

In differentiated rodent hippocampal neurons, a FRET-based activity reporter reveals distinct spatiotemporal activity patterns of BRSK and AMPK, two homologous kinases that play important roles in neuronal polarity. AMPK exhibits maximal stimulated activity, whereas BRSK displays polarized basal activity in the distal region of the axon and axon tips.
Abstract
5′-Adenosine monophosphate–activated protein kinase (AMPK) is a master metabolic regulator that has been shown to inhibit the establishment of neuronal polarity/axogenesis under energy stress conditions, whereas brain-specific kinase (BRSK) promotes the establishment of axon-dendrite polarity and synaptic development. However, little information exists regarding the localized activity and regulation of these kinases in developing neurons. In this study, using a fluorescence resonance energy transfer (FRET)-based activity reporter that responds to both AMPK and BRSK, we found that BRSK activity is elevated in the distal region of axons in polarized hippocampal neurons before any stimulation and does not respond to either Ca2+ or 2-deoxyglucose (2-DG) stimulation. In contrast, AMPK activity is stimulated by either Ca2+ or 2-DG in the soma, dendrites, and axons of hippocampal neurons, with maximal stimulated activity observed in the distal region of the axon. Our study shows that the activities of both AMPK and BRSK are polarized in developing hippocampal neurons, with high levels in the distal region of extended axons.
INTRODUCTION
5′-Adenosine monophosphate–activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine kinase that serves as a sensor of cellular and organismal energy balance and stress (Carling et al., 2012; Hardie et al., 2012). AMPK is activated by physiological (hormones) or pathophysiological (cellular stress) signals that either elevate intracellular Ca2+ or produce decreases in cellular ATP levels, resulting in an increase in AMP and ADP levels. AMPK is maximally activated when threonine 172 (T172) in the activation loop is phosphorylated and when AMP or ADP is bound (Hardie et al., 2011). Two upstream kinases—the tumor suppressor LKB1 (liver kinase B1) and (Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ)—phosphorylate T172 of the α subunit. It has been proposed that phosphorylation by LKB1 is AMP dependent, whereas phosphorylation by CaMKKβ is AMP independent and enhanced by elevated intracellular Ca2+ levels (Witters et al., 2006). On its activation, AMPK restores energy balance by activating energy-producing catabolic pathways and inhibiting energy-consuming anabolic pathways, as well as by regulating transcription to achieve both short- and long-term adaptive responses in a wide range of tissues (Ronnett et al., 2009; Mihaylova and Shaw, 2011). In the context of neuronal development, one group using a genetic loss-of-function approach showed that under normal conditions, AMPK catalytic activity is not required for neurogenesis, polarization, or survival in vivo (Williams et al., 2011). However, during neuronal polarization, metabolic stress results in the activation of AMPK, leading to the inhibition of axon formation and growth in a mechanistic target of rapamycin– and Akt-dependent manner (Amato et al., 2011; Williams et al., 2011).
The AMPK kinase complexes (denoted by the catalytic isoforms α1 or α2) are members of a larger family of 13 AMPK-related kinases that are also activated by the upstream LKB1 kinase (Manning et al., 2002; Lizcano et al., 2004). Among the AMPK-related kinases, the brain-specific kinases 1 and 2 (BRSK1 and BRSK2, also known as SAD-B and SAD-A, respectively) share significant homology with AMPK α1 and α2 in their kinase domains (Alessi et al., 2006). The BRSKs are highly expressed in neurons, and it has been demonstrated that activation of LKB1 and BRSK promotes the establishment of neuronal axon–dendrite polarity and synaptic development both in vivo and in vitro (Kishi et al., 2005; Inoue et al., 2006; Barnes et al., 2007; Shelly et al., 2007; Choi et al., 2008).
Although both AMPK and BRSK play important roles in developing neurons, their regulation in developing neurons is not completely understood. For example, in addition to LKB1, there is conflicting evidence that the Ca2+/CaMKK and cAMP/protein kinase A (PKA) pathways stimulate BRSK activity (Guo et al., 2006; Bright et al., 2008; Fujimoto et al., 2008; Thornton et al., 2011). Furthermore, although both AMPK and BRSK are involved in the regulation of cell polarity in neurons and other cell types, little information exists regarding their spatiotemporal activation patterns in polarized cell types (Hardie, 2011).
Given the complexity of AMPK and BRSK signaling and the important roles they play in neurons as well as in a multitude of other cell types, our understanding would benefit significantly by methods that allow monitoring of kinase activity in a native context. Genetically encodable fluorescence resonance energy transfer (FRET)–based reporters are powerful tools that can be used to visualize signaling dynamics with high spatiotemporal resolution. A FRET-based AMPK activity reporter (AMPKAR; Tsou et al., 2011) has been developed based on the general design of genetically encoded kinase activity reporters (Oldach and Zhang, 2014). In this reporter, a fluorescent protein FRET pair flanks a kinase activity–dependent molecular switch comprising an AMPK substrate sequence optimized using a positional scanning peptide library screen (Turk et al., 2006) and forkhead-associated domain 1 (FHA1), a protein module that binds to the phosphorylated substrate. Activation of AMPK and consequent phosphorylation of the substrate motif within the reporter drive a conformational change in the molecular switch, which alters the relative distance and orientation of the two fluorophores and resulting in an increase in FRET. In this study, we report the improvement and further characterization of this FRET-based kinase activity reporter. Using the improved reporter, we investigated AMPK and BRSK activation pathways in discrete subcellular locations in primary cultures of developing rat hippocampal neurons and discovered distinct spatiotemporal activity patterns for these two kinases.
