

Vimentin intermediate filaments-structures that were considered extremely stable-are in fact very dynamic. Filaments are actively transported throughout the cell along microtubules. In addition, filaments undergo constant rearrangement by robust severing and reannealing but never completely disassemble and reassemble.
Abstract
We studied two aspects of vimentin intermediate filament dynamics—transport of filaments and subunit exchange. We observed transport of long filaments in the periphery of cells using live-cell structured illumination microscopy. We studied filament transport elsewhere in cells using a photoconvertible-vimentin probe and total internal reflection microscopy. We found that filaments were rapidly transported along linear tracks in both anterograde and retrograde directions. Filament transport was microtubule dependent but independent of microtubule polymerization and/or an interaction with the plus end–binding protein APC. We also studied subunit exchange in filaments by long-term imaging after photoconversion. We found that converted vimentin remained in small clusters along the length of filaments rather than redistributing uniformly throughout the network, even in cells that divided after photoconversion. These data show that vimentin filaments do not depolymerize into individual subunits; they recompose by severing and reannealing. Together these results show that vimentin filaments are very dynamic and that their transport is required for network maintenance.
INTRODUCTION
The best-known function of intermediate filaments is to provide mechanical integrity to cells (Janmey et al., 1991). However, intermediate filaments also participate in a variety of other cell functions, including cell signaling, cell migration, and wound healing (Ivaska et al., 2007). In addition, vimentin intermediate filaments have been implicated in many pathological processes, including the ability of cancer cells to form metastases (Satelli and Li, 2011). Despite these important functions, the dynamics of intermediate filaments, including vimentin filaments, is poorly understood. The dynamics of filaments determines how filament networks are organized, which is likely important for maintaining mechanical integrity of cells, especially during cell migration and mechanical stress.
Vimentin filaments in cells form networks that typically extend throughout the entire cytoplasm. Immature forms, consisting of nonfilamentous “particles” that are one or a few unit-length filaments (ULFs; the smallest polymerized form of intermediate filaments) and short filament “squiggles” produced by particles that have joined together, are found in the cell periphery (Yang et al., 1985; Yoon et al., 1998). Particles and squiggles combine and become compact along the filament radius to form the mature filaments that make up the rest of the network (Kirmse et al., 2007). Mature filaments are often tightly packed and bundled around the nucleus and radiate out to the periphery.
A relationship between the vimentin network and microtubules has been well established. Vimentin network distribution corresponds to that of the microtubule network (Ball and Singer, 1981), and the depolymerization of microtubules results in the dramatic retraction of the vimentin network to the perinuclear region of the cell (Goldman, 1971). In addition, inhibition of the microtubule-based motors kinesin (Gyoeva and Gelfand, 1991) and dynein (Helfand et al., 2002) also rearranges the vimentin network. Together these data suggest that vimentin filaments are distributed in cells by transport along microtubules.
Transport of another type of intermediate filament—neurofilaments—has been observed in axons of cultured neurons (Wang et al., 2000). These filaments are also transported by microtubule-based motors (Theiss et al., 2005; Uchida et al., 2009). In addition, microtubule-dependent transport of immature forms of vimentin filaments has been observed at the cell periphery (Prahlad et al., 1998; Yoon et al., 1998) and in cells expressing a mutant form of vimentin that only forms ULFs (Robert et al., 2014). However, it is difficult to study transport of mature vimentin filaments due to their small diameter and very dense packing in cells, and it remains to be determined whether they move and, if so, whether their transport is dependent on microtubules.
Here we used two imaging modalities to observe transport of vimentin filaments. We used live-cell structured illumination microscopy (SIM) of vimentin tagged with the fluorescent protein mEmerald to study filament transport at the cell periphery, and we used a recently developed photoconvertible optical probe, mEos3.2 (Zhang et al., 2012), fused to vimentin (mEos3.2-vimentin) combined with total internal reflection microscopy (TIRFM) to study filament transport in dense network regions. We found that mature vimentin filaments were rapidly transported in multiple cell lines, and this transport was microtubule dependent, likely driven by microtubule-based motors.
We also used mEos3.2-vimentin to study subunit exchange and assembly/disassembly of filaments. In agreement with earlier studies of both neurofilaments and vimentin, we found evidence for severing and annealing of filaments (Colakg˘ lu and Brown, 2009; Uchida et al., 2013). Even with long-term imaging, we found no evidence of complete disassembly and reassembly of filaments or subunit exchange along filaments with a soluble vimentin pool. Instead, we found that filaments exchange in polymerized blocks by the severing and reannealing of preexisting filaments. Together these results demonstrate that vimentin filaments, similar to microtubules and actin microfilaments, are highly dynamic cytoskeletal structures. Like microtubules (Jolly et al., 2010), intermediate filaments undergo robust transport along cytoplasmic microtubules. However, unlike other cytoskeletal structures, intermediate filaments are also recomposed by severing and annealing. These dynamic properties are likely important for remodeling the intermediate filament cytoskeleton maintaining the mechanical stability of cells as they change shape, such as during cell migration.
RESULTS
Rapid transport of vimentin filaments observed by superresolution microscopy
Vimentin filament networks typically consist of tightly packed and bundled filaments in the perinuclear region that radiate outward to the cell edge, where the filament precursors—particles and squiggles—are often found (Yang et al., 1985; Yoon et al., 1998). To observe the network in live cells, we performed time-lapse imaging of RPE cells expressing vimentin tagged with the fluorescent protein mEmerald. Expression of mEmerald-vimentin (Supplemental Figure S1B) did not aggregate, bundle, or otherwise change vimentin network organization (Supplemental Figure S1A). Because filaments have diameters of only ∼10 nm, they cannot be observed in detail using conventional microscopy. Therefore, to determine whether filaments, like filament precursors, are capable of transport, we used the live-cell superresolution microscopy technique of TIRF-SIM, which permits high-speed image acquisition with minimal phototoxicity. TIRF-SIM time-lapse sequences over a 1-min imaging period showed that although most thin filaments remained stationary, some of them underwent rapid transport (Figure 1 and Supplemental Video S1). This included the motility of short squiggles, consistent with previous observations (Prahlad et al., 1998; Yoon et al., 1998), in addition to the movement of longer filaments. Filaments moved toward, away from, and parallel to the cell edge (Supplemental Video S1). When it was possible to follow the end of a moving filament, we measured the distance it traveled between frames. From this, we calculated that filaments moved at an average rate of ∼0.29 (±0.14) μm/s.
FIGURE 1:
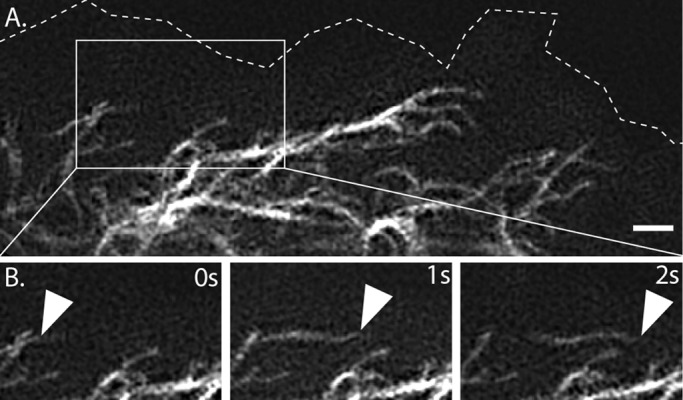
Rapid motility of mature filaments at cell periphery revealed by TIRF-SIM. (A) Edge of Emerald-vimentin–expressing RPE cell. (B) Time-lapse at 1-s intervals shows rapid transport of filaments in the boxed region. Arrow points to the end of a filament that could be followed between frames. Scale bar, 1 μm. Corresponds to Supplemental Video S1, first sequence.
