

Abstract
MET (MNNG HOS transforming gene) is one of the receptor tyrosine kinases whose activities are frequently altered in human cancers, and it is a promising therapeutic target. MET is normally activated by its lone ligand, hepatocyte growth factor (HGF), eliciting its diverse biological activities that are crucial for development and physiology. Alteration of the HGF-MET axis results in inappropriate activation of a cascade of intracellular signaling pathways that contributes to hallmark cancer events including deregulated cell proliferation and survival, angiogenesis, invasion, and metastasis. Aberrant MET activation results from autocrine or paracrine mechanisms due to overexpression of HGF and/or MET or from a ligand-independent mechanism caused by activating mutations or amplification of MET. The literature provides compelling evidence for the role of MET signaling in cancer development and progression. The finding that cancer cells often use MET activation to escape therapies targeting other pathways strengthens the argument for MET-targeted therapeutics. Diverse strategies have been explored to deactivate MET signaling, and compounds and biologics targeting the MET pathway are in clinical development. Despite promising results from various clinical trials, we are still waiting for true MET-targeted therapeutics in the clinic. This review will explore recent progress and hurdles in the pursuit of MET-targeted cancer drugs and discuss the challenges in such development.
Keywords: MET, Hepatocyte growth factor, Targeted therapy, Receptor tyrosine kinase, Cancer therapeutics
Core tip: Aberrant activation of MET receptor tyrosine kinase signaling is frequently observed in many human cancers. Such activation not only affects cancer development and progression, but it also contributes to resistance against other cancer drugs. The inhibition of MET signaling is an attractive approach for cancer intervention, and pursuit of MET-targeted cancer therapeutics is underway. Even though promising results have been reported from various clinical trials, many challenges remain to be addressed before and even after the arrival of such drugs in the clinic.
INTRODUCTION
It has been three decades since the discoveries of MET (MNNG HOS transforming gene) and its ligand, hepatocyte growth factor [HGF; also known as scatter factor (SF)][1-5]. MET, encoded by the proto-oncogene MET on chromosome 7 (7q31), is a receptor tyrosine kinase (RTK). Under physiological conditions, it is stimulated by HGF, mainly through a paracrine mechanism, which triggers a cascade of intracellular signaling networks. The signaling driven by this ligand-receptor pair is involved in mitogenesis, motility, and morphogenesis, and it is essential for many developmental and physiological processes[6-8]. Like that of many RTKs, MET signaling is tightly regulated, and its timely attenuation is crucial for proper regulation of its activities[9-11]. Inappropriate activation of this signaling cascade can cause hallmark cancer events that include deregulated cell proliferation, survival, transformation, angiogenesis, and invasion[12,13]. Several different mechanisms can lead to an aberrant MET signaling, including autocrine or paracrine activation resulting from overexpression of MET and or of HGF, and a ligand-independent mechanism caused by activating mutations or amplification of the MET gene[12].
Alteration of MET signaling has been reported in almost all types of human cancers and is often associated with poor prognosis[7,12] (http://www.vai.org/met/). The evidence provides a compelling rationale for targeting this pathway, and the rationale is strengthened by the fact that cancer cells often use the HGF-MET axis to escape therapies targeting other RTKs or signaling molecules[12,14-16]. Cancer treatment has been revolutionized by targeted therapy since the success of Gleevec (imatinib) for treating chronic myelogenous leukemia by inhibiting BCR-Abl tyrosine kinase activity[17]. Other targeted therapies include drugs targeting epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR)[11]. Targeted therapies are the trend in cancer treatment, even though not all such drugs have lived up to their promise, in part due to the complexity of the cancer genome[18-21]. Relative to how much we know about the molecular mechanisms of cancers and the numbers of suitable therapeutic targets that have been identified, the targeted therapies available in the clinic are quite limited. MET is one of the targetable molecules that are still lacking effective drugs for cancer treatment.
Over the years, diverse strategies have been explored to inhibit MET pathway activation, from blocking either ligand access or receptor dimerization to inhibiting MET kinase activity or preventing downstream signaling activation[12,22,23]. These efforts have led to the discovery of many MET inhibitors possessing distinct specificities and efficacies (Figure 1). There are hundreds of clinical trials aiming to bring MET inhibitors from the bench to bedside[12,23-25] (https://ccrod.cancer.gov/confluence/display/CCRHGF/Home). While many MET inhibitor trials have shown promising results, various challenges remain. For instance, which patient will benefit from MET-targeted therapeutics and what companion diagnostics will be needed for patient stratification? How can MET-targeted therapeutics be effectively used for tailoring a patient-oriented treatment plan?
Figure 1.
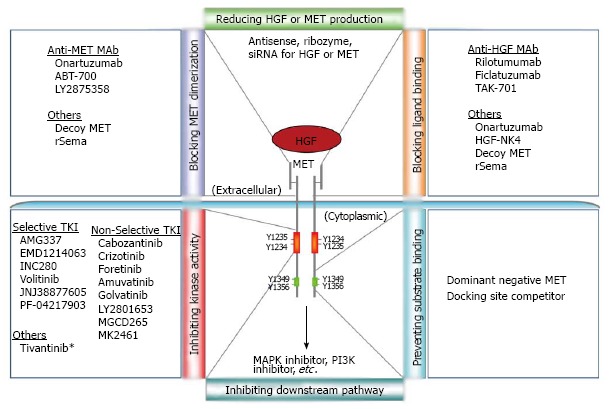
MET-targeted therapeutic strategies and inhibitors. Many strategies have been explored to inhibit MET signaling: blocking ligand binding or receptor dimerization; inhibiting MET kinase activity; preventing substrate binding; reducing hepatocyte growth factor (HGF) or MET production; and inhibiting downstream pathways. Shown in the diagram are representative inhibitors in each category, many of which are in clinical trials. Y1234 and Y1235 are two essential tyrosine residues in the TK-domain of MET, and Y1349 and Y1356 in its docking site are key tyrosine residues for substrate binding. Tivantinib is a non-ATP-competitive, selective MET inhibitor, but it also exhibits MET-independent antitumor activity by inhibiting microtubules. rSema: Recombinant MET Sema domain fragment.
