9:e1003781. doi: 10.1371/journal.pgen.1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr DM. Cooperation of the molecular chaperone Ydj1 with specific Hsp70 homologs to suppress protein aggregation. FEBS Lett. 1995;359:129–132. doi: 10.1016/0014-5793(95)00024-4. [DOI] [PubMed] [Google Scholar]
- Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA. 2008;105:7206–7211. doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele F, Wolf DH. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582:4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Escusa-Toret S, Vonk WI, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol. 2013;15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts SJ, Karnitz LM, Toft DO. Functioning of the Hsp90 machine in chaperoning checkpoint kinase I (Chk1) and the progesterone receptor (PR) Cell Stress Chaperones. 2007;12:353–363. doi: 10.1379/CSC-299.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flom GA, Langner E, Johnson JL. Identification of an Hsp90 mutation that selectively disrupts cAMP/PKA signaling in Saccharomyces cerevisiae. Curr Genet. 2012;58:149–163. doi: 10.1007/s00294-012-0373-7. [DOI] [PubMed] [Google Scholar]
- Flom GA, Lemieszek M, Fortunato EA, Johnson JL. Farnesylation of Ydj1 is required for in vivo interaction with Hsp90 client proteins. Mol Biol Cell. 2008;19:5249–5258. doi: 10.1091/mbc.E08-04-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevaux P, Georgopoulos C, Kelley WL. The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol Microbiol. 2007;66:840–857. doi: 10.1111/j.1365-2958.2007.05961.x. [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Goloubinoff P, De Los Rios P. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends Biochem Sci. 2007;32:372–380. doi: 10.1016/j.tibs.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Gowda NK, Kandasamy G, Froehlich MS, Dohmen RJ, Andreasson C. Hsp70 nucleotide exchange factor Fes1 is essential for ubiquitin-dependent degradation of misfolded cytosolic proteins. Proc Natl Acad Sci USA. 2013;110:5975–5980. doi: 10.1073/pnas.1216778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MM, Brown AS, Corwin LM, Troxler RF, Franzblau C. Effect of L-azetidine 2-carboxylic acid on growth and proline metabolism in Escherichia coli. Biochim Biophys Acta. 1975;404:180–187. doi: 10.1016/0304-4165(75)90324-4. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J Biol Chem. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liu Y, Chang A. Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J Biol Chem. 2007;282:26140–26149. doi: 10.1074/jbc.M701969200. [DOI] [PubMed] [Google Scholar]
- Han W, Christen P. Mutations in the interdomain linker region of DnaK abolish the chaperone action of the DnaK/DnaJ/GrpE system. FEBS Lett. 2001;497:55–58. doi: 10.1016/s0014-5793(01)02435-8. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2013;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck SA, Singh S, Cyr DM. Cellular responses to misfolded proteins and protein aggregates. Methods Mol Biol. 2012;832:455–461. doi: 10.1007/978-1-61779-474-2_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Nie L, Xu M, Sun XH. Notch-induced E2A degradation requires CHIP and Hsc70 as novel facilitators of ubiquitination. Mol Cell Biol. 2004;24:8951–8962. doi: 10.1128/MCB.24.20.8951-8962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Koren J, O'Leary JC, Jones JR, Abisambra JF, Dickey CA. Hsp70 ATPase modulators as therapeutics for Alzheimer's and other neurodegenerative diseases. Mol Cell Pharmacol. 2010a;2:43–46. [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Miyata Y, Koren J, III, Jones JR, Trotter JH, Chang L, O'Leary J, Morgan D, Lee DC, Shults CL, et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, O'Leary JC, III, Borysov SI, Jones JR, Li Q, Koren J, III, Abisambra JF, Vestal GD, Lawson LY, Johnson AG, et al. Hsc70 rapidly engages tau after microtubule destabilization. J Biol Chem. 2010b;285:16798–16805. doi: 10.1074/jbc.M110.113753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanelakis KC, Shewach DS, Pratt WB. Nucleotide binding states of hsp70 and hsp90 during sequential steps in the process of glucocorticoid receptor.hsp90 heterocomplex assembly. J Biol Chem. 2002;277:33698–33703. doi: 10.1074/jbc.M204164200. [DOI] [PubMed] [Google Scholar]
- Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell. 2012;48:863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- Kriegenburg F, Jakopec V, Poulsen EG, Nielsen SV, Roguev A, Krogan N, Gordon C, Fleig U, Hartmann-Petersen R. A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS Genet. 2014;10:e1004140. doi: 10.1371/journal.pgen.1004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Sherman MY, Goldberg AL. Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4773–4781. doi: 10.1128/mcb.16.9.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Pandey P, Murphy ME, George DL. HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol Cancer Res. 2011;9:936–947. doi: 10.1158/1541-7786.MCR-11-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Mandal AK, Gibney PA, Nillegoda NB, Theodoraki MA, Caplan AJ, Morano KA. Hsp110 chaperones control client fate determination in the hsp70-Hsp90 chaperone system. Mol Biol Cell. 2010;21:1439–1448. doi: 10.1091/mbc.E09-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal AK, Nillegoda NB, Chen JA, Caplan AJ. Ydj1 protects nascent protein kinases from degradation and controls the rate of their maturation. Mol Cell Biol. 2008;28:4434–4444. doi: 10.1128/MCB.00543-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal AK, Theodoraki MA, Nillegoda NB, Caplan AJ. Role of molecular chaperones in biogenesis of the protein kinome. Methods Mol Biol. 2011;787:75–81. doi: 10.1007/978-1-61779-295-3_6. [DOI] [PubMed] [Google Scholar]
