

Abstract
Non-GIST soft tissue sarcomas are a heterogeneous grouping of mesenchymal tumors that comprise less than 1% of adult malignancies. Treatment continues to be based on cytotoxic chemotherapy regimens. However, characterization of the molecular pathway deregulations that drive these tumors has led to the emergence of more customized treatment options. In this review, we focus on the multitude of molecular inhibitors targeting angiogenesis and cell cycle pathways being tested in clinical trials.
Keywords: Non-GIST soft tissue sarcoma, targeted therapy, angiogenesis inhibitors, cell cycle inhibitors
OVERVIEW
Sarcomas are mesenchymal in origin and comprise less than 1% of adult cancers. The term sarcoma encompasses a wide gamut of diseases originating from bone, cartilage, adipose, muscular, vascular, or hematopoietic tissues. In this review, we focus on soft tissue sarcomas (STS) other than gastrointestinal stromal tumors (GIST). Traditionally, non-GIST soft tissue sarcomas have been classified based on their tissue of origin. An evolving understanding of the associated genetic and molecular pathway aberrations may provide more reproducible classifications and lead to more efficacious treatment options.
Currently, treatment options for patients with non-GIST STS are limited. In the locally advanced and metastatic setting, anthracycline-based cytotoxic chemotherapy, either as a single agent or in combination with other cytotoxic agents, has remained the mainstay of the majority of clinical treatment regimens with modest responses of up to 25% for single agent therapy and 30–40% when used in combination[1]. New treatment options are therefore crucial to improve clinical response.
Sarcomas, as characteristic of cancers as a whole, often activate oncogenic pathways and suppress tumor suppressor pathways to sustain their growth. Likewise, a multitude of dysregulated molecular pathways are implicated in the oncogenesis of STS. A summary of major known pathways is illustrated in Figure 1 below. Knowledge of the specific STS subtypes and their propensity for specific pathway alterations is critical to developing more effective therapeutic combinations. In particular, this review focuses on phase 2 or higher clinical trials incorporating agents that target aberrant angiogenic and cell cycle pathways (highlighted boxes Figure 1). The pharmacologic agents referenced in this review are listed in Table 1 and our recommendations are in Table 2.
Figure 1. Dysregulated pathways associated with non-GIST STS oncogenesis.
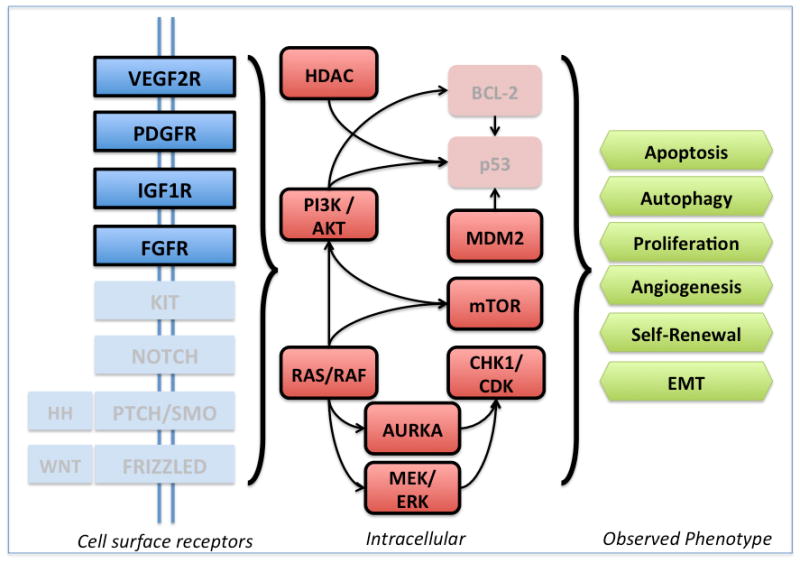
Highlighted boxes indicate molecular deregulations that are covered in this review.
Table 1.
Single-agent activity of agents targeting angiogenesis and cell cycle pathway in non-GIST STS.
| Agent | Phase/Total patients | Selected Single-agent Clinical Trials of targeted therapies in non-GIST STS Disease Subtype (number of patients) | Response |
|---|---|---|---|
|
| |||
| VEGF – Monoclonal Antibody | |||
|
| |||
| Bevacizumab | 2/30 | AS (23), epithelioid hemangioendothelioma (7) | 13% PR (2 from each group); 50% SD after 2 cycles (11 AS, 4 EA); median PFS 12.4w |
|
| |||
| VEGF – Tyrosine Kinase Inhibitors | |||
|
| |||
| Pazopanib | 2/142 | LS (19), LMS (42), SS (38), FS (1), FHS (4), RMS (1), AS (1), tumor of uncertain differentiation (7), MPNST (5), MSFT (3), undifferentiated sarcoma NOS (12) | 9 PR (LMS (1), SS (5), other (3)) |
| 3/246 | Non-liposarcoma, non-GIST STS | 6% PR, 67%SD | |
|
| |||
| Sunitinib | 2/53 | LS (11), hemangiopericytoma (3), AS (3), intimal sarcoma (1), desmoid (1), sarcoma NOS (5), SS (4), CaS (3), DRSCT (3), LS (2), alveolar soft-part sarcoma (1), clear cell sarcoma (1), extraskeletal myxoid chondrosarcoma, giant cell tumor of bone (1) | 1 PR (DSRCT); 10 SD (hemangiopericytoma (2), giant cell tumor of the bone (1), AS (1), LS (1), SS (1), chordoma (4)) |
| 2/48 | LS, LMS, MFH | Median PFS LS 3.9m, LMS 4.2m, MFH 2.5m Median OS LS 18.6m, LMS 10.1m, MFH 13.6m 3m PFR in pretreated patients LS 69%, LMS 62.5%, MFH 44.4% |
|
|
| |||
| Sorafenib | 2/145 | LMS (37), AS (37), MPNST (12), SS (12), MFH (12), other (12) | 1CR (AS), 14% AS patients had PR; median PFS 3.2m, OS 14.3m; 3m PFR AS 64%, LMS 54% |
| 2/26 | LMS (6), spindle cell (5), GIST (3), FS (3), LS (2), chondrosarcoma (2), clear cell (1), Ewings (1), osteosarcoma (1) | 1PR, 3 MR; 5 SD | |
| 2/37 | Advanced VS (8) (AS, malignant hemangiosarcoma, solitary fibrous tumor), LS (10), LMS (19) | Median PFS 3m and OS 17m; 6/8 patients with advanced VS had SD or better with PFS of 5m | |
|
| |||
| PDGFR inhibitors | |||
|
| |||
| Imatinib | 2/31 | Osteosarcoma(2), Ewing/PNET (2), RMS (1), LPS (1), FS (4), SS (6), MFH (1), Epithelioid Sarcoma (2), CaS (3), others (1) | 1 PR (SS) and 9 SD (SS (1), Ewing (1), FS (1), Epithelioid sarcoma (2), CS (2), DFSP (2)) |
|
| |||
| IMC-3G3 | 1b-2/NA | NA | NA |
|
| |||
| IGF-1R inhibitors | |||
|
| |||
| Cixutumumab | 2/111 | RMS (17), LMS (24), adipocytic sarcoma (37), SS (17), Ewings (18) | PFS 12.1w in LS, double that of patients with other non- GIST STS subtypes |
|
| |||
| R1507 | 2/163 | Osteosarcoma(38), RMS (36), SS (23), other (66) | ORR 2.5%, 4 PR |
|
| |||
| FGFR inhibitors | |||
|
| |||
| Brivanib | 2/251 | LMS (60), LS (61), AS (20), other (110) | 12w PFS significantly prolonged in FGF2+ patients treated with Brivanib (vs placebo); 3% ORR, 7 PR (3 PR in AS patients) |
|
| |||
| JNJ-042756493 | 1/37 | NA | 1 SD (CS) |
|
| |||
| mTOR inhibitors | |||
|
| |||
| Everolimus | 2/38 | STS patients that failed prior anthracycline-based therapy | 10 SD at 4m, 1 PR (AS), PFS 1.9m |
|
| |||
| Sirolimus | R/3 | Vascular epithelioid cell tumors(PEComas) | 100% radiographic response |
|
| |||
| Temsirolimus | 2/40 | STS patients without previous treatment for metastatic disease | 2 PR (undifferentiated FS, uterine LMS); median PFS 2m |
|
| |||
| Ridaforolimus | 3/711 | Advanced STS patients previously benefiting from cytotoxic therapy | 40.6% clinical benefit at 4m; 28% reduction in risk of progression or death versus placebo |
|
| |||
| MEK inhibitors | |||
|
| |||
| RO5126766 | 1/12 | 5 patients with advanced sarcoma | PD in 4 patients, SD for 65din the other |
|
| |||
| Trametinib | 1/1 | NA | SD, length unknown |
|
| |||
| CDK inhibitors | |||
|
| |||
| Flavopuridol | 2/18 | AS (2), Clear cell sarcoma (1), Epithelial Sarcoma (1), FS (1), GIST (2), Malignant hemangiopericytoma (1), LMS (2), LS (2), MFH (1), SS (1), NOS (3), malignant schwannoma (1) | No objective responses, SD in 47% patients for a median of 4.3m; 3m PFS 44% |
|
| |||
| Palbociclib | 2/29 | Purely well-(5) and de-+/-well-differentiated (24) LS | 3m PFS 66%, median PFS 18.7w; 1 PR at w74 |
|
| |||
| PD0332991 | 2/29 | Locally advanced or metastatic WDLS or DDLS | 3-m PFS 52%, median PFS 17.9w |
|
| |||
| P1446A-05 | 1/39 | NA | 1SD > 11 cycles(ASPS) |
|
| |||
| Aurora Kinase inhibitors | |||
|
| |||