MET SIGNAL TRANSDUCTION AND ACTIVITIES
MET is normally expressed in cells of epithelial or endothelial origin, while its ligand HGF is predominantly produced by mesenchymal cells[26]. This decoupling enables a tight regulation of MET signaling in tissues and cells where its activity is required, through the response of MET to gradients of HGF. When HGF binds, the MET receptor undergoes dimerization and a conformational change, leading to phosphorylation of its key tyrosine residues and recruitment of downstream signaling molecules. This recruitment triggers activation of important intracellular signaling pathways such as Ras-MAPK, PI3K-AKT, Src, STAT3, PLC-γ or Cdc42/Rac. Such activation is either mediated by scaffolding adaptors like Gab1 and Grb2 or by direct binding of signaling molecules to the multisubstrate-docking sites in the MET cytoplasmic region[27-30]. MET signaling is subject to timely attenuation that is regulated by several mechanisms, including phosphatase-mediated dephosphorylation, receptor turnover, and negative feedback inhibition[27,31-34]. Such regulation determines the duration and threshold of the MET signal output, how cells will respond, and what biological activities will be induced.
Diverse biological activities, from cell proliferation and survival to cell motility and invasion, can be induced by MET signaling, but it is still vague how the overall response to this signaling is produced. For instance, under what circumstances stimulated cells will proliferate rather than migrate or invade. Among many signaling pathways downstream of MET, activation of Ras-MAPK/ERK is crucial for cell proliferation; PI3K-Akt contributes more to cell survival; Cdc42/Rac activation induces cell motility; and STAT3 has been implicated in cell transformation and tubulogenesis[6-8,34]. Nonetheless, these intracellular pathways often have crossover activities, and they interplay to carry out many complicated biological roles driven by MET, such as invasion and branching morphogenesis[6-8,34]. The signal output and cellular response can be further complicated by crosstalk between the HGF-MET axis and many other cell-surface receptors or signaling pathways[12,34].
MET-mediated biological activities are part of many developmental and physiological processes. For example, the HGF-MET axis is essential for the developing placenta, embryonic liver, and limb muscles during embryogenesis[35-37], and it may be involved in the early development of the lungs, kidneys, and mammary glands[38-42]. HGF promotes formation of blood vessels and lymphatic vessels[43-45], and it plays a role in developing neurons[46,47]. Physiologically, MET signaling is indispensable for liver regeneration and repair[48-50] and for skin wound healing[51]; it also contributes to insulin secretion and glucose metabolism[52,53]. A significant role of this signaling axis in the regulation of stem cell activity has also been found[54]. HGF can stimulate the migration and differentiation of mesenchymal stem cells (MSCs) while inhibiting their proliferation[55,56], and it can induce differentiation of bone marrow stem cells into hepatocytes[57,58].
MET SIGNALING IN CANCERS
The link of MET to cancer can be traced back thirty years. MET was originally cloned from a carcinogen-induced chromosome rearrangement in a human osteosarcoma cell line as part of the TPR-MET fusion[1,59], an oncogenic product which was also observed in human gastric carcinoma[60]. The most decisive evidence of MET signaling in cancers came from the identification of its germline and somatic mutations in papillary renal cell carcinomas (RCC)[61]. These mutations mainly locate in the tyrosine kinase (TK) domain of MET, resulting in constitutive activation of its signaling[62]. Such mutations have also been sporadically identified in childhood hepatocellular carcinoma (HCC) and head neck squamous cell carcinoma (HNSCC)[63,64]. Interestingly, MET mutations identified in other human solid cancers (such as lung and gastric cancers, and melanoma and thyroid carcinomas) are mostly in the extracellular semaphorin (Sema) domain and the juxtamembrane (JM) domain[65-69]. In lung adenocarcinoma, it is estimated that 4% of the tumors have exon 14 skipping in the MET mRNA due to splicing site mutations, and thus have JM-domain defect[20]. These non-TK-domain mutations likely affect ligand binding or CBL-mediated turnover of MET, thereby altering MET signal transduction. Besides genetic abnormalities, MET signaling is mostly altered through a paracrine or autocrine activation mechanism by inappropriate increases in MET and/or HGF expression. Evidence of such alterations has been documented in almost all types of human cancers, and high MET and HGF expressions are often correlated with invasive phenotype and poor prognosis[7,12]. Alternatively, aberrant MET activation in cancer cells can be the result of amplification of the MET gene, which is found in gastric, esophageal, lung, colorectal, and breast cancers[70-76].
Another important aspect of MET signaling in cancers emerged from studies of drug resistance. Activation of the HGF-MET axis has become one of the most crucial mechanisms that cancer cells adapt to bypass therapies targeting other oncogenic pathways. The first such evidence came from the analysis of non-small-cell lung cancer (NSCLC) patients treated with gefitinib (an EGFR inhibitor), which revealed amplification of MET as a mechanism for gefitinib resistance[14]. This mechanism accounts for acquired resistance to EGFR-targeted therapies (gefitinib and erlotinib) in about 5% of NSCLCs that harbor EGFR-activating mutations and are primarily sensitive to the treatment[77-80]. MET amplification is also associated with acquired resistance to cetuximab or panitumumab (both EGFR-targeted monoclonal antibodies) in patients with metastatic colorectal cancer (mCRC)[81]. An alternative mechanism of resistance to EGFR-targeted therapies in lung cancer via MET signaling may be through up-regulation of HGF expression[82-84]. This mechanism could have a widespread impact on drug resistance to various anticancer kinase inhibitors[16]. For instance, stroma-derived HGF contributes to innate and acquired resistance to vemurafenib (RAF inhibitor) treatment of melanomas[15], and it may trigger resistance to ALK inhibitors in EML4-ALK lung cancer cells[85]. MET activation also confers tumor resistance to chemotherapy or radiotherapy.
Aberrant MET activation can elicit a multitude of biological consequences, ultimately leading to tumorigenesis and metastasis[6-8,12]. It causes oncogenic transformation and provides growth and survival signals to cancer cells by overactivating numerous downstream pathways (RAS-MAPK, PI3K-AKT, and STAT3, to name a few). In animal models, overexpression of MET in the liver results in HCC[86], while targeted expression of mutant MET in mammary epithelium leads to the development of breast cancers[87,88]. In parallel, MET causes invasive behavior of cancer cells, leading to metastasis; this is achieved by its abilities to up-regulate multiple extracellular matrix-degrading proteases, inducing the epithelial-to-mesenchymal transition and activating cell-mobilizing machinery[6-8,12]. The HGF-MET axis has also been implicated in the regulation of cancer stem cell activities in colon cancer and glioblastoma[89-91]. Besides direct contribution to the pathogenesis of cancer cells, MET signaling can enhance angiogenesis to strengthen tumor-supporting circuitry for promoting growth and survival[43,92,93].
