

Abstract
An understanding of the role of autophagic processes in the management of cardiac metabolic stress responses is advancing rapidly and progressing beyond a conceptualization of the autophagosome as a simple cell recycling depot. The importance of autophagy dysregulation in diabetic cardiomyopathy and in ischemic heart disease - both conditions comprising the majority of cardiac disease burden - has now become apparent. New findings have revealed that specific autophagic processes may operate in the cardiomyocyte, specialized for selective recognition and management of mitochondria and glycogen particles in addition to protein macromolecular structures. Thus mitophagy, glycophagy, and macroautophagy regulatory pathways have become the focus of intensive experimental effort, and delineating the signaling pathways involved in these processes offers potential for targeted therapeutic intervention. Chronically elevated macroautophagic activity in the diabetic myocardium is generally observed in association with structural and functional cardiomyopathy; yet there are also numerous reports of detrimental effect of autophagy suppression in diabetes. Autophagy induction has been identified as a key component of protective mechanisms that can be recruited to support the ischemic heart, but in this setting benefit may be mitigated by adverse downstream autophagic consequences. Recent report of glycophagy upregulation in diabetic cardiomyopathy opens up a novel area of investigation. Similarly, a role for glycogen management in ischemia protection through glycophagy initiation is an exciting prospect under investigation.
Keywords: autophagy, heart, cardiac, diabetes, ischemia, cardiomyocyte
sugar isn't always sweet,
When your heart is worn and skips a beat
Membranes keep it in or out
And insulin gives a special route
Glut transporters form a pore
Bringing sugar in the open door
Too much is bad-too little, too:
Cells need the proper fuel.
Inside the cell sugar's stored
Glycogen the sweetest hoard.
Two enzymes live to break it down:
A neutral enzyme can be found
In cytosol where granules roam,
But in the acidic lysosome
Another waits on bended knee
To play its role in glycophagy.
Excess carbs are bad: this much is clear.
So consider maltose when quaffing beer!
Roberta A. Gottlieb
Energy Stress and Autophagy Induction Overview
Autophagy is an intracellular degradation and recycling function to support energy homeostasis and/or to eliminate defective cell components. Autophagy is upregulated with cellular stress and in the heart was first identified four decades ago to be induced by ischemia (37) and starvation (8), and later determined to be critical for neonatal cardiac tissues during the period of temporary nutrient deprivation accompanying postnatal transition (45). Although upregulated expression of autophagy mediators can be associated with acute performance improvement post-ischemia (24, 35), a sustained elevation of autophagy has been linked with transition to cardiac failure in vivo (112). The general consensus is that autophagy can be beneficial and pro-survival as a short-term strategy to deal with acute stress, but when chronically elevated or constitutive, excess autophagic activity is linked to cell death, stimulating cytokine-mediated collagen infill responses (31, 66, 81). The signaling complexity regulating autophagy in acute and chronic conditions under various trophic and endocrine settings is yet to be fully elucidated. Involvement from PI3K/Akt/mTOR (60, 77) and AMPK (100) pathways in regulating autophagy has been identified in diabetic cardiomyopathy and may play a role in other cardiac pathological settings.
The term autophagy has been considered synonymous with the term macroautophagy (implying a single macromolecular degradative pathway). Macro-autophagy machinery has been relatively well characterized and involves assembly of a membrane structure and parallel enzymatic conjugations analogous to ubiquitin ligation, resulting in conjugation of Atg12 onto Atg5, and subsequent conjugation of Atg8 proteins (e.g. LC3, GABARAPL1, GABARAP) onto phosphatidylethanolamine in the phagophore membrane (additional details provided in Fig. 1). Engulfment of selective cargo requires adaptor proteins, which bridge Atg8 proteins and the target. More recently, a new understanding of selective autophagy processing has emerged, with specific protein machinery recognized for autophagic degradation of mitochondria [mitophagy (22, 33)], endoplasmic reticulum [reticulophagy or ERphagy (26)], peroxisomes [pexophagy (89)], lipid droplets [lipophagy (52)], and glycogen [glycophagy (39, 76)]. In this context, the term macroautophagy is best used to denote the process of general autophagic degradation of heterogeneous macromolecules. A case can now be made that the autophagic processes delineated by the commonly used markers LC3B and p62 (and possibly Beclin-1) are predominantly those involved in macroautophagic breakdown of protein aggregates and some organelles (including mitochondria). Identification of the machinery specific to each autophagic pathway has enabled new tools for investigation of selective autophagy, and new insights are emerging in relation to their differential roles in cardiac pathology.