| Alisertib | 2/72 | LS, LMS, US, MPNST, other | 3m PFS was 73% (LS), 44% (LMS), 23% (US), 57% (MPNST) and 39% (other); 1 PR (AS) |
|
| |||
| ENMD-2076 | 2/10 | Advanced/metastatic STS >1previous line of therapy | 2 PR, 1 SD > 6m |
|
| |||
| MDM2 inhibitor | |||
|
| |||
| RG7112 | 1/20 | WDLS and DDLS | 14 SD, 1 PR |
|
| |||
| RG7338 | 1/NA | NA | SD in unreported number of sarcoma patients |
|
| |||
| HDAC inhibitor | |||
|
| |||
| Panabinostat | 2/48 | Myxoid LS (4), SS (6), ESS (3), ASPS (1), WDLS (4), DDLS(5), pleomorphic LS (2), LMS (10), MPNST (6), NOS (3), other (3) | 6 SD (SFT (1), LPS (1), LMS (2), ESS (2)); median PFS 1.7m |
AS = angiosarcoma, LS = liposarcoma, LMS = leiomyosarcoma, SS = synovial sarcoma, FS = fibrosarcoma, FHS = fibrous histiocytoma ,RMS = rhabdomyosarcoma, MPNST = malignant peripheral nerve sheath tumor, MSFT = malignant solitary fibrous tumor, NOS = not otherwise specified, GIST = gastrointestinal stromal tumor, STS = soft tissue sarcoma, CaS = carcinosarcoma, DRSCT = desmoplastic round small cell tumor, MFH = malignant fibrous histiocytoma, VS = vascular saroma, EA = epithelioid hemangioendothelioma, CS = chondrosarcoma; NA = not available; PR = partial response, SD = stable disease, PFS = progression free survival, OS = overall survival, PFR = progression free survival rate, CR = complete response, MR = minor response, ORR = overall response rate; TSH = thyroid stimulating hormone; NOS = not otherwise specified; m = month, w = week
Table 2.
Table of summary statements from each section.
| Target | Recommendations |
|---|---|
| VEGFR | Anti-VEGF therapies are most effective against highly vascularized STS and have moderate activity in LMS. |
| PDGFR | Aside from DFSP, the lack of correlative PDGF-related analyses in these trials do not allow for definitive conclusions to be made about the role of anti-PDGF activity in STS patients. |
| IFGR1 | Patients with adipocytic sarcoma may benefit from treatment with single agent cixutumumab. |
| FGFR | Appears promising particularly in AS however there are no FGFR only directed treatments so extrapolation of clinical benefit due to this pathway specifically must be made carefully. |
| HDAC | No data to support single agent usage. Potential use in combination with other agents as an anti-autophagy modality. |
| mTOR | Single-agent mTOR inhibition shows a very modest PFS benefit but no survival advantage. |
| MDM2 | Too soon to tell; may have utility in liposarcoma. |
| CDK / CHK1 | Too soon to tell; potential therapeutic role in patients with CDK- amplified liposarcoma. |
| MEK | Too soon to tell; may have utility in LMS and in sarcomas with NF1 deletion. |
| AURKA | Too soon to tell; may have utility in angiosarcoma. |
TARGETING ANGIOGENESIS
Angiogenesis is defined as the process of forming new blood vessels from pre-existing vessels and has been suggested as one of the hallmarks of cancer [2]. Vascular endothelial growth factors (VEGF) and their receptors (VEGFR) are key components of endothelial cell proliferation during new blood vessel formation. Concurrently, platelet-derived growth factors (PDGF) and their receptors (PDGFR) are critical regulators of the tumor stroma. Activation leads to pericyte recruitment and stabilization of the newly formed blood vessels vascular smooth muscle [3]. More recently, it has been elucidated that insulin-like growth factor (IGF) and its receptor IGF-R1 play a role in VEGF stimulation of angiogenesis and that inhibition of IGF-R1 can inhibit angiogenesis [4]. Similarly, our understanding of the fibroblast growth factor (FGF) pathway has lead to the discovery that its signaling not only leads to cell differentiation and pro-survival but tumor angiogenesis and VEGF inhibition resistance as well [5].
VEGFR
STS, like other proliferating malignancies, are dependent on the formation of new blood vessels to support their growth, invasion and metastasis. This process is complex and not fully understood, but the interplay between numerous factors, including oncogenic mutations, mechanical stress, and tumoral and microenvironmental hypoxia are thought to shift tumors into a pro-angiogenic state [6]. Endothelial cell proliferation and tube formation is mediated through VEGF (VEGF-A) signaling with VEGFR-2. VEGFR-1 regulates VEGFR-2 mediated angiogenesis, and VEGF-C and -D induce lymphangiogenesis via VEGFR-3 signaling [7,8]. In STS, numerous strategies – either as single agents or in combination – have been employed to block VEGF-activity and are associated with anti-tumor activity.
Analyses of non-GIST STS patient samples utilizing immunohistochemistry and ELISA have been instrumental in identifying tumor subtypes with the highest likelihood of susceptibility to anti-VEGF therapies [9–18]. Collectively, evidence indicates that VEGF levels are increased in malignancy compared to non-malignant controls, and that increased VEGF expression is associated with higher tumor grade [9–11,14,18]. Conversely, VEGF expression does not consistently correlate with disease-free or overall survival, and occurrence of metastasis [9,12,13,16,18]. Interestingly, no consistent association between microvessel density (MVD) and VEGF expression or histologic subtype has been reported, but MVD itself has been shown to correlate with tumor grade and tumor size [14,16,18]. Though these studies do not depict a prognostic role of VEGF/VEGFR in non-GIST STS sarcomas, they do highlight numerous potential STS subtypes that may be targetable with anti-VEGF therapies.
Several strategies targeting the VEGF pathway have been developed and tested in various phase clinical trials in patients with non-GIST STS, including recombinant humanized antibody against VEGF, VEFGR tyrosine kinase inhibitors (TKI) [19–25] and combined treatment with target and chemotherapy agents [26–28].
Bevacizumab (Avastin), a humanized recombinant antibody against VEGF has shown clinical efficacy in combinatorial regimens in numerous malignancies. Considering the dependence of advanced vascular sarcomas on the VEGF pathway for proliferation, growth and metastasis, Agulnik et al. (2012) evaluated its use in the first, prospective, multicenter phase 2 trial evaluating target therapy in a subset of vascular sarcoma patients[29]. Thirty patients with metastatic or locally advanced angiosarcoma (AS) or epithelioid hemangioendothelioma were enrolled to evaluate the safety and efficacy of single-agent bevacizumab. Single-agent treatment was well tolerated and no unexpected adverse events were experienced. Two patients from each group had evidence of partial response (PR), 50% of patients had stable disease (SD) after two cycles of treatment, and the overall median progression-free survival (PFS) was 12.4 weeks. In patients with AS, median PFS was 12 weeks and overall survival (OS) was 52.7 weeks. Vascular sarcomas are rare, aggressive and have shown minimal response to chemotherapy. This trial indicated that bevacizumab is safe with modest single-agent efficacy in subset of this population.