DEVELOPMENT OF MET-TARGETED THERAPEUTICS: THE PROMISE
The indisputable role of MET signaling in cancer has made it a promising target for cancer intervention. Many approaches have been taken to try to effectively inhibit MET signaling activation in cancer cells[12,22-23]. Resulting from these efforts is a spectrum of targeted inhibitors having diverse biochemical and biological properties, including neutralizing antibodies to MET or HGF, small-molecule tyrosine kinase inhibitors (TKIs) of MET, and others (Figure 1). To date, more than two dozen MET-targeted inhibitors are in clinical development, with hundreds of trials conducted or underway, either as a single agent or in combination with other cancer drugs[12,23-25].
Ficlatuzumab (AV-299), rilotumumab (AMG102), and TAK-701 are humanized anti-HGF monoclonal antibodies that block HGF-dependent paracrine/autocrine MET activation. Ficlatuzumab, as a single agent for patients with advanced solid tumors, showed a partial benefit of stable disease in phase I trials. Favorable responses were observed in a subgroup of NSCLC patients who had low MET expression when ficlatuzumab was used in combination with an EGFR inhibitor in a phase II study[94-97]. Patients with refractory advanced solid tumors had a response of stable disease when treated with rilotumumab alone[98]. Recent phase II clinical trials of rilotumumab in combination with chemotherapy extended progression-free survival (PFS) in patients with gastric cancer[99]. A benefit of combining rilotumumab with panitumumab was reported in a randomized phase II trial of patients with mCRC who carry wild-type KRAS[100]. TAK-701, which has been tested in a phase I trial, inhibits HGF-mediated resistance to gefitinib in an NSCLC tumor model[101,102].
Unlike HGF blockers, onartuzumab (MetMAb, a humanized monovalent antibody to MET) neutralizes MET by inhibiting HGF binding and receptor dimerization[103,104]. In a phase I dose-escalation study, onartuzumab, as a single agent and in combination with bevacizumab, was well tolerated in patients with advanced solid tumors[105], while in a preclinical model it enhanced the antitumor efficacy of anti-VEGF biologics[106]. Several phase II/III trials of onartuzumab in combination with bevacizumab and or chemotherapeutic agents have been initiated for treating cancers such as mCRC, glioblastoma, NSCLC, breast cancer, and gastric cancer. In a randomized phase II trial, onartuzumab and erlotinib in combination had a favorable outcome in MET-positive NSCLC patients, with MET expression of 2+ or 3+ scores based on immunohistochemical (IHC) staining[107]. This group of patients had a significant improvement of PFS (median 2.9 mo vs 1.5 mo) and overall survival (OS; median 12.6 mo vs 3.8 mo) relative to the placebo plus erlotinib controls. However in the MET-negative group, a worse OS was observed relative to the control group[107]. This result has led to a phase III trial of onartuzumab plus erlotinib in MET-positive advanced NSCLC patients[108]. ABT-700 and LY2875358, two other antagonist antibodies against MET, are also being evaluated in early-phase trials[109,110].
While the anti-HGF and anti-MET biologics provide unique target specificity and long-lasting efficacy, the majority of potent MET inhibitors in clinical development are small-molecule TKIs. These are either selective or non-selective inhibitors of MET, and they mostly compete for the ATP-binding pocket in the TK-domain. Examples of selective inhibitors include AMG337, EMD1214063 (MSC2156119J), INC280 (INCB028060), and volitinib (HMPL-504); no significant safety concerns have been reported from early-phase studies of these oral inhibitors[111-114]. EMD1214063 is potent in suppressing the activities of both wild-type MET and its activating mutants in preclinical models[112,115,116], and ongoing phase I/II trials will evaluate its safety/efficacy in NSCLC and HCC. Several phase I/II trials of INC280 are recruiting patients with cancers including advanced cases of HCC, NSCLC, glioblastoma, or melanoma. A global phase II study of volitinib has been initiated for papillary RCC[114]. PF-04217903, another potent selective TKI of MET, has been discontinued from trials due to a strategic development decision[117].
Non-selective MET TKIs represent a different class of blockers; among them are crizotinib, cabozantinib, and foretinib. Crizotinib (Xalkori, PF-02341066), which was primarily designed for MET inhibition and displayed antitumor effects in MET-amplified NSCLC, is a multikinase inhibitor of MET, ALK, and ROS1[118-120]. Crizotinib is FDA-approved for treatment of ALK-fusion NSCLC patients, and it has shown antitumor activity in advanced ROS1-rearranged NSCLC[120]. A durable response to crizotinib was reported in an NSCLC patient with MET amplification but no ALK rearrangement[121]. Patients with MET-amplified esophagogastric adenocarcinoma or recurrent glioblastoma have also shown clinical responses to crizotinib treatment[122,123]. A cross-tumoral phase II trial of crizotinib has been launched in patients with locally advanced and/or metastatic tumors that carry ALK and/or MET alteration. Ongoing trials also include crizotinib in combination with erlotinib or other inhibitors.
Cabozantinib (XL184) inhibits multiple molecular targets, including MET, VEGFR2, RET, AXL KIT, and FLT3, and it suppresses tumor growth, angiogenesis and metastasis[124]. Cabozantinib (Cometriq) was approved by the FDA as an orphan product for treating medullary thyroid cancer (MTC) in late 2012. A phase III trial for this rare human cancer showed a response rate of 28% for cabozantinib versus none for placebo (median PFS of 11.2 mo vs 4.0 mo), most likely due to its inhibitory activity on RET; it showed significant but manageable toxicity[125]. Cabozantinib phase II trials displayed clinical activity in metastatic CRPC, resulting in improvement in PFS, bone scans, and pain, and a reduction of soft tissue lesions[126,127]. Clinical trials of cabozantinib are under way for several other cancer types, including NSCLC, glioblastoma, breast, ovarian, and urothelial cancers. Its combination with erlotinib has been tested in advanced NSCLC in an early-phase trial[128]; many other combination studies involving cabozantinib are under way.
Foretinib (XL880, EXEL-2880, and GSK1363089) is a multikinase inhibitor targeting MET, VEGFR2, RON, TIE-2, PDGFRβ, KIT, FLT3, and AXL. It has shown antitumor activity in xenograft tumors and in a phase I trial of metastatic or unresectable solid tumors[129,130]. With a manageable toxicity profile, it demonstrated in a phase II trial a high response rate in advanced papillary RCC patients who had germline MET mutations[131]. Phase II trials are studying foretinib in metastatic gastric cancer, recurrent/metastatic HNSCC, and triple-negative breast cancer. Phase I/II trials also test combinations of foretinib with erlotinib against locally advanced/metastatic NSCLC or with lapatinib against HER2-overexpressing metastatic breast cancer. Other non-selective TKIs of MET include amuvatinib (MP470), golvatinib (E7050), LY2801653, MGCD265, and MK-2461, all of these inhibit both MET and other targets[132-136].