RESULTS
Improvement of a FRET-based reporter
In the previously described AMPKAR (Tsou et al., 2011), the FRET pair consists of an enhanced cyan fluorescent protein (ECFP) as the donor fluorophore and a yellow fluorescent protein variant, cpVenusE172, as the acceptor fluorophore (Herbst et al., 2009). The substrate sequence within this reporter is MRRVATLVDL, in which the threonine is phosphorylated by AMPK (Tsou et al., 2011). To facilitate the detection of subtle changes in AMPK activity, we set out to further improve the reporter. We previously showed that kinase activity reporters benefit from using brighter variants of CFP and yellow fluorescent protein (YFP; Allen and Zhang, 2006; Depry et al., 2011). We replaced the ECFP from the reporter with a brighter cyan variant, cerulean3 (Figure 1A; Markwardt et al., 2011). Similar to the original reporter, Cos7 cells transfected with the new reporter displayed a uniform distribution of fluorescence throughout the cell (Figure 1B). When these cells were stimulated with 20 mM 2-deoxyglucose (2-DG), an inhibitor of glycolysis and activator of AMPK that exerts its effects through LKB1, we observed a robust and sustained response of 39.4 ± 2.1% (Figure 1C). This response was measured as an increase in the ratio of YFP emission to CFP emission upon CFP excitation. On comparison to the original reporter, which showed a response of 18.3 ± 1.7% in Cos7 cells, the new reporter exhibited twofold-enhanced response (Figure 1D). To test its response to AMPK activation mediated by CaMKKβ, we treated Cos7 cells with 1 μM ionomycin, a calcium ionophore. This reporter showed a rapid and transient response of 51.5 ± 3.1% (Figure 1E), doubling the original reporter response of 24.4 ± 5.1% (Figure 1F). To ensure that the observed responses were due to phosphorylation of the threonine in the substrate motif of the reporter, we mutated this threonine to an alanine and tested the mutant reporter. Cos7 cells expressing this mutant reporter did not respond to either 2-DG (Figure 1C) or ionomycin (Figure 1E), suggesting that the responses seen with the reporter were indeed dependent on phosphorylation of the designated threonine.
FIGURE 1:

Development of an improved AMPK activity reporter and its characterization in Cos7 cells. (A) Domain structure of the original and improved reporter, composed of a CFP variant, FHA1 phospho–amino acid–binding domain, AMPK substrate motif, and a variant of YFP. (B) YFP direct images of the original and improved reporter show uniform and similar expression patterns throughout the cell. (C) Time course of average response of the improved activity reporter (black curve; number of cells n = 8) and phosphorylation-deficient T/A mutant (red curve; n = 4) to 20 mM 2-DG stimulation. Reporter responses are normalized to the ratio before the point of drug addition. (D) Bar graph response comparison of original and improved activity reporters to 2-DG stimulation. (E) Time course of average responses of the improved reporter to 1 μM ionomycin and 10 mM Ca2+ stimulation (black curve; n = 9) and T/A mutant (red curve; n = 3). (F) Bar graph response comparison of original and improved activity reporters to ionomycin stimulation.
Visualizing AMPK activity in developing rat hippocampal neurons
Next we set out to analyze the activity and regulation of AMPK in developing neurons. It has been shown that AMPK plays a crucial role in the regulation of axon initiation, as its activation has been shown to halt the process of axon specification and growth, leading to a loss of neuronal polarization (Amato et al., 2011; Williams et al., 2011). Although AMPK is expressed throughout the neuron, it is not known whether it is differentially active in axons versus dendrites in polarized neurons (Turnley et al., 1999). In the first series of experiments, we examined primary rat undifferentiated hippocampal neurons (HNs) characterized by multiple neurites but lacking distinct axon–dendrite polarity (Figure 2). When these neurons at 1 d in vitro (DIV 1) expressing the FRET-based reporter were stimulated with 2-DG to activate AMPK (Supplemental Figure S1), we observed a sustained response of 9.0 ± 1.1% in the cell body and 9.7 ± 0.5% in the neurites (Figure 2C). Addition of ionomycin resulted in an emission ratio change of 29.30 ± 3.7% in the cell body and 31.0 ± 2.3% in the neurites (Figure 2D). The lower response to 2-DG than with ionomycin could be due to the fact that the majority of the upstream kinase LKB1 is in the nucleus at this stage (Shelly et al., 2007). These data suggest that both energetic stress (through 2-DG stimulation) and calcium influx (through ionomycin stimulation) uniformly activate AMPK in undifferentiated neurons.
FIGURE 2:

Undifferentiated rat hippocampal neurons show uniform stimulated AMPK activity. (A) Pseudocolor images of undifferentiated neurons showing the response to ionomycin followed by Ca2+. Scale bar, 10 μm. (B) Bar graphs representing starting ratios in the cell body and neurites (n = 9). (C) Representative response curves of the improved reporter to 40 mM 2-DG stimulation in the cell body (black curve) and neurites (gray curve). (D) Representative response curves of the improved reporter to 1 μM ionomycin and 10 mM Ca2+ in cell body (black curve) and neurites (gray curve). The data are from at least five independent hippocampal neuron culture preparations.
In the next series of experiments, we investigated whether the reporter responses displayed any spatial differences in differentiated rat HNs on DIV 5 that have a discrete axon–dendrite polarity (Supplemental Figure S2) and also high expression levels of AMPK subunits (Ramamurthy et al., 2014). For the purposes of the analysis of the responses, the neurons were segmented into the cell body and dendrites, and the axons were divided into three equal regions of interest up to the point of branching into the axon terminal (Supplemental Figure S2B). On stimulation of these cells, the cell body, the dendrites, and the axonal region immediately proximal to the cell body showed transient but robust Ca2+-induced responses, but, intriguingly, the distal region of the axon did not respond (Figure 3C). When we analyzed the starting emission ratios—the ratio of the acceptor fluorophore emission to the donor fluorophore emission in resting cells—we found that the distal region of the axon displayed high starting emission ratios, suggesting that the probe could be basally phosphorylated (Figure 3B). In contrast, the starting ratios within neurites at DIV 1 were uniform and similar to that of the cell body (Figure 2B). In addition, in a separate experiment, differentiated neurons expressing the T/A mutant reporter showed no Ca2+-mediated responses or spatial difference in starting ratios, indicating that the observed high starting ratio in the distal region of the axon was due to basal phosphorylation of the probe (Figure 3D). When differentiated neurons expressing the wild-type reporter were probed for AMPK responses via 2-DG stimulation (Supplemental Figure S1), we found a similar trend of high starting ratios and no 2-DG–stimulated responses in the distal region of the axon (Figure 3E). Figure 4 summarizes the distinct reporter responses at each subcellular location of differentiated (Figure 4A) and undifferentiated neurons (Figure 4B). The key spatial difference was apparent in DIV 5 HNs when the neurites/dendrites and proximal regions of the axon were compared to the distal region of the axon, where 2-DG or Ca2+-mediated activation did not elicit a response, but maximum basal activity was observed.