Based on the previously described phase 2 trial of single-agent bevacizumab, numerous trials have evaluated the role of bevacizumab in combination regimens in patients with STS. D’Adamo et al. (2005) evaluated the combination of bevacizumab and doxorubicin in a single-arm phase 2 trial in 17 patients with metastatic STS (11 with leiomyosarcoma (LMS)) [26]. Treatment was not associated with any complete responses, but 65% of patients had SD, the overall response rate (ORR) was 12%, median OS was 16 months, and two patients with uterine LMS had a PR. Toxicities did not differ greatly from that reported for single-agent doxorubicin; however, a greater than expected decline in cardiac function was noted. Specifically, 35% of patients were removed from the study for grade 2 or higher cardiac toxicity; one patient each with grade 3 and 4 toxicities following 11 and 8 cycles of treatment respectively, and 4 patients with grade 2 reductions in the left ventricular ejection fraction (LVEF) after one, two, and 4 cycles (2 patients) of treatment. At follow-up, 5 of these 6 patients had improvement in LVEF. Verschraegan et al. (2011) conducted a dose-finding study of combination docetaxel-gemcitabine-bevacizumab in 38 patients with STS (15 in the neoadjuvant setting and 20 for metastatic disease) [27]. One patient had bowel perforation attributable to bevacizumab and later died from a pulmonary embolism. Further, grade 4 events included one patient each with pneumothorax and necrotizing pneumonia. The overall response rate was 31.4%, clinical benefit rate was 82.8%, and patients treated in the neoadjuvant setting had an OS rate of 69% at 4 years. All 4 patients with AS and 40% of the patients with undifferentiated high-grade sarcomas had partial response or better. PRs were also observed in patients with myxoid liposarcoma (LS), synovial sarcoma (SS), and adenosarcoma of the uterus. These results suggest that a 3-drug regimen improves response, albeit at the expense of additional risk for adverse events. Recently, preliminary results from the ANGIOTAX-PLUS trial were presented at the 2014 American Society of Clinical Oncology (ASCO) Conference [30]. This was a randomized phase 2 trial assessing the activity of weekly paclitaxel plus or minus bevacizumab in patients with advanced AS. Despite promising results in prior studies in patients with AS, this randomized trial showed no difference in ORR, PFS, or OS with the addition of bevacizumab.
Currently, pazopanib is the only anti-VEGF FDA-approved targeted molecular therapy in STS. This 2nd generation VEGF inhibitor TKI has high affinity against VEFGR 1/2/3 and lower affinity for PDGFR-α/β, FGFR-1/2 and stem cell factor receptor (c-Kit). The European Organisation for Research and Treatment of Cancer (EORTC) 62043 phase 2 trial in 142 patients with advanced STS [23] included 4 cohorts based on disease subtype: LS, LMS, SS, and ‘other’. Treatment was generally well tolerated and associated with no complete responses, but 9 patients had evidence of partial response (SS (5), other (3), and LMS (1)). The 3 month PFS was 44%, 49%, and 39% for LMS, SS, and ‘other’, respectively. The OS was 354 days for patients with LMS and 310 days for patients with SS. Importantly, the LPS cohort was closed early due to evidence of insufficient activity. The subsequent pivotal PALETTE (Pazopanib Explored in Soft-tissue Sarcoma) trial was a phase 3 multinational, randomized, double-blinded, placebo controlled study that further defined the role of pazopanib in non-GIST STS [24]. This study enrolled 372 patients with non-adipocytic STS, and included patients with LMS (115), SS (30), and ‘other’ (101). Six percent of patients had PR and 67% had SD that were sufficient to obtain its FDA indication. Although generally well tolerated, 39% of patients necessitated dose reduction of the 800 mg starting dose and 34% of patients withdrew from the trial due to drug toxicity.
Suntinib is a multi-targeted tyrosine kinase inhibitor with activity against VEGF receptors 1/2/3, PDGFR- α/β, c-Kit, FLT3, RET, and CSF-1. Similar to pazopanib, sunitinib has multiple effector sites that make it a plausible antitumor agent in patients with non-GIST STS, and 3 different phase 2 trials have evaluated its safety and clinical efficacy. George et al. (2009) enrolled 53 patients with non-GIST sarcoma (LMS (11), malignant fibrous histiocytoma (MFH) (5) and SS (4)) in a phase 2 trial investigating single-agent sunitinib [19]. Twenty percent of patients had evidence of stable disease, and one patient with desmoplastic round cell tumor had a PR and remained on therapy for 56 weeks. Mahmood et al. (2011) evaluated patients with LS, LMS, and MFH in a phase 2 single agent trial [20]. Fifty percent of patients required dose reduction. The median PFS was 3.9, 4.2, and 2.5 months, median OS was 18.6, 10.1, and 13.6 months, and 3-month PFS was 69%, 62.5%, and 44.4% for LPS, LMS and MFH, respectively. It should be noted that these promising results were not recapitulated in a uterine LMS cohort of 23 patients [31].
Sorafenib inhibits BRAF, VEGFR-1 – 3, PDGFR-β, fms-like tyrosine kinase 3, and c-Kit. Evidence from four different clinical trials indicate that single-agent sorafenib has variable efficacy in non-GIST STS, but that similar to other TKIs, it may have therapeutic efficacy in the treatment of vascular tumors. Maki et al. (2009) conducted a phase 2, multicenter trial in 122 patients [21]. Sixty-two of the enrolled patients had SD, 14% of patients with had evidence of PR, and one patient with AS had a complete response. The median PFS was 3.2 months, median OS was 14.3, and the 3-month PFS was highest in AS (64%) and LMS (54%). These findings led to the conclusion that sorafenib is active in metastatic AS and is associated and modest activity in LMS. In a randomized phase 2 trial in 26 patients non-GIST STS, 3 patients had evidence of tumor regression (SS, spindle cell sarcoma, and GIST), 5 patients had SD, and time to progression was 23, 47, and 47 weeks in fibrosarcoma (FS), LMS, and chondrosarcoma, respectively [22]. Further, in the cohort of patients with vascular tumors (AS and hemangiopericytoma), 7 of 9 patients had stable disease and two remained on treatment for over 12 months, suggesting an improved anti-VEGF effect in more these more vascular tumors. Ray-Coquard et al. (2012) further define the role of sorafenib in highly vascularized tumors in a phase 2 trial in 41 patients with AS, 73% of which had received prior chemotherapy [32]. Chemotherapy-naïve patients gained no clinical benefit from treatment, but previously treated patients had modest antitumor efficacy with an associated 40% tumor control and 23% response rate (RR). Similar results were evident in a 2012 phase 2 trial investigating sorafenib in 51 patients who had received 0 – 1 prior therapies for advanced disease [25]. In this study, 6 of 8 patients with advanced vascular sarcoma had SD or better and had an associated median PFS of 5 months, which was 2 – 3 months longer than in those patients with LS and LMS.
Vincenzi et al. (2013) combined dacarbazine with sorafenib in a phase 1 trial in 17 patients with STS [28]. Of the 14 evaluable patients, no complete responses were seen, but 3 patients had a PR, and 6 had SD. The median OS was 43 weeks and clinical benefit was evident in 64% of patients. Five patients required dose reduction of both agents and three patients required dose reduction of sorafenib only due to dermatologic toxicities.