Fig. 1.

Schematic of autophagy. 1) Formation of the nucleating membrane is initiated by Ulk1 followed by Beclin1 and Vps34. Atg12 is conjugated onto Atg5 by the action of ubiquitin ligase-like enzymes Atg7 and Atg10. Atg8 (e.g., LC3, GABARAP, GABARAPL1) is activated by Atg4 cleavage and then ligated onto membrane phosphatidylethanolamine by the action of Atg3 and Atg7 with the participation of Atg5-12 and Atg16Lcomplex, to form a lipidated Atg8 moiety (e.g., LC3B-II). 2) Specific cargo is tagged with bifunctional adaptor proteins that interact with Atg8 or homologs to facilitate engulfment. 3) Atg5-12 is released from the outer membrane when the phagophore closes upon itself. 4) A lysosome fuses with the autophagosomal outer membrane and delivers its contents. 5) The vacuolar proton ATPase lowers the pH, causing activation of proteases, lipases, nucleases, and glycohydrolases to degrade cargo to single amino acids, fatty acids, nucleotides, and monosaccharides, which are exported across the membrane to the cytosol.
This review examines the role of autophagy in myocardial energy stress settings, most particularly in diabetic/insulin resistant and ischemic states. We focus on current questions relating to macroautophagy/glycophagy signaling pathways in these contrasting settings, and consider the interpretation of disparate findings of autophagy occurrence in similar disease models. Finally, prospects for targeted manipulation of specific autophagic processes of potential therapeutic value are highlighted.
Diabetes: An Energy Access Denial in the Myocardium
In diabetes, cardiac complications are evident even in the absence of vascular abnormalities and represent a primary manifestation of the disease. Diastolic abnormality is an early sign of diabetic cardiomyopathy, linked with altered metabolism and energetics (25, 51, 74, 91). At a molecular level, suppression of the PI3K/Akt insulin signaling pathway, either due to insulin ligand deficiency (type 1 diabetes, or T1D) or due to tissue insulin resistance (type 2 diabetes, or T2D), results in reduced GLUT4 glucose transporters in the plasma membrane and limited glucose uptake (10). These glucose handling abnormalities may underlie the observed metabolic shift decreasing carbohydrate oxidation with associated increase in fatty acid oxidation for energy supply (95). Paradoxically, diabetic cardiomyocytes exhibit an accumulation of glycogen, despite suppressed glucose uptake. In contrast with other tissues (i.e., skeletal muscle, liver), the heart uniquely increases glycogen content in periods of energy deprivation (e.g., fasting) (76), perhaps as a protective mechanism to mobilize glucose from nonessential tissues and elevate cardiac stores. In diabetes, the intracellular glucose deprivation conferred by impaired glucose transport (albeit in the context of extracellular fuel overabundance) may trigger a pseudo fasted heart phenotype. Energy stress is also a potent activator of cardiac autophagy in vivo and in isolated cardiomyocytes (3, 42, 76). The role of autophagy in the diabetic heart is emerging as an important research question. Reports to date provide interesting and discrepant findings, and these are evaluated in detail below.
New insights into the role of autophagy in the diabetic heart.