Tivantinib (ARQ197), unlike other MET TKIs, is a non-ATP-competitive small-molecule inhibitor of MET that has a broad spectrum of antitumor activity[137]. It selectively binds to inactive/unphosphorylated MET and inhibits its autophosphorylation[138]. Clinical development of tivantinib is being actively pursued. Examples from completed phase II trials include tivantinib as a second-line treatment for advanced HCC, as well as its combination with erlotinib for previously treated NSCLC[139,140]. These results have led to the launches of phase III trials of tivantinib either as a monotherapy or in combination, among many other ongoing clinical studies.
CHALLENGES OF MET-TARGETED THERAPEUTICS
Despite promising results, there have also been setbacks to MET-targeted therapeutics development, and numerous challenges remain to be addressed. For instance, rilotumumab in several phase II trials showed no or limited efficacy in patients with metastatic RCC, castration-resistant prostate cancer (CRPC), or recurrent glioblastoma or ovarian cancer[141-144]. Foretinib as a single agent lacked efficacy in unselected patients with metastatic gastric cancer in a phase II study[145]. The COMET-1 phase III trial of cabozantinib in men with metastatic CRPC failed to meet its primary endpoint of demonstrating a statistically significant improvement of overall survival relative to prednisone treatment[146]. Further, the MARQUEE phase III trial of tivantinib plus erlotinib and the MetLung phase III trial of onartuzumab plus erlotinib in patients with advanced NSCLC were terminated following independent review board examination. Both trials failed to demonstrate any meaningful efficacy of combination compared with erlotinib alone[25,108,147]. Also, recent studies have demonstrated that tivantinib has cytotoxic activity independent of MET[148,149], raising the question of whether the clinical antitumor efficacy of this selective MET inhibitor was solely due to MET inhibition.
Such setbacks lead to the question of whether those clinical trials targeted the right groups of cancer patients for MET inhibition. One major challenge in patient selection is a lack of reliable biomarkers for companion diagnosis. IHC staining of MET has been widely used for assessing its protein status in tumors; patients with MET-IHC-positive gastric cancer displayed the greatest survival benefit from rilotumumab in combination with chemotherapy[99]. However, such staining unexpectedly failed in selecting patients for the MetLung phase III trial, even though this biomarker was able to identify responders (MET-IHC score 2+ or 3+) for onartuzumab plus erlotinib treatment in a phase II study[25,107,108,150]. It remains to be seen why MET-IHC biomarker failed in the latter trial and whether improvements in sensitivity and specificity would make it more reliable for patient stratification.
Other potential biomarkers include MET mutations and MET amplification (gains in gene copy number or chromosome 7 polysomy). Germline MET TK-domain mutations are highly predictive for papillary RCC patients’ response to foretinib treatment[131]. Nonetheless, MET mutation as a biomarker in general has its limitations, because a majority that are found in broader cancers are non-TK-domain mutations[20,65-69] whose value in predicting response to MET inhibitors remains undetermined. Fluorescence in-situ hybridization (FISH) of MET has been evaluated for a link between MET amplification and patient response in MET inhibitor trials[99,111,122,131,145,150]. Even though the sample sizes were mostly small for interpretation, patients with MET-amplified tumors usually have better response, indicating the value of MET-FISH as a predictive biomarker. The power of this biomarker will be further defined when results from several ongoing trials enrolling MET-amplified cancer patients become available. In addition, serum/plasma HGF and soluble MET concentrations have also been used in exploratory biomarker analyses[95,99,150].
While improved biomarkers are vital for better design and success of MET inhibitor trials, developing the proper therapeutic strategy to maximize drug efficacy in the clinic is equally important. The latter can be challenging, because alterations of MET signaling found in the majority of human cancers are not activating mutations or amplifications of MET, and the alterations are often accompanied by the activation of other pathways[7,12]. The use of MET-targeted drugs as a monotherapy has its own merit if MET alteration is the oncogenic driver; however, it might not perform well when different oncogenic pathway(s) is co-activated. The existence of crosstalk between MET and other pathways such as EGFR and VEGFR also complicates the outcome of MET-targeted monotherapy, providing further rationale for drug combination[12,34]. Clinical trials of various combination therapies have aimed to simultaneously inhibit MET and other molecular targets. However, determining what drug combination is best suited to which patient in the clinic demands a comprehensive platform of multiplexed molecular diagnoses. On the other hand, several targeted therapies, including those of EGFR and B-RAF, may theoretically benefit from combination with MET inhibitors to prevent MET signaling-mediated drug resistance[14-16,77-84]. Such combination strategies, nevertheless, are practically challenging because this resistance mechanism only accounts for a fraction of the resistant cases.
CONCLUSION
MET signaling is highly important in cancer malignancy, making it a promising molecular target for cancer intervention. The development of MET-targeted drugs is in full gear at multiple fronts, but numerous challenges remain. The failure of several late-stage clinical trials has put such development under scrutiny, but encouraging results are coming from other trials and new developments. Future trials should also put the activity of MET inhibitors on cancer invasion and metastasis in perspective. Inevitably, drug resistance will be expected for MET-targeted therapeutics, and such events have been observed in in vivo studies[151-154]. To enhance the performance of MET inhibition and to prevent cross-drug resistance, engineering bi-specific or multi-specific antibodies against MET and other cell surface receptors may provide a solution[155-157]. Many clinical benefits can be expected from the eventual approval of MET-targeted drugs.
ACKNOWLEDGMENTS
Thanks to Dr. George Vande Woude and Dafna Kaufman for critical reading, and David Nadziejka for manuscript editing.