PDGFR
Platelet-derived growth factor (PDGF) ligand binding to kinase receptors (PDGFR-α and PDGFR-β) results in activation of signaling pathways (Eg. Ras, PI3K) and transcription factors that function, in part, to stimulate cell growth and survival, and regulate tissue homeostasis. Mutational changes involving the PDGF family can lead to overactive autocrine and paracrine signaling ultimately associated with tumorigenesis and the development of progressive disease and metastasis [33]. Evidence from in vitro and clinical correlative studies demonstrate an association between aberrant PDGF family signaling and numerous STS subtypes, including dermatofibrosarcoma protuberans (DFSP), AS, endometrial stromal sarcoma (ESS), LMS, rhabdomyosarcoma (RMS), LS, and SS.
We have previously reviewed the multi-targeted tyrosine kinase inhibitors pazopanib, sunitinib, and sorafenib in the treatment non-GIST STS, and thus refer you to the prior section. Positive preclinical and clinical data has led to the FDA approval of multi-targeted tyrosine kinase receptor inhibitors in STS subtypes including DFSP (imatinib) and non-GIST STS (pazopanib). An association between aberrant PDGF activity and imatinib responsiveness has been described in a subset of DFSP patients [34]. In this population, DFSP is associated with a chromosome 17 and 22 translocation and production of the COL1A1-PDGF-β fusion protein that drives an autocrine PDGFR signaling loop and promotes tumorigenesis [35]. In these tumors, imatinib inhibits the growth and induces apoptosis of DFSP cells via PDGFR inhibition, likely accounting for the 50% RR in patients treated with this agent [36]. In other non-DFSP and -GIST sarcomas, PGFR aberrancy has not been shown to correlate with response to anti-PDGF agents, as was shown by Sugiura et al. 2010 in a phase 2 trial of 25 patients with non-GIST STS (osteosarcoma (OS), Ewing, RMS, LPS, FS, SS, MFH, epithelioid sarcoma and others) treated with imatinib [37]. Aside from DFSP, the lack of correlative PDGF-related analyses in these trials do not allow for definitive conclusions to be made about the role of anti-PDGF activity in STS patients. That being said, there is evidence of preclinical efficacy of IMC-3G3 (Olaratumab), a fully human IgG1 monoclonal antibody that specifically binds to PDGFR-α and blocks ligand binding and receptor activation, in LMS [38] and OS cell lines. As such, a randomized phase 1b/2 trial investigating doxorubicin with or without IMC-3G3 in patients with advanced STS (NCT01185964) is currently underway, but preliminary data is not yet available.
IGF-1R
Insulin-like growth factor (IGF) signaling is a critical factor in malignant transformation and resistance to cytotoxic, hormonal and radiation therapies [39]. IGF-1R is a tyrosine kinase receptor, and is a glycoprotein composed of two extracellular alpha subunits that preferentially bind IGF-1 and with a lesser affinity to IGF-2 and insulin. Activation promotes survival and cell growth via the PI3K/AKT/mTOR and Ras/Raf/MAPK pro-angiogenic pathway [40], and approximately 50% of LMS, UPS, and sarcomatoid non-GIST STS tumors express IGF-1R [41].
Cixutumumab (IMC-A12) is an IgG 1/λ monoclonal antibody that binds to the IGF-IR with high affinity. Phase 2 studies investigating single-agent cixutumumab have shown modest clinical benefit in LPS, with an associated PFS of 12.1 weeks, double that of patients with other non-GIST STS subtypes [42]. Another monoclonal antibody, R1507, was associated with similar modest clinical benefit, such that in a phase 2 clinical trial investigating 163 eligible patients with osteosarcoma (n = 38), RMS (n = 36), SS (n = 23), and other sarcomas (n = 66), the overall ORR was 2.5%, including 4 partial responses [43].
FGFR
The fibroblast growth factor receptor (FGFR) family is composed of 4 receptor tyrosine kinases, including FGFR 1, 2, 3, and 4. FGFR signaling plays a critical role in cell migration, proliferation, survival and differentiation in normal cells. Along with VEGF and PDGF, FGF signaling is associated with angiogenesis, tumorigenesis, lymphangiogenesis and metastasis in non-GIST STS [5,44]. Utilizing next generation sequencing, Jour et al, 2014 showed that in 25 patients with STS, that 40% of patients had tumors associated with a FGFR copy number gain and amplification, specifically in LMS and clear cell sarcomas [45]. Further, FRS2, a FGFR substrate that plays a critical role in FGFR signaling, along with CDK4 and MDM2, is a characteristic feature of high grade or DDLS [46]. Aberrant FGF signaling has also been implicated in other non-GIST STS subtypes including, but not limited to, AS, CS, OS, SS, and RMS.
Based on this preclinical data, aberrant FGF signaling likely has a tumorigenic role in STS. The clinical efficacy of both pazopanib and dasatinib (discussed above) may be at least partially attributable to their non-selective nature that results in numerous off-target effects, including inhibition of FGFR. More selective anti-FGF agents are also currently being developed and investigated in early phase clinical trials. Schwartz et al (2011) conducted a randomized phase 2 discontinuation trial evaluating Brivanib (BMS-582664), an oral FGF and VEGF signaling inhibitor, in 251 patients with advanced STS (LMS, LS, AS, other)[47]. Patients with stable disease were randomized (n = 76) to receive the study drug or placebo. Patients with immunohistochemical evidence of FGF2 expression were shown to have a significantly increased 3-month PFS (2.8 months versus 1.4 months), and the study was not able to exclude the benefit of this agent even in FGF2-negative patients. At 12 weeks, 7 patients achieved a partial response, 3 of which were in patients with AS. Though data is limited, Bahleda et al. (2014) conducted a phase 1 trial investigating JNJ042756493, a pan-fibroblast growth factor receptor (1 – 4) inhibitor in 37 patients with advanced solid tumors [48]. Ten patients on this trial had grade 3 or higher adverse events, and AST/ALT elevation represented the only grade 3 dose-limiting toxicity and occurred at the highest tested dose. Incomplete data is available pertaining to the number of STS patients enrolled, but one patient with chondrosarcoma had evidence of stable disease, the duration of which is unknown. Considering the importance of FGF signaling in the pathogenesis of numerous malignancies, there are a number of ongoing early phase trials investigating the role of novel agents in patients with advanced STS, including BGJ398 (NCT01004224) and TEN-010 (NCT01987362).
TARGETING CELL CYCLE
The mammalian target of rapamycin (mTOR) assimilates upstream nutrient and mitogen signals to regulate cell growth and cell division. Like the mTOR pathway, the MEK/ERK signaling cascade is one of the key signaling pathways that integrates extracellular pressures (i.e. nutrient starvation). MEK activates ERK1 and ERK2 isoforms resulting in cell proliferation. Once cell cycle begins, cyclin-dependent kinases (CDKs), along with cyclins, are responsible for regulating the progression of cells through the cell cycle. The cell cycle pathway is further controlled by the p53 tumor suppressor which mediates the transition from G1 to S and is negatively regulated by the murine double minute 2 (MDM2). Next, aurora kinases such as Aurora Kinase A (AURKA) that mediate the transition of cell cycle from G2 to M. Similarly, histone deacetylases (HDACs) regulate gene expression by deacetylating histones and modulate the acetylation of a number of non-histone proteins including cell division – and indeed have been shown to also be critical for the G1 to S transition [49].
mTOR / PI3K / AKT
The mTOR pathway integrates the upstream pathway information from insulin, growth factors and amino acids. The centrality of the mTOR pathway with regard to sensing cellular nutrient, oxygen, and energy levels explains its integral role in oncogenesis. Multiple studies have shown that the PI3K/Akt/mTOR pathway is activated in and mediates translational control of various sarcoma subtypes (53–57). In vitro expression analyses of 140 bone and soft tissue sarcomas revealed Akt activation in 55% of cases (61% in malignant and 27% in benign), and mTOR expression in 61% (66% in malignant and 39% in benign) of cases, most notably in RMS and SS [50]. To note, mTOR pathway activation is associated with a higher likelihood of metastases, adverse prognosis, and decreased survival [51–54].
Everolimus, ridaforolimus, sirolimus and temsirolimus have been evaluated in single-agent clinical trials. A multi-center phase 2 trial investigated everolimus in 38 patients with STS who failed prior standard anthracycline based therapy. One partial response was noted in a patient with AS, 10 patients had SD at 16 weeks, and the PFS was 1.9 months [55]. Further, three patients with peripheral vascular epithelioid cell tumors (PEComas) were treated with sirolimus and all demonstrated radiographic responses [56]. Temsirolimus, which is converted by CYP3A4 enzyme to sirolimus, was investigated in a phase 2 trial in 40 evaluable patients that had not previously received treatment for metastatic disease. Partial responses were evident in one patient each with undifferentiated FS and uterine LMS, and the median PFS was 2.0 months [57].