FIGURE 3:

Differentiated rat hippocampal neurons show spatial differences in stimulated AMPK activity. (A) Pseudocolor images of differentiated neurons treated with ionomycin. Scale bar, 20 μm. (B) Bar graphs depicting starting ratios in different regions of the neuron (n = 10). High starting ratios are observed in the distal region of the axon. Starting ratio comparisons between different regions and the cell body in differentiated neurons were performed using one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. (C) Representative response curve of the reporter to 1 μM ionomycin in cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost). The data are from at least five independent hippocampal neuron culture preparations. (D) Time course response of a phosphorylation-deficient version of the improved activity reporter in differentiated neurons. No responses were observed in cell body (black curve), neuritis (gray curve), and axon purple curves) upon ionomycin stimulation. (E) Representative response curve of the reporter to 40 mM 2-DG stimulation in the cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost).
FIGURE 4:
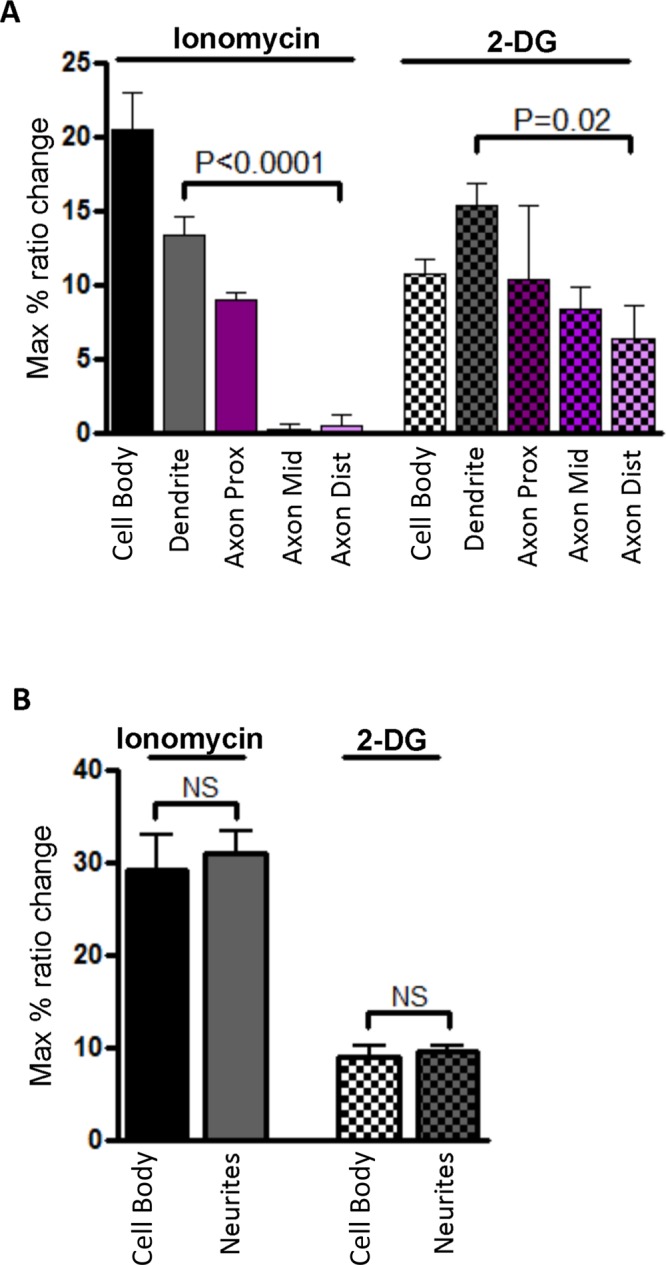
Quantification of multiple independent experiments for compartmentalized responses of the reporter in developing neurons shows spatial differences in differentiated neurons. (A) Bar graph of responses in differentiated neurons (DIV 5) depicting stimulated activity in the cell body, dendrites, and axon (n = 3 for ionomycin and n = 4 for 2-DG). Owing to similar levels of reporter expression, reporter responses in dendrites were compared with the distal region of the axon by performing unpaired t test followed by Welch's correction. (B) Bar graph of responses in undifferentiated neurons (DIV 1) depicting stimulated activity in the cell body and neurites (n = 6 for ionomycin and n = 5 for 2-DG). Reporter responses were compared by unpaired t tests followed by Welch's correction. The data are from three to five independent experiments per stimulated condition.
To probe further this unique response pattern, we used short hairpin RNAs (shRNAs) to knock down the AMPK α1 and α2 catalytic subunits in HNs. Although the shRNA constructs efficiently knocked down both AMPK subunits (Figure 5A), the DIV 5 HNs continued to exhibit high basal activity, as visualized by the elevated starting ratios (Figure 5B). However, these cells were largely unresponsive to either 1 μM ionomycin or 40 mM 2-DG (Supplemental Figure S3), indicating that stimulated responses to 2-DG or ionomycin depend on the presence of AMPK. In similar experiments, we also knocked down the AMPK α1 subunit in HNs from AMPK α2–knockout mice and monitored the reporter responses. Stimulation with either 2-DG or ionomycin did not induce an emission ratio change in any region of the neuron (Figure 5, C and D). Collectively these data demonstrate that the stimulated reporter responses, that is, changes in the emission ratios, are due to stimulated increases in AMPK activity. However, the polarized activity in the basal state as indicated by the high starting emission ratios is not dependent on AMPK activity.
FIGURE 5:

Knockdown of AMPK α1 subunits in α2-knockout hippocampal neurons abolishes stimulated response. (A) Western blot showing knockdown efficiency of the AMPK α1/2 shRNA constructs in rat hippocampal neurons. (B) Bar graphs of starting ratios in different subcellular compartments of differentiated neurons (n = 4). Knockdown of α1/2 subunits does not abolish high starting ratios in the distal region of the axon. Starting ratio comparisons between different regions and the cell body in differentiated neurons were performed using one-way ANOVA followed by Dunnett's multiple comparison test. (C) Representative response curves of the improved reporter to 40 mM 2-DG stimulation in the cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost) upon AMPKα1 knock down in α2 knockout hippocampal neurons. (D) Representative response curves of the improved reporter to 1 μM ionomycin and 10 mM Ca2+ in cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost). The data are from at least five independent hippocampal neuron culture preparations.