Footnotes
P- Reviewer: Chui YL, Utkin YN, Wang QE S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: There is no potential conflict of interest relevant to this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 20, 2015
First decision: February 7, 2015
Article in press: April 18, 2015
References
- 1.Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 2.Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–242. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 4.Rosen EM, Goldberg ID, Kacinski BM, Buckholz T, Vinter DW. Smooth muscle releases an epithelial cell scatter factor which binds to heparin. In Vitro Cell Dev Biol. 1989;25:163–173. doi: 10.1007/BF02626174. [DOI] [PubMed] [Google Scholar]
- 5.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 6.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 7.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YW, Vande Woude GF. HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88:408–417. doi: 10.1002/jcb.10358. [DOI] [PubMed] [Google Scholar]
- 9.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 10.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 11.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 12.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 15.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Chen ZS. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34:1255–1268. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 18.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang YW, Graveel C, Shinomiya N, Vande Woude GF. Met decoys: will cancer take the bait? Cancer Cell. 2004;6:5–6. doi: 10.1016/j.ccr.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 24.Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–2214. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 25.Garber K. MET inhibitors start on road to recovery. Nat Rev Drug Discov. 2014;13:563–565. doi: 10.1038/nrd4406. [DOI] [PubMed] [Google Scholar]
- 26.Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- 27.Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–5589. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- 28.Naldini L, Vigna E, Ferracini R, Longati P, Gandino L, Prat M, Comoglio PM. The tyrosine kinase encoded by the MET proto-oncogene is activated by autophosphorylation. Mol Cell Biol. 1991;11:1793–1803. doi: 10.1128/mcb.11.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene. 1994;9:2019–2027. [PubMed] [Google Scholar]
- 30.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77:261–271. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 31.Clague MJ. Met receptor: a moving target. Sci Signal. 2011;4:pe40. doi: 10.1126/scisignal.2002422. [DOI] [PubMed] [Google Scholar]
- 32.Peschard P, Park M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene. 2007;26:1276–1285. doi: 10.1038/sj.onc.1210201. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YW, Vande Woude GF. MIG-6 and SPRY2 in the regulation of receptor tyrosine kinase signaling: Balancing act via negative feedback loops. Future Aspects of Tumor Suppressor Gene. USA: InTech Open Science; 2013. pp. 199–221. [Google Scholar]
- 34.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 35.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- 37.Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- 38.Ohmichi H, Koshimizu U, Matsumoto K, Nakamura T. Hepatocyte growth factor (HGF) acts as a mesenchyme-derived morphogenic factor during fetal lung development. Development. 1998;125:1315–1324. doi: 10.1242/dev.125.7.1315. [DOI] [PubMed] [Google Scholar]
- 39.Santos OF, Barros EJ, Yang XM, Matsumoto K, Nakamura T, Park M, Nigam SK. Involvement of hepatocyte growth factor in kidney development. Dev Biol. 1994;163:525–529. doi: 10.1006/dbio.1994.1169. [DOI] [PubMed] [Google Scholar]
- 40.Soriano JV, Pepper MS, Nakamura T, Orci L, Montesano R. Hepatocyte growth factor stimulates extensive development of branching duct-like structures by cloned mammary gland epithelial cells. J Cell Sci. 1995;108(Pt 2):413–430. doi: 10.1242/jcs.108.2.413. [DOI] [PubMed] [Google Scholar]
- 41.Woolf AS, Kolatsi-Joannou M, Hardman P, Andermarcher E, Moorby C, Fine LG, Jat PS, Noble MD, Gherardi E. Roles of hepatocyte growth factor/scatter factor and the met receptor in the early development of the metanephros. J Cell Biol. 1995;128:171–184. doi: 10.1083/jcb.128.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Spitzer E, Meyer D, Sachs M, Niemann C, Hartmann G, Weidner KM, Birchmeier C, Birchmeier W. Sequential requirement of hepatocyte growth factor and neuregulin in the morphogenesis and differentiation of the mammary gland. J Cell Biol. 1995;131:215–226. doi: 10.1083/jcb.131.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grant DS, Kleinman HK, Goldberg ID, Bhargava MM, Nickoloff BJ, Kinsella JL, Polverini P, Rosen EM. Scatter factor induces blood vessel formation in vivo. Proc Natl Acad Sci USA. 1993;90:1937–1941. doi: 10.1073/pnas.90.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kajiya K, Hirakawa S, Ma B, Drinnenberg I, Detmar M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005;24:2885–2895. doi: 10.1038/sj.emboj.7600763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao R, Björndahl MA, Gallego MI, Chen S, Religa P, Hansen AJ, Cao Y. Hepatocyte growth factor is a lymphangiogenic factor with an indirect mechanism of action. Blood. 2006;107:3531–3536. doi: 10.1182/blood-2005-06-2538. [DOI] [PubMed] [Google Scholar]
- 46.Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997;11:3341–3350. doi: 10.1101/gad.11.24.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maina F, Klein R. Hepatocyte growth factor, a versatile signal for developing neurons. Nat Neurosci. 1999;2:213–217. doi: 10.1038/6310. [DOI] [PubMed] [Google Scholar]
- 48.Borowiak M, Garratt AN, Wüstefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci USA. 2004;101:10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phaneuf D, Moscioni AD, LeClair C, Raper SE, Wilson JM. Generation of a mouse expressing a conditional knockout of the hepatocyte growth factor gene: demonstration of impaired liver regeneration. DNA Cell Biol. 2004;23:592–603. doi: 10.1089/dna.2004.23.592. [DOI] [PubMed] [Google Scholar]
- 51.Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai C, Huh CG, Thorgeirsson SS, Liu Y. Beta-cell-specific ablation of the hepatocyte growth factor receptor results in reduced islet size, impaired insulin secretion, and glucose intolerance. Am J Pathol. 2005;167:429–436. doi: 10.1016/s0002-9440(10)62987-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roccisana J, Reddy V, Vasavada RC, Gonzalez-Pertusa JA, Magnuson MA, Garcia-Ocaña A. Targeted inactivation of hepatocyte growth factor receptor c-met in beta-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of beta-cell mass. Diabetes. 2005;54:2090–2102. doi: 10.2337/diabetes.54.7.2090. [DOI] [PubMed] [Google Scholar]