The largest studies incorporating mTOR inhibition have been conducted with ridaforolimus (previously known as deforolimus). Based on promising phase 2 data, SUCCEED, a multinational, randomized, double-blinded, placebo-controlled, phase 3 trial of ridaforolimus versus placebo in 711 patients with advanced sarcoma who had achieved clinical benefit with prior cytotoxic therapy was completed. Clinical benefit at four months was achieved more often with ridaforolimus than placebo (40.6% vs 28.6%; p<0.001). The ridaforolimus group had a 28% reduction in the risk of progression or death (p<0.001), but the median PFS benefit was small (17.7 weeks for ridaforolimus versus 14.6 weeks for placebo). Overall survival at 24 months was not significant [58]. With these modest results, ridaforolimus did not receive FDA approval.
Given the small responses of mTOR inhibition and IGF-R1 alone, combination pathway inhibition therapy has been explored. This may permit compensatory activation of the PI3K/AKT pathway post mTORC1 inhibition can occur through an IGF-1R-dependent mechanism [59]. Furthermore, inhibition of IGF-1R may abrograte some AKT activation in a TORC1 independent manner [60]. This and other preclinical observations have spurred a phase 2 trial combining temsirolimus with cixutumumab[61]. In this open-label trial, patients were stratified into 3 arms IGF-1R-positive soft-tissue sarcoma, IGF-1R-positive bone sarcomas, or IGF-1R-negative bone and soft-tissue sarcoma. 31% (17/54) of the IGF-1R-positive soft-tissue sarcoma group, 35% (19/54) IGF-1R-positive bone sarcoma group, and 39% (21/54) of the IGF-1R-negative group were progression free at 12 weeks. Median overall survival was over 14 months for all patients in the study. In the pediatric population presented in abstract form, of 43 evaluable patients, no objective responses were observed with only 16% patients progression free at 12 weeks[62]. Thus this combination, failed to improve upon single agents alone.
Combination mTOR clinical trials have been or are being performed with multiple therapeutic partners. Regarding cytotoxics, liposomal doxorubicin [63], cyclophosphamide [64], irinotecan [65] have all been attempted with modest results. In addition, there is an ongoing study with gemcitabine (NCT01684449). Nevertheless, whether any of these mTOR-cytotoxic combinations outperform single agent mTOR inhibition is still in question. Combinations with different molecular pathway inhibition are also underway. There are Phase 2 studies of everolimus with bevacizumab (anti-VEGF monoclonal antibody) in MPNST (NCT01661283), everolimus with sorafenib (anti-VEGF TKI) in OS (NCT01804374). Perhaps most interesting are the combinations that address feedback activation of PI3K/AKT pathways after mTORC1 inhibition with RAS/MEK pathway inhibitors. Indeed, dual inhibition of RAS/MEK and mTOR appears to be a promising modality in vitro [66].
MEKi
The RAS pathway is involved in cell proliferation, differentiation, angiogenesis and survival. The mitogen-activated protein kinase (MAPK) signaling pathway, also known as the extracellular signal-regulated pathway (ERK), occurs downstream of RAS and consists of three different kinases, including ERK, and the serine-threonine kinases RAF and MEK. The constitutive activation of this pathway is associated with proliferation and metastasis in numerous malignancies (pancreas, colon, lung, ovary, prostate, and kidney). Aberrant activation of the MAPK pathway has been demonstrated in numerous STS subtypes and preclinical studies have demonstrated antitumor efficacy of numerous MEK inhibitors in sarcoma cell lines and mouse models. Specific STS subtypes that have been studied include SS, malignant peripheral nerve sheath tumor (MPNST), MFH, FS, AS, and RMS.
Despite preclinical evidence suggesting the efficacy of MEK inhibition in the treatment of certain STS subtypes, relatively little clinical data substantiating its therapeutic efficacy is available. Honda et al. (2013) conducted a small phase 1 study evaluating the oral MEK inhibitor RO5126766 in 12 patients with advanced solid tumors [67]. Among the patients enrolled, 5 patients had sarcoma. Of these, treatment was associated with progressive disease in 4 patients, and SD for 65 days in the other. Trametinib (GSK1120212) has been relatively well studied in advanced solid tumors, but very little data is available in regard to STS. Infante et al. (2012) conducted a phase 1 trial in patients with advanced solid tumors [68]. In this trial, one patient with STS was enrolled and was found to have SD, although the specific disease subtype and length of disease stability is unknown.
In the NCI-sponsored Phase 2 trial of temsirolimus and the MEK inhibitor, selumetinib, (NCT01206140), 69 subjects were randomized to selumetinib and allowed to crossover upon progression, or to selumetinib plus temsirolimus weekly (n=35). There was no difference in PFS between the single agent vs. combination arm for the overall cohort but an improved median PFS was observed in the combination arm (N=11) over single agent (N=10) in the pre-specified leiomyosarcoma stratum (median 3.7 vs. 1.8 months; p=0.01) [69]. In addition, recent preclinical work suggests that NF1 deletion may predispose STS to MEKi sensitivity[70].
CDKi
Cyclin-dependent kinases (CDKs), along with cyclins, are responsible for regulating the progression of cells through the cell cycle. Disruption of this process is a common phenomenon in malignancy, and agents that manipulate aberrant cell cycle activation are being tested in early phase clinical trials.
Flavopiridol, a non-selective CDK 1, 2, 4, 6, 7, and 9 inhibitor, is an analogue of a naturally occurring flavonoid isolated from the bark of Dysoxylum binectariferum, a plant that is indigenous to India [71]. Morris et al. (2006) evaluated single-agent flavopuridol in a phase 2 trial in 18 patients with untreated advanced STS [71]. In this trial, no objective responses were seen, and 47% of the patients had SD for a median of 4.3 months. Luke et al. (2012) evaluated flavopiridol in combination with doxorubicin in a phase 1 trial in 31 patients with advanced STS[72]. Combination treatment was associated with neutropenia, thrombocytopenia, and febrile neutropenia, but despite 2 DLTs in the highest dose cohort, the maximum tolerated dose was not achieved. In total, there were two PR, and 16 patients had SD. Subset analysis revealed that 8 of 12 patients with well- or de-differentiated liposarcoma (WDLS or DDLS) had stable disease for over 3 months.
Based on the finding that up to 90% of WDLS and DDLS are associated with CDK4 amplification, Schwartz et al. (2011) investigated the CDK4/6 selective inhibitor palbociclib in a phase 1 trial in 33 patietns with retinoblastoma-positive advanced tumors [73]. Treatment was well tolerated and all 4 patients with WDLS and DDLS had SD, two of which for > 10 cycles. A follow-up phase 2 trial in 29 patients with CDK4 amplified advanced STS revealed that treatment was well tolerated and associated with a single PR and a median PFS of 18 months [74]. More recently, a phase 2 trial investigated the CDK4 inhibitor PD0332991 in 29 patients with locally advanced or metastatic WDLS and DDLS. Patients in this trial had a 3-month PFS of 52% and median PFS of 17.9 weeks; one patient had a confirmed PR lasting over one year [74].
A phase 1 PK study of continuous oral administration of P1446A-05, a novel oral inhibitor of CDK4-D1, CDK1-B, and CDK9-T enrolled 39 patients with advanced malignancies. Treatment was associated with dose-limiting diarrhea, and one patient with alveolar STS whose disease was progressing at the time of enrollment remained on treatment with SD evident after 11 cycles [75].