Since our first report of increased cardiac autophagy in the type 2 diabetic fructose-fed mouse (59), the experimental literature in this field has expanded considerably, but it is not yet possible to synthesize a comprehensive understanding. Relying on a variable selection of molecular tools, states of increased (4, 12, 49, 50, 59, 61, 78, 93, 102), unchanged (47, 48, 57, 62), and decreased (6, 23, 28, 29, 73, 82, 101, 103, 104, 110) basal cardiac autophagy activity have all been reported in diabetic/insulin resistant contexts (see Table 1). These discrepancies are not necessarily attributable to the different models of diabetes, since contrasting findings have been observed within the same diabetic model [e.g., STZ-mouse: increased LC3BII (12) vs. decreased LC3BII (103)]. Duration/severity of diabetes is also unable to explain the variable findings [e.g., high fat-fed mouse 18–20 wk duration: increased LC3BII (102) vs. decreased LC3BII (82)]. New work involving evaluation of autophagic flux will be important in gaining mechanistic understanding to resolve these variable findings, and we have recently highlighted the importance of flux measurement in relation to interpreting data from a dietary intervention model (21). Suppressed insulin signaling through the PI3K/Akt/mTOR pathway is a common observation in most diabetic models due to either lower insulin ligand stimulation of the receptor (T1D) or tissue insulin resistance (T2D). Downregulation of this pathway would be expected to be linked to upregulated autophagy as inhibition via mTOR would be relieved (40). Conversely, downregulation of AMPK signaling, also commonly observed in the diabetic heart, would be expected to suppress autophagy activity (11, 27, 108). Thus the autophagy outcome may reflect the net effect of these opposing autophagy regulators and depend on the extent of downregulation of Akt versus AMPK signaling (as shown in Fig. 2). As summarized in Table 1, suppressed cardiac autophagy in diabetes is often observed coincident with decreased AMPK activity, despite variable findings relating to Akt (29, 82, 103, 110). The absence of both Akt and AMPK data in many of these diabetes-autophagy studies limits these conclusions, and new studies are required to elucidate the role of autophagy in the diabetic heart.
Table 1.
Literature reports of autophagy in the insulin resistant or diabetic heart
| Autophagy | Model |
Autophagy Measurement | Duration of Diabetes | Akt Activity | AMPK Activity | Reference | |
|---|---|---|---|---|---|---|---|
| ↑ | Fructose-fed mouse | T2D | ↑ LC3BII:I ratio, Beclin1, p62 protein | 12 wk | ↓ | ↔* | 59 |
| ↑ | Goto-Kakizaki rat | T2D | ↑ LC3BI, LC3BII, LC3BII:I ratio, Beclin1, Atg12-Atg5, Atg7 protein | 18 wk old | nr | ↑ | 50 |
| ↑ | STZ-mouse (x5 inj) | T1D | ↑ LC3II:I ratio#, p62, Beclin1 protein | 16 wk | nr | nr | 93 |
| ↑ autophagosomes (EM) | |||||||
| ↑ Atg5, Atg12, Beclin1 mRNA | |||||||
| ↔ LC3#, p62 mRNA | |||||||
| ↑ | STZ rat (x1 inj) | T1D | ↑ LC3BII:I ratio | 8 wk | ↓ | nr | 61 |