- 54.Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc Natl Acad Sci USA. 2000;97:11307–11312. doi: 10.1073/pnas.97.21.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neuss S, Becher E, Wöltje M, Tietze L, Jahnen-Dechent W. Functional expression of HGF and HGF receptor/c-met in adult human mesenchymal stem cells suggests a role in cell mobilization, tissue repair, and wound healing. Stem Cells. 2004;22:405–414. doi: 10.1634/stemcells.22-3-405. [DOI] [PubMed] [Google Scholar]
- 56.Forte G, Minieri M, Cossa P, Antenucci D, Sala M, Gnocchi V, Fiaccavento R, Carotenuto F, De Vito P, Baldini PM, et al. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23–33. doi: 10.1634/stemcells.2004-0176. [DOI] [PubMed] [Google Scholar]
- 57.Oh SH, Miyazaki M, Kouchi H, Inoue Y, Sakaguchi M, Tsuji T, Shima N, Higashio K, Namba M. Hepatocyte growth factor induces differentiation of adult rat bone marrow cells into a hepatocyte lineage in vitro. Biochem Biophys Res Commun. 2000;279:500–504. doi: 10.1006/bbrc.2000.3985. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz RE, Reyes M, Koodie L, Jiang Y, Blackstad M, Lund T, Lenvik T, Johnson S, Hu WS, Verfaillie CM. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109:1291–1302. doi: 10.1172/JCI15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park M, Dean M, Cooper CS, Schmidt M, O’Brien SJ, Blair DG, Vande Woude GF. Mechanism of met oncogene activation. Cell. 1986;45:895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- 60.Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci USA. 1991;88:4892–4896. doi: 10.1073/pnas.88.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 62.Jeffers M, Fiscella M, Webb CP, Anver M, Koochekpour S, Vande Woude GF. The mutationally activated Met receptor mediates motility and metastasis. Proc Natl Acad Sci USA. 1998;95:14417–14422. doi: 10.1073/pnas.95.24.14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, Kim BJ, Bae JH, Hong YK, Lee KS, et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999;59:307–310. [PubMed] [Google Scholar]
- 64.Di Renzo MF, Olivero M, Martone T, Maffe A, Maggiora P, Stefani AD, Valente G, Giordano S, Cortesina G, Comoglio PM. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19:1547–1555. doi: 10.1038/sj.onc.1203455. [DOI] [PubMed] [Google Scholar]
- 65.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–6281. [PubMed] [Google Scholar]
- 66.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K, Lader A, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 67.Lee JH, Han SU, Cho H, Jennings B, Gerrard B, Dean M, Schmidt L, Zbar B, Vande Woude GF. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–4953. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 68.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R, Salgia R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–2253. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 69.Wasenius VM, Hemmer S, Karjalainen-Lindsberg ML, Nupponen NN, Franssila K, Joensuu H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol. 2005;29:544–549. doi: 10.1097/01.pas.0000156103.37756.e2. [DOI] [PubMed] [Google Scholar]
- 70.Houldsworth J, Cordon-Cardo C, Ladanyi M, Kelsen DP, Chaganti RS. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. 1990;50:6417–6422. [PubMed] [Google Scholar]
- 71.Tong CY, Hui AB, Yin XL, Pang JC, Zhu XL, Poon WS, Ng HK. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J Neurosurg. 2004;100:187–193. doi: 10.3171/ped.2004.100.2.0187. [DOI] [PubMed] [Google Scholar]
- 72.Miller CT, Lin L, Casper AM, Lim J, Thomas DG, Orringer MB, Chang AC, Chambers AF, Giordano TJ, Glover TW, et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene. 2006;25:409–418. doi: 10.1038/sj.onc.1209057. [DOI] [PubMed] [Google Scholar]
- 73.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008;3:331–339. doi: 10.1097/JTO.0b013e318168d9d4. [DOI] [PubMed] [Google Scholar]
- 75.Jardim DL, Tang C, Gagliato Dde M, Falchook GS, Hess K, Janku F, Fu S, Wheler JJ, Zinner RG, Naing A, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin Cancer Res. 2014;20:6336–6345. doi: 10.1158/1078-0432.CCR-14-1293. [DOI] [PubMed] [Google Scholar]
- 76.de Melo Gagliato D, Jardim DL, Falchook G, Tang C, Zinner R, Wheler JJ, Janku F, Subbiah V, Piha-Paul SA, Fu S, et al. Analysis of MET genetic aberrations in patients with breast cancer at MD Anderson Phase I unit. Clin Breast Cancer. 2014;14:468–474. doi: 10.1016/j.clbc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Workman P, Clarke PA. Resisting targeted therapy: fifty ways to leave your EGFR. Cancer Cell. 2011;19:437–440. doi: 10.1016/j.ccr.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 78.Soria JC, Mok TS, Cappuzzo F, Jänne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev. 2012;38:416–430. doi: 10.1016/j.ctrv.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 79.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 83.Yamada T, Takeuchi S, Kita K, Bando H, Nakamura T, Matsumoto K, Yano S. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J Thorac Oncol. 2012;7:272–280. doi: 10.1097/JTO.0b013e3182398e69. [DOI] [PubMed] [Google Scholar]
- 84.Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32:3846–3856. doi: 10.1038/onc.2012.396. [DOI] [PubMed] [Google Scholar]
- 85.Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. 2012;18:3592–3602. doi: 10.1158/1078-0432.CCR-11-2972. [DOI] [PubMed] [Google Scholar]
- 86.Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. 2001;153:1023–1034. doi: 10.1083/jcb.153.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D, Andrulis IL, Bull SB, Chughtai N, Zuo D, Souleimanova M, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci USA. 2009;106:12903–12908. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Graveel CR, DeGroot JD, Su Y, Koeman J, Dykema K, Leung S, Snider J, Davies SR, Swiatek PJ, Cottingham S, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci USA. 2009;106:12909–12914. doi: 10.1073/pnas.0810403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 90.De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R, Luraghi P, Reato G, D’Ambrosio A, Porrati P, et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72:4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- 91.Jun HJ, Bronson RT, Charest A. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells. 2014;32:338–348. doi: 10.1002/stem.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci USA. 2003;100:12718–12723. doi: 10.1073/pnas.2135113100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Patnaik A, Weiss GJ, Papadopoulos R, Tibes R, Tolcher AW, Payumo FC. Phase I study of SCH 900105, an anti-hepatocyte growth factor monoclonal antibody, as a single agent and in combination with erlotinib in patients with advanced solid tumors. J Clin Oncol. 2010;28:Abstract 2525. [Google Scholar]
- 95.Tabernero J, Elez ME, Herranz M, Rico I, Prudkin L, Andreu J, Mateos J, Carreras MJ, Han M, Gifford J, et al. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases. Clin Cancer Res. 2014;20:2793–2804. doi: 10.1158/1078-0432.CCR-13-1837. [DOI] [PubMed] [Google Scholar]
- 96.Tan E, Park K, Lim WT. Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. J Clin Oncol. 2011;29:Abstract 7571. [Google Scholar]