AURKA inhibition
Microarray studies indicate that Aurora Kinase A (AURKA) is commonly overexpressed in sarcoma [76]. AURKA is a serine/threonine kinase that regulates cell entry into mitosis, and thus is an important regular of cell cycle. In preclinical models, inhibition of AURKA by shRNA or by a specific AURKA inhibitor blocks in vitro proliferation of multiple sarcoma subtypes. Multiple clinical trials are underway examining whether AURKA inhibition. Alisertib is a novel, oral, ATP-competitive, selective small-molecule inhibitor of AURKA. A cooperative group study (A091102) of MLN8237 (alisertib) was recently reported at the 2014 ASCO Conference. In this trial, 72 patients at 24 sites were evaluated. One patient with AS had a confirmed PR, and the 3-month PFS was 73%, 44%, 23%, 57%, and 39% for patients with LPS, LMS, US, MPNST, and other, respectively [77]. A smaller, single-institution, phase 2 trial of the AURKA inhibitor ENMD-2076 enrolled 10 patients with advanced/metastatic STS who had received ≤1 line of prior therapy. Two patients had confirmed PR and one patient had a confirmed SD of > 6 months.
MDM2
p53 is a transcription factor that responds to intra- and extracellular stresses via activation of numerous genes associated with DNA repair, metabolism, cell-cycle arrest, cellular senescence, metabolism, and apoptosis [78]. It is tightly controlled by the negative regulators human double minute 2 (MDM2 or HDM2) and human double minute 4 (MDM4 or HDMX) [79,80]. Augmentation of this signaling pathway occurs in over 50% of malignancies [81] and is caused either by suppression of p53 activity via MDM2 +/- MDM4 amplification in p53 wild-type alleles or by p53 mutational events that result in loss of p53 tumor suppression. Numerous therapeutic strategies targeting aberrant p53 signaling are being developed, including MDM2 inhibitors in wild type p53 tumors, dual HDM2/HDMX antagonists, p53-binding antagonists, and agents that restore the activity of mutant p53 [82]. Altered p53 signaling is evident in sarcomagenesis [83], and these therapeutic strategies have great potential in the treatment in a variety of STS subtypes, especially in chemoresistant tumors.
In STS, well-differentiated (WDLPS) and dedifferentiated liposarcomas (DDLPS) are closely associated with amplification of the chromosomal region 12q13–15 [84]; mutations that ultimately result in suppression of p53 activity and tumorigenesis. In a phase 1 study in 20 patients with WDLPS and DDLPS treated with RG7112, a MDM2 inhibitor; 30% developed significant hematologic toxicities. Treatment resulted in one confirmed PR and SD in 14 patients, results that suggest that inhibition of MDM2 is associated with p53 pathway activation and decreased cell proliferation in MDM2-amplified liposarcomas [85]. A phase 1b dose-escalation trial evaluated the combination of RG7112 and doxorubicin in 23 patients with advanced STS. Treatment was associated with grade 3 or 4 neutropenia, thrombocytopenia, and febrile neutropenia in 12, 9 and 5 of the 20 evaluable patients, respectively. At the time of publication, interim analysis indicated that 50% of patients had evidence of stable disease (disease subsets not presented) [86]. RG7388, a second generation MDM2 inhibitor, also recently reported stable disease in sarcoma patients in their phase I trial [87]. Other MDM2 inhibitors, including MI-773 (SAR205838) and DS-3032b are undergoing clinical development.
HDACi
Aberrant histone acetylase (HAT) and/or histone deacetylase modification of gene expression has been implicated in the pathogenesis of various malignancies. As such, HDAC inhibitors (HDACi) are being actively explored in a number of other solid organ and hematologic malignancies. These agents induce the transcription of positive and negative regulators that alter cell signaling and cause differentiation, growth arrest and/or apoptosis of tumor cells [88,89]. Preclinical studies in STS models highlight a potential role for these agents in specified sarcoma subsets, including endometrial stromal sarcoma, rhabdomyosarcoma, and synovial sarcoma.
HDACi’s are currently being evaluated in early phase clinical trials in patients with advanced STS [90,91]. Cassier et al. (2013) evaluated single-agent panabinostat in anthracycline-refractory patients with advanced STS [90]. This trial enrolled 48 patients, including 14 patients with translocation-related sarcoma (myxoid LS, SS, ESS, and alveolar soft part sarcoma), and non-translocation related sarcoma (WDLS, DDLS, pleomorphic LS, LMS, MPNST, and sarcoma NOS). No objective responses were evident, but 36% of the patients had stable disease, 20% had a 3-month PFS, and 6 patients were progression free at 6 months. The study did not reach its primary endpoint (3-month PFS), but subset analyses revealed that 6 patients had prolonged SD (SFT, well differentiated LPS, LMS, and ESS).
Attia et al. (2011) evaluated the combination of vorinostat and bortezomib in a phase 1 trial in patients with advanced malignancies [91]. One of the two patients with partial response had MFH, findings that suggest clinically relevant synergism between these 2 agents in patients with advanced refractory STS. More recently preclinical data suggests that a combination of HDAC and VEGF inhibition may be synergistic in overcoming autophagy [92].
CONCLUSION
No single targeted agent or existing combination therapy was effective for more than a minority of sarcoma patients. Yet, inhibition of angiogenic and cell cycle pathways has demonstrated remarkable responses in specific non-GIST STS subtypes and in individual patients. Thus, the future of targeted therapy in sarcoma will require further deciphering of the molecular heterogeneity to allow better personalization of these targeted therapies.
Acknowledgments
DWS is supported by the National Cancer Institute of the National Institutes of Health under Award Number T32CA165998 (PI Miguel Villalona and Steven Devine). The content is solely the responsibility of the authors and does not represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Disclosure: Both authors are without relevant conflicts of interest.
Contributor Information
Douglas Sborov, Email: douglas.sborov@osumc.edu.
James L Chen, Email: james.chen@osumc.edu.
References
- 1.Spira AI, Ettinger DS. The use of chemotherapy in soft-tissue sarcomas. Oncologist. 2002;7:348–359. doi: 10.1634/theoncologist.7-4-348. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15:275–286. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Bid HK, Zhan J, Phelps DA, et al. Potent inhibition of angiogenesis by the IGF-1 receptor-targeting antibody SCH717454 is reversed by IGF-2. Mol Cancer Ther. 2012;11:649–659. doi: 10.1158/1535-7163.MCT-11-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieu C, Heymach J, Overman M, et al. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res. 2011;17:6130–6139. doi: 10.1158/1078-0432.CCR-11-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonini T, Rossi F, Claudio PP. Molecular basis of angiogenesis and cancer. Oncogene. 22:6549–6556. doi: 10.1038/sj.onc.1206816. 0000. [DOI] [PubMed] [Google Scholar]
- 7.McColl BK, Stacker SA, Achen MG. Molecular regulation of the VEGF family – inducers of angiogenesis and lymphangiogenesis. APMIS. 2004;112:463–480. doi: 10.1111/j.1600-0463.2004.apm11207-0807.x. [DOI] [PubMed] [Google Scholar]
- 8.Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovascular Research. 2005;65:550–563. doi: 10.1016/j.cardiores.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Chao C, Al-Saleem T, Brooks J, et al. Vascular Endothelial Growth Factor and Soft Tissue Sarcomas: Tumor Expression Correlates With Grade. Ann Surg Oncol. 2001;8:260–267. doi: 10.1007/s10434-001-0260-9. [DOI] [PubMed] [Google Scholar]
- 10.Graeven U, Andre N, Achilles E, et al. Serum levels of vascular endothelial growth factor and basic fibroblast growth factor in patients with soft-tissue sarcoma. J Cancer Res Clin Oncol. 1999;125:577–581. doi: 10.1007/s004320050319. [DOI] [PubMed] [Google Scholar]
- 11.Hayes AJ, Mostyn-Jones A, Koban MU, et al. Serum vascular endothelial growth factor as a tumour marker in soft tissue sarcoma. The British journal of surgery. 2004;91:242–247. doi: 10.1002/bjs.4398. [DOI] [PubMed] [Google Scholar]
- 12.Itakura E, Yamamoto H, Oda Y, Tsuneyoshi M. Detection and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomas. Journal of surgical oncology. 2008;97:74–81. doi: 10.1002/jso.20766. [DOI] [PubMed] [Google Scholar]