- Masison DC, Kirkland PA, Sharma D. Influence of Hsp70s and their regulators on yeast prion propagation. Prion. 2009;3:65–73. doi: 10.4161/pri.3.2.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem. 2013;288:21399–21411. doi: 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39:321–331. doi: 10.1016/j.molcel.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Meimaridou E, Gooljar SB, Chapple JP. From hatching to dispatching: the multiple cellular roles of the Hsp70 molecular chaperone machinery. J Mol Endocrinol. 2009;42:1–9. doi: 10.1677/JME-08-0116. [DOI] [PubMed] [Google Scholar]
- Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J. The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol Cell Biol. 2003;23:3141–3151. doi: 10.1128/MCB.23.9.3141-3151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Michaelis S. Analysis of quality control substrates in distinct cellular compartments reveals a unique role for Rpn4p in tolerating misfolded membrane proteins. Mol Biol Cell. 2009;20:1006–1019. doi: 10.1091/mbc.E08-02-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Theodoraki MA, Mandal AK, Mayo KJ, Ren HY, Sultana R, Wu K, Johnson J, Cyr DM, Caplan AJ. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol Biol Cell. 2010;21:2102–2116. doi: 10.1091/mbc.E10-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Salmonowicz H, Brown R, Turkowska J, Sredniawa W, Pattabiraman S, Amen T, Abraham AC, Eichler N, Lyakhovetsky R, Kaganovich D. Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc Natl Acad Sci USA. 2014;111:8049–8054. doi: 10.1073/pnas.1324035111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Bolender N, Eisele F, Kostova Z, Takeuchi J, Coffino P, Wolf DH. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol Biol Cell. 2007;18:153–165. doi: 10.1091/mbc.E06-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Prasad R, Kawaguchi S, Ng DT. A nucleus-based quality control mechanism for cytosolic proteins. Mol Biol Cell. 2010;21:2117–2127. doi: 10.1091/mbc.E10-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya S, Sharma SK, Goloubinoff P. Molecular chaperones as enzymes that catalytically unfold misfolded polypeptides. FEBS Lett. 2013;587:1981–1987. doi: 10.1016/j.febslet.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Schroder H, Langer T, Hartl FU, Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Wegele H, Buchner J, Morano KA. The yeast Hsp110 Sse1 functionally interacts with the Hsp70 chaperones Ssa and Ssb. J Biol Chem. 2005;280:41262–41269. doi: 10.1074/jbc.M503614200. [DOI] [PubMed] [Google Scholar]
- Sharma D, Masison DC. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept Lett. 2009;16:571–581. doi: 10.2174/092986609788490230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell. 2013;24:2076–2087. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A, Ravid T. Chaperoning proteins for destruction: diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome. Biomolecules. 2014;4:704–724. doi: 10.3390/biom4030704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht S, Miller SB, Mogk A, Bukau B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol. 2011;195:617–629. doi: 10.1083/jcb.201106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M, Kaganovich D. Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep. 2012;2:738–747. doi: 10.1016/j.celrep.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9:1937–1954. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- Summers DW, Wolfe KJ, Ren HY, Cyr DM. The Type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PLoS One. 2013;8:e52099. doi: 10.1371/journal.pone.0052099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoraki MA, Nillegoda NB, Saini J, Caplan AJ. A network of ubiquitin ligases is important for the dynamics of misfolded protein aggregates in yeast. J Biol Chem. 2012;287:23911–23922. doi: 10.1074/jbc.M112.341164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton KA, Verges KJ, Weissman JS. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol Cell. 2008;32:584–591. doi: 10.1016/j.molcel.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 2009;23:451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SJ, Lyakhovetsky R, Werdiger AC, Gitler AD, Soen Y, Kaganovich D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc Natl Acad Sci USA. 2012;109:15811–15816. doi: 10.1073/pnas.1205829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Washburne M, Stone DE, Craig EA. Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2568–2577. doi: 10.1128/mcb.7.7.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol. 2012a;179:152–160. doi: 10.1016/j.jsb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Winkler J, Tyedmers J, Bukau B, Mogk A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol. 2012b;198:387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LQ, Wu S, Buell AK, Cohen SI, Chen LJ, Hu WH, Cusack SA, Itzhaki LS, Zhang H, Knowles TP, et al. Influence of specific HSP70 domains on fibril formation of the yeast prion protein Ure2. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110410. doi: 10.1098/rstb.2011.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang K, Guo Y, Shi Q, Tian C, Chen C, Gao C, Zhang BY, Dong XP. Heat shock protein 70 selectively mediates the degradation of cytosolic PrPs and restores the cytosolic PrP-induced cytotoxicity via a molecular interaction. Virol J. 2012;9:303. doi: 10.1186/1743-422X-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Qian SB. Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol Biol Cell. 2011;22:3277–3288. doi: 10.1091/mbc.E11-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zietkiewicz S, Krzewska J, Liberek K. Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J Biol Chem. 2004;279:44376–44383. doi: 10.1074/jbc.M402405200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.