- 97.Mok TSK, Park K, Geater SL, Agarwal S, Han M, Credi M, McKee K, Kuriyama N, Slichenmyer W, Tan EH. A randomized phase (Ph) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory MAB in combination with gefitinib (G) versus G in Asian patients (pts) with lung adenocarcinoma (LA) Ann Oncol. 2012;23:Abstract 1198P. [Google Scholar]
- 98.Gordon MS, Sweeney CS, Mendelson DS, Eckhardt SG, Anderson A, Beaupre DM, Branstetter D, Burgess TL, Coxon A, Deng H, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16:699–710. doi: 10.1158/1078-0432.CCR-09-1365. [DOI] [PubMed] [Google Scholar]
- 99.Iveson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, Nirni S, Lakshmaiah K, Thomas A, Jiang Y, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014;15:1007–1018. doi: 10.1016/S1470-2045(14)70023-3. [DOI] [PubMed] [Google Scholar]
- 100.Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, Davidenko I, Stephenson J, Elez E, Prenen H, et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res. 2014;20:4240–4250. doi: 10.1158/1078-0432.CCR-13-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Okamoto W, Okamoto I, Tanaka K, Hatashita E, Yamada Y, Kuwata K, Yamaguchi H, Arao T, Nishio K, Fukuoka M, et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther. 2010;9:2785–2792. doi: 10.1158/1535-7163.MCT-10-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones SF, Cohen RB, Bendell JC, Denlinger CS, Harvey RD, Parasuraman S, Chi X, Scholz C, Wyant T, Kauh J. Safety, tolerability and pharmacokinetics of TAK-701 in patient with advanced nonhematological malignancies, first-in-human phase 1 dose escalation study. J Clin Oncol. 2010;28:Abstract 3081. [Google Scholar]
- 103.Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R, Westphal M, Lamszus K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12:6144–6152. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- 104.Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, Huang A, Yang NY, Nishimura M, Greve J, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci USA. 2013;110:E2987–E2996. doi: 10.1073/pnas.1302725110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Salgia R, Patel P, Bothos J, Yu W, Eppler S, Hegde P, Bai S, Kaur S, Nijem I, Catenacci DV, et al. Phase I dose-escalation study of onartuzumab as a single agent and in combination with bevacizumab in patients with advanced solid malignancies. Clin Cancer Res. 2014;20:1666–1675. doi: 10.1158/1078-0432.CCR-13-2070. [DOI] [PubMed] [Google Scholar]
- 106.Merchant M, Zhang YW, Su Y. MetMAb significantly enhances anti-tumor activity of anti-VEGF and/or erlotinib in several animal tumor models. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2008. p. Meeting Abstract 556. [Google Scholar]
- 107.Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH, Blumenschein GR, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:4105–4114. doi: 10.1200/JCO.2012.47.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Spigel DR, Edelman MJ, Mok T, O’Byrne K, Paz-Ares L, Yu W, Rittweger K, Thurm H. Treatment Rationale Study Design for the MetLung Trial: A Randomized, Double-Blind Phase III Study of Onartuzumab (MetMAb) in Combination With Erlotinib Versus Erlotinib Alone in Patients Who Have Received Standard Chemotherapy for Stage IIIB or IV Met-Positive Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2012;13:500–504. doi: 10.1016/j.cllc.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 109.Strickler JH, LoRusso P, Yen CJ, Lin CC, Kang YK, Kaminker P, Ansell P, Bhathena A, Wong S, Dudley MW, et al. Phase 1, open-label, dose-escalation, and expansion study of ABT-700, an anti-C-met antibody, in patients (pts) with advanced solid tumors. J Clin Oncol. 2014;32:5s (suppl; abstr 2507). [Google Scholar]
- 110.Goldman JW. First-in-human dose escalation study of LY2875358 (LY), a bivalent MET antibody, as monotherapy and in combination with erlotinib (E) in patients with advanced cancer. ASCO Annual Meeting. 2013:Abstract 8093. [Google Scholar]
- 111.Hong DS, LoRusso P, Hamid O. First-in-human study of AMG 337, a highly selective oral inhibitor of MET, in adult patients with advanced solid tumors. J Clin Oncol. 2014;32:Abstract 2508. [Google Scholar]
- 112.Falchook GS, Hong DS, Amin HM. Results of the first-in-human phase I trial assessing MSC2156119J (EMD 1214063), an oral selective c-Met inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014;32:Abstract 2521. [Google Scholar]
- 113.Donehower RC, Scardina A, Hill M. A phase I dose-escalation study of INCB028060, an inhibitor of c-MET receptor tyrosine kinase, in patients with advanced solid tumors. J Clin Oncol. 2011;29:Abstract 3091. [Google Scholar]
- 114.Hutchison China Meditech, AstraZeneca. Initiation of AZD6094 (HMPL-504/volitinib) global phase II study in papillary renal cell carcinoma. Available from: http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=509205.
- 115.Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, Grädler U, Meyring M, Dorsch D, Jaehrling F, et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19:2941–2951. doi: 10.1158/1078-0432.CCR-12-3247. [DOI] [PubMed] [Google Scholar]
- 116.Medová M, Pochon B, Streit B, Blank-Liss W, Francica P, Stroka D, Keogh A, Aebersold DM, Blaukat A, Bladt F, et al. The novel ATP-competitive inhibitor of the MET hepatocyte growth factor receptor EMD1214063 displays inhibitory activity against selected MET-mutated variants. Mol Cancer Ther. 2013;12:2415–2424. doi: 10.1158/1535-7163.MCT-13-0151. [DOI] [PubMed] [Google Scholar]
- 117.Liu X, Wang Q, Yang G, Marando C, Koblish HK, Hall LM, Fridman JS, Behshad E, Wynn R, Li Y, et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res. 2011;17:7127–7138. doi: 10.1158/1078-0432.CCR-11-1157. [DOI] [PubMed] [Google Scholar]
- 118.Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 119.Tanizaki J, Okamoto I, Okamoto K, Takezawa K, Kuwata K, Yamaguchi H, Nakagawa K. MET tyrosine kinase inhibitor crizotinib (PF-02341066) shows differential antitumor effects in non-small cell lung cancer according to MET alterations. J Thorac Oncol. 2011;6:1624–1631. doi: 10.1097/JTO.0b013e31822591e9. [DOI] [PubMed] [Google Scholar]
- 120.Rothschild SI, Gautschi O. Crizotinib in the treatment of non--small-cell lung cancer. Clin Lung Cancer. 2013;14:473–480. doi: 10.1016/j.cllc.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 121.Ou SH, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, Camidge DR, Solomon BJ, Maki RG, Bang YJ, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6:942–946. doi: 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- 122.Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, Lauwers GY, Christensen JG, Wilner KD, Haber DA, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29:4803–4810. doi: 10.1200/JCO.2011.35.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chi AS, Batchelor TT, Kwak EL, Clark JW, Wang DL, Wilner KD, Louis DN, Iafrate AJ. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J Clin Oncol. 2012;30:e30–e33. doi: 10.1200/JCO.2011.38.4586. [DOI] [PubMed] [Google Scholar]