- 13.Kilvaer TK, Valkov A, Sorbye S, et al. Profiling of VEGFs and VEGFRs as prognostic factors in soft tissue sarcoma: VEGFR-3 is an independent predictor of poor prognosis. PloS one. 2010;5:e15368. doi: 10.1371/journal.pone.0015368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.PAKOS EE, GOUSSIA AC, TSEKERIS PG, et al. Expression of Vascular Endothelial Growth Factor and its Receptor, KDR/Flk-1, in Soft Tissue Sarcomas. Anticancer Research. 2005;25:3591–3596. [PubMed] [Google Scholar]
- 15.Tokuyama W, Mikami T, Masuzawa M, Okayasu I. Autocrine and paracrine roles of VEGF/VEGFR-2 and VEGF-C/VEGFR-3 signaling in angiosarcomas of the scalp and face. Human pathology. 2010;41:407–414. doi: 10.1016/j.humpath.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 16.West CC, Brown NJ, Mangham DC, et al. Microvessel density does not predict outcome in high grade soft tissue sarcoma. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2005;31:1198–1205. doi: 10.1016/j.ejso.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Yoon SS, Segal NH, Olshen AB, et al. Circulating angiogenic factor levels correlate with extent of disease and risk of recurrence in patients with soft tissue sarcoma. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2004;15:1261–1266. doi: 10.1093/annonc/mdh309. [DOI] [PubMed] [Google Scholar]
- 18.Yudoh K, Kanamori M, Ohmori K, et al. Concentration of vascular endothelial growth factor in the tumour tissue as a prognostic factor of soft tissue sarcomas. British journal of cancer. 2001;84:1610–1615. doi: 10.1054/bjoc.2001.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3154–3160. doi: 10.1200/JCO.2008.20.9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahmood ST, Agresta S, Vigil CE, et al. Phase II study of sunitinib malate, a multitargeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on three prevalent histologies: leiomyosarcoma, liposarcoma and malignant fibrous histiocytoma International journal of cancer Journal international du cancer. 2011;129:1963–1969. doi: 10.1002/ijc.25843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maki RG, D'Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pacey S, Ratain MJ, Flaherty KT, et al. Efficacy and safety of sorafenib in a subset of patients with advanced soft tissue sarcoma from a Phase II randomized discontinuation trial. Investigational new drugs. 2011;29:481–488. doi: 10.1007/s10637-009-9367-9. [DOI] [PubMed] [Google Scholar]
- 23.Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 24.van der Graaf WTA, Blay J-Y, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet. 379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 25.von Mehren M, Rankin C, Goldblum JR, et al. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer. 2012;118:770–776. doi: 10.1002/cncr.26334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Adamo DR, Anderson SE, Albritton K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:7135–7142. doi: 10.1200/JCO.2005.16.139. [DOI] [PubMed] [Google Scholar]
- 27.Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23:785–790. doi: 10.1093/annonc/mdr299. [DOI] [PubMed] [Google Scholar]
- 28.Vincenzi B, Silletta M, Schiavon G, et al. Sorafenib and dacarbazine in soft tissue sarcoma: a single institution experience. Expert opinion on investigational drugs. 2013;22:1–7. doi: 10.1517/13543784.2013.742886. [DOI] [PubMed] [Google Scholar]
- 29.Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2013;24:257–263. doi: 10.1093/annonc/mds237. [DOI] [PubMed] [Google Scholar]
- 30.Nicolas Penel J-YB, Mir Olivier, Tresch Emmanuelle, Bompas Emmanuelle, Domont Julien, Cassier Philippe Alexandre, Rolland Frederic, Piperno-Neumann Sophie, Italiano Antoine, Chevreau Christine, Cupissol Didier, Bay Jacques-Olivier, Collard Olivier, Saada Esma, Bertucci François, Isambert Nicolas, Delcambre Corinne, Clisant Stephanie Isabelle Laure Ray-Coquard and French Sarcoma Group (GSF/GETO) ANGIOTAX-PLUS trial: A randomized phase II trial assessing the activity of weekly paclitaxel (WP) plus or minus bevacizumab (B) in advanced angiosarcoma (AS) Journal of Clinical Oncology, 2014 ASCO Annual Meeting Abstracts. 2014 May 20;32(15_suppl) Supplement [Google Scholar]
- 31.Hensley ML, Sill MW, Scribner DR, Jr, et al. Sunitinib malate in the treatment of recurrent or persistent uterine leiomyosarcoma: a Gynecologic Oncology Group phase II study. Gynecologic oncology. 2009;115:460–465. doi: 10.1016/j.ygyno.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II Trial from the French Sarcoma Group (GSF/GETO) The oncologist. 2012;17:260–266. doi: 10.1634/theoncologist.2011-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietras K, Sjöblom T, Rubin K, et al. PDGF receptors as cancer drug targets. Cancer cell. 2003;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 34.Rutkowski P, Van Glabbeke M, Rankin CJ, et al. Imatinib Mesylate in Advanced Dermatofibrosarcoma Protuberans: Pooled Analysis of Two Phase II Clinical Trials. Journal of Clinical Oncology. 2010;28:1772–1779. doi: 10.1200/JCO.2009.25.7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Human pathology. 2008;39:184–193. doi: 10.1016/j.humpath.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Malhotra B, Schuetze SM. Dermatofibrosarcoma protruberans treatment with platelet-derived growth factor receptor inhibitor: a review of clinical trial results. Current opinion in oncology. 2012;24:419–424. doi: 10.1097/CCO.1090b1013e328353d328378d. [DOI] [PubMed] [Google Scholar]
- 37.Sugiura H, Fujiwara Y, Ando M, et al. Multicenter Phase II trial assessing effectiveness of imatinib mesylate on relapsed or refractory KIT-positive or PDGFR-positive sarcoma. J Orthop Sci. 2010;15:654–660. doi: 10.1007/s00776-010-1506-9. [DOI] [PubMed] [Google Scholar]
- 38.MolCancerTher-2005-Loizos-369-79-1.pdf
- 39.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 40.Peruzzi F, Prisco M, Dews M, et al. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol Cell Biol. 1999;19:7203–7215. doi: 10.1128/mcb.19.10.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lasota J, Wang Z, Kim SY, et al. Expression of the receptor for type i insulin-like growth factor (IGF1R) in gastrointestinal stromal tumors: an immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am J Surg Pathol. 2013;37:114–119. doi: 10.1097/PAS.0b013e3182613c86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoffski P, Adkins D, Blay JY, et al. An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur J Cancer. 2013;49:3219–3228. doi: 10.1016/j.ejca.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Pappo AS, Vassal G, Crowley JJ, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014 doi: 10.1002/cncr.28728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kilvaer TK, Valkov A, Sorbye SW, et al. Fibroblast growth factor 2 orchestrates angiogenic networking in non-GIST STS patients. Journal of translational medicine. 2011;9:104. doi: 10.1186/1479-5876-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Human pathology. 2014;45:1563–1571. doi: 10.1016/j.humpath.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Zhang K, Chu K, Wu X, et al. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res. 2013;73:1298–1307. doi: 10.1158/0008-5472.CAN-12-2086. [DOI] [PubMed] [Google Scholar]
- 47.Ratain M, Schwartz G, Oza A, et al. Brivanib (BMS-582664) in advanced solid tumors (AST): Results of a phase II randomized discontinuation trial (RDT) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:3079. [Google Scholar]
- 48.Bahleda R, Dienstmann R, Adamo B, et al. Phase 1 study of JNJ-42756493, a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients with advanced solid tumors. ASCO Meeting Abstracts. 2014;32:2501. doi: 10.1200/JCO.2014.60.7341. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi T, Cubizolles F, Zhang Y, et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24:455–469. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobashi Y, Suzuki S, Sato E, et al. EGFR-dependent and independent activation of Akt/mTOR cascade in bone and soft tissue tumors. Mod Pathol. 2009;22:1328–1340. doi: 10.1038/modpathol.2009.104. [DOI] [PubMed] [Google Scholar]
- 51.Setsu N, Yamamoto H, Kohashi K, et al. The Akt/mammalian target of rapamycin pathway is activated and associated with adverse prognosis in soft tissue leiomyosarcomas. Cancer. 2012;118:1637–1648. doi: 10.1002/cncr.26448. [DOI] [PubMed] [Google Scholar]
- 52.Petricoin EF, 3rd, Espina V, Araujo RP, et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007;67:3431–3440. doi: 10.1158/0008-5472.CAN-06-1344. [DOI] [PubMed] [Google Scholar]
- 53.Setsu N, Kohashi K, Fushimi F, et al. Prognostic impact of the activation status of the Akt/mTOR pathway in synovial sarcoma. Cancer. 2013;119:3504–3513. doi: 10.1002/cncr.28255. [DOI] [PubMed] [Google Scholar]
- 54.Conti A, Espina V, Chiechi A, et al. Mapping protein signal pathway interaction in sarcoma bone metastasis: linkage between rank, metalloproteinases turnover and growth factor signaling pathways. Clin Exp Metastasis. 2013 doi: 10.1007/s10585-013-9605-6. [DOI] [PubMed] [Google Scholar]
- 55.Yoo C, Lee J, Rha SY, et al. Multicenter phase II study of everolimus in patients with metastatic or recurrent bone and soft-tissue sarcomas after failure of anthracycline and ifosfamide. Invest New Drugs. 2013;31:1602–1608. doi: 10.1007/s10637-013-0028-7. [DOI] [PubMed] [Google Scholar]
- 56.Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010;28:835–840. doi: 10.1200/JCO.2009.25.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okuno S, Bailey H, Mahoney MR, et al. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas: a study of the Mayo phase 2 consortium (P2C) Cancer. 2011;117:3468–3475. doi: 10.1002/cncr.25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demetri GD, Chawla SP, Ray-Coquard I, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol. 2013;31:2485–2492. doi: 10.1200/JCO.2012.45.5766. [DOI] [PubMed] [Google Scholar]
- 59.Wan X, Harkavy B, Shen N, et al. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 60.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Research. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartz GK, Tap WD, Qin LX, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14:371–382. doi: 10.1016/S1470-2045(13)70049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagner Lars MMF, Ahmed Atif, Krailo Mark D, Weigel Brenda, DuBois Steven G, Doyle Austin, Chen Helen X, Blaney Susan. Phase II trial of cixutumumab in combination with temsirolimus in pediatric patients with recurrent or refractory sarcoma: A report from the Children’s Oncology Group. ASCO Annual Meeting; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thornton KA, Chen AR, Trucco MM, et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int J Cancer. 2013;133:997–1005. doi: 10.1002/ijc.28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuetze SM, Zhao L, Chugh R, et al. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur J Cancer. 2012;48:1347–1353. doi: 10.1016/j.ejca.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 65.Verschraegen CF, Movva S, Ji Y, et al. A phase I study of the combination of temsirolimus with irinotecan for metastatic sarcoma. Cancers (Basel) 2013;5:418–429. doi: 10.3390/cancers5020418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renshaw J, Taylor KR, Bishop R, et al. Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res. 2013;19:5940–5951. doi: 10.1158/1078-0432.CCR-13-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Honda K, Yamamoto N, Nokihara H, et al. Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2013;72:577–584. doi: 10.1007/s00280-013-2228-4. [DOI] [PubMed] [Google Scholar]
- 68.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncology. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 69.Zeynep Eroglu HA-HT, Hu James, Guan Min, Frankel Paul Henry, Ruel Nora, Wilczynski Sharon, Christensen Scott, Gandara David R, Chow Warren Allen. NCI #8412: A randomized phase II trial of AZD6244 alone and AZD6244 plus temsirolimus for soft-tissue sarcomas. ASCO Annual Meeting; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dodd RD, Mito JK, Eward WC, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013;12:1906–1917. doi: 10.1158/1535-7163.MCT-13-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morris DG, Bramwell VHC, Turcotte R, et al. A Phase II Study of Flavopiridol in Patients With Previously Untreated Advanced Soft Tissue Sarcoma. Sarcoma. 2006;2006 doi: 10.1155/SRCM/2006/64374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luke JJ, D'Adamo DR, Dickson MA, et al. The Cyclin-Dependent Kinase Inhibitor Flavopiridol Potentiates Doxorubicin Efficacy in Advanced Sarcomas: Preclinical Investigations and Results of a Phase I Dose-Escalation Clinical Trial. Clinical Cancer Research. 2012;18:2638–2647. doi: 10.1158/1078-0432.CCR-11-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwartz GK, LoRusso PM, Dickson MA, et al. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) British journal of cancer. 2011;104:1862–1868. doi: 10.1038/bjc.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dickson MA, Tap WD, Keohan ML, et al. Phase II Trial of the CDK4 Inhibitor PD0332991 in Patients With Advanced CDK4-Amplified Well-Differentiated or Dedifferentiated Liposarcoma. Journal of Clinical Oncology. 2013;31:2024–2028. doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hao D, Chu Q, Welch S, et al. A phase I and pharmacokinetic (PK) study of continuous daily administration of P1446A-05, a potent and specific oral Cdk4 inhibitor. ASCO Meeting Abstracts. 2012;30:3013. [Google Scholar]
- 76.Taylor BS, Barretina J, Maki RG, et al. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer. 2011;11:541–557. doi: 10.1038/nrc3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mark Andrew Dickson MRM, Tap William D, D'Angelo Sandra P, Keohan Mary Louise, Van Tine Brian Andrew, Agulnik Mark, Horvath Laura E, Schwartz Gary K. Alliance A091102: Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. ASCO Annual Meeting; Chicago. 2014. [Google Scholar]
- 78.Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nature Reviews Drug Discovery. 2014;13:217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 79.Huang L, Yan Z, Liao X, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proceedings of the National Academy of Sciences. 2011;108:12001–12006. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3:228. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 82.Biswas S, Killick E, Jochemsen AG, Lunec J. The clinical development of p53-reactivating drugs in sarcomas-charting future therapeutic approaches and understanding the clinical molecular toxicology of Nutlins. Expert opinion on investigational drugs. 2014;23:629–645. doi: 10.1517/13543784.2014.892924. [DOI] [PubMed] [Google Scholar]
- 83.Ito M, Barys L, O'Reilly T, et al. Comprehensive mapping of p53 pathway alterations reveals an apparent role for both SNP309 and MDM2 amplification in sarcomagenesis. Clinical Cancer Research. 2011;17:416–426. doi: 10.1158/1078-0432.CCR-10-2050. [DOI] [PubMed] [Google Scholar]
- 84.Coindre J-M, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Archiv. 2010;456:167–179. doi: 10.1007/s00428-009-0815-x. [DOI] [PubMed] [Google Scholar]
- 85.Ray-Coquard I, Blay J-Y, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with< i> MDM2</i>-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The lancet oncology. 2012;13:1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 86.Chawla Sant PJ-YB, Italiano Antoine, Gutierrez Martin, Le Cesne Axel, Gomez-Roca Carlos Alberto, Gouw Launce G, von Mehren Margaret, Wagner Andrew, Maki Robert G, Higgins Brian, Middleton Steven, Nichols Gwen L, Geho David, Blotner Steven, Zhi Jianguo, Chen Lin Chi. Phase Ib study of RG7112 with doxorubicin (D) in advanced soft tissue sarcoma (ASTS) Journal of Clinical Oncology. 2013:31. [Google Scholar]
- 87.Siu LLIA, Miller WH, Blay J-Y, Gietema JA, Bang Y-J, Mileshkin LR, Hirte HW, Reckner M, Higgins B, Jukofsky L, Blotner S, Zhi J, Middleton S, Nichols GL, Chen LC. Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. ASCO Annual Meeting; 2014. [Google Scholar]
- 88.Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Butler LM, Agus DB, Scher HI, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- 90.Cassier PA, Lefranc A, Amela YE, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. British journal of cancer. 2013;109:909–914. doi: 10.1038/bjc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schelman W, Traynor A, Holen K, et al. A phase I study of vorinostat in combination with bortezomib in patients with advanced malignancies. Investigational new drugs. 2013;31:1539–1546. doi: 10.1007/s10637-013-0029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tavallai S, Hamed HA, Grant S, et al. Pazopanib and HDAC inhibitors interact to kill sarcoma cells. Cancer Biol Ther. 2014;15:578–585. doi: 10.4161/cbt.28163. [DOI] [PMC free article] [PubMed] [Google Scholar]