BRSK1/2 phosphorylate the FRET-based reporter
Although this reporter was developed based on a consensus AMPK substrate sequence (Tsou et al., 2011), the possibility existed that other, related kinases could phosphorylate this reporter in a cellular context. Therefore we considered that the high basal starting ratios observed in the distal region of DIV 5 neuron axon could derive from an AMPK-related kinase. Among the AMPK-related kinases, BRSK1 and BRSK2 share significant homology with AMPKα within their kinase domains and substrate recognition regions. BRSK2 has 65% homology with the α1 subunit of AMPK, and BRSK2 is 49% homologous with α2. Both BRSK1 and 2 are highly expressed in the brain and have been implicated in establishing neuronal polarity and promoting synaptogenesis (Kishi et al., 2005; Inoue et al., 2006; Barnes et al., 2007). Although the regulation of BRSK1/2 is not fully understood, LKB1 has been established as an upstream kinase (Lizcano et al., 2004; Bright et al., 2008). However, whereas one report provided evidence that CaMKKα can also activate BRSK1/2 (Fujimoto et al., 2008), another study showed that BRSK1/2 are not responsive to ionomycin stimulation and increases in cytosolic calcium (Thornton et al., 2011). Initially, to determine whether the reporter is indeed a substrate of BRSK1 and BRSK2, we performed experiments in HeLa cells, which do not natively express the BRSK1/2-activating kinase LKB1. To avoid nonspecific effects of AMPK or other related kinases, we generated constitutively active and kinase-deficient mutants of BRSK1 and BRSK2 (Lizcano et al., 2004). We cotransfected cells with constitutively active BRSK1 (T189E), kinase- deficient BRSK1 (K63R), constitutively active BRSK2 (T175E), or kinase-deficient BRSK2 (K49R) along with the FRET reporter. Subsequently, cells were harvested and lysates probed with a phospho-AMPK substrate antibody that was raised against the consensus AMPK substrate motif also found in this reporter. Both active BRSK1 and BRSK2 produced a threefold increase in the levels of phosphorylated reporter (Figure 6, A and B) compared with the kinase-deficient variants. In contrast, microtubule affinity–regulated kinase (MARK), another AMPK-related kinase (Drewes et al., 1997; Lizcano et al., 2004; Li and Guan, 2013) that has also been shown to be present in neurons and regulate axon formation (Biernat et al., 2002), did not phosphorylate the reporter (Supplemental Figure S4). These results provide evidence that in addition to AMPK, this reporter can also report on BRSK1/2 but not MARK activity in cells. We therefore named it AMPK and BRSK dual-activity reporter (ABKAR).
FIGURE 6:
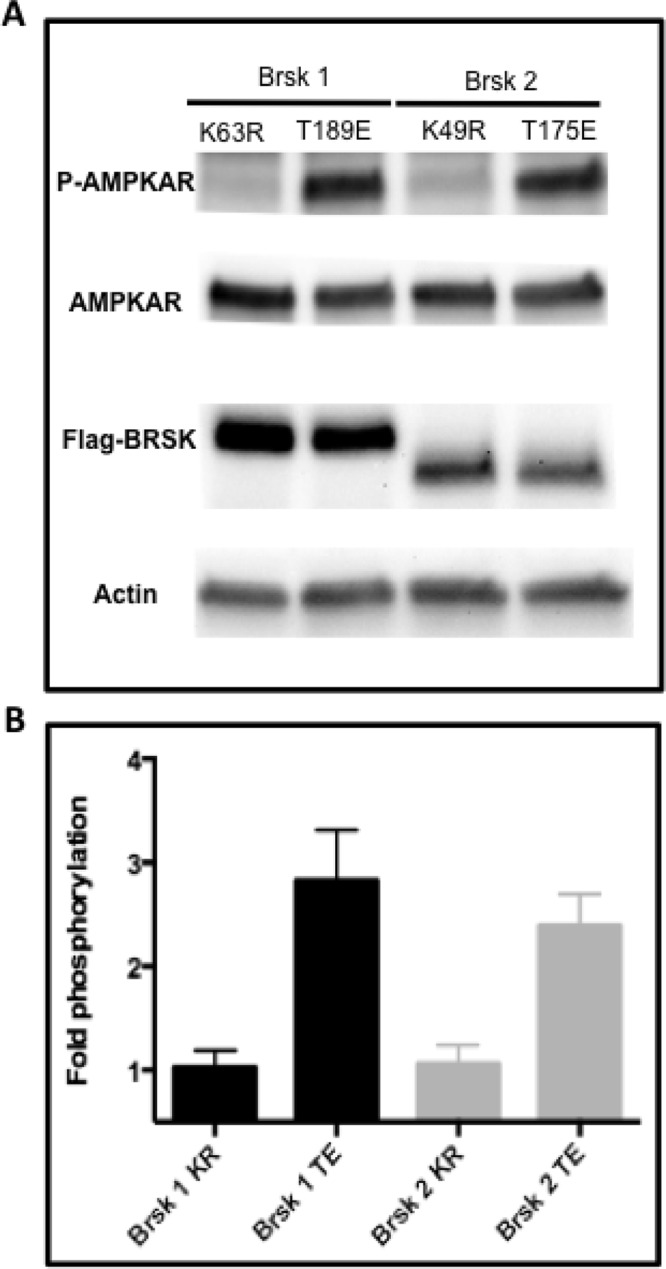
The activity reporter is a substrate for BRSK1 and BRSK2 and therefore named the AMPK and BRSK dual-activity reporter (ABKAR). (A) Western blot showing that ABKAR is a substrate for BRSK1/2. (B) Immunoblot analysis using a phospho-AMPKAR substrate antibody shows increased levels of phosphorylation of the reporter in the presence of constitutively active BRSK1 (T189E) and BRSK2 (T175E) compared with catalytically inactive BRSK1 (K63R) and BRSK2 (K49R). HeLa cells coexpressing BRSK1/2 along with the reporter were used for Western blotting. The data are from four independent experiments.
Polarized BRSK and AMPK activities in growing axons
In the next series of experiments, we investigated whether it is BRSK1/2 that contribute to the unique basal response pattern of ABKAR in the distal region of axons in differentiated HNs. Genetic ablation of BRSK1/2 in an undifferentiated neuron results in a cell that fails to form distinct axons and dendrites (Kishi et al., 2005). Therefore, to determine whether BRSK basally phosphorylated ABKAR in a polarized neuron, we transfected HNs with shRNAs for BRSK1 and BRSK2 along with ABKAR at DIV 3, when axon–dendrite polarity is already established. When observed on DIV 5, although the expressions of BRSK1 and BRSK2 were reduced (Figure 7A), these neurons exhibited normal polarization (Supplemental Figure S5). In these cells, the spatial difference in the starting ratios was completely abolished (Figure 7B), indicating that the phosphorylation level of the probe was now comparable between different compartments of differentiated neurons. These data suggest that BRSK1/2 are basally active, with highest activity observed in the distal region of the axon.