- 124.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 125.Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–3646. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, Vogelzang NJ, Small EJ, Harzstark AL, Gordon MS, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31:412–419. doi: 10.1200/JCO.2012.45.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith MR, Sweeney CJ, Corn PG, Rathkopf DE, Smith DC, Hussain M, George DJ, Higano CS, Harzstark AL, Sartor AO, et al. Cabozantinib in chemotherapy-pretreated metastatic castration-resistant prostate cancer: results of a phase II nonrandomized expansion study. J Clin Oncol. 2014;32:3391–3399. doi: 10.1200/JCO.2013.54.5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wakelee HA, Gettinger SN, Engelman JA, et al. A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib (E) in patients (pts) with non-small cell lung cancer (NSCLC) J Clin Oncol. 2010;28:Abstract 3017. [Google Scholar]
- 129.Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 130.Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, Cancilla B, Chu F, Hitchcock-Bryan S, Sherman L, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16:3507–3516. doi: 10.1158/1078-0432.CCR-10-0574. [DOI] [PubMed] [Google Scholar]
- 131.Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31:181–186. doi: 10.1200/JCO.2012.43.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol. 2009;4:69. doi: 10.1186/1748-717X-4-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakagawa T, Tohyama O, Yamaguchi A, Matsushima T, Takahashi K, Funasaka S, Shirotori S, Asada M, Obaishi H. E7050: a dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010;101:210–215. doi: 10.1111/j.1349-7006.2009.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2013;31:833–844. doi: 10.1007/s10637-012-9912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Besterman JM, Fournel M, Dupont I, Bonfils C, Dubay M, Ste-Croix H, Maroun CR. Potent preclinical antitumor activity of MGCD265, an oral Met/VEGFR kinase inhibitor in phase II clinical development, in combination with taxanes or erlotinib. J Clin Oncol. 2010;28:e13595. [Google Scholar]
- 136.Pan BS, Chan GK, Chenard M, Chi A, Davis LJ, Deshmukh SV, Gibbs JB, Gil S, Hang G, Hatch H, et al. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 2010;70:1524–1533. doi: 10.1158/0008-5472.CAN-09-2541. [DOI] [PubMed] [Google Scholar]
- 137.Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, Hill J, Moussa MM, Leggett DS, Li CJ. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544–1553. doi: 10.1158/1535-7163.MCT-09-1173. [DOI] [PubMed] [Google Scholar]
- 138.Eathiraj S, Palma R, Volckova E, Hirschi M, France DS, Ashwell MA, Chan TC. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J Biol Chem. 2011;286:20666–20676. doi: 10.1074/jbc.M110.213801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs FT, Weiss A, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14:55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- 140.Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29:3307–3315. doi: 10.1200/JCO.2010.34.0570. [DOI] [PubMed] [Google Scholar]
- 141.Schöffski P, Garcia JA, Stadler WM, Gil T, Jonasch E, Tagawa ST, Smitt M, Yang X, Oliner KS, Anderson A, et al. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int. 2011;108:679–686. doi: 10.1111/j.1464-410X.2010.09947.x. [DOI] [PubMed] [Google Scholar]
- 142.Ryan CJ, Rosenthal M, Ng S, Alumkal J, Picus J, Gravis G, Fizazi K, Forget F, Machiels JP, Srinivas S, et al. Targeted MET inhibition in castration-resistant prostate cancer: a randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin Cancer Res. 2013;19:215–224. doi: 10.1158/1078-0432.CCR-12-2605. [DOI] [PubMed] [Google Scholar]
- 143.Wen PY, Schiff D, Cloughesy TF, Raizer JJ, Laterra J, Smitt M, Wolf M, Oliner KS, Anderson A, Zhu M, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011;13:437–446. doi: 10.1093/neuonc/noq198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martin LP, Sill M, Shahin MS, Powell M, DiSilvestro P, Landrum LM, Gaillard SL, Goodheart MJ, Hoffman J, Schilder RJ. A phase II evaluation of AMG 102 (rilotumumab) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2014;132:526–530. doi: 10.1016/j.ygyno.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe DC, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8:e54014. doi: 10.1371/journal.pone.0054014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Exelixis , Inc Exelixis announces results from the COMET-1 phase 3 pivotal trial of cabozantinib in men with metastatic castration-resistant prostate cancer. Available from: http://ir.exelixis.com/phoenix.zhtml?c=120923&p=irol-newsArticle&ID=1962549.
- 147.Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, von Pawel J, Schwartz B, Von Roemeling R, Sandler AB. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13:391–395. doi: 10.1016/j.cllc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 148.Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A, Valdembri D, Serini G, Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19:2381–2392. doi: 10.1158/1078-0432.CCR-12-3459. [DOI] [PubMed] [Google Scholar]
- 149.Katayama R, Aoyama A, Yamori T, Qi J, Oh-hara T, Song Y, Engelman JA, Fujita N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73:3087–3096. doi: 10.1158/0008-5472.CAN-12-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Koeppen H, Yu W, Zha J, Pandita A, Penuel E, Rangell L, Raja R, Mohan S, Patel R, Desai R, et al. Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib±onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res. 2014;20:4488–4498. doi: 10.1158/1078-0432.CCR-13-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70:1625–1634. doi: 10.1158/0008-5472.CAN-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Corso S, Ghiso E, Cepero V, Sierra JR, Migliore C, Bertotti A, Trusolino L, Comoglio PM, Giordano S. Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol Cancer. 2010;9:121. doi: 10.1186/1476-4598-9-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cepero V, Sierra JR, Corso S, Ghiso E, Casorzo L, Perera T, Comoglio PM, Giordano S. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70:7580–7590. doi: 10.1158/0008-5472.CAN-10-0436. [DOI] [PubMed] [Google Scholar]
- 154.Petti C, Picco G, Martelli ML, Trisolini E, Bucci E, Perera T, Isella C, Medico E. Truncated RAF kinases drive resistance to MET inhibition in MET-addicted cancer cells. Oncotarget. 2015;6:221–233. doi: 10.18632/oncotarget.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31:753–758. doi: 10.1038/nbt.2621. [DOI] [PubMed] [Google Scholar]
- 156.Choi HJ, Kim YJ, Lee S, Kim YS. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol Cancer Ther. 2013;12:2748–2759. doi: 10.1158/1535-7163.MCT-13-0628. [DOI] [PubMed] [Google Scholar]
- 157.Hu S, Fu W, Xu W, Yang Y, Cruz M, Berezov SD, Jorissen D, Takeda H, Zhu W. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res. 2015;75:159–170. doi: 10.1158/0008-5472.CAN-14-1670. [DOI] [PubMed] [Google Scholar]