Photoconversion reveals rapid vimentin intermediate filament transport
We imaged mEmerald-vimentin at the cell edge because the vimentin network is so dense and complex in the central region of cells that, even with TIRFM, it is difficult to tease apart the behavior of individual filaments or small bundles. To observe filament transport in regions of the cell with a high density of vimentin filaments, we created a fusion of vimentin with the photoconvertible protein mEos3.2 (mEos3.2-vimentin) and another with photoactivatable green fluorescent protein (PA-GFP-vimentin). These proteins change fluorescent properties when exposed to ultraviolet (UV) light, mEos3.2 from green to red, and PA-GFP from nonfluorescent to green. Like tagging vimentin with mEmerald, tagging vimentin with these constructs did not affect vimentin organization (Supplemental Figure S1, C and D). We restricted photoconversion to a circular area of ∼16 μm in diameter. This manipulation produced fiduciary marks on filaments so that we could monitor filament transport in the red channel (for mEos3.2-vimentin) or green channel (for PA-GFP-vimentin) using time-lapse imaging with TIRFM or spinning-disk confocal microscopy. Because both PA-GFP and mEos3.2 bleach quickly, we collected ≤10 frames during image sequence acquisition after photoactivation/conversion to limit photobleaching to 5–10% of the initial frame and avoid effects of phototoxicity.
First, we expressed mEos3.2 vimentin in RPE cells, photoconverted in a circular region as described, and imaged over a period of 3 min. Remarkably, whereas several converted filaments remained within the region of photoconversion for the duration of imaging, many moved rapidly away from the photoconverted area (Figure 2A and Supplemental Video S2, first sequence). Most of the transported filaments were at least as long as the region of conversion (Figure 2A, inset), indicating that long, mature filaments can be efficiently transported. In addition, sometimes shorter photoconverted pieces also moved, but it is not known whether these were immature vimentin squiggles, converted sections of longer filaments, or a part of longer converted filaments otherwise located outside the TIRFM imaging plane (since TIRFM selectively illuminates only fluorophores within ∼100 nm of the coverslip; Mattheyses et al., 2010).
FIGURE 2:

Photoconvertible-vimentin reveals the rapid transport of mature vimentin intermediate filament (IF) in multiple cell types. (A) Left, Eos3.2-vimentin RPE cell immediately after a circular region was exposed to UV light. All converted filaments were initially confined to the region of exposure (see inset). Right, same cell after 3 min. Many filaments had moved outside of the initial region of conversion, including long filaments (inset). Scale bars, 5 μm. Corresponds to Supplemental Video S2, first sequence. (B) Vimentin IF transport occurred in many cell types. Several filaments had moved outside the region of photoconversion, marked with a circle, within 3 min in (from left to right) NIH3T3, SW13, and primary MEF cells. Scale bars, 5 μm. Corresponds to Supplemental Video S3.
Filaments moved from converted regions in every direction, although mostly along the long axis of an elongated cell (Supplemental Video S2, first sequence). Although some moved progressively throughout the 3-min imaging period, many filaments paused, occasionally resuming rapid motility after pausing for several seconds. Filaments sometimes moved in looping paths but typically did not reverse direction along the same track.
To determine whether the dramatic filament motility we observed in RPE cells was a universal property of vimentin filaments and not cell-type specific, we expressed mEos3.2-vimentin in several other cell types. We chose cell lines that differed in species of origin, morphology, and migratory properties. We also transfected primary cells in addition to immortalized lines. Despite these differences, vimentin filaments moved away from the initial region of photoconversion in every cell type tested, including mouse fibroblast NIH-3T3 cells, human adenocarcinoma SW13 cells, and primary mouse embryonic fibroblasts (MEFs; Figure 2B and Supplemental Video S3). However, we did find some differences in vimentin filament motility between cell types. For example, the proportion of converted filaments that moved outside the region of conversion within the 3-min imaging window varied by cell type. The greatest amount of motile converted filaments was observed in RPE cells, whereas NIH-3T3 and MEF had slightly fewer, and SW13 had the fewest. SW13 also differed from the other cell types in terms of the lengths of motile filaments. Although it was possible to see long filaments move in NIH-3T3, MEF, and RPE cells, the motile filaments in SW13 were shorter than the size of the converted region. However, due to the limitations of TIRFM, it is not clear whether the filaments in SW13 cells are indeed shorter than filaments in other cell types or whether SW13 filaments are not able to lie flat against the bottom of the cell.
Vimentin intermediate filament transport depends on microtubules
To test whether vimentin filament transport was microtubule dependent, we depolymerized microtubules with 10 μM nocodazole. Normally, when microtubules are depolymerized with nocodazole at 37°C, vimentin filaments are retracted to the perinuclear region (Goldman, 1971). Therefore we changed the protocol to include incubation at 4˚C before rewarming cells to 37°C for imaging. The 4°C incubation slows vimentin retraction in the absence of microtubules, so that vimentin filament redistribution in the absence of microtubules is minimized at the time of imaging. Microtubules repolymerize at 37°C in control but remain depolymerized if cells are transferred to 37°C in the presence of nocodazole (Figure 3D). Therefore we were able to compare the motility of similar vimentin networks at 37˚C by varying only the presence versus absence of microtubules (Figure 3B, left).
FIGURE 3:
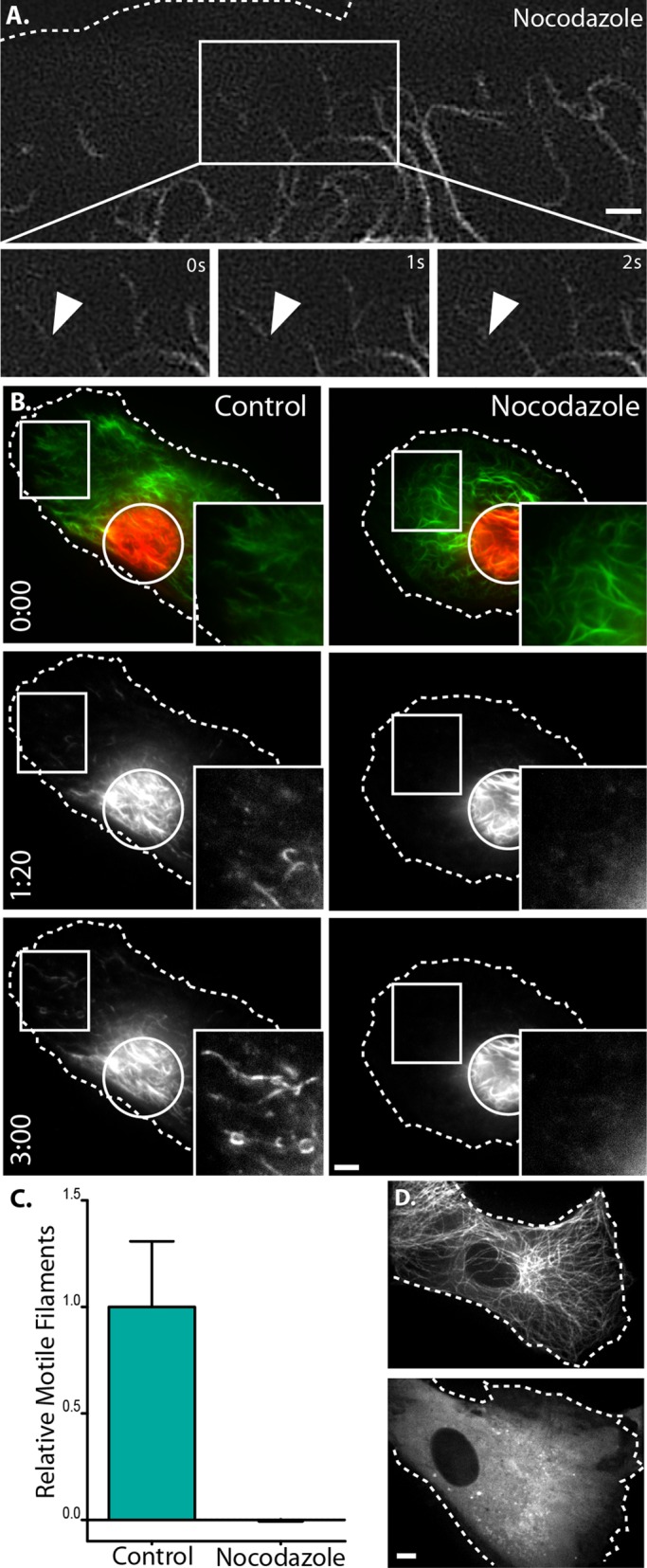
Vimentin IF transport depends on microtubules. (A) Two-dimensional TIRF-SIM at the cell periphery of an emerald-vimentin–expressing RPE cell treated with nocodazole (top) shows that filaments remained stationary without microtubules (time lapse; bottom). Scale bar, 1 μm. Corresponds to Supplemental Video S1, second sequence. (B) Control (left) and nocodazole-treated (right) cells immediately after conversion and at 1 min, 20s and 3 min. Scale bar, 5 μm. Corresponds to Supplemental Video S2, second sequence. (C) Quantification of filament motility in control (n = 23) vs. nocodazole-treated (n = 19) cells. The 95% confidence interval is represented by error bars. (D) mtagRFPt-cells under control conditions (top) and after nocodazole treatment (bottom). Scale bar, 5 μm.
First, we investigated the effect of microtubule depolymerization on the movements of filaments at the cell periphery imaged with TIRF-SIM. We found that, in contrast to control, mEmerald-vimentin filaments remained stationary after microtubule depolymerization (Figure 3A and Supplemental Video S1, second sequence). Next we tested whether the filament motility revealed by conversion of mEos3.2-vimentin in the central region of cells was also microtubule dependent. In control cells, many filaments moved away from the region where they were initially activated, as in Figure 2, but converted filaments in the absence of microtubules remained within the region of conversion (Figure 3B and Supplemental Video S2, second sequence).
Although there was an obvious qualitative difference in filament transport between cells with and without microtubules, we sought to quantify this difference. To quantify filament transport, we identified filament segments in the TIRFM images and reconstructed the filament network as binary representations (Figure 4B). For each frame, we measured filament transport as the number of filaments outside the zone of photoconversion. To account for any differences in the initial number of filaments activated between cells, we normalized filament transport to the sum intensity inside the photoconversion zone of the first frame. For each time-lapse sequence, we took the slope of normalized filament transport over time (Figure 4E). Using this method to quantify the effect of microtubules, we found that depolymerizing microtubules significantly impaired transport compared with controls (p < 0.0001; Figure 3C). These results demonstrate that vimentin filaments require microtubules for their movement throughout the cell.
FIGURE 4:
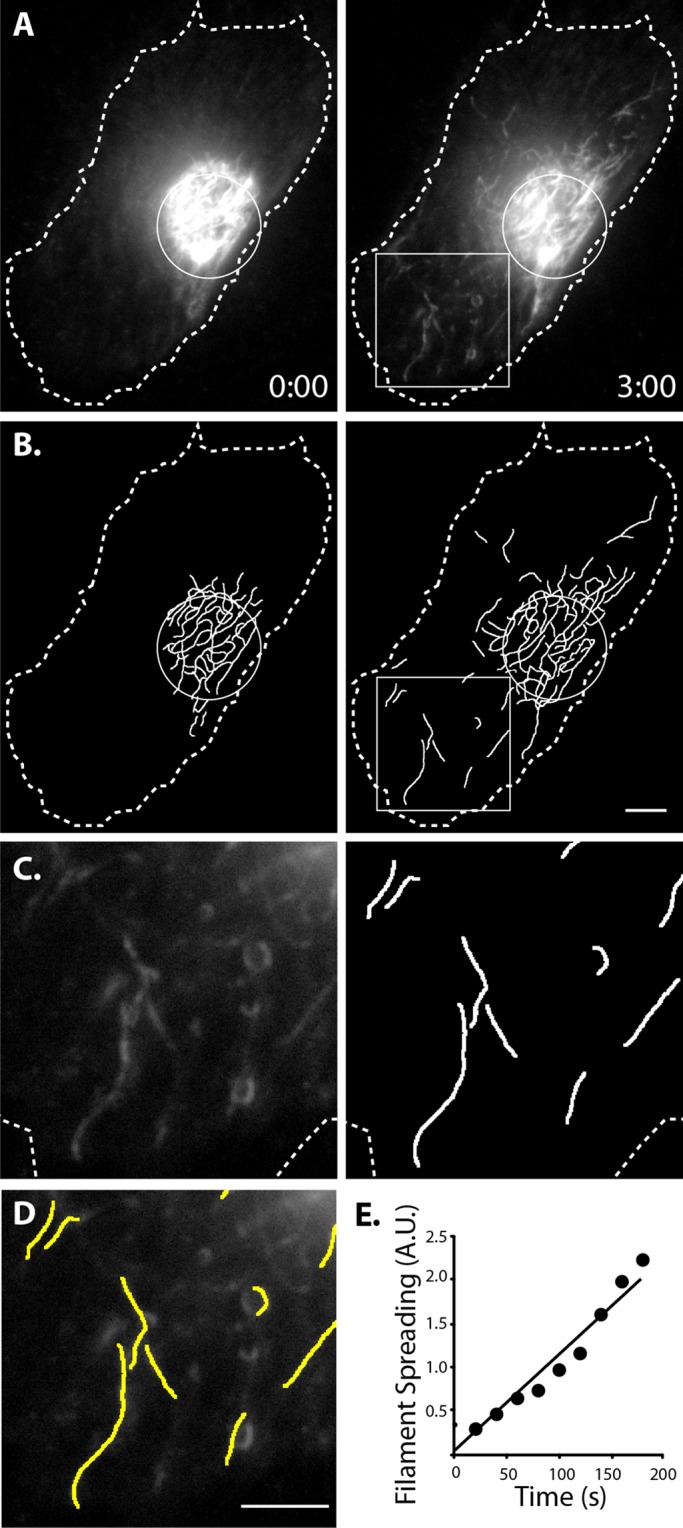
Method to quantify vimentin filament motility. (A) Control cell 0 and 3 min after photoconversion. (B) Filaments detected using custom software to detect linear segments. (C) Enlargement of boxed regions in A and B. (D) The overlay of boxed regions. Scale bars, 5 μm. (E) Plot of filament spreading from cell represented in A–D.
Vimentin transport is independent of microtubule dynamics
Because vimentin filament motility depended on microtubules, and microtubules are highly dynamic structures undergoing constant polymerization and depolymerization, we next tested whether microtubule polymerization contributes to vimentin filament transport. This possibility was recently underscored by the finding that vimentin directly binds the microtubule plus end–binding protein adenomatous polyposis coli (APC; Sakamoto et al., 2013). Therefore vimentin filaments could theoretically be pulled along as microtubules polymerize, with APC serving as a link to the growing microtubule tip.
First, we tested whether blocking microtubule dynamics affects filament transport. To this end, we treated cells with 10 nM vinblastine and assessed microtubule dynamics by expressing mTagRFPT-end-binding protein 3 (RFPT-EB3), a protein that binds to plus ends of polymerizing microtubules. Under control conditions, fluorescently tagged EB3 produces comet-like structures that move in the cytoplasm as microtubule plus ends elongate (Stepanova et al., 2003; Figure 5A and Supplemental Video S4, first sequence). Treatment of cells with 10 nM vinblastine dramatically diminished these comets, indicating that there was little growth at microtubule ends under this condition (Figure 5A and Supplemental Video S4, second sequence). Of importance, 10 nM vinblastine left the microtubule network intact, as the concentration was too low to cause microtubule depolymerization (Figure 5B). When we photoconverted mEos3.2-vimentin in the presence versus the absence of 10 nM vinblastine, we found that suppression of microtubule dynamics had no effect on vimentin filament motility (p = 0.338 in Welch's t test; Figure 5, C and D). This result shows that vimentin transport is independent of microtubule polymerization.
FIGURE 5:

Blocking microtubule dynamics does not affect vimentin IF transport. (A) mtagRFPT-EB3–labeled growing microtubule plus ends. Frames from time-lapse sequences were individually pseudocolored and superimposed. Differences in frames result in the appearance of rainbows; where frames overlap, colors merge and appear white. Comets can be seen in control (left; see also first sequence of Supplemental Video S4) but not in the presence of 10 nM vinblastine (right; see also second sequence of Supplemental Video S4). Scale bar, 10 μm; color scale, 16 s. (B) Microtubule network is indistinguishable between control (left) and the presence of 10 nM vinblastine (right). Scale bar, 5 μm. (C) Examples of photoconverted Eos3.2-vimentin RPE cells after 3 min in the absence (left) and presence (right) of 10 nM vinblastine shows filament motility in both conditions. (D) Quantification of filament motility in control (n = 16) vs. vinblastine-treated (n = 13) cells. The 95% confidence interval is represented by error bars.
Second, we directly tested whether vimentin transport could be mediated by its association with APC (Sakamoto et al., 2013). We used small interfering RNA (siRNA)–mediated knockdown to deplete APC in mEos3.2-vimentin cells. We were able to efficiently reduce protein levels of APC in cells transfected with APC-targeted siRNA compared with cells transfected with a scrambled-sequence siRNA (Supplemental Figure S2B). However, vimentin filament motility was not changed by APC depletion (p = 0.442 in Welch's t test; Supplemental Figure S2, A and C). Therefore vimentin filament transport is not mediated by the interaction between vimentin and APC or by microtubule dynamics.
Vimentin intermediate filaments are transported bidirectionally along microtubules
Because filament transport depended on microtubules but not microtubule polymerization, filaments likely move along preexisting microtubules. Therefore we used live-cell, two-color TIRF-SIM of RPE coexpressing mEmerald-vimentin and tag-RFPT-tubulin to relate vimentin filament transport to the microtubule network. We observed that vimentin filaments moved along microtubules, sometimes in opposite directions on the same microtubule track (Figure 6 and Supplemental Video S5). These results, together with the sensitivity of transport to microtubule depolymerization, show that vimentin filaments use microtubule tracks for bidirectional transport in the cytoplasm.
FIGURE 6:

Vimentin filament transport along microtubules revealed by live-cell, two-color TIRF-SIM. Frames from time-lapse imaging show vimentin filaments (left) moving in the periphery of a cell. Arrowheads indicate the ends of two filaments in each frame. The microtubule network was also captured at each time point (middle). The merged images show that vimentin filaments are translocating along microtubules (right). Note that the filaments marked with yellow and blue arrowheads are traveling in opposing directions along the same microtubule. Scale bar, 1 μm.
To demonstrate further the directionality of vimentin filament transport along microtubules, we photoconverted filaments in the periphery of cells, where the majority of microtubule plus ends are located in RPE cells. We already showed that filaments move away from centrally converted regions, demonstrating that filaments are capable of plus end–directed, anterograde transport (Figure 2A). After conversion at the periphery, vimentin filaments were transported in a retrograde manner toward the cell center (Supplemental Figure S3A). These results additionally support that vimentin filaments undergo bidirectional transport along microtubules. However, microtubules are not strictly polarized in the periphery of RPE cells, as some microtubules loop back inward when they reach the cell edge. Thus filaments moving toward the cell center after conversion at the edge could have been moving toward plus ends of looping-back microtubules. Therefore we decided to examine the directionality of vimentin filament transport in a cell type in which microtubule orientation is strictly uniform. To do this, we expressed PA-GFP-vimentin or mEos3.2-vimentin in CAD neuroblastoma cells, which extend neurites containing uniformly oriented (plus-end-out) microtubules. We confirmed the uniform polarity of microtubules in CAD neurites by expressing GFP-EB3. Comets moved toward the tips of the neurites, indicating that microtubules in neurites are indeed oriented with their plus ends out (Supplemental Figure S3B). We directed UV light to vimentin filaments in sections of CAD neurites and found that activated or converted filaments (in PA-GFP-vimentin or Eos3.2-vimentin CAD, respectively) moved both toward and away from CAD neurite tips in 87.5% of neurites (Supplemental Figure S3C and Supplemental Video S6), corroborating our finding that vimentin filament transport is bidirectional. Filaments moved only in an anterograde direction in 6.25% of neurites, only retrograde in 3.125%, and remained stationary in 3.15% of neurites.
Vimentin filament transport is regulated by histone deacetylase inhibition
Next we addressed whether the microtubule tracks could regulate vimentin filament transport. Because vimentin preferentially colocalizes with detyrosinated, stable microtubules (Gurland and Gundersen, 1995), we tested whether increasing stable microtubules has an effect on vimentin filament transport. We hypothesized that making more stable microtubules could increase the number of vimentin filaments that localize to microtubule tracks and thereby make more vimentin filaments available for transport. We used the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) to increase acetylated microtubules (Supplemental Figure S4A) and compared filament transport in these cells to controls. To our surprise, we found that vimentin transport was significantly decreased by HDAC inhibition (p = 0.0089 in Welch's t test) (Supplemental Figure S4, B and C). This suggests that vimentin filaments may be transported preferentially along newer, dynamic microtubules.
Serum starvation and stimulation have no effect on vimentin filament transport
In fibroblasts, serum starvation and activation regulate the phosphorylation state, form (mature filaments vs. particles and squiggles) at the cell edge, and affect recovery of fluorescence in bleached regions of GFP-vimentin–expressing cells (Helfand et al., 2011). Therefore we compared vimentin filament transport in Eos3.2-vimentin RPE cells after 72 h of serum starvation, after 72 h of serum starvation followed by serum addition, and in untreated controls. We found no difference between these groups (p = 0.171 in one-way analysis of variance; Supplemental Figure S5), demonstrating that serum starvation and stimulation do not affect vimentin filament transport.
Vimentin filaments are recomposed by severing and reannealing but not by disassembly and reassembly
We determined that vimentin filaments were highly dynamic in terms of their transport. Because filament turnover can also contribute to vimentin network organization, we next turned to investigate this aspect of vimentin dynamics. We used the same photoconversion strategy, but rather than imaging over a few minutes, we imaged photoconverted cells for several hours (Figure 7 and Supplemental Video S7). For these experiments, we expressed mEos3.2-vimentin in SW13 cells, which remain mostly stationary, in order to image converted filaments in the same cell over long periods of time.
FIGURE 7:
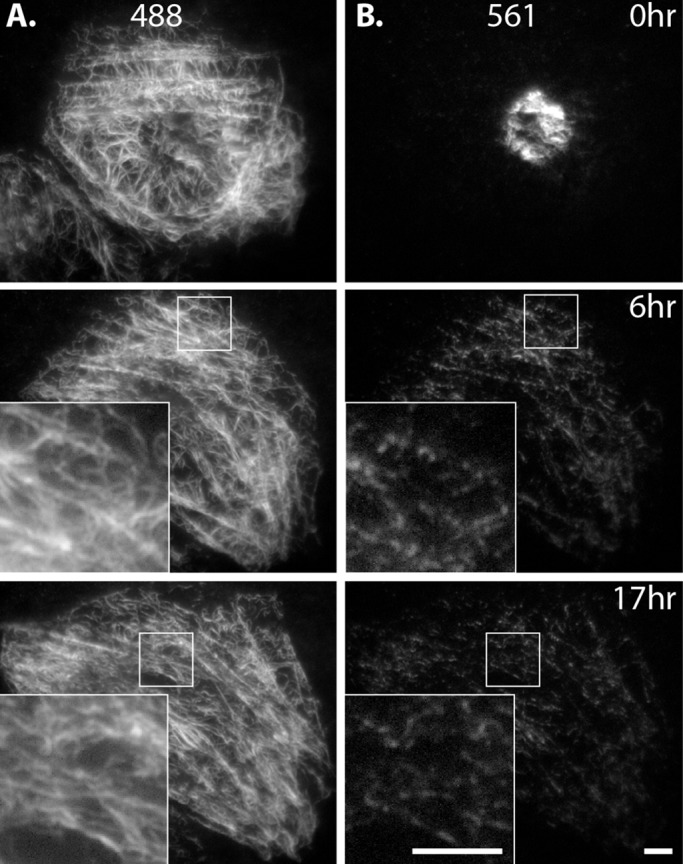
Long-term imaging after photoconversion reveals extensive severing and reannealing of filaments. Eos3.2-vimentin–expressing SW13 cell immediately after photoconversion and 6 and 17 h later. (A) The 488 channel shows that nonconverted and incompletely converted labeling of filaments was uniform throughout the network. (B) The 561 channel shows filaments distributed in segments throughout the network. Even at 17 h, 561 labeling was restricted to patches along filaments (inset, bottom right). Scale bars, 5 μm. Corresponds to Supplemental Video S7.
We saw that converted filaments were dispersed throughout the cytoplasm within the first 1–2 h after photoconversion, further confirming that, even in SW13 cells, where we saw the least filament motility compared with the other cell types tested, the network is highly dynamic. We observed the pattern of fluorescence within the network over several hours to study dynamics of filament assembly and disassembly. We reasoned that there were three possibilities: 1) All the red fluorescence in converted filaments would remain unchanged; red segments would be the same intensity and length throughout the duration of imaging. This would indicate that, other than transport, filaments are not dynamic. 2) Converted red segments would decrease in intensity as the intensity of the entire network would increase. Eventually, the entire network would be uniformly red. This result would indicate that the size of the exchangeable unit is below the resolution limit of the light microscopy (individual vimentin polypeptides or small oligomers). We did not see either of these two possibilities. 3) Instead, we observed that photoconverted filaments appeared in segmented form (Figure 7B), often occurring as long or short red puncta along a filament (Figure 7B insets). This result supports the model of filament severing and reannealing but suggests that complete disassembly and reassembly of filaments, or any subunit exchange along the length of a filament, must be either extremely slow or beyond the sensitivity of live-cell imaging (see Discussion). In contrast to the pattern in the red channel, vimentin filaments maintained uniform green fluorescence for the duration of imaging, since photoconversion was not complete, and even converted regions of filaments retained green signal (Figure 7A). Thus the green channel demonstrates that the segmented appearance of converted filaments in the red channel was not simply an artifact of filaments going into and out of the plane of TIRF. Finally, we tested whether microtubule-dependent transport of vimentin filaments contributes to their severing and reannealing. To do this, we repeated the experiment described so far but prevented vimentin filament transport by depolymerizing microtubules with nocodazole. Again, we saw a segmented pattern of converted vimentin along filaments in the network after several hours (Supplemental Figure S6). Further, we saw severing and reannealing persisted even in serum-starved cells (Supplemental Figure S7), showing that, like filament transport, severing and reannealing is also unaffected by serum starvation.
Finally, vimentin organization is known to reorganize dramatically during cell division, including complete disassembly and reassembly during mitosis in some cell types (Rosevear et al., 1990). We captured time-lapse sequences of SW13 cells as they divided after mEos3.2-vimentin photoconversion. We found that converted vimentin filaments remained in segmented, punctate form at all time points, just as we observed in interphase cells (Figure 8 and Supplemental Video S8). This observation demonstrates that vimentin filaments are not completely disassembled and reassembled during cell division in SW13 cells.
FIGURE 8:

Filaments remain in polymerized form throughout cell division in SW13 cells. Eos3.2-vimentin–expressing SW13 cell immediately after photoconversion (left), rounding and lifting from the coverslip in preparation for division at 4 h (middle), and after division (right). (A) Green (nonconverted and incompletely converted) labeling of filaments is uniform. (B) Converted, red filaments remain in segmented form throughout the network and in daughter cells. Scale bar, 5 μm. Corresponds to Supplemental Video S8.
DISCUSSION
Using advanced live-cell imaging techniques and recently developed optical probes, we were able to demonstrate that vimentin intermediate filaments form a highly dynamic network in the cytoplasm of many types of mammalian cells. Vimentin dynamics includes the robust transport of filaments in the cytoplasm, as well as filament recomposition by severing and reannealing (but not by polymerization and depolymerization). We found that mature vimentin filaments moved at rates similar to transport rates of squiggles and particles of vimentin (Prahlad et al., 1998; Yoon et al., 1998). In addition, we found that the mechanism of filament transport by microtubule-based motors is consistent with that of precursors (Prahlad et al., 1998; Helfand et al., 2002; Robert et al., 2014), as well as with transport of another type of mature filament, neurofilaments (Theiss et al., 2005; Uchida et al., 2009). Further, our findings have direct implications for results that show that vimentin and the microtubule plus end–binding protein APC directly interact (Sakamoto et al., 2013). We demonstrated that neither inhibition of polymerization nor APC depletion from cells has any effect on transport of vimentin filament transport along microtubules. Therefore the APC-vimentin interaction observed by Sakamoto et al. (2013) is not important for microtubule-dependent vimentin filament transport.
Of interest, at every moment, only a small fraction of filaments was moving in a cell, suggesting that vimentin transport in the cell, like transport of many other types of cargoes (Barlan et al., 2013), is tightly regulated. Here we tested two mechanisms that could regulate vimentin filament transport. First, we found that microtubule acetylation negatively regulated filament transport, although previous work showing that vimentin preferentially colocalizes to stable, detyrosinated microtubules led us to expect the opposite (Gurland and Gundersen, 1995). This suggests that vimentin filaments are preferentially transported on dynamic microtubules, but it is also possible that our HDAC inhibition resulted in some off-target effect that affected filament transport by an unseen mechanism. Future studies will be needed to determine whether and which posttranslational modifications of microtubules are able to regulate vimentin filament transport. The second mechanism of regulation of filament transport that we tested was serum starvation and stimulation. Although starvation has been shown to decrease recovery after photobleaching of GFP-vimentin filaments in fibroblasts, we were unable to detect any change in vimentin filament dynamics in RPE cells.
Several observations suggest the major factor restricting vimentin filament motility is the interaction between vimentin and polymerized actin. First, analysis of vimentin distribution in the cell shows that vimentin filaments are often coaligned with actin cables (Mott and Helmke, 2007; Murray et al., 2014; see also how vimentin filaments concentrate along parallel structures [presumable actin stress fibers] that run along the long axis of the cell in Figure 8A). Second, transport of vimentin filament precursors is inhibited by actin microfilaments (Robert et al., 2014). Finally, vimentin interacts directly with actin (Esue et al., 2006) and indirectly through linkers, including plectin (Svitkina et al., 1996), fimbrin (Correia et al., 1999), and filamin A (Kim and McCulloch, 2011). In addition, phosphorylation has been shown to regulate the vimentin–plectin interaction (Foisner et al., 1996), suggesting that vimentin filament anchorage to actin might be relieved by phosphorylation of a cross-linker. It will be important for future work to determine whether the same mechanisms regulate transport of precursors as do mature filaments or whether mature filaments can be selectively affected. For example, a mature filament may need only a small number of actin–vimentin cross-links to restrict its motility, making it more sensitive to regulatory signals than movement of small filament precursors.
Vimentin expression and network organization are particularly important for processes involving change in cell shape, including cell migration (Ivaska et al., 2007). For example, vimentin-null fibroblasts migrate into wounds more slowly than vimentin-expressing cells (Eckes et al., 1998), and vimentin expression in epithelial cells induces them to break cell–cell contacts and become migratory (Mendez et al., 2010). Vimentin transport is likely important for redistributing the vimentin network in motile cells. Rearranging the network by filament transport is probably more efficient than localized disassembly and reassembly, especially given the finding that filaments are extremely stable.
Our finding that filaments did not completely disassemble/reassemble or substantially exchange subunits with a soluble pool may seem surprising, given previous results (Vikstrom et al., 1992; Yoon et al., 1998). These studies used fluorescence recovery after photobleaching to demonstrate that vimentin is dynamic. However, recovery of fluorescence was attributed to subunit exchange, and the contribution of motile filaments was not taken into account. Therefore recovery of fluorescence in these studies was probably due to transport of filaments into bleached zones rather than by exchange of subunits in individual filaments. The lack of individual vimentin subunit exchange represents a fundamental difference between intermediate filaments and other cytoskeletal structures. Whereas microtubules and actin microfilaments in a cell are constantly polymerizing and depolymerizing, the major form of exchange in the case of vimentin filaments was their severing and reannealing instead of exchange with a soluble pool. However, we cannot completely exclude that soluble vimentin tetramers are able to exchange within mature filaments. Recent work shows that exchange occurs between polymerized filaments in vitro, although the rate of exchange in the case that mimics filaments in vivo is only ∼6 tetramers/10 μm of filament per hour (Nöding et al., 2014); thus evidence of exchange within mature filaments is likely beyond the current limit of detection in live-cell imaging.
Our data are consistent with reports that show end-to-end annealing of vimentin filaments (Colakg˘ lu and Brown, 2009) and cyclical severing and annealing of neurofilaments (Uchida et al., 2013). Future investigations will be necessary to determine the mechanism for breaking and reannealing. One possibility is that filaments are mechanically broken during either cell movement or intracellular transport. For example, F-actin can mechanically break microtubules during cell migration (Gupton et al., 2002). However, it is also possible that cells have specific mechanisms for severing filaments, which might include selective targeting and regulation. Severing proteins have been described for microtubules (Roll-Mecak and McNally, 2010) and actin microfilaments (Ono, 2007). It will also be interesting to elucidate the mechanism and regulation of filament annealing, including how filament annealing compares to the process of filament assembly. Furthermore, it is known that transfected and injected forms of intermediate filaments become incorporated into endogenous networks (Albers and Fuchs, 1989; Vikstrom et al., 1989; Yoon et al., 1998). Future studies will need to address how and where newly synthesized vimentin gets incorporated into preexisting networks.
Taken together, our data show that the dynamics of vimentin filaments can be described by two processes—active transport along microtubules, and constant severing and reannealing of filaments (Figure 9). These processes determine the organization of the vimentin network, which is important for several processes that require cells to change their shape, including cell migration. Future work will be required to address whether the contributions of transport versus severing and annealing can be teased apart and how each process is regulated.
FIGURE 9:
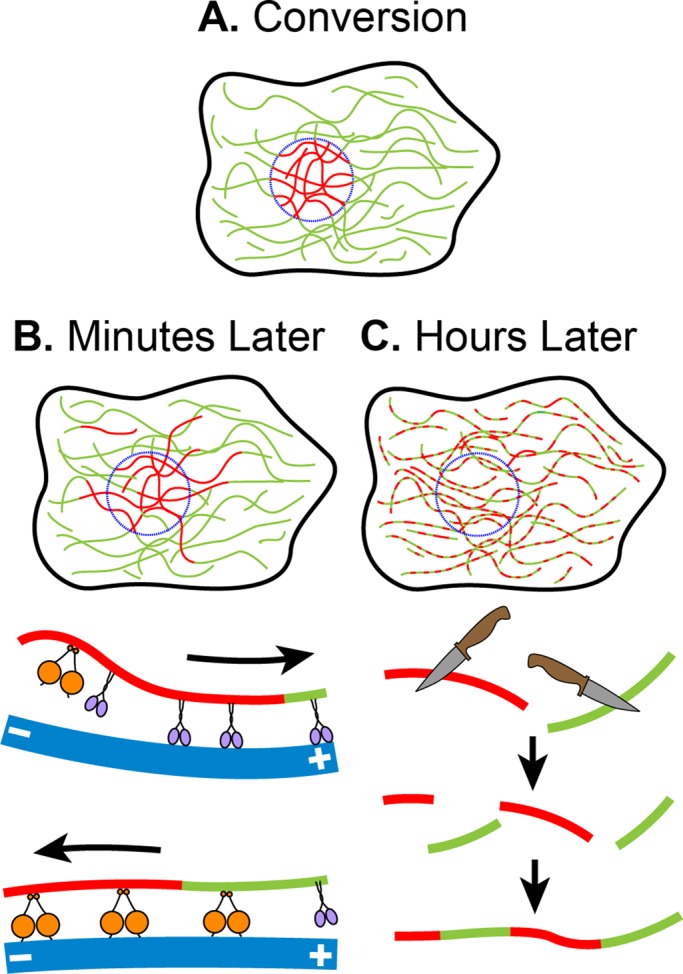
Model of vimentin filament dynamics. (A) Filament dynamics can be followed by producing fiduciary marks on them using photoconversion. (B) Within only minutes, filaments can be seen to be transported outside the region of conversion, likely mediated by microtubule motors kinesin (purple) and dynein (orange). (C) After several hours, converted filaments appear in a patch-like pattern as a result of filament severing and reannealing.
MATERIALS AND METHODS
DNA constructs
PA-GFP-vimentin.
PA-GFP plus linker and human vimentin were amplified by PCR from two separate donor vectors. The amplicons were then inserted into pQCXIP (Clontech, Mountain View, CA) using Clontech In-Fusion cloning kit as recommend by the manufacturer. Next PA-GFP was PCR amplified from this vector. This amplicon and the PA-GFP-vimentin vector were digested with EcoRI, and the digestion products were ligated together. The resulting vector had two copies of PA-GFP separated by TCCGGACTCAGATCTCGAGCTCAAGCTTCGAATTCC placed on the N-terminus of vimentin with the linker TCCGGACTCAGATCTCGAGCTCAAGCTTCGAATTCTGCAGTCGACGGTACCGCGGGCCCGGGATCCATG between the last tag and vimentin.
mEos3.2-vimentin.
Human vimentin was cloned into a pEGFP-C1 vector (Clontech) in which enhanced GFP had been replaced by mEos3.2, creating the linker TCCGGACTCAGATCTGGCAGCGGTGGAGGCAGCGCATCCGGCGGAAGCGGAAGC between mEos3.2 and vimentin. The mEos3.2-vimentin portion of this vector was then amplified by PCR. Next pQCXIP and the PCR amplicon were digested with AgeI and BamHI, and the products were ligated together to produce mEos3.2-vimentin in a viral vector. EB3-GFP was a gift from the Akhmanova lab (Stepanova et al., 2003). mTagRFP-EB3 was cloned as reported previously (Robert et al., 2014).
Cell lines
All cells were maintained at 37°C in 5% CO2. h-TERT RPE cells were maintained in DMEM plus 1 mM sodium pyruvate plus 10% fetal bovine serum (FBS); SW13 cells in Leibovitz's L-15 plus 10% FBS; NIH-3T3, MEF (primary cells at passage 4), and HEK 293-FT in DMEM plus 10% FBS; and CAD cells in 1:1 DMEM/F-12 plus 10% FBS. CAD cells were switched to DMEM/F-12 (without FBS) 16–24 h before imaging to induce differentiation and neurite outgrowth. mEmerald-vimentin RPE cells used in SIM experiments expressed mEmerald-vimentin under the endogenous vimentin promotor. mEmerald-vimentin and mTagRFPT-tubulin coexpressing RPE used in the two-color TIRF-SIM experiments were the mEmerald RPE cells also genome edited to express mTagRFPT-tubulin under the endogenous tubulin promotor. All genome-edited cells were a gift of the Danuser lab. Vimentin cell lines (PA-GFP-vimentin and mEos3.2-vimentin) were created by viral transduction of the wild-type cell lines listed, except MEF and HEK 293-FT. Virus was produced by transfection of HEK 293-FT cells with PA-GFP-vimentin or mEos3.2-vimentin and helper plasmids pVSVG (Clontech) and pCL-Eco (Imgenex, San Diego, CA). Supernatant from transfected cells plus 8 μg/ml Polybrene was filtered and applied to target cell lines for 6–8 h for two consecutive days. Target cells were then selected using puromycin at 0.5 μg/ml (SW13), 3 μg/ml puromycin (CAD and NIH-3T3), or 2 μg/ml (RPE) for 3–5 d. MEF cells were transfected with Eos3.2-vimentin using the Lipofectamine 2000 (Invitrogen) transfection agent per manufacturer's instructions.
APC RNA interference knockdown
A duplex scrambled sequence control and two APC siRNA duplex target sequences (5′-rCrGrA rCrArA rGrArG rCrUrA rGrArA rGrArU rArArU rUrCC A-3′ + 5′-rUrGrG rArArU rUrArU rCrUrU rCrUrA rGrCrU rCrUrU rGrUrC rGrArA-3′ and 5′-rUrGrA rCrArA rUrArA rArGrC rArGrA rGrGrA rArGrG rUrGA T-3′ + 5′-rArUrC rArCrC rUrUrC rCrUrC rUrGrC rUrUrU rArUrU rGrUrC rArUrC-3′) were ordered from Integrated DNA Technologies (Coralville, IA). RPE cells were transfected for two consecutive days for 14–16 h with either a combination of 10 nM of each APC sequence (APC condition) or 20 nM of scrambled sequence (control condition) using HiPerfect (Invitrogen, Carlsbad, CA). Cells were imaged in FluroBrite DMEM (Invitrogen) plus 10% FBS (17 cells in each group across two experiments; also see TIRFM and wide-field fluorescence microscopy) and harvested for Western blotting on the same day, 48 h after the last transfection. Knockdown efficiency was assessed using Western blot analysis using the APC-M2 antibody at 1:5000 (generously gifted to us by Kristi Neufeld, University of Kansas, Lawrence, KS) and Coomassie stain as loading control.
Structured illumination microscopy
Cells were plated on glass coverslips 14–16 h before imaging in FluoroBrite DMEM supplemented with 10% FBS and 1 mM sodium pyruvate. Live-cell TIRF-SIM images were collected on a Nikon Ti-E inverted microscope (Nikon Instruments, Melville, NY) with a SIM illuminator and SIM enclosure equipped with an Apo TIRF 100×/numerical aperture (NA) 1.49 oil objective and electron-multiplying charge-coupled device (EMCCD) camera (iXon DU897; Andor, Belfast, United Kingdom) at the Nikon Center at the Northwestern University Feinberg School of Medicine Center for Advanced Microscopy. Cells were maintained at 37°C during imaging using a Tokai-Hit stage-top incubator (Tokai-Hit, Fujinomiya City, Japan). Images were collected at 1-s intervals and reconstructed using Nikon Elements software. For nocodazole experiments, FluoroBrite medium supplemented as before ± 10 μM nocodazole (Sigma-Aldrich, St. Louis, MO) was placed on cells, and cells were incubated at 4°C for 30 min, warmed to 37°C for 30 min, and then imaged at 37°C. Filament ends were tracked (14 filaments for 7–20 s across four cells) in reconstructed images using the Manual Tracking plug-in in Fiji (ImageJ processing package).
Two-color TIRF-SIM experiments were conducted at the Advanced Imaging Center at the Janelia Research Campus. Cells were prepared as described for the other SIM experiments and kept at 37°C by regulated hot air blown into a plastic enclosure around the microscope stand. Briefly, this system offered the speed necessary to capture transport dynamics in two channels by using a spatial light modulator in place of a rotating diffraction grating. Details of the system have been described (Kner et al., 2009).
TIRFM and wide-field fluorescence microscopy
Live-cell TIRFM images were collected on a Nikon Eclipse U2000 inverted microscope equipped with a Plan-Apo TIRF 100×/1.45 NA objective and a Hamamatsu CMOS Orca Flash 4.0 camera (Hamamatsu Photonics, Hamamatsu, Japan), controlled by MetaMorph 7.7.7.0 software (Molecular Devices, Downingtown, PA). Cells were maintained at 37°C plus 5% CO2 during imaging using a Tokai-Hit stage-top incubator (Tokai-Hit, Fujinomiya, Japan) and Okolab gas mixer (Okolab, Naples, Italy). The angle of a 561-nm laser was manually adjusted until near total internal reflection was reached, as judged by imaging of photoconverted mEos3.2-vimentin–expressing cells. To photoconvert, cells were exposed to UV light from a Hg+ light source for 10 s through a pinhole in the light path. Time-lapse sequences were acquired at 20-s intervals for 3 min using the 561-nm laser. For all experiments except the serum starvation experiment, cells were plated on glass coverslips ∼16 h before imaging.
For nocodazole experiments, cells were treated as described in Structured illumination microscopy (23 control and 19 nocodazole-treated cells). For vinblastine experiments, the medium for cells was switched to 37°C FluoroBrite DMEM plus 10% FBS ± 10 nM vinblastine (Sigma-Aldrich) for 10 min before imaging (16 control and 13 vinblastine-treated cells). For APC experiments, cells were treated as described in APC RNA interference knockdown and imaged in DMEM plus 10% FBS (17 cells in each group). For serum starvation experiments, cells were split to coverslips (in their maintenance medium), left unchanged (control, 10 cells), changed to serum-free medium 12–16 h later, and incubated in the serum-free medium for 72 h before imaging (serum-starved group, 19 cells) or imaged 2–10 min after the serum-free medium was replaced by that containing 10% FBS (serum stimulation group, 15 cells). For TSA experiments, cells were incubated in DMEM plus 10% FBS ± 500 nM TSA for ∼16 h (28 control, 28 TSA treated).
For filament motility, time-lapse sequences were analyzed as described later. For display purposes (but not for analysis), TIRFM images of mEos3.2-vimentin RPE were modified using the Log function in Fiji imaging software, and gamma was adjusted to between 1.3 and 1.4. Similarly, images of mEos3.2-vimentin SW13 cells were logged, but gamma remained at 1.0.
RPE cells were transfected using X-Treme Gene (Roche, Indianapolis, IN) with mTagRFPt-EB3. mTagRFPt-EB3–transfected RPE cells were also imaged at near-TIRF using the 561-nm laser at 2-s intervals for 16 s. These cells were treated ± 10 nM vinblastine as described for mEos3.2-vimentin cells. To display comets, image sequences were color coded using Temporal Color Code in Fiji, which assigns a single color to each frame and superimposes all colored frames. CAD cells were transfected with Lipofectamine 2000 with GFP-EB3. GFP-EB3–transfected CAD cells were imaged using a Plan Apo 100×/1.40 NA objective, Hg+ light source, and Pixis 1024B CCD camera (Princeton Instruments, Roper Industries, Sarasota, FL) at intervals of 1 s for 1 min. An EB3 kymograph was created of a 5- pixel-wide section of GFP-EB3–expressing CAD neurite using the Reslice command in Fiji.
Confocal microscopy
Confocal images were collected on a Nikon Eclipse U2000 inverted microscope equipped with a Yokogawa CSU10 spinning-disk confocal head (Yokogawa Electric Corporation, Sugar Land, TX), a Plan Apo 100×/1.45 NA objective, an Agilent MLC 400 laser set (including 488- and 561-nm lasers; Agilent Technologies, Wood Dale, IL), 89 North Heliophor pumped phosphor light engine at 405 nm (Chroma Technology, Bellows Falls, VT) to drive photoactivation/photoconversion, and an Evolve EMCCD (Photometrics, Tucson, AZ) driven by Nikon Elements software. Cells were maintained at 37°C with 5% CO2 as described earlier. Sections of PA-GFP-vimentin or mEos3.2-vimentin CAD cell neurites were photoconverted with 405-nm light restricted by a pinhole in the light path. Time-lapse sequences in the photoactivated (488 nm, PA-GFP-vimentin CAD) or photoconverted (561 nm, mEos3.2-vimentin CAD) channel were acquired for 3 min at 4 frames/min. Sequences from 32 neurites were visually inspected to determine the direction of movement of photoactivated/photoconverted filaments. For display only, images of PA-GFP-vimentin CAD were adjusted using the Log function in Fiji imaging software. mTagRFPt-tubulin RPE cells were treated ± nocodazole or vinblastine as described for mEos3.2-vimentin cells and then fixed with 0.5% glutaraldehyde plus 0.1% Triton for 5 min. Confocal images of these cells were gathered to record microtubule distribution. Similarly, RPE fixed with 0.5% glutaraldehyde plus 0.1% Triton and immunolabeled with 1:800 anti-vimentin antibody PCK-594P (Covance, Princeton, NJ), mEmerald-vimentin RPE, and mEos3.2-vimentin RPE were imaged on the confocal system to record vimentin distributions in these cells.
Quantification of filaments
Custom software was created in Matlab to process images for quantification of filament transport. Photomicrographs were first flattened with nonlinear mapping to boost the filament in the dimmer region. A Gaussian filter was then applied to reduce noise from the background. Multiple-scale steerable filtering was used to enhance the line features in the image. The non–maximum suppression image of the steerable filtering results were used to segment all possible filaments on their centerline locations. Based on geometrical and imaging features of filaments—the length, curvature, intensity, and steerable filtering response value—filaments were identified out of the whole set. From this, an output filament network was made by creating a binary image of the filament network with 1-pixel-wide filaments. For each image, the total length of filaments was counted by summing the identified pixels. Filament transport was defined as this amount outside the initial region of photoconversion. To account for variations in the initial number of photoconverted filaments between cells, filament transport for each frame was normalized to the sum intensity measured in the region of photoconversion in the first frame. These normalized values were then plotted against time, and slopes were calculated. Finally, these values were normalized to control average, groups were compared using Welch's t test analysis, and bar graphs were plotted using Prism software (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
We thank Kristi Neufeld (University of Kansas, Lawrence, KS) for gifting us the APC-M2 antibody, Christoph Burckhardt (University of Texas Southwestern Medical Center, Dallas, TX) for providing us with the genome-edited cells, Lin Shao and Teng-Leong Chew (Janelia Research Campus, Ashburn, VA) for assistance with two-color TIRF SIM, Michelle A. Baird, Edward R. Kuczmarski, and Amélie Robert for critical reading of the manuscript, and Robert D. Goldman for sharing his everlasting enthusiasm and expertise in the field of intermediate filaments. Research reported in this publication was supported by the National Institute of General Medical Science of the National Institutes of Health under Award P01GM09697. The work of Michael W. Davidson was performed at the National High Magnetic Field Laboratory, supported by National Science Foundation Cooperative Agreement DMR-1157490 and the State of Florida. Single-color TIRF-SIM was performed at the Northwestern University Center for Advanced Microscopy using a Nikon N-SIM system purchased through the support of National Institutes of Health Grant 1S10OD016342, and two-color TIRF-SIM imaging was produced in collaboration with the Advanced Imaging Center, a facility jointly supported by the Gordon and Betty Moore Foundation and the Howard Hughes Medical Institute at Howard Hughes Medical Institute's Janelia Research Campus.
Abbreviations used:
- EB3
end-binding protein 3
- PA-GFP
photoactivatable green fluorescent protein
- SIM
structured illumination microscopy
- TIRFM
total internal reflection fluorescence microscopy.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-09-1398) February 25, 2015.
REFERENCES
- Albers K, Fuchs E. Expression of mutant keratin cDNAs in epithelial cells reveals possible mechanisms for initiation and assembly of intermediate filaments. J Cell Biol. 1989;108:1477–1493. doi: 10.1083/jcb.108.4.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball EH, Singer SJ. Association of microtubules and intermediate filaments in normal fibroblasts and its disruption upon transformation by a temperature-sensitive mutant of Rous sarcoma virus. Proc Natl Acad Sci USA. 1981;78:6986–6990. doi: 10.1073/pnas.78.11.6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlan K, Rossow MJ, Gelfand VI. The journey of the organelle: teamwork and regulation in intracellular transport. Curr Opin Cell Biol. 2013;25:483–488. doi: 10.1016/j.ceb.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colakog˘ lu G, Brown A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J Cell Biol. 2009;185:769–777. doi: 10.1083/jcb.200809166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia I, Chu D, Chou YH, Goldman RD, Matsudaira P. Integrating the actin and vimentin cytoskeletons. adhesion-dependent formation of fimbrin-vimentin complexes in macrophages. J Cell Biol. 1999;146:831–842. doi: 10.1083/jcb.146.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckes B, Dogic D, Colucci-Guyon E, Wang N, Maniotis A, Ingber D, Merckling A, Langa F, Aumailley M, Delouvée A, et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J Cell Sci. 1998;111:1897–1907. doi: 10.1242/jcs.111.13.1897. [DOI] [PubMed] [Google Scholar]
- Esue O, Carson AA, Tseng Y, Wirtz D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J Biol Chem. 2006;281:30393–30399. doi: 10.1074/jbc.M605452200. [DOI] [PubMed] [Google Scholar]
- Foisner R, Malecz N, Dressel N, Stadler C, Wiche G. M-phase-specific phosphorylation and structural rearrangement of the cytoplasmic cross-linking protein plectin involve p34cdc2 kinase. Mol Biol Cell. 1996;7:273–288. doi: 10.1091/mbc.7.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD. The role of three cytoplasmic fibers in BHK-21 cell motility. I. Microtubules and the effects of colchicine. J Cell Biol. 1971;51:752–762. doi: 10.1083/jcb.51.3.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Salmon WC, Waterman-Storer CM. Converging populations of f-actin promote breakage of associated microtubules to spatially regulate microtubule turnover in migrating cells. Curr Biol. 2002;12:1891–1899. doi: 10.1016/s0960-9822(02)01276-9. [DOI] [PubMed] [Google Scholar]
- Gurland G, Gundersen GG. Stable, detyrosinated microtubules function to localize vimentin intermediate filaments in fibroblasts. J Cell Biol. 1995;131:1275–1290. doi: 10.1083/jcb.131.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoeva FK, Gelfand VI. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Helfand BT, Mendez MG, Murthy SN, Shumaker DK, Grin B, Mahammad S, Aebi U, Wedig T, Wu YI, Hahn KM, et al. Vimentin organization modulates the formation of lamellipodia. Mol Biol Cell. 2011;22:1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Mikami A, Vallee RB, Goldman RD. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J Cell Biol. 2002;157:795–806. doi: 10.1083/jcb.200202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Pallari H-M, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313:2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Euteneuer U, Traub P, Schliwa M. Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J Cell Biol. 1991;113:155–160. doi: 10.1083/jcb.113.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly AL, Kim H, Srinivasan D, Lakonishok M, Larson AG, Gelfand VI. Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc Natl Acad Sci USA. 2010;107:12151–12156. doi: 10.1073/pnas.1004736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, McCulloch CA. Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 2011;585:18–22. doi: 10.1016/j.febslet.2010.11.033. [DOI] [PubMed] [Google Scholar]
- Kirmse R, Portet S, Mücke N, Aebi U, Herrmann H, Langowski J. A quantitative kinetic model for the in vitro assembly of intermediate filaments from tetrameric vimentin. J Biol Chem. 2007;282:18563–18572. doi: 10.1074/jbc.M701063200. [DOI] [PubMed] [Google Scholar]
- Kner P, Chhun BB, Griffis ER, Winoto L, Gustafsson MG L. Super-resolution video microscopy of live cells by structured illumination. Nat Methods. 2009;6:339–342. doi: 10.1038/nmeth.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheyses AL, Simon SM, Rappoport JZ. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG, Kojima S-I, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott RE, Helmke BP. Mapping the dynamics of shear stress-induced structural changes in endothelial cells. Am J Physiol Cell Physiol. 2007;293:C1616–C1626. doi: 10.1152/ajpcell.00457.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray ME, Mendez MG, Janmey PA. Substrate stiffness regulates solubility of cellular vimentin. Mol Biol Cell. 2014;25:87–94. doi: 10.1091/mbc.E13-06-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nöding B, Herrmann H, Köster S. Direct observation of subunit exchange along mature vimentin intermediate filaments. Biophys J. 2014;107:2914–2922. doi: 10.1016/j.bpj.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int Rev Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Herrmann H, Davidson MW, Gelfand VI. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases. FASEB J. 2014;28:2879–2890. doi: 10.1096/fj.14-250019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, McNally FJ. Microtubule-severing enzymes. Curr Opin Cell Biol. 2010;22:96–103. doi: 10.1016/j.ceb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosevear ER, McReynolds M, Goldman RD. Dynamic properties of intermediate filaments: disassembly and reassembly during mitosis in baby hamster kidney cells. Cell Motil Cytoskeleton. 1990;17:150–166. doi: 10.1002/cm.970170303. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Boeda B, Etienne-Manneville S. APC binds intermediate filaments and is required for their reorganization during cell migration. J Cell Biol. 2013;200:249–258. doi: 10.1083/jcb.201206010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss C, Napirei M, Meller K. Impairment of anterograde and retrograde neurofilament transport after anti-kinesin and anti-dynein antibody microinjection in chicken dorsal root ganglia. Eur J Cell Biol. 2005;84:29–43. doi: 10.1016/j.ejcb.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20:4997–5006. doi: 10.1091/mbc.E09-04-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Colakg˘ lu G, Wang L, Monsma PC, Brown A. Severing and end-to-end annealing of neurofilaments in neurons. Proc Natl Acad Sci USA. 2013;110:E2696–E2705. doi: 10.1073/pnas.1221835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom KL, Borisy GG, Goldman RD. Dynamic aspects of intermediate filament networks in BHK-21 cells. Proc Natl Acad Sci USA. 1989;86:549–553. doi: 10.1073/pnas.86.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikstrom KL, Lim SS, Goldman RD, Borisy GG. Steady state dynamics of intermediate filament networks. J Cell Biol. 1992;118:121–129. doi: 10.1083/jcb.118.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RK, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- Yang HY, Lieska N, Goldman AE, Goldman RD. A 300,000-mol-wt intermediate filament-associated protein in baby hamster kidney (BHK-21) cells. J Cell Biol. 1985;100:620–631. doi: 10.1083/jcb.100.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Moir RD, Prahlad V, Goldman RD. Motile properties of vimentin intermediate filament networks in living cells. J Cell Biol. 1998;143:147–157. doi: 10.1083/jcb.143.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.