FIGURE 7:

BRSK1 and 2 knockdown in differentiated rat hippocampal neuron does not abolish the stimulated response of the reporter and reveals polarized AMPK activity. (A) Western blot showing knockdown of BRSK1 and BRSK2. (B) Bar graphs of starting ratios in different regions of differentiated neurons (n = 3). Knockdown of BRSK results in abolition of high starting ratios in the distal region of the axon. (C) Representative response curve of diffusible ABKAR to 40 mM 2-DG stimulation in the cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost). (D) Representative response curves of diffusible ABKAR to 1 μM ionomycin in cell body (black curve), neurites (gray curve), and axon (purple curves, rightmost). The data are from at least three independent hippocampal culture preparations.
In these cells with BRSK1/2 knockdown, the AMPK activities stimulated by 2-DG (Figure 7C) and ionomycin (Figure 7D) were clearly observed. As shown in Figure 7, the distal region of the axon exhibited maximal AMPK responses to 2-DG and ionomycin stimulation. Thus, although AMPK is known to be ubiquitously present throughout neurons, these data demonstrate that in developing neurons, stimulated AMPK activity is polarized, with elevated activity in the distal region of axons.
DISCUSSION
AMPK has been designated a master energy sensor and has a well-established role in the regulation of metabolism. Evidence has emerged to suggest that AMPK also has critical functions in the regulation of cell growth, autophagy, and cellular morphology (Carling et al., 2012). These effects involve signaling from the upstream kinase LKB1, a tumor suppressor and established regulator of cell polarity (Alessi et al., 2006). Studies have demonstrated that the LKB1-AMPK pathway also plays a role in neuronal polarization. Although AMPK is not required for normal CNS development or neuronal polarization, it regulates axogenesis under cellular stress conditions (Amato et al., 2011; Williams et al., 2011). It was determined that stress-activated AMPK prevents the establishment of proper neuronal morphology through the phosphorylation of the kinesin light chain of the Kif5 motor protein and mislocalization of phosphatidylyinositol-4,5-bisphosphate 3-kinase from the growing axon tip. Two other AMPK-related kinases, BRSK1 and BRSK2, are also highly expressed in the mammalian brain and are crucial for neuronal development. Mammalian BRSK1 and BRSK2 are evolutionarily conserved kinases with orthologues in Caenorhabditis elegans and Drosophila (Crump et al., 2001; Kishi et al., 2005; Shelly et al., 2007). Several studies reported that mutant or knockout animals exhibit impaired synaptic vesicle clustering, defective synaptogenesis, and absence of discrete axon–dendrite polarity (Crump et al., 2001; Kishi et al., 2005; Lilley et al., 2014).
Although it is known that AMPK, BRSK, and their upstream kinases are highly expressed in neurons, controversy exists regarding how this pathway is activated in these cells. Initial in vitro studies in heterologous systems showed that LKB1 is an upstream kinase for both AMPK and BRSK (Lizcano et al., 2004). However, whereas one group demonstrated LKB1-mediated phosphorylation of AMPK in neuronal cells, another study showed no reduction in AMPK phosphorylation but an absence of BRSK phosphorylation in LKB1-deficient brains (Barnes et al., 2007; Bright et al., 2008). In addition, numerous studies have established a role for CaMKKβ in the Ca2+-mediated activation of AMPK in the brain and other tissues (Racioppi and Means, 2012). With regard to Ca2+-mediated BRSK activation, one study identified CaMKKα to be an upstream activator of BRSK, whereas other studies provided evidence that neither CaMKKβ nor Ca2+ leads to BRSK activation (Bright et al., 2008; Fujimoto et al., 2008; Fogarty et al., 2010; Thornton et al., 2011). Furthermore, PKA-dependent activation of BRSK2 was suggested in one study; however, this finding could not be demonstrated in neuronal cells (Guo et al., 2006; Bright et al., 2008). In this work, using a FRET-based kinase activity reporter, we examined kinase activities in either the basal or the stimulated state in undifferentiated and differentiated rodent hippocampal neurons. We demonstrated that acute stimuli, such as changes in ATP or Ca2+ levels, induce AMPK activation, presumably through LKB1 and CaMKKβ, respectively. In contrast, BRSK is basally active but not sensitive to changes in ATP or Ca2+ levels in neurons.
Substrate-specific regulatory mechanisms may account for the observed differences. AMPK is under tighter intrinsic and extrinsic regulatory control than BRSK1/2. This regulation involves an autoinhibitory sequence within the catalytic domain that binds the catalytic site in the inactive state, as well as phosphatase-mediated dephosphorylation of T172 in the activation loop, which imparts minimal activity in the basal state (Pang et al., 2007; Chen et al., 2009; Hardie et al., 2012). This regulation pattern is consistent with the idea that basally active BRSK is required for neuronal development and polarization, whereas AMPK remains inactive until it is activated to regulate axogenesis under cellular stress conditions. Thus, under the stimulated conditions of energetic stress, axonal LKB1 activates AMPK and inhibits polarization and axon growth (Amato et al., 2011; Williams et al., 2011), consistent with our observations of maximal AMPK activity after 2-DG stimulation. Future studies will focus on using ABKAR in AMPK α double-knockout mice to determine precisely the onset of BRSK activity in developing neurons.