| ↑ | STZ rat (x1 inj) | T1D | ↑LC3# puncta | 4 wk | nr | nr | 49 |
| ↑ LC3II:I ratio#, Beclin1 protein | |||||||
| ↑ | High-fat fed mouse | ins res | ↑ Beclin1, LAMP2A mRNA | 24 wk | nr | nr | 4 |
| ↑ | High-fat fed mouse | ins res | ↑ LC3B, Beclin1 mRNA | 18 wk | nr | nr | 78 |
| ↑ LC3B puncta | |||||||
| ↑ | STZ mouse (x1 inj) | T1D | ↑LC3BII, CathepsinD protein | 7 wk | nr | nr | 12 |
| ↓ p62 protein | |||||||
| ↔ | T2D humans | T2D | ↔ LC3II:I ratio# | 7–20 yr | nr | nr | 62 |
| ↔ | High-fat fed rat | ins res | ↔ LC3II:I ratio#, Beclin1, LAMP1 protein | 10 wk | nr | nr | 57 |
| ↔ | High fat-STZ rat | T2D | ↔ LC3II:I ratio#, Beclin1, LAMP1 protein | 8 wk | nr | nr | 57 |
| ↔ | High-fat fed mouse (female) | ins res | ↔ LC3II:I ratio#, ↔ Atg5 mRNA | 10–12 wk | nr | nr | 47 |
| ↔ | pancreatectomy rats | T1D | ↔LC3II:I ratio#, p62 protein | 11 wk | ↔ | nr | 48 |
| ↓ | High-fat fed mouse | ins res | ↓ LC3BII:I ratio, LC3BII, Beclin1 protein | 20 wk | ↑ | nr | 104 |
| ↑ p62 protein | |||||||
| ↑ pULK1/ULK1, ↔ LC3BI | |||||||
| ↓ | High-fat fed mouse | ins res | ↓ LC3BII:I ratio, LC3BII protein | 22 wk | nr | ↓ | 23 |
| ↑ p62 protein | |||||||
| ↔ LC3BI, Beclin1 protein | |||||||
| ↓ | High-fat fed mouse | ins res | ↓ LC3II:I ratio# | 8 wk | nr | ↔ | 6 |
| ↓ | High-fat fed mouse | ins res | ↓ LC3II:I ratio (ns)#, ↑ p62 protein | 12 wk | nr | nr | 28 |
| ↓ | db/db mouse | T2D | ↓ LC3II:I ratio# | 14–18 wk old | ↓ | ↔ | 73 |
| ↔Beclin1, Atg12-Atg5 protein | |||||||
| ↓ | High-fat fed mouse | ins res | ↓ LC3II# protein | 18–20 wk | ↔ | ↓ | 82 |
| ↓ | STZ mouse (x1 inj) | T1D | ↓ LC3II#, Atg12, Atg12-Atg5 protein | 6, 9, 12 wk | ↑ | ↓ | 103 |
| ↓ LC3 protein (Baf-Con) | |||||||
| ↓ LC3 puncta (Baf-Con) | |||||||
| ↓ | STZ mouse (x5 inj) | T1D | ↓ LC3II#, Beclin1 protein | 8 wk | ↓ | ↓ | 110 |
| ↓ | STZ mouse (x5 inj) | T1D | ↓ LC3BII protein | 16 wk | ↓ | ↓ | 29 |
| ↓ | STZ mouse (x5 inj) | T1D | ↓ LC3II protein# | 16 wk | nr | ↓ | 101 |
| ↓ | OVE26 mouse | T1D | ↓ LC3II protein# | 40 wk old | nr | ↓ | 101 |
| ↓ autophagic vacuoles (EM) | |||||||
| ↓ Beclin1 staining (IHC) | |||||||
| ↓ | db/db mouse | T2D | ↓ LC3II# (+Baf, sample blot) | 10–14 wk old | nr | nr | 57 |
| Ambiguous | High-fat fed mouse | ins res | ↑ LC3BI, LC3BII, p62 protein | 20 wk | ↑tAkt2 | ↓ | 102 |
| ↑ LC3, p62 mRNA | |||||||
| ↑ double membrane-bound vacuole/cell | |||||||
| ↔LC3BII:I ratio, Beclin1 protein | |||||||
| CQ-induced LC3BII increase abrogated | |||||||
nr, not reported; STZ, streptozotocin; T2D, type 2 diabetes; T1D, type 1 diabetes; Atg, autophagy; ns, not significant; ins res, insulin resistance; LAMP, lysosome associated membrane protein 1; Baf-Con, bafilomycin-control; EM, electron microscopy; CQ, chloroquine. ↑, Diabetes-induced increase; ↓, diabetes-induced decrease; ↔, no effect of diabetes.
Personal communication; #LC3 isoform nonselective antibody used. All data presented are obtained in the absence of lysosomal inhibition unless otherwise noted.
Fig. 2.
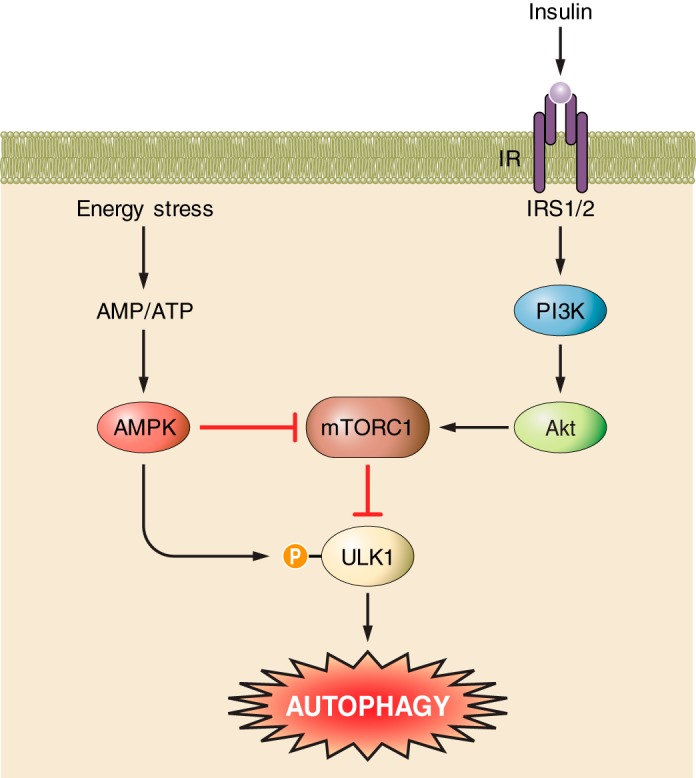
Key signaling regulation of autophagy in the heart. Energy stress increases the AMP-to-ATP ratio and activates AMPK. AMPK has dual action to promote autophagy via inhibition of mTORC1 and activation of ULK1 via phosphorylation. The insulin signaling pathway inhibits autophagy via activation of PI3K/Akt/mTORC1 to inhibit ULK1.
A role for glycophagy in the heart.
Recently we have demonstrated that the autophagic/lysosomal glycogen degradation pathway (glycogen-specific autophagy, ‘glycophagy’) is operational in the adult heart and is upregulated by diabetes in vivo (61, 76). Glycophagy is well characterized in the newborn liver and heart where a transient upregulation of lysosomal acid α-glucosidase mediates an increase in autophagic glycogen degradation (43). In this setting autophagic glycogen breakdown is thought to provide glycolytic substrate support during the placental-lactational transition. This bulk glycogen degradation pathway is understood to operate in parallel with the well-described conventional kinase-regulated process of glycogen breakdown by glycogen phosphorylase. In that pathway, Akt via GSK3β mediates glycogen synthase activation, whereas PKA activation via glycogen phosphorylase kinase mediates glycogen phosphorylase activation (as shown in Fig. 3). Thus glycogenolysis may take place in the cytosol at neutral pH or in the lysosome at acidic pH, mediated by distinct enzymes. Lysosomal glycogen degradation is mediated by the acid α-glucosidase, which possesses glycogen, maltose, and isomaltose hydrolyzing activities (43). Glucose liberated from glycogen degradation exits lysosomes via the lysosomal membrane glucose carrier (55). The glycogen hydrolyzing activity of acid α-glucosidase is enhanced by Ca2+, which in the setting of increased cytosolic Ca2+ in the ischemic myocardium, in concert with cAMP-mediated augmented lysosomal Ca2+ uptake, may prime lysosomes for accelerated glycogen degradation (41, 79). There is strong evidence from glycogen storage diseases that autophagic/lysosomal glycogen breakdown is important for maintaining normal cardiac glycogen levels and does not simply constitute a redundant alternative breakdown route for glycogen (68). Deficiency of functional lysosomal acid α-glucosidase as observed in Pompe disease results in the accumulation of glycogen-filled lysosomes in multiple tissues including the heart (75), indicating that lysosomal degradation of glycogen is essential in normal physiology. Glycophagy may provide a quantitatively different route of glycogen breakdown in comparison with phosphorylase-mediated degradation, facilitating large glucose release in settings of high energy demand. Glycogen destined for autophagic/lysosomal degradation may also be qualitatively different to that degraded in the cytosol, thus necessitating a different mode of degradation. For example, abnormally branched, insoluble, and/or hyperphosphorylated glycogen may impede phosphorylase action and favor recruitment to the glycophagosome.
Fig. 3.

Regulation of glycogen synthesis and degradation in the heart. Akt signaling promotes glycogen synthesis via relieving the GSK3β-mediated inhibition of glycogen synthase. PKA signaling promotes glycogen degradation via activation of glycogen phosphorylase kinase and subsequent activation of glycogen phosphorylase. Phosphorylase removes a glycosyl residue from the glycogen strand to release glucose-1-phosphate. STBD1 tags glycogen for degradation via glycophagy. Glycogen is engulfed by the glycophagosome, which may involve STBD1 binding to GABARAPL1 in the forming glycophagosome membrane. The glycophagosome fuses with a lysosome, and the glycogen is degraded to free glucose by acid α-glucosidase (black dots).
Starch binding domain protein 1 (STBD1) is a likely candidate for facilitating glycogen sequestration into the glycophagosome. STBD1 contains both a carbohydrate binding domain and an Atg8 interacting motif (39). Proteomics analysis has demonstrated that STBD1 is localized to the glycogen particle (86). In cell lines overexpressing HA-tagged STBD1, colocalization (via immunostaining) and interaction (via co-immunoprecipitation) of STBD1 and the autophagy (Atg8) protein, GABARAPL1, has been observed (38, 39). STBD1 colocalization with the lysosomal protein, LAMP1, has also been reported (38). Thus STBD1 may play a role in intracellular trafficking of glycogen to the autophagosome by binding to GABARAPL1 on the autophagosome membrane, constituting specific autophagy machinery for glycophagy. We have demonstrated that STBD1 does not colocalize with the macroautophagy Atg8 protein, LC3B, in cultured cardiomyocytes, suggesting that the glycophagy process is distinct from conventional macroautophagy (Fig. 4) (61). Visualization of STBD1 and GABARAPL1 colocalization has been demonstrated in a noncardiac cell line, and further information is required regarding the direct/indirect interaction of these two proteins in vitro in cardiomyocytes. It is likely that, in pathological settings, selective autophagy pathways play differential roles and identification/characterization of these specific pathways may lead to more targeted intervention opportunities.
Fig. 4.
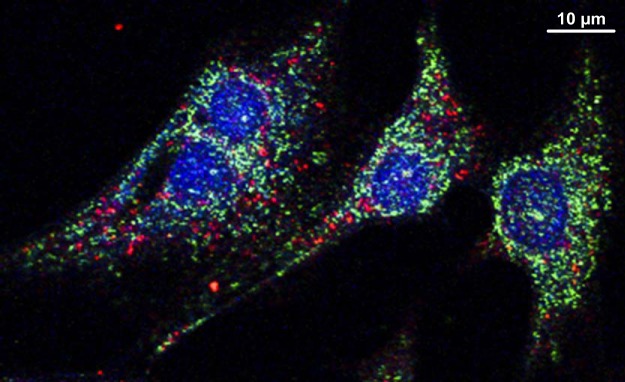
Distinct cardiomyocyte immunohistologic localization of glycogen autophagic trafficking cargo in vitro. STBD1 (green) and LC3B (red) do not colocalize in neonatal ventricular rat myocytes. Confocal image, x63 mag: DAPI (blue) shows nuclei. Reprinted from Ref. 53 with permission.
Glycogen flux in the diabetic myocardium.
Glycogen pathology in human diabetic myocardium was first reported over 80 years ago (96). This is a replicated finding in many diabetic experimental settings (2, 46, 84) and occurs in parallel to a depletion of glycogen stores in the liver and skeletal muscle (30, 85, 87). The fundamental bases for differential glycogen handling responses in distinct muscle types are still not yet understood. Advances in the understanding of the gene/protein origins of various genetically determined glycogen storage diseases have provided considerable new insight into the role of disrupted glycogen handling in the etiology of cardiac pathology. In these diseases, glycogen disturbance results in contractile dysfunction, derangement of myofibrillar and cytoskeletal proteins, arrhythmia, and hypertrophy (63). Presentation of vacuole-filled glycogen granules in cardiomyocytes supports a role for glycophagy in these settings (32). Glycogen handling dysregulation in diabetes may similarly reflect a storage anomaly.
In diabetes, disturbances in insulin signaling and glucose handling may underlie cardiac glycogen pathology. In cultured neonatal rat cardiomyocytes, protein expression of the glycophagy marker STBD1 was increased by extracellular insulin concentration linked with activation of the insulin-regulated PI3K/Akt signaling pathway (61). Similarly, fasting-induced upregulation of STBD1 and GABARAPL1 protein content was associated with activation of Akt in vivo (76). In cultured cardiomyocytes, it has been demonstrated using chromatin immunoprecipitation that FoxO1 and FoxO3, which are inhibited by Akt, directly bind to the GABARAPL1 promoter in the nucleus and regulate transcription of this glycophagy marker (83). Thus Akt activation may lead to inhibition of FoxO-mediated GABARAPL1 transcription, but such a response is difficult to reconcile with the experimental observations that there is parallel upregulation of glycophagy and Akt activity with fasting (76). A more detailed dissection of the signaling nexus of PI3K/Akt and AMPK pathways in mediating glycophagy induction is required to resolve these contrasting observations, in both physiological and pathophysiological settings.
Ischemia-Reperfusion: An Energy Supply Crisis
Role of autophagy in the ischemic and reperfused heart.
Autophagy is powerfully upregulated as part of the response to ischemic stress, and it has been suggested that macroautophagy induction is beneficial during ischemia but deleterious during reperfusion (58). We have shown that autophagy is essential for cardioprotection observed with ischemic preconditioning (35), and others have shown its importance in postconditioning (98). In a Langendorff model of ischemia/reperfusion, inhibition of autophagy with the cell-permeable inhibitor (TAT-Atg5K130R) neither increased nor reduced infarct size, suggesting that in the absence of pre- or post-conditioning, the beneficial and deleterious effects of autophagy may be balanced (34). Inhibition of autophagic flux increases hypoxia/reoxygenation injury (109) and blocks the beneficial effects of sevoflurane postconditioning (107). Ischemia/reperfusion injury disrupts autophagosome-lysosome fusion (54), accompanied by a significant increase in Beclin-1. Beclin-1 effects are complex. Beclin-1 plays an essential role in autophagosome initiation for both canonical and noncanonical macroautophagy pathways, but it also partners with Rubicon to interfere with autophagosome-lysosome fusion (111), and binds to Bcl-2, thereby fostering mitochondria-dependent apoptosis (36). The preponderance of evidence supports the notion that autophagy is a protective response to acute ischemia/reperfusion injury, and new studies are required to elucidate the mechanism by which autophagy confers benefit in this setting.
Mitochondrial clearance (mitophagy) and mitochondrial sequestration (lysosomal regulation).
Mitochondria represent both a target of injury during ischemia, and a source of tissue damage, via production of reactive oxygen species (ROS), during reperfusion. Clearance of damaged mitochondria by autophagy (mitophagy) may play a beneficial role in the setting of myocardial ischemia/reperfusion. We have demonstrated that ischemic preconditioning ameliorates reperfusion injury by triggering Parkin-dependent mitophagy (33). Given the role of autophagy in liberating metabolic substrates (amino acids, carbohydrates, lipids) during nutrient deprivation, it has been generally assumed that both autophