

Most chemoattractants rely on activation of the heterotrimeric G-protein Gαi to regulate directional cell migration, but few links from Gαi to chemotactic effectors are known. Homer3 is a novel Gαi2-interacting protein that spatially organizes actin assembly to support efficient polarity and motility in neutrophils.
Abstract
Most chemoattractants rely on activation of the heterotrimeric G-protein Gαi to regulate directional cell migration, but few links from Gαi to chemotactic effectors are known. Through affinity chromatography using primary neutrophil lysate, we identify Homer3 as a novel Gαi2-binding protein. RNA interference–mediated knockdown of Homer3 in neutrophil-like HL-60 cells impairs chemotaxis and the establishment of polarity of phosphatidylinositol 3,4,5-triphosphate (PIP3) and the actin cytoskeleton, as well as the persistence of the WAVE2 complex. Most previously characterized proteins that are required for cell polarity are needed for actin assembly or activation of core chemotactic effectors such as the Rac GTPase. In contrast, Homer3-knockdown cells show normal magnitude and kinetics of chemoattractant-induced activation of phosphoinositide 3-kinase and Rac effectors. Chemoattractant-stimulated Homer3-knockdown cells also exhibit a normal initial magnitude of actin polymerization but fail to polarize actin assembly and intracellular PIP3 and are defective in the initiation of cell polarity and motility. Our data suggest that Homer3 acts as a scaffold that spatially organizes actin assembly to support neutrophil polarity and motility downstream of GPCR activation.
INTRODUCTION
Directed cell migration plays a central role in many physiological and pathological processes from development to homing of immune cells such as neutrophils, to cancer metastasis. Most chemoattractant receptors mediate activation of motility effectors through Gαi-family heterotrimeric G-proteins (Neptune and Bourne, 1997; Rickert et al., 2000). Genetic ablation of Gαi2 or pharmacological inhibition of Gαi signaling blocks chemotaxis toward most agonists for neutrophils (Spangrude et al., 1985; Wiege et al., 2012) and other cells (Kumagai et al., 1991).
On binding chemoattractant, G-protein–coupled receptors (GPCRs) trigger GDP to be exchanged for GTP on Gαi2, thereby inducing Gαi to dissociate from Gβγ. Both Gαi2-GTP and Gβγ interact with downstream signaling partners (Oldham and Hamm, 2008). Many potential links from Gβγ to downstream chemotactic effectors are known, including phosphatidylinositol 3-kinase (PI3K)-γ (which stimulates of phosphatidylinositol 3,4,5-triphosphate [PIP3] production; Stephens et al., 2008), P-Rex1 and Dock2 (activators of Rac1 and Rac2 GTPases; Welch et al., 2002; Dong et al., 2005; Kunisaki et al., 2006), and phospholipase β (which hydrolyzes phosphatidylinositol 4,5-bisphosphate into diacylglycerol and inositol trisphosphate; Tang et al., 2011).
Chemoattractant stimulation generates several intracellular signaling asymmetries that organize filamentous actin (F-actin) at the leading edge and actomyosin at the trailing edge (Wang, 2009; Berzat and Hall, 2010). For example, activation of the small GTPase Rac is localized to the leading edge and is necessary and sufficient for actin assembly and migration in neutrophils (Gardiner et al., 2002; Sun et al., 2004; Zhang et al., 2009; Yoo et al., 2010) and other cells (Allen et al., 1998; Chung et al., 2000; Levskaya et al., 2009; Wu et al., 2009). Rac activates the WAVE2 complex, which promotes actin polymerization through the Arp2/3 complex (Weiner et al., 2006; Lebensohn and Kirschner, 2009; Chen et al., 2010; Koronakis et al., 2011). The phospholipid PIP3 also accumulates at the leading edge (Servant et al., 2000) and plays a role in neutrophil migration in vivo (Yoo et al., 2010). How these molecules achieve their polarized distribution during chemotaxis is not known.
Whether Gαi2 has its own suite of distinct chemotactic effectors or whether it is simply a handle to release Gβγ is only beginning to be understood. Recent studies have begun to identify Gαi2-specific effectors in chemotaxis, such as mInsc, which indirectly binds Gαi2-GDP at the leading edge to direct neutrophil migration through the recruitment of polarity effectors (Kamakura et al., 2013). In addition, Dock180, a Rac activator homologous to Dock2, has also been shown to be a potential Gαi2 effector (Li et al., 2013).
Here we identify Homer3 as a novel Gαi2-interacting protein that regulates actin organization in neutrophils. Homer3, a member of the Homer family of scaffold proteins, has been shown to play a role in actin dynamics after stimulation in neurons and T-cells (Ishiguro and Xavier, 2004; Shiraishi-Yamaguchi and Furuichi, 2007; Shiraishi-Yamaguchi et al., 2009), but its role in chemotaxis is unknown. Here we show that Homer3 spatially organizes actin assembly to support efficient polarity and motility in neutrophils.
RESULTS
We previously used affinity chromatography to identify novel Gα effectors in Dictyostelium (Kataria et al., 2013) and sought to use a similar approach to identify Gαi effectors in neutrophils. Glutathione S-transferase (GST)–tagged Gαi2 was purified and incubated with neutrophil lysate harvested from cavitated pig leukocytes. The GST-Gαi2 and associated proteins were then isolated and separated by SDS–PAGE. The protein bands were analyzed by mass spectrometry. A total of four independent pull-down screens (two GST-Gαi2-GDP, one GST-Gαi2-Gpp(NH)p, and one GST-Gαi2-GDP-AlF4) identified several known Gαi-interacting proteins, including RASA3, TNFAIP8, Gβγ, RGS3, and RIC8A (Figure 1A; Neptune and Bourne, 1997; Anger et al., 2007; Nafisi et al., 2008; Laliberté et al., 2010; Kataria et al., 2013). We identified new potential targets as well. From this list of Gαi2-interacting proteins, we were most interested in proteins that have functional domains predicted to regulate the cytoskeleton or for which genetic evidence exists in other systems implicating them in actin assembly or cell motility. From this prioritized list of Gαi2 interactors, we tested each candidate for a role in directed cell migration with a follow-up short hairpin RNA (shRNA)–based chemotaxis screen in neutrophil-like HL-60 cells (Supplemental Figure S1).
FIGURE 1:

Identification of Homer3 as a neutrophil protein that binds Gαi2. (A) Network analysis of the proteins captured by one or more Gαi2 baits after affinity chromatography in pig leukocyte lysates. The baits are shown in gray, and the proteins are shown in blue. For each protein–bait combination, the numbers at the arrows show the counts, and the width of the lines represents the ZP score. (B) Affinity chromatography with GST-Gαi2 or GST alone for neutrophil lysate containing FLAG-Homer3. Eluted GST-tagged bait plus associated proteins and final wash fraction were subjected to SDS–PAGE and analyzed by immunoblot with anti-FLAG antibody. (C) Affinity chromatography–based test for direct interaction between purified, bacterially expressed Homer3 (prey) and GST-Gαi2 or GST alone (baits). Homer3 directly binds to both GST-Gαi2-GDP and GST-Gαi2-Gpp(NH)p with similar affinity (n = 5; not significantly different). “Beads” refer to baits without Homer3 (prey). Samples were analyzed with SDS–PAGE and stained with CBB. Arrows indicate GST-Gαi2 (66 kDa), Homer3 (47 kDa), and GST (26 kDa).
Homer3, a novel Gαi interactor, was identified in both the Gαi2 interaction screen and the follow-up genetic screen. Homer3 is part of a family of scaffolds that binds a variety of proteins relevant to chemotaxis signaling, including actin and Rac1 (Shiraishi et al., 1999; Shiraishi-Yamaguchi and Furuichi, 2007; Shiraishi-Yamaguchi et al., 2009). Homer proteins have been primarily studied in neurons, where they localize to the synapse and participate in calcium signaling, axon guidance, and dendritic spine morphology (Foa et al., 2001; Sala et al., 2001; Fagni et al., 2002; Hwang et al., 2003; Moutin et al., 2012). In our affinity chromatography experiments, Homer3 bound to GDP-loaded GST-Gαi2 and GST-Gαi2 loaded with GTP analogues GDP-AlF4 and Gpp(NH)p with relatively similar peptide counts (Supplemental Figure S1 and Supplemental Tables S1–S4). The interaction between Homer3 and Gαi2 was confirmed via GST pull-down assays in neutrophil lysate (Figure 1B). The binding between Gαi2 and Homer3 is direct, as shown with GST pull-down assays using proteins purified from bacteria, and independent of whether Gαi2 is loaded with GDP or Gpp(NH)p (Figure 1C).
Since we were most interested in novel Gαi2 interactors relevant to chemotactic signaling, we assayed whether Homer3 was necessary for neutrophil chemotaxis. Using the neutrophil-like differentiated HL-60 cell line, we knocked down the expression of Homer3 with lentiviral shRNAs (Figure 2A and Supplemental Figure S2; Hauert et al., 2002). Differentiated HL-60s infected with nonsense (control) or Homer3 shRNA were assessed with a Transwell chemotaxis assay in which cells migrate through a microporous filter toward a source of chemoattractant. When presented with a gradient of the Gαi-coupled GPCR ligand formyl-methionyl-leucyl-phenylalanine (fMLP), Homer3-knockdown cells exhibited a sixfold decrease in migration (Figure 2B). The chemotaxis defect was observed in two independent lines, each expressing a different shRNA against Homer3. The magnitude of the defect scaled with the degree of Homer3 knockdown as measured by real-time PCR (Figure 2A). We chose the line with the higher knockdown efficiency (shRNA 1) for all of our subsequent experiments.
FIGURE 2:
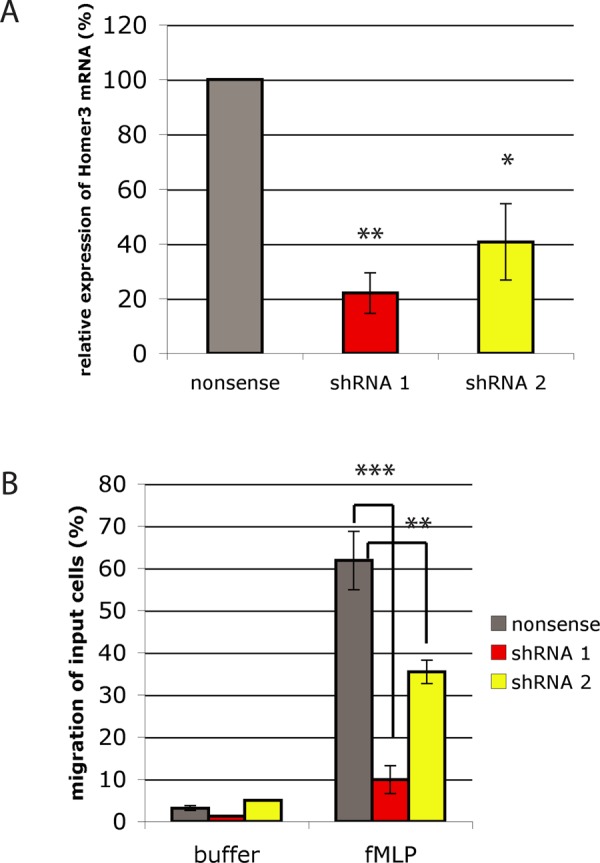
Homer3 knockdown impairs HL-60 chemotaxis. (A) RNA was isolated from control cells (nonsense shRNA) and HL-60 cell lines expressing one of two different Homer3 shRNAs (shRNA 1 and shRNA 2). Relative expression of Homer3 was quantified by quantitative real-time PCR using GAPDH as a reference gene. Results represent the mean with SD of three replicates. (B) Migration of control or Homer3-knockdown differentiated HL-60 cells in response to 10 nM fMLP measured via Transwell chemotaxis chambers after 2 h. Results are a representative example of five independent experiments. *p < 0.05, **p < 0.005, ***p < 0.0005 by unpaired t test.
Although Transwell assays can uncover a defect in chemotaxis, this device does not allow for direct visualization of cells during their migration. This makes it difficult to determine whether the chemotaxis defect represents an impairment in speed, directionality, or persistence. To address this question, we used time-lapse microscopy to visualize Homer3-knockdown cells during random cell migration after stimulation with uniform chemoattractant. We used a “chimney assay” (Malawista and de Boisfleury Chevance, 1997) in which cells are resuspended into a small volume of liquid sandwiched between two coverslips. In this context, migration is not dependent on cellular adhesion, enabling us to screen for cells whose lack of movement is not a consequence of a failure to adhere to the substrate. A substantial fraction of the Homer3-knockdown cells fail to move in this context (Figure 3A and Supplemental Movies S1–S3). These nonmotile cells either extended short protrusions that were quickly retracted or completely failed to protrude.
FIGURE 3:

Homer3 knockdown impairs the initiation of HL-60 migration. (A) Percentage of nonmotile cells in time-lapse migration assays in uniform 10 nM fMLP, expressed as mean with SE. Results are from three independent experiments with two replicates each. ***p < 0.0005 by unpaired t test. Corresponds to Supplemental Movies S1 and S2. Representative cell tracks of nonsense and Homer3-knockdown cells. Corresponds to Supplemental Movie S3. (B) Length of pauses in migration tracks, as defined in Materials and Methods. **p < 0.005 by Mann–Whitney test. Corresponds to Supplemental Movie S4. (C) Speed of control (nonsense shRNA) and motile Homer3-knockdown cells was assayed via time-lapse microscopy. Dot plot shows the overall population distribution; box and whiskers plots show quartiles. (D) Persistence index, defined as (final distance from start)/(total distanced traveled).
Homer3-knockdown cells not only exhibited a significant increase in the proportion of nonmotile cells, but they also exhibited subtle defects in the motile population of cells. The Homer3-knockdown cells showed a significant increase in the length of pauses between migratory events (Figure 3B and Supplemental Movie S4), consistent with a general defect in initiation of migration. However, Homer3-knockdown cells have a normal overall persistence and speed of cell movement (Figure 3, C and D). Therefore Homer3 appears to play a prominent role in initiation of migration but does not seem to affect the maintenance of migration.
Does the motility defect for Homer3-knockdown cells represent a general lack of activation of heterotrimeric G-protein effectors, as observed for the Ric8 protein in Dictyostelium (Kataria et al., 2013)? To investigate whether there is a general defect in signaling, we assayed calcium release from Homer3-knockdown cells after stimulation. Calcium release was assessed by loading cells with the cell-permeable calcium indicator dye fluo-4 AM and measuring fluorescence intensity in individual cells before and after stimulation with fMLP (Dandekar et al., 2013). A similar proportion of cells responded in both the control (407 of 451 cells, 90%) and Homer3-knockdown cells (465 of 546 cells, 85%; Supplemental Figure S3A). Since undifferentiated HL-60s do not express the fMLP receptor, this suggests that Homer3 knockdown does not block cell differentiation. To confirm cell differentiation in Homer3-knockdown cells, we used a phycoerythrin (PE)-conjugated antibody for Cd11b, a receptor expressed exclusively on differentiated cells, and measured fluorescence via fluorescence-activated cell sorting (FACS). Nonsense shRNA cells are 97 ± 2% differentiated after dimethyl sulfoxide (DMSO) addition, and Homer3 shRNA cells are 92 ± 5% differentiated (average ± SD of four measurements; Supplemental Figure S3B). Homer3 knockdown did not affect cell growth or viability through 10 consecutive cell passages (Supplemental Figure S3, C and D). Thus Homer3 knockdown does not prevent cell signaling in response to chemoattractant, nor does it affect differentiation or viability.