Kinase activities are often spatially compartmentalized, and this spatial activation pattern can provide hints about their regulatory roles (Fuller et al., 2008; Lim et al., 2008). Available activation-specific antibodies have limited specificity in immunocytochemistry. Hence visualization of the activation patterns of AMPK and BRSK in neurons has been difficult. In this study, we used a FRET-based biosensor, ABKAR, for analyzing the dynamic and spatially compartmentalized signaling activities of AMPK and BRSK in developing neurons. The dual specificity of ABKAR, when combined with specific knockdown via shRNA, allowed the elucidation of unique activity patterns of both these kinases within one cell type. In our study, we found that the activities of both AMPK and BRSK are polarized but under different conditions. In differentiated cells, significant basal ABKAR phosphorylation that was not present in undifferentiated neurons was detected in the distal region of the axon in neurons exhibiting distinct axon–dendrite morphologies. This basal ABKAR phosphorylation was abolished by the knockdown of BRSK1/2 but not AMPK1/2. Under stimulated conditions, whereas undifferentiated neurons exhibited uniform reporter responses, differentiated neurons displayed higher responses in the distal region of the axon compared with other regions of the axon and dendrites. Knockdown experiments revealed that this polarized, stimulated activity was due to AMPK1/2 but not BRSK1/2. This ability to visualize both AMPK and BRSK activity with ABKAR in differentiated neurons presents a unique platform to further dissect the molecular mechanisms regulating two related yet nonredundant kinases at the distal region of the axon, a hub for neuronal activity. The asymmetric basal activity of BRSK in axons may contribute to effects of LKB1 during axon initiation and extension. Furthermore, studies (Inoue et al., 2006; Lilley et al., 2014) suggest that BRSKs play a role in nerve terminal differentiation and neurotransmitter release. Thus, polarized BRSK activity may be involved in BRSK's roles in the axon at multiple stages of development. Although less is known regarding the stimulated asymmetric AMPK activity, it is possible that AMPK activation in axons may modulate growth, path finding, or synaptogenesis under normal or pathological conditions.
Regarding the mechanisms underlying polarized activity patterns of BRSK and AMPK, it has been shown that CaMKK, LKB1, AMPK, and BRSK are not spatially restricted and are expressed in both the somatodendritic and axonal regions in neurons (Turnley et al., 1999; Sakagami et al., 2000; Kishi et al., 2005; Barnes et al., 2007; Shelley et al., 2007; Supplemental Figure S6). In contrast, phosphorylated LKB1 (at serine 431) was localized to growing axons. Although it is controversial whether PKA or other kinase-mediated phosphorylation at serine 431 regulates LKB1 activity (Sapkota et al., 2001; Fogarty and Hardie, 2009), it is evident that pSer 431 promotes accumulation of the LKB1/STRAD/MO25 complex in growing axons, and mutation of serine 431 to alanine abolishes axon formation (Barnes et al., 2007). Thus, polarized distribution of phosphorylated LKB1 may contribute to the polarized BRSK1/2 activity observed here. However, the previous model proposes uniform accumulation of active LKB1 throughout the length of the growing axon (Shelly et al., 2007). Yet our studies indicate that in a differentiated neuron, there is a gradient of basal BRSK activity or stimulated AMPK activity, the two major downstream substrates of LKB1. Future studies will examine directly LKB1 activity under these conditions to further our understanding of the establishment of these gradients of activity. Together the findings in this study lead to a better understanding of the spatiotemporal activation patterns of the two related kinases that play pivotal roles in neuronal polarity and energy regulation. In polarized neurons, BRSK is basally active, with higher activity in the axon tip. In contrast, energetic stress or Ca2+ influx results in AMPK activation throughout the neuron, with maximal activity in the distal region of the axon. Thus two homologous kinase family members, AMPK and BRSK1/2, are distinctly regulated in a coordinated manner.
MATERIALS AND METHODS
All animal experiments in this study were conducted in accordance with the National Institutes of Health Guide for the care and use of laboratory animals, and all protocols were approved by the Johns Hopkins University Institutional Animal Care and Use Committee. All cell culture materials were from Invitrogen (Carlsbad, CA), and other reagents were from either Invitrogen or Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
ABKAR gene construction
Cerulean3 was PCR amplified using primers containing BamHI and SphI cut sites. ABKAR was generated by ligating the PCR product into the BamHI and SphI sites of the bacterial pRSETB (Invitrogen) AMPKAR vector and subcloning into the pcDNA3 mammalian expression vector (Invitrogen; Tsou et al., 2011).
Kinase gene construction
Flag-tagged mouse BRSK1 (NM_001003920) and BRSK2 (NM_001009929) were generated by PCR cloning. Total RNA was isolated from the BALB/c mouse cortex using the TRIzol reagent (Invitrogen). Primers containing 5′-BamHI and 3′-EcoRI restriction sites were used to amplify full-length BRSK1 and BRSK2 using the Superscript One-Step RT-PCR kit (Invitrogen). Full-length BRSK1 and BRSK2 were ligated into the BamHI and EcoRI sites of the pCMVTag4A vector (Agilent, Santa Clara, CA), which contains an N-terminal Flag tag. Constitutively active BRSK1-T189E (threonine 189 → glutamate), kinase-deficient BRSK1-K63R (lysine 63 → arginine), constitutively active BRSK2-T175E (threonine 175 → glutamate), and kinase-deficient BRSK2-K49R (lysine 49 → arginine; Lizcano et al., 2004) were generated from wild-type BRSK1 and BRSK2 using the QuikChange II Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer's instructions. The clone for MARK4 (NM_031417) was obtained from the Invitrogen Gateway Ultimate ORF set and subcloned into the 5′-Not1 and 3′-EcoRI sites of pCMVTag4A. Constitutively active MARK4-T214E (threonine 214 → glutamate) and kinase-deficient MARK4-K88R (lysine 88 → arginine) were generated using the QuikChange kit. Constitutively active truncated AMPK1-132 and kinase-deficient AMPK K63R (lysine 63 → arginine) in the pEBG vector were obtained (Crute et al., 1998) and subcloned into the pCMVTag4A vector.
Cell culture
Cos7 cells were maintained in high-glucose DMEM containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (pen-strep), and 1 mM sodium pyruvate at 37°C and 5% CO2. For imaging, cells were plated onto custom-made, sterilized, glass-bottom 35-mm dishes and transfected with reporter plasmid DNA using the Lipofectamine 2000 reagent (Invitrogen) at a confluency of 50–60%. The cells were allowed to grow for 18–24 h before imaging. HeLa or HEK293T cells were maintained in low-glucose (HeLa) or high-glucose (HEK293T) DMEM containing 10% FBS, 1% pen-strep, and 1 mM sodium pyruvate at 37°C and 5% CO2. Cells were plated in Nunclon six-well dishes and transfected at 40x50% confluency with ABKAR and either BRSK1-T189E, BRSK1-K63R, BRSK2-T175E, BRSK2-K49R, MARK4-T214E, MARK4-K188R, AMPK1-312, or AMPK K63R vectors. The cells were harvested after 60–66 h for Western blotting analysis.
Rat primary hippocampal neuronal culture
Hippocampi were dissected from E17 Sprague–Dawley rat pups (Harlan, Indianapolis, IN) in ice-cold 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–buffered Hank's balanced salt solution (HBSS; containing 20 mM HEPES, 5 mM d-glucose, 0.23 mM sodium pyruvate, and 0.5% pen-strep, at pH 7.4) and were dissociated using the Papain Dissociation System (Worthington, Lakewood, NJ) according the manufacturer's instructions. Dissociated HNs were resuspended in Neurobasal-A medium (05-0128DJ; Invitrogen) supplemented with 2% B27 supplement, 0.5% pen-strep, 2 mM glutaMAX, 3 mM d-glucose, and 0.23 mM sodium pyruvate. Cells were plated at a density of 4.2 × 104 cells/cm2 on poly-d-lysine (PDL)–coated Nunclon six-well plates for Western blot analysis. One-half of the medium was replaced at DIV 3 with 2 μM cytosine arabinoside, and cells were given a 25–50% medium change every 3–4 d to restore glucose levels to 3 mM as previously described (Kleman et al., 2008). Cells were cultured in an atmosphere-controlled, humidified incubator at 5% CO2 and 5% O2. AMPK α1 and α2–deficient mice were generated as previously described (Jorgensen et al., 2003; Viollet et al., 2003). Wild-type littermates served as controls in all experiments. Genotyping was performed as previously described. Mouse HNs were isolated as described. For imaging experiments, isolated mouse or rat HNs (2.0 × 106/transfection) were electroporated with 5 μg of cDNA using the Amaxa Nucleofector device (Lonza, Walkersville, MD) according to the manufacturer's instructions and plated onto 35-mm glass-bottom imaging dishes. Cells were imaged after at least 18 h posttransfection.
Live-cell imaging
Cells were washed twice with and maintained in low-glucose HBSS buffer. After identification of transfected cells, the cells were treated with 2-DG, ionomycin, calyculin A, or CaCl2 as indicated. Dual emission ratio imaging was performed on a Zeiss Axiovert 200M microscope with a MicroMAX BFT512 cooled charge-coupled device camera (Roper Scientific, Trenton, NJ) controlled by METAFLUOR 6.2 software (Universal Imaging, Downingtown, PA). Emission ratios were obtained using a 420DF20 excitation filter, a 450DRLP dichroic mirror, and two emission filters (475DF40 for ECFP and Cerulean and 535DF25 for cpVenus) alternated by a Lambda 10-2 filter changer (Sutter Instruments, Novato, CA). Images were taken every 15–30 s with an exposure time of 100–500 ms. Fluorescence images were background corrected by subtracting autofluorescence intensities of untransfected cells (or background with no cells) from the emission intensities of fluorescent cells expressing the reporter.
shRNA-mediated gene knockdown
Individual clones for lentiviral shRNA constructs from the TRC RNAi Library (Broad Institute, Boston, MA) were purchased from Sigma-Aldrich. For knockdown of AMPK α1, clones TRCN00000360842 and TRCN00000360770 were used. For knockdown of AMPK α2, clones TRCN00000360775 and TRCN00000360848 were used. BRSK1 knockdown was achieved using clones TRCN00000368711 and TRCN00000360985. BRSK2 knockdown was achieved using clones TRCN0000363494 and TRCN0000363520. The pLKO.1 scrambled shRNA vector (plasmid 1864; Addgene, Cambridge, MA) was used as a control, as previously described (Sarbassov et al., 2005). shRNAs (2.5 μg/clone) were transfected as lentiviral cDNA vectors as described.
Western blot analysis
Cells were washed once with ice-cold Dulbecco's phosphate-buffered saline and lysed directly in 1× protein sample buffer (62.5 mM Tris-Cl, pH 6.8, 10% glycerol, 2% SDS, 1% β-mercaptoethanol, and trace amounts of bromophenol blue), boiled, and stored at −80°C. Proteins were separated by SDS–PAGE on a 4–15% gradient Tris-HCl polyacrylamide gel (Bio-Rad, Hercules, CA) and transferred to a polyvinylidene difluoride membrane (Bio-Rad). Blots were probed with the following primary antibodies overnight in Tris-buffered saline solution, pH 7.5, containing 0.1% Tween-20, 5% protease-free bovine serum albumin (BSA), and 50 mM NaF (TBST): pAMPK, pACC, tAMPK, tACC, pAMPK substrate, AMPK α1, AMPK α2, BRSK1, BRSK2 (1:1000; Cell Signaling; Danvers MA), and actin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA). Blots were incubated for 2 h with the following secondary antibodies in 5% nonfat dry milk in TBST: anti-goat and anti-rabbit horseradish peroxidase (1:5000; EMD Millipore, Billerica, MA). The enhanced chemiluminescence signal was detected with the FluorChem Q imaging system (Protein Simple, Santa Clara, CA).
Immunocytochemistry
Neurons were cultured on PDL-coated glass-bottom dishes or coverslips for 3 DIV before transfection with ABKAR and BRSK shRNAs or 5 DIV for untransfected cells. Cells were fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) for 20 min at room temperature. Free PFA was quenched with 100 mM glycine in HBSS. Cells were then permeabilized and preincubated in blocking buffer (5% goat serum, 0.2% BSA, 0.1% Triton X-100, and 0.01% NaN3 in HBSS) for 30 min. The cells were incubated at 4°C overnight with the following primary antibodies in blocking buffer: mouse MAP2 MAB3148 (1:1000; EMD Millipore, Darmstadt, Germany), mouse Tau1 ab64193 (1:100; Abcam, Cambridge, United Kingdom), mouse NST ab14545 (1:1000; Abcam), rabbit CaMKKβ (1:500, Novus Biologicals, Littleton, CO), or mouse LKB1 (1:100, EMD Millipore). On the following day, the cells were washed three times in HBSS and incubated with goat anti-mouse Alexa Fluor 568 or goat anti-rabbit Alexa Fluor 488 secondary antibodies (1:500; Life Technologies, Carlsbad, CA) for 2 h at room temperature in the dark. Cells were then washed with HBSS three times and visualized by fluorescence microscopy. For ABKAR detection in fixed cells, the intrinsic fluorescence of the reporter was imaged using a 488/535 excitation/emission filter set. All imaging was done on the Axiovert 200M inverted epifluorescence microscope (Zeiss, Oberkochen, Germany). All image processing was done using ImageJ software (National Institutes of Health, Bethesda, MD).