To determine whether other GPCR effectors are activated normally in Homer3-knockdown cells, we next assayed the downstream heterotrimeric G-protein effectors Rac and PI3K, both of which contribute to regulation of neutrophil chemotaxis (Sun et al., 2004; Yoo et al., 2010). We used phosphorylation of p21-activated kinase (Pak) as a downstream readout for Rac activation (Knaus et al., 1995; Weiss-Haljiti et al., 2004). We used phosphorylation of Akt to read out signaling through the PI3K cascade (Burgering and Coffer, 1995; Franke et al., 1995; Stokoe et al., 1997). Homer3 cells have normal stimulation kinetics and general magnitude of Akt and Pak phosphorylation (Figure 4). This is in contrast to the Transwell migration assay, in which the majority of Homer3-knockdown cells failed to migrate (Figure 2B).
FIGURE 4:

Homer3 depletion does not affect levels of Pak phosphorylation (a readout of Rac activation) or Akt phosphorylation (a readout of PIP3 generation). (A, C) Time course of Akt (A) or Pak (C) phosphorylation, measured by Western blot, after uniform 10 nM fMLP stimulation. Total Akt (A) or total Pak (C) was used as loading control. (B, D) Quantification of phosphorylated Akt (B) or Pak (D) for three independent runs of the experiment shown in A and C , respectively. Phosphorylated Akt (B) or Pak (D) was normalized to total Akt (B) or Pak (D).
Because Homer proteins have been associated with regulation of the actin cytoskeleton via their N-terminal enabled/vasodilator-stimulated phosphoprotein homology 1 (EVH1)–like domain (Ishiguro and Xavier, 2004; Shiraishi-Yamaguchi et al., 2009), we investigated whether Homer3 plays a similar role in neutrophils. Using purified proteins in an actin cosedimentation assay, we showed that Homer3's N-terminus is necessary and sufficient to bind to actin filaments (Figure 5A). This supports a role for Homer3 in regulating actin in cells, which we explored by analyzing how Homer3 knockdown affects actin polymerization. Exposure to fMLP induces transient global F-actin accumulation, which peaks around 1 min and later organizes into a polarized pseudopod at later time points. In control cells, 80% of the stimulated cells have an organized F-actin pseudopod at 7 min, compared with 50% of Homer3 cells (Figure 5, B and C). In contrast to the significant polarity defect, Homer3-knockdown cells show normal kinetics and overall magnitude of actin assembly (Figure 5D). This suggests that although Homer3- knockdown cells retain their ability to polymerize actin in response to stimulus, they lack the ability to organize the actin into a polarized distribution.
FIGURE 5:

Homer3 is an actin-binding protein necessary for persistent actin and PIP3 polarization. (A) A cosedimentation assay reveals that the N-terminal portion of Homer3 is necessary and sufficient to directly bind F-actin. Purified, bacterially expressed GST–N-terminal (N-term) and GST–C-terminal (C-term) fragments of Homer3 were incubated with (+) and without (–) F-actin and then centrifuged. Equal amounts of the supernatant (S) and pellet (P) fractions were separated by SDS–PAGE and stained with CBB. Arrows indicate C-term (53 kDa), actin (42 kDa), and N-term (40 kDa). Schematic of Homer3 and the truncated proteins. (B–D) Differentiated control (nonsense shRNA) or Homer3- knockdown HL-60 cells were stimulated in suspension with 10 nM fMLP. At the time points indicated, cells were fixed and stained with rhodamine-phalloidin to visualize F-actin. (B) Representative epifluorescence images before stimulation (0 min), at peak response (1 min), and after polarization (7 min). (C) Quantification of proportion of polarized cells at the 7-min time point for control (n = 577) and Homer3-knockdown (n = 754) cells. Results are the mean and SE of three independent experiments. Asterisk represents p < 0.05 by unpaired t test. (D) Average fluorescence intensity of the whole-cell population, as quantified by FACS, was measured and normalized to the unstimulated control population to correct for FACS and staining variation between experiments. Results are the mean and SE of three independent experiments. (E) Polarization of actin nucleation was assessed by TIRF imaging of a fluorescent component of the WAVE complex (Hem1-YFP) for cells exposed to uniform 100 nM fMLP in a squeeze chamber. Images are representative of at least 10 cells. Arrowheads indicate regions of increased Hem1 intensity. Corresponds to Supplemental Movies S4 and S5. (F) Persistence of Hem1-YFP polarization was quantified as described in Materials and Methods for nonsense (n = 5) and Homer3-knockdown (n = 6) cells. *p < 0.05 by unpaired t test. (G) Line-scan analysis of the ratio images shown in Supplemental Figure S4. Differentiated HL-60 cells (nonsense shRNA or Homer3 shRNA) expressing PH-Akt-Citrine and labeled with CellMask Orange were stimulated with fMLP released from a micropipette. The normalized ratio values between PH-Akt-Citrine and CellMask Orange within the cell periphery were calculated, with each cell normalized such that the average of all ratios along each cell edge is 1. The cell edge of each cell was divided into 10 angular sectors, and values were averaged within each angular sector. Internalized CellMask Orange vesicles were excluded from analysis. Error bars are SE (n = 5 for each line). The difference between nonsense and Homer3 knockdown (motile) is not significant by F test, whereas the difference between nonsense and Homer3 knockdown (nonmotile) is significant (p < 0.005) by F test. (H) Cytoplasmic depletion of PH-Akt-Citrine after addition of 100 nM fMLP. Normalized average fluorescence in a cytoplasmic region was measured at the time points indicated (100% is the maximum depletion in the control line). Error bars are SE (n = 10 for each line). No significant differences by t test.
To follow the spatial and temporal dynamics of actin nucleation in live cells, we visualized Hem1–yellow fluorescent protein (YFP), an immune cell–specific subunit of the WAVE2 complex (Hromas et al., 1991; Weiner et al., 2006). Hem1-YFP shows strong leading-edge localization in neutrophils and serves as a dynamic marker of actin polymerization in these cells (Weiner et al., 2006, 2007). Hem1-YFP persistently localized to the leading edge of control and motile Homer3-knockdown cells (Figure 5, E and F, and Supplemental Movie S5). In nonmotile Homer3-knockdown cells, Hem1-YFP exhibited transient flashes that wandered throughout the cell, indicating a requirement for Homer3 to sustain persistent polarized actin assembly (Figure 5, E and F, and Supplemental Movie S6).
Similarly, Homer3-knockdown cells failed to generate a polarized PIP3 response to a chemoattractant gradient set up by a micropipette (Figure 5G). We measured the membrane localization of PH-Akt-Citrine, which binds PIP3, in response to a micropipette containing chemoattractant. Whereas control cells displayed an asymmetric accumulation of PH-Akt-Citrine toward chemoattractant, immotile Homer3 knockdown cells showed no preferential accumulation. As a control, we measured PH-Akt-Citrine depletion from the cytoplasm and recruitment to the membrane in response to uniform chemoattractant. The initial response in control and Homer3-knockdown cells was the same (Figure 5, G and H), consistent with our previous phospho-Akt assays (Figure 4). In immotile Homer3-knockdown cells, PH-Akt-Citrine returns to the cytoplasm more quickly after stimulation. Homer3 knockdown may affect the persistence of PIP3 on the membrane, in addition to PIP3 polarization. This suggests that Homer3 knockdown interferes with the spatial organization of key regulators of actin organization. This result is consistent with the actin assembly polarization defect with Homer3 knockdown (Figure 5, E and F) and suggests that Homer3 functions with Gαi2 to spatially regulate the downstream cytoskeletal polarization necessary for efficient neutrophil chemotaxis.
DISCUSSION
Most chemoattractants act through Gαi heterotrimeric proteins to mediate directional movement, but only a few chemotaxis-relevant effectors of Gαi are known. Recent work from other groups has identified two such effectors, mInsc and the Dock180/Elmo1 complex. mInsc indirectly binds Gαi2-GDP via LGN/AGS3 and helps maintain directionality in neutrophils. The Dock180/Elmo1 complex, a Rac GEF, associates with Gαi2 upon stimulation of breast cancer cells (Kamakura et al., 2013; Li et al., 2013) and may organize Rac activity downstream of GPCR activation. Our work adds to this suite of effectors with the identification of Homer3 as a Gαi2-binding protein that is essential for efficient cell polarity and motility in neutrophils. Homer3 does not act upstream of Gαi2, since the magnitude of Gαi effectors (calcium influx, Rac activity, PIP3 production, bulk actin assembly) are unaffected after Homer3 knockdown. This distinguishes Homer3 from Gαi2-interacting proteins that control chemotaxis by regulating the magnitude of Gαi activity, such as the Gα GEF Ric8A (Kataria et al., 2013).
Our work shows that Homer3 is necessary for the organization of polarity but does not significantly affect overall magnitude of chemotactic effector stimulation. Importantly, the effect of Homer3 knockdown is different from that of most chemotaxis mutants, the bulk of which affect the magnitude of activation of core chemotactic effectors (Weiner et al., 2006). Deletion of components at all levels of the cascade—Gαi2, Gβγ, Rac, and WAVE—affects both the magnitude and spatial localization of signals such as Rac activation and actin polymerization (Neptune and Bourne, 1997; Glogauer et al., 2003; Weiner et al., 2006; Wiege et al., 2012; Yan et al., 2012). Deletion of downstream effectors of Gβγ, such as Rac GEFs P-Rex1 and Dock2, also significantly inhibit the magnitude of Rac activation and cell migration (Kunisaki et al., 2006; Lawson et al., 2011).
The shRNA phenotypes suggest a role for Homer3 in organizing polarized, persistent actin assembly. A large fraction of Homer3-knockdown cells form either transient protrusions or no protrusions in uniform chemoattractant. In addition, the motile Homer3-knockdown cells pause longer, supporting a role for initiation of protrusions. This behavior is in contrast to that in wild-type cells, which form persistent pseudopodia in uniform chemoattractant. Because chemoattractant-stimulated neutrophils can polarize in suspension but Homer3-knockdown cells exhibit a significant defect in this context, this defect is not due to misregulation of substrate adhesion. If the actin nucleation machinery does not localize in one spot long enough to organize a pseudopod, this could explain the transient or absent protrusions in Homer3-knockdown cells.
How might Homer3 organize cell polarity? Consistent with previous studies (Shiraishi et al., 1999), we show that Homer3 directly binds F-actin. Furthermore, previous studies showed that Homer3 binds directly to active Rac1-GTP and not inactive Rac1-GDP (Shiraishi et al., 1999). We also find that Rac2 interacts with Gαi2 in our pull-down screen, possibly through association with Homer3. Thus it is possible that Homer3 could integrate signals from actin, Rac-GTP, and Gαi2 to enable neutrophils to organize a persistent leading edge.
We find that Homer3 binds Gαi2-GTP and Gαi2-GDP with similar affinity. However, this in vitro association is likely to be dependent on chemoattractant-induced dissociation of Gαi and Gβγ in vivo. Gβγ sequesters nonsignaling Gαi2-GDP within cells, interrupting the interaction of Gαi2-GDP with binding partners. Of note, mInsc, another recently discovered Gαi2 effector, does not show a preference for Gαi2-GTP (Kamakura et al., 2013) but still plays a role in organizing cell polarity and colocalizes with free Gαi2 at the leading edge. However, although both mInsc and Homer3 interact with Gαi2 and are required for efficient chemotaxis, our biochemical and genetic data suggest that they act in different pathways. We observe a very different migration phenotype with Homer3 knockdown than what was observed with mInsc knockout. Whereas Homer3 knockdown produces rounded, immotile cells, mInsc knockout produces multiple pseudopodia and lack of persistence (Kamakura et al., 2013). Moreover, we do not find mInsc in our mass spectrometry data. Taken together, these data suggest that Homer3 and mInsc represent separate pathways from Gαi2 to the cell migration machinery.
In summary, we found that Homer3 associates with Gαi2 and is necessary for the initiation of cell migration. Whereas nonmotile Homer3-knockdown cells can form transient protrusions, they cannot sustain a leading edge and are unable to generate morphological polarity or polarized F-actin accumulation. Similarly, Gαi2-knockout macrophages fail to polarize in chemoattractant (Wiege et al., 2012). Homer3 may enhance the initiation of migration by scaffolding signaling proteins such as active Rac and actin that regulate early steps in leading-edge organization. From our study, we find that the overall magnitude of actin accumulation and its upstream regulators (Rac, PI3K) is not dependent on Homer3. However, persistent and polarized PIP3 accumulation and actin assembly depend on Homer3. Our previous work (Ku et al., 2012) demonstrated that there may be distinct control of the intensity versus polarity of the actin cytoskeleton, and our present work on Homer3 provides the first insight into the pathways that specifically control actin polarity. By characterizing Homer3's role in chemotaxis and linking it to Gαi2 and actin polymer, this study enhances our understanding of how Gαi2 contributes to cell migration and actin organization.
MATERIALS AND METHODS
Cell lines and culture
HL-60 cells were cultured as described previously (Dandekar et al., 2013). Briefly, cells were grown at 37°C/5% CO2, in RPMI 1640 medium containing l-glutamine, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES;10-041-CM; Mediatech, Manassas, VA), and 10% heat-inactivated fetal bovine serum (FBS). Cell differentiation was initiated by adding 1.5% DMSO (endotoxin-free, hybridoma-tested; D2650; Sigma-Aldrich, St. Louis, MO) to cells in growth media. Cells were used at 2–4 d after differentiation. Differentiation was confirmed with PE-conjugated anti-CDllb antibody (BD Biosciences, Franklin Lakes, NJ).
Cell differentiation
Two hundred thousand cells were pelleted and incubated in 7 μl of CD11b/MAC-I R-PE–conjugated fluorescence antibody (BD PharMingen, San Diego, CA) on ice for 30 min, washed with ice-cold modified Hank's buffered saline solution (mHBSS) with 2% BSA, and resuspended in the same buffer at 106 cells/ml for analysis. mHBSS with 2% BSA was used to establish background signal with unstained cells and undifferentiated cells. Cells with fluorescent signal above background were considered differentiated.
Imaging and analysis
Total internal reflection fluorescence (TIRF) images were acquired on a Nikon Ti Eclipse inverted microscope with a 60× Apo TIRF 1.49 numerical aperture (NA) objective and an electron-multiplying charge-coupled device (EM-CCD) camera (Evolve; Photometrics, Tucson, AZ) controlled by NIS-Elements (Nikon, Melville, NY). Sample drift was minimized using an autofocus system (Perfect Focus; Nikon). Laser lines (514, 561 nm; all 200 mW) were supplied from a Spectral Applied Research LMM5 Laser Merge Module (Richmond Hill, Canada). This laser launch uses acousto-optic tunable filters (AOTFs) to control laser output to a single-mode TIRF fiber for imaging. TIRF imaging was performed with ≤50 mW laser power, achieved through AOTF and neutral density–based laser attenuation.
Confocal images were acquired in a custom-built environmental chamber with temperature and CO2 control (In Vivo Scientific, St. Louis, MO) on a Nikon Ti Eclipse inverted microscope equipped with a Yokogawa CSU-X1 spinning disk confocal, a 60× Apo TIRF 1.49 NA objective, and a Clara interline CCD (Andor, Belfast, Ireland). The 405-, 488-nm, and 561-nm laser wavelengths (MLC400B; Agilent, Santa Clara, CA) were used for excitation.
Calcium assays and time-lapse migration assays were imaged using a CCD camera (Cool Snap HQ; Photometrics) and a Nikon TE-2000 inverted microscope with a 20× PlanFluor 0.5 NA objective in an In Vivo Scientific microscope incubator to create a 37°C climate.
NIS-Elements was used for image acquisition, and NIS-Elements, ImageJ (National Institutes of Health), and Excel (Microsoft, Redmond, WA) were used for data analysis. Graphing and statistical analyses were performed using Prism 6 (GraphPad, San Diego, CA). All p values were calculated using t test (populations were of equal variance) or, where indicated, F test, paired t test, or Mann–Whitney test.
Micropipette experiments
Glass capillaries were pulled as described (Dandekar et al., 2013). Needles were backfilled with a solution containing 1 μM fMLP (F3506; Sigma-Aldrich) and 100 nM Alexa 430 succinimidyl ester (A-10169; Invitrogen, Carlsbad, CA) and held by a micromanipulator (MM-89; Narishige, East Meadow, NY). Agonist flow rate from the pipette was controlled as described (Dandekar et al., 2013). Cells were labeled with CellMask Orange (Life Technologies, Carlsbad, CA) according to manufacturer's instructions.
Knockdown-line generation
Lentiviral Homer3 and nonsense shRNA control in pLKO.1 were purchased from Sigma-Aldrich. Sequences used in this study were as follows: Homer3 shRNA 1, 5′-CGGCTAAAGAAGATGTTGTCT-3′; Homer3 shRNA 2, 5′-GAACAGCATCTGACACAGTTT-3′; and control shRNA, 5′-GCGCGATAGCGCTAATAATTT-3′. HEK293T cells were grown to 70% confluency in a six-well plate for each lentiviral target and transfected using 0.5 μg of Homer3 or nonsense shRNA, 50 ng of vesicular stomatitis virus-G, and 0.5 μg of cytomegalovirus 8.91 with TransIT-293T (Mirus Bio, Madison, WI) according to the manufacturer's instructions. Medium was changed at 18 h after transfection, and viral supernatant was collected at 42 and 66 h posttransfection. A 4-ml amount of combined viral supernatant was used to infect 106 undifferentiated HL-60 cells by spinfection in the presence of 8 μg/ml polybrene. Stable cell lines were generated with 1 μg/ml puromycin selection for 2 wk.
Quantitative real-time PCR
Total RNA was extracted from differentiated HL-60 cells using RNEasy (Qiagen, Mississauga, Canada) according to the manufacturer's instructions. Next 1 μg of total RNA was reverse transcribed with the QuantiTect reverse transcription kit (Qiagen). An equal amount of cDNA from each cell line was amplified by real-time PCR (RT-PCR) using SYBR Green QPCR Master Mix (Applied Biosystems, Foster City, CA). Homer3 RT-PCR primers were 5′-CAGGGAGCAGCCAATCTTCA-3′ (forward) and 5′-GGGAGTGACAGTGCTGTTGA-3′ (reverse). Expression levels were normalized to a housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]), and the relative expression value between the samples was calculated based on the threshold cycle (CT) value using the standard curve method.
Transwell chemotaxis assay
Transwell chemotaxis assays were performed using 24-well Fluoroblok Transwell chambers (pore size, 3.0 μm; Corning, Tewksbury, MA) as previously described (Park et al., 2014). Briefly, cells were stained with the membrane dye DiD (V-22887; Life Technologies), and 300,000 cells in mHBSS with 0.2% BSA were loaded to each top well. Cells were allowed to migrate toward the bottom well containing 10 nM fMLP for 2 h at 37°C. The migrated cells were measured by fluorescence from the bottom of the insert, and the opaque filter prevented excitation of cells on top of the filter. Analysis was performed with a FlexStation 3 Microplate Reader (Molecular Devices, Sunnyvale, CA). The percentage of migrating neutrophils was calculated by dividing the fluorescence reading from each well by the fluorescence reading of the total input cells.
Preparation of high-speed cytosol from pig leukocytes
Pig leukocyte cytosol was prepared essentially as described previously (Weiner et al., 2006). Pig blood was obtained from Rancho Veal (Petaluma, CA). A 40-l amount of blood was collected into five polypropylene jugs containing a total of 9 l of 1× sterile acid–citrate–dextrose anticoagulant (80 mM sodium citrate, 15 mM NaH2PO4, 160 mM glucose, 17 mM citric acid, and 2 mM adenine). Blood was transported to the laboratory at room temperature. At the laboratory, 250 ml of 154 mM NaCl/3% polyvinylpyrrolidone (molecular weight, 360,000) was added per liter of blood plus anticoagulant, mixed thoroughly, poured into 2-l of polypropylene containers, and allowed to settle into two phases for 30-45 min. The upper phase (containing leukocytes and contaminating red blood cells) was decanted and pelleted at 1500 × g for 15 min at room temperature in an IEC swingout bucket rotor. The supernatant was poured off, and the pellets were resuspended in calcium-free mHBSS containing 0.2% BSA. Cells were pelleted at 1500 × g for 15 min. Cells were resuspended in a minimum volume of mHBSS, and then 10× volume of double-distilled H2O was added for 20 s to lyse contaminating red blood cells. Then 1.1× volume of 10× mHBSS was added to regain an isotonic solution. Cells were pelleted, washed, and then resuspended in freshly prepared 3 mM diisopropylfluorophosphate in mHBSS to inactivate serine proteases, then allowed to sit for 20 min on ice. Cells were pelleted and resuspended in cavitation buffer (50 mM NaCl, 50 mM Tris, pH 7.5, at 4°C, 5 mM MgCl2, 5 mM dithiothreitol [DTT], 1× EDTA-free protease inhibitor tablets [Roche, Basel, Switzerland] per 50 ml of solution). Cells were cavitated in a nitrogen Parr bomb (350 psi, 20 min) into a collection vessel containing ethylene glycol tetraacetic acid (EGTA) for a final concentration of 2 mM EGTA. Disrupted cells were spun at 1500 × g for 15 min to remove nuclei and unbroken cells and then 96,000 × g for 60 min to remove membranes. High-speed supernatant was carefully removed without disturbing the pellet.
Affinity-based chromatography
Rat Gαi2, bacterially expressed as a GST-fusion protein, was purified with glutathione-Sepharose FF (GE Healthcare, Little Chalfont, UK) as previously described (Ghosh et al., 2008). GST-Gαi2 was loaded with GDP or the nonhydrolyzable GTP analogue Gpp(NH)p using the alkaline phosphatase protocol (Kataria et al., 2013). Nucleotide loading was assessed with high-performance liquid chromatography (HPLC). For GDP-AlF4 loading, 50 μM AlCl3 and 30 mM NaF were added to GST-Gαi2 and incubated at 30°C for 30 min. For the mass spectrometry screen, 3–5 mg of GST or GST-Gαi2 bound to glutathione-Sepharose FF in pull-down buffer (50 mM NaCl, 50 mM Tris, pH 7.5, at 4°C, 5 mM MgCl2, 5 mM DTT) was incubated with 50 ml of 3 mg/ml leukocyte lysate overnight with recirculation. The column was washed with three column volumes of pull-down buffer, and bait and bound proteins were eluted with 20 mM glutathione. Peak protein fractions were pooled and concentrated before running an SDS–PAGE gel and cutting bands for mass spectrometry.
For preparation of FLAG–Homer3, HEK293T cells were transfected with this expression construct. At 48 h posttransfection, the culture medium was removed, and the cells were washed once with phosphate-buffered saline and lysed with 0.4% NP-40 in pull-down buffer. The 293T lysate was clarified by centrifugation and combined with leukocyte lysate prepared as described. GST-Gαi2 bound to glutathione-Sepharose FF was incubated with FLAG-Homer3 and leukocyte lysate overnight. Proteins were eluted with glutathione and subjected to SDS–PAGE, followed by staining with Coomassie brilliant blue (CBB) or processing for Western blot with 1:1000 anti-FLAG antibody overnight (F1804; Sigma-Aldrich).
For preparation of purified Homer3, bacterially expressed GST-Homer3 was purified and cleaved using thrombin as described previously (Shiraishi-Yamaguchi et al., 2009). One milligram of GST-Gαi2 or GST was bound to glutathione-Sepharose FF beads and then loaded with GDP or Gpp(NH)p using the alkaline phosphatase protocol (Smith and Rittinger, 2002). Each condition was incubated with 500 μg of Homer3 overnight in pull-down buffer (with 0.1 mM of either GDP or Gpp(NH)p). Beads were washed in pull-down buffer and analyzed with SDS–PAGE and CBB staining. A t test was used to compare the Homer3 fraction in GDP and Gpp(NH)p conditions. Molecular weights were calculated by fitting to a standard curve generated using Precision Plus Protein All Blue Standards (161-0373; Bio-Rad, Hercules, CA).
Protein identification by mass spectrometry
Protein samples were concentrated and separated by one-dimensional SDS–PAGE. After Coomassie staining, each lane was cut into 24 slices and subjected to in-gel digestion with 100 ng of trypsin (Trypsin Gold; Promega, Madison, WI) before reduction with 10 mM DTT and alkylation with 55 mM iodoacetamide. Peptide mixtures were trapped on a C18 reversed-phase EASY-Column and separated on a 100-mm C18 reversed-phase column (75 μm × 100 mm, 3-μm particle size; Thermo Scientific, Waltham, MA) using a linear gradient from 0 to 35% (vol/vol) acetonitrile in 0.1% formic acid over 70 min at a constant flow rate of 300 nl/min. Nanoflow liquid chromatography–tandem mass spectrometry (LC-MS/MS) was performed on an EASYII LC system (Thermo Scientific) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific) operating in positive mode. MS scans were acquired in the Orbitrap in the range from 350 to 1800 m/z, with a resolution of 60,000 (full-width at half-maximum). The seven most intense ions per scan were submitted to MS/MS fragmentation (35% Normalized Collision Energy) and detected in the linear ion trap. Peak lists were obtained from raw data files using the Proteome Discoverer, version 1.3, software. Mascot (version 2.1; MatrixScience, Boston, MA) was used for searching against a sequence database obtained by combining the Escherichia coli with the Homo sapiens proteome sequences. The peptide tolerance was set to 40 ppm and the fragment ion tolerance to 2.0 Da, using semitrypsin as protease specificity and allowing for up to two missed cleavages. Oxidation of methionine residues, deamidation of asparagine and glutamine, and carboamidomethylation of cysteines were specified as variable modifications. Peptide and protein identifications were further validated with the program Scaffold (version 3.2; Proteome Software, Portland, OR). Protein identifications based on at least two unique peptides identified by MS/MS, each with a confidence of identification probability >95%, were accepted.
Mass spectrometry data analysis
The spectral counts as obtained from the proteomics analysis were used for further data analysis. For each sample, the counts were standardized by calculating the standardized score (ZP) for each protein–bait combination according to
where XP is the spectral count for protein P, μ is the average count in the sample of the specific bait, and σ is the SD of the counts in the sample. Both the ZP score and the counts were used to estimate the strength of the connection between the protein and the bait. Ranked lists of proteins for a specific bait were constructed by ordering the proteins on the basis of the ratio between the spectral counts in the sample with the bait compared with the control sample. To establish a lower level of the threshold for the counts, the counts of a set of scrambled protein sequences were used. This scrambled set had a median count of 1 and a maximum count of 5. Based on this, all proteins with a count <6 were regarded as false positives and not included in the ranked protein lists. Gene annotations for the Swiss-Prot accession numbers were obtained from the HGNC database (www.genenames.org). Data analysis and graph drawing were done with the statistical package R (www.r-project.org).
Calcium assay
Cells were incubated in medium containing 1 μM fluo-4 AM (F14201; Invitrogen) for 30 min at 37°C. Cells were plated on fibronectin and stimulated by gently adding 100 nM fMLP. Neutral density filters were used to attenuate the intensity of the fluorescence excitation light to prevent spontaneous calcium release. Positive calcium release was scored for cells exhibiting at least a threefold increase in fluorescence intensity.
Phospho-Pak and phospho-Akt assay
Cells were resuspended to a concentration of 2 million/ml in RPMI with 0.2% FBS. We stimulated cells with 10 nM fMLP and quenched the reaction at the indicated time points by adding aliquots of the cell mixture to ice-cold 20% trichloroacetic acid (TCA) containing the phosphatase inhibitors 40 mM NaF and 20 mM β-glycerol phosphate (50020; Fluka, St. Gallen, Switzerland). The samples were spun at 20,000 × g for 15 min to pellet. The sample pellets were washed with 0.5% TCA and resuspended in Laemmli protein sample buffer (161-0737; Bio-Rad) containing 5% β-mercaptoethanol. Protein bands were separated by SDS–PAGE gel electrophoresis, transferred to nitrocellulose, blocked with Odyssey block, and incubated at 4°C overnight with 1:1000 dilutions of anti–phospho-PAK (2605S; Cell Signaling, Danvers, MA) and anti-Pak2 (4825S; Cell Signaling) or anti–phospho-Akt (4060S; Cell Signaling) and anti-Akt (40D4; Cell Signaling). The blot was developed with the fluorescent secondary antibodies, and protein bands were imaged using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Time-lapse migration assays
Three hundred thousand cells were centrifuged at 400 × g for 2 min, resuspended in 12.5 μl of mHBSS containing 2% BSA and 10 nM fMLP, and plated on 5 μg/ml fibronectin-coated coverslips previously blocked with BSA. A coverslip was placed over the cell suspension, and the edges were sealed with a melted mixture of Vaseline, lanolin, and paraffin, forming a squeeze chamber. Starting 10 min after plating, cells were imaged by phase contrast every 10 s for 1 h on a Nikon TE-2000 with a 20×/0.5 numerical aperture objective. The cells were kept at 37°C using a thermostatic chamber.
Cell tracking was performed manually using the MTrackJ plug-in of ImageJ. Cells were scored as “motile” if the maximum displacement from the origin was ≥5 μm (approximately one-half a cell radius) over a given time frame. Error associated with manual rendering of images can result in low speeds in otherwise stationary cells, as well as in fluctuation in instantaneous velocity from any given frame. To determine pauses, a moving average of the instantaneous velocity from the current frame and three previous frames was calculated for each frame of each track. If the moving average fell one SD below the mean velocity, the cell was scored as paused.
Actin cosedimentation
Actin cosedimentation was carried out as described previously (Shiraishi et al., 1999) with slight modifications. For preparation of purified Homer proteins, bacterially expressed GST proteins were purified with 1 ml of GSTrap FF (GE Healthcare) and precleared by centrifugation for 1 h at room temperature (TLA100 rotor, 80,000 rpm). Purified monomeric rabbit skeletal muscle actin was provided by the Mullins lab (University of California, San Francisco, San Francisco, CA). To obtain F-actin, 10 μM of monomeric actin was polymerized for at least 1 h at room temperature in polymerization buffer (10 mM Tris-HCl, pH 7.0, 100 mM KCl, 1 mM MgCl2, and 1 mM ATP). F-actin and 10 μM purified protein were incubated together for 45 min at room temperature before centrifugation for 1 h at room temperature (TLA100 rotor, 80,000 rpm). The pellets were then resuspended to the initial assay volume. Equal amounts of supernatant and resuspended pellet were analyzed by SDS–PAGE and Coomassie brilliant blue stain.
F-actin staining and live-cell imaging
Cells were resuspended in mHBSS with 0.2% BSA. Cells were stimulated with addition of 10 nM fMLP, fixed with 3.7% paraformaldehyde in intracellular buffer (140 mM KCl, 1 mM MgCl2, 2 mM EGTA, 320 mM sucrose, 20 mM HEPES, pH 7.5), and incubated on ice for 20 min. After centrifugation at 400 × g for 2 min, the cell pellet was resuspended in intracellular buffer containing 0.2% Triton X-100 and 1:500 rhodamine-phalloidin and stained for 20 min. The cells were centrifuged, resuspended in mHBSS, and analyzed on a FACSAria (BD Biosciences). Size correction for fluorescence intensity was derived from the ratio of background fluorescence from unstained control and Homer3-knockdown cells. Data analysis was performed on FlowJo (TreeStar, Ashland, OR).
Hem1-YFP–expressing HL-60s with nonsense shRNA or Homer3 shRNA were plated using squeeze chambers in RPMI containing 10% FBS and uniform 100 nM fMLP. Cells were imaged with TIRF and analyzed with ImageJ. The polarization of Hem1 localization was calculated by finding the vector between the cell center of mass and the center of mass of the Hem1 signal. A moving average of the angle of polarization was calculated from the current frame and two previous frames. If the moving average was different from the current angle of polarization by <0.3 rad, that frame was considered to have persistent polarization.
Supplementary Material
Acknowledgments
We thank Leo Meyerovich for coding assistance for calculating moving averages, Elizabeth Zhang for assistance with initial image data analysis, Johnny Rodriguez and the Mullins lab for purified actin, and Marilyn Farquhar, Delquin Gong, and Doug Tischer for constructs. We also thank Grace Peng and members of the Weiner lab for helpful discussion and critical reading of the manuscript. This work was supported by National Institutes of Health Grant R01-GM084040 to O.D.W. and an American Heart Association Predoctoral Fellowship to J.T.W.
Abbreviations used:
- AOTFs
acousto-optic tunable filters
- BSA
bovine serum albumin
- CBB
Coomassie brilliant blue
- CT
threshold cycle
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- EM-CCD
electron-multiplying charge-coupled device
- EVH1
enabled/vasodilator-stimulated phosphoprotein homology 1
- F-actin
filamentous actin
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- fMLP
formyl-methionyl-leucyl-phenylalanine
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GDP
guanosine diphosphate
- Gpp(NH)p
5′-guanylyl imidodiphosphate
- GTP
guanosine triphosphate
- GPCR
G-protein coupled receptor
- GST
glutathione S-transferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HGNC
HUGO Gene Nomenclature Committee
- HPLC
high-performance liquid chromatography
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- mHBSS
modified Hank's buffered saline solution
- NA
numerical aperture
- Pak
p21-activated kinase
- PE
phycoerythrin
- PH
pleckstrin homology
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- RPMI
Roswell Park Memorial Institute
- RNAi
RNA interference
- RT-PCR
real-time PCR
- shRNA
short hairpin RNA
- TCA
trichloroacetic acid
- TIRF
total internal reflection fluorescence
- YFP
yellow fluorescent protein
- ZP
standardized score.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1197) on March 4, 2015.
REFERENCES
- Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anger T, Klintworth N, Stumpf C, Daniel WG, Mende U, Garlichs CD. RGS protein specificity towards Gq- and Gi/o-mediated ERK 1/2 and Akt activation, in vitro. J Biochem Mol Biol. 2007;40:899–910. doi: 10.5483/bmbrep.2007.40.6.899. [DOI] [PubMed] [Google Scholar]
- Berzat A, Hall A. Cellular responses to extracellular guidance cues. EMBO J. 2010;29:2734–2745. doi: 10.1038/emboj.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Lee S, Briscoe C, Ellsworth C, Firtel RA. Role of Rac in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc Natl Acad Sci USA. 2000;97:5225–5230. doi: 10.1073/pnas.97.10.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar SN, Park JS, Peng GE, Onuffer JJ, Lim WA, Weiner OD. Actin dynamics rapidly reset chemoattractant receptor sensitivity following adaptation in neutrophils. Philos Trans R Soc B Biol Sci. 2013;368:20130008. doi: 10.1098/rstb.2013.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Fagni L, Worley PF, Ango F. Homer as both a scaffold and transduction molecule. Sci Signal. 2002;2002:re8. doi: 10.1126/stke.2002.137.re8. [DOI] [PubMed] [Google Scholar]
- Foa L, Rajan I, Haas K, Wu GY, Brakeman P, Worley P, Cline H. The scaffold protein, Homer1b/c, regulates axon pathfinding in the central nervous system in vivo. Nat Neurosci. 2001;4:499–506. doi: 10.1038/87447. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Gardiner EM, Pestonjamasp KN, Bohl BP, Chamberlain C, Hahn KM, Bokoch GM. Spatial and temporal analysis of Rac activation during live neutrophil chemotaxis. Curr Biol. 2002;12:2029–2034. doi: 10.1016/s0960-9822(02)01334-9. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Garcia-Marcos M, Bornheimer SJ, Farquhar MG. Activation of Gαi3 triggers cell migration via regulation of GIV. J Cell Biol. 2008;182:381–393. doi: 10.1083/jcb.200712066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- Hauert AB, Martinelli S, Marone C, Niggli V. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. Int J Biochem Cell Biol. 2002;34:838–854. doi: 10.1016/s1357-2725(02)00010-9. [DOI] [PubMed] [Google Scholar]
- Hromas R, Collins S, Raskind W, Deaven L, Kaushansky K. Hem-1, a potential membrane protein, with expression restricted to blood cells. Biochim Biophys Acta. 1991;1090:241–244. doi: 10.1016/0167-4781(91)90109-y. [DOI] [PubMed] [Google Scholar]
- Hwang S-Y, Wei J, Westhoff JH, Duncan RS, Ozawa F, Volpe P, Inokuchi K, Koulen P. Differential functional interaction of two Vesl/Homer protein isoforms with ryanodine receptor type 1: a novel mechanism for control of intracellular calcium signaling. Cell Calcium. 2003;34:177–184. doi: 10.1016/s0143-4160(03)00082-4. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Xavier R. Homer-3 regulates activation of serum response element in T cells via its EVH1 domain. Blood. 2004;103:2248–2256. doi: 10.1182/blood-2003-08-2671. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Nomura M, Hayase J, Iwakiri Y, Nishikimi A, Takayanagi R, Fukui Y, Sumimoto H. The cell polarity protein mInsc regulates neutrophil chemotaxis via a noncanonical G protein signaling pathway. Dev Cell. 2013;26:292–302. doi: 10.1016/j.devcel.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Kataria R, Xu X, Fusetti F, Keizer-Gunnink I, Jin T, van Haastert PJM, Kortholt A. Dictyostelium Ric8 is a nonreceptor guanine exchange factor for heterotrimeric G proteins and is important for development and chemotaxis. Proc Natl Acad Sci USA. 2013;110:6424–6429. doi: 10.1073/pnas.1301851110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. Regulation of human leukocyte p21-activated kinases through G protein–coupled receptors. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- Koronakis V, Hume PJ, Humphreys D, Liu T, Hørning O, Jensen ON, McGhie EJ. WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc Natl Acad Sci USA. 2011;108:14449–14454. doi: 10.1073/pnas.1107666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku C-J, Wang Y, Weiner OD, Altschuler SJ, Wu LF. Network crosstalk dynamically changes during neutrophil polarization. Cell. 2012;149:1073–1083. doi: 10.1016/j.cell.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Hadwiger JA, Pupillo M, Firtel RA. Molecular genetic analysis of two G alpha protein subunits in Dictyostelium. J Biol Chem. 1991;266:1220–1228. [PubMed] [Google Scholar]
- Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, Fukui Y. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol. 2006;174:647–652. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laliberté B, Wilson AM, Nafisi H, Mao H, Zhou YY, Daigle M, Albert PR. TNFAIP8: a new effector for Galpha(i) coupling to reduce cell death and induce cell transformation. J Cell Physiol. 2010;225:865–874. doi: 10.1002/jcp.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CD, Donald S, Anderson KE, Patton DT, Welch HCE. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine–dependent neutrophil responses. J Immunol. 2011;186:1467–1476. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yang L, Fu H, Yan J, Wang Y, Guo H, Hao X, Xu X, Jin T, Zhang N. Association between Gαi2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat Commun. 2013;4:1706. doi: 10.1038/ncomms2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malawista SE, de Boisfleury Chevance A. Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes (PMN) in the presence of EDTA: PMN in close quarters require neither leukocyte integrins nor external divalent cations. Proc Natl Acad Sci USA. 1997;94:11577–11582. doi: 10.1073/pnas.94.21.11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutin E, Raynaud F, Roger J, Pellegrino E, Homburger V, Bertaso F, Ollendorff V, Bockaert J, Fagni L, Perroy J. Dynamic remodeling of scaffold interactions in dendritic spines controls synaptic excitability. J Cell Biol. 2012;198:251–263. doi: 10.1083/jcb.201110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafisi H, Banihashemi B, Daigle M, Albert PR. GAP1(IP4BP)/RASA3 mediates Galphai-induced inhibition of mitogen-activated protein kinase. J Biol Chem. 2008;283:35908–35917. doi: 10.1074/jbc.M803622200. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Bourne HR. Receptors induce chemotaxis by releasing the βγ subunit of Gi, not by activating Gq or Gs. Proc Natl Acad Sci USA. 1997;94:14489–14494. doi: 10.1073/pnas.94.26.14489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Park JS, Rhau B, Hermann A, McNally KA, Zhou C, Gong D, Weiner OD, Conklin BR, Onuffer J, Lim WA. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. Proc Natl Acad Sci USA. 2014;111:5896–5901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert P, Weiner OD, Wang F, Bourne HR, Servant G. Leukocytes navigate by compass: roles of PI3Kγ and its lipid products. Trends Cell Biol. 2000;10:466–473. doi: 10.1016/s0962-8924(00)01841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y, Mizutani A, Bito H, Fujisawa K, Narumiya S, Mikoshiba K, Furuichi T. Cupidin, an isoform of Homer/Vesl, interacts with the actin cytoskeleton and activated Rho family small GTPases and is expressed in developing mouse cerebellar granule cells. J Neurosci. 1999;19:8389–8400. doi: 10.1523/JNEUROSCI.19-19-08389.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi-Yamaguchi Y, Furuichi T. The Homer family proteins. Genome Biol. 2007;8:1–12. doi: 10.1186/gb-2007-8-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi-Yamaguchi Y, Sato Y, Sakai R, Mizutani A, Knöpfel T, Mori N, Mikoshiba K, Furuichi T. Interaction of Cupidin/Homer2 with two actin cytoskeletal regulators, Cdc42 small GTPase and Drebrin, in dendritic spines. BMC Neurosci. 2009;10:25. doi: 10.1186/1471-2202-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Rittinger R. Preparation of GTPases for structural and biophysical analysis. Methods Mol Biol. 2002;189:13–24. doi: 10.1385/1-59259-281-3:013. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Sacchi F, Hill HR, Van Epps DE, Daynes RA. Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J Immunol 1950. 1985;135:4135–4143. [PubMed] [Google Scholar]
- Stephens L, Milne L, Hawkins P. Moving towards a better understanding of chemotaxis. Curr Biol. 2008;18:R485–R494. doi: 10.1016/j.cub.2008.04.048. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Sun CX, Downey GP, Zhu F, Koh ALY, Thang H, Glogauer M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood. 2004;104:3758–3765. doi: 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- Tang W, Zhang Y, Xu W, Harden TK, Sondek J, Sun L, Li L, Wu D. A PLCβ/PI3Kγ-GSK3 signaling pathway regulates cofilin phosphatase Slingshot2 and neutrophil polarization and chemotaxis. Dev. Cell. 2011;21:1038–1050. doi: 10.1016/j.devcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb Perspect Biol. 2009;1:a002980. doi: 10.1101/cshperspect.a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner OD, Marganski WA, Wu LF, Altschuler SJ, Kirschner MW. An actin-based wave generator organizes cell motility. PLoS Biol. 2007;5:e221. doi: 10.1371/journal.pbio.0050221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner OD, Rentel MC, Ott A, Brown GE, Jedrychowski M, Yaffe MB, Gygi SP, Cantley LC, Bourne HR, Kirschner MW. Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biol. 2006;4:e38. doi: 10.1371/journal.pbio.0040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss-Haljiti C, Pasquali C, Ji H, Gillieron C, Chabert C, Curchod ML, Hirsch E, Ridley AJ, Hooft van Huijsduijnen R, Camps M, Rommel C. Involvement of phosphoinositide 3-kinase gamma, Rac, and PAK signaling in chemokine-induced macrophage migration. J Biol Chem. 2004;279:43273–43284. doi: 10.1074/jbc.M402924200. [DOI] [PubMed] [Google Scholar]
- Welch HCE, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Wiege K, Le DD, Syed SN, Ali SR, Novakovic A, Beer-Hammer S, Piekorz RP, Schmidt RE, Nürnberg B, Gessner JE. Defective macrophage migration in Gαi2- but not Gαi3-deficient mice. J Immunol. 2012;189:980–987. doi: 10.4049/jimmunol.1200891. [DOI] [PubMed] [Google Scholar]
- Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Mihaylov V, Xu X, Brzostowski JA, Li H, Liu L, Veenstra TD, Parent CA, Jin T. A Gβγ effector, ElmoE, transduces GPCR signaling to the actin network during chemotaxis. Dev Cell. 2012;22:92–103. doi: 10.1016/j.devcel.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, Huttenlocher A. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 2010;18:226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun C, Glogauer M, Bokoch GM. Human neutrophils coordinate chemotaxis by differential activation of Rac1 and Rac2. J Immunol. 2009;183:2718–2728. doi: 10.4049/jimmunol.0900849. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.