Supplementary Material
Acknowledgments
We thank Lewis Cantley (Weill Cornell Medical College, New York, NY) for the generous gift of AMPKAR. This work was supported by National Institutes of Health Grant RO1 DK073368 (to J.Z.) and DGIST Convergence Science Center Grant 11-BD-04 (to G.V.R.).
Abbreviations used:
- 2-DG
2-deoxyglucose
- ABKAR
AMPK BRSK dual-activity reporter
- AMPK
5′-adenosine monophosphate–activated protein kinase
- BRSK
brain-specific protein kinase
- CaMKKβ
Ca2+/calmodulin-dependent protein kinase β
- LKB1
liver kinase B1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-02-0764) on March 18, 2015.
*These authors contributed equally to this work.
V.S., S.R., G.V.R., and J.Z. designed the experimental aspects of the project. V.S. and S.R. performed experiments and analyzed data. K.G. performed some of the immunostaining experiments. V.S., S.R., G.V.R., and J.Z. wrote the manuscript.
REFERENCES
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332:247–251. doi: 10.1126/science.1201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Biernat J, Wu YZ, Timm T, Zheng-Fischhöfer Q, Mandelkow E, Meijer L, Mandelkow EM. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell. 2002;13:4013–4028. doi: 10.1091/mbc.02-03-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright NJ, Carling D, Thornton C. Investigating the regulation of brain-specific kinases 1 and 2 by phosphorylation. J Biol Chem. 2008;283:14946–14954. doi: 10.1074/jbc.M710381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles. Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- Chen L, Jiao ZH, Zheng LS, Zhang YY, Xie ST, Wang ZX, Wu JW. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature. 2009;459:1146–1149. doi: 10.1038/nature08075. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron. 2001;29:115–129. doi: 10.1016/s0896-6273(01)00184-2. [DOI] [PubMed] [Google Scholar]
- Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- Depry C, Allen MD, Zhang J. Visualization of PKA activity in plasma membrane microdomains. Mol Biosystems. 2011;7:52–58. doi: 10.1039/c0mb00079e. [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandel-kow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Fogarty S, Hardie DG. C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J Biol Chem. 2009;284:77–84. doi: 10.1074/jbc.M806152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, Hardie DG. Calmodulin-dependent protein kinase kinase-b activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, Yurimoto S, Hatano N, Nozaki N, Sueyoshi N, Kameshita I, Mizutani A, Mikoshiba K, Kobayashi R, Tokumitsu H. Activation of SAD kinase by Ca2+/calmodulin-dependent protein kinase kinase. Biochemistry. 2008;47:4151–4159. doi: 10.1021/bi702528r. [DOI] [PubMed] [Google Scholar]
- Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–1136. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Tang W, Yuan J, Chen X, Wan B, Gu X, Luo K, Wang Y, Yu L. BRSK2 is activated by cyclic AMP-dependent protein kinase A through phosphorylation at Thr260. Biochem Biophys Res Commun. 2006;347:867–871. doi: 10.1016/j.bbrc.2006.06.178. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP. Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst KJ, Ni Q, Zhang J. Dynamic visualization of signal transduction in living cells: from second messengers to kinases. IUBMB Life. 2009;61:902–908. doi: 10.1002/iub.232. [DOI] [PubMed] [Google Scholar]
- Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, Inoue M, Yao I, Takeuchi K, Kitajima I, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron. 2006;50:261–275. doi: 10.1016/j.neuron.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha-2 but not alpha-1 5'AMP-activated protein kinase isoform abolishes AICAR- but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 31, 216–219. 2003 doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–932. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- Kleman AM, Yuan JY, Aja S, Ronnett GV, Landree LE. Physiological glucose is critical for optimized neuronal viability and AMPK responsiveness in vitro. J Neurosci Methods. 2008;167:292–301. doi: 10.1016/j.jneumeth.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guan KL. Microtubule-associated protein/microtubule affinity-regulating kinase 4 (MARK4) is a negative regulator of the mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2013;288:703–708. doi: 10.1074/jbc.C112.396903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci USA. 2014;111:1138–1143. doi: 10.1073/pnas.1321990111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CJ, Kain KH, Tkachenko E, Goldfinger LE, Gutierrez E, Allen MD, Groisman A, Zhang J, Ginsberg MH. Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol Biol Cell. 2008;11:4930–4941. doi: 10.1091/mbc.E08-06-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Markwardt ML, Kremers GJ, Kraft CA, Ray K, Cranfill PJ, Wilson KA, Day RN, Wachter RM, Davidson MW, Rizzo MA. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One. 2011;6:e17896. doi: 10.1371/journal.pone.0017896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldach L, Zhang J. Genetically encoded fluorescent biosensors for live-cell visualization of protein phoshorylation. Chem Biol. 2014;21:186–197. doi: 10.1016/j.chembiol.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J. Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J Biol Chem. 2007;282:495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287:31658–31665. doi: 10.1074/jbc.R112.356485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy S, Chang E, Cao Y, Zhu J, Ronnett GV. AMPK activation regulates neuronal structure in developing hippocampal neurons. Neuroscience. 2014;259:13–24. doi: 10.1016/j.neuroscience.2013.11.048. [DOI] [PubMed] [Google Scholar]
- Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109((Suppl 1)):17–23. doi: 10.1111/j.1471-4159.2009.05916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami H, Umemiya M, Saito S, Kondo H. Distinct immunohistochemical localizations of two isoforms of Ca2+/calmodulin-dependent protein kinase kinases in the adult rat brain. Eur J Neurosci. 2000;12:89–99. doi: 10.1046/j.1460-9568.2000.00883.x. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys433, is essential for LKB1 to supress cell growth. J Biol Chem. 2001;276:19469–19482. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J. 2011;434:503–512. doi: 10.1042/BJ20101485. [DOI] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk BE, Hutti JE, Cantley LC. Determining protein kinase substrate specificity by parallel solution-phase assay of large numbers of peptide substrates. Nat Protoc. 2006;1:375–379. doi: 10.1038/nprot.2006.57. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci USA. 2011;108:5849–5854. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters LA, Kemp BE, Means AR. Chutes and ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31:13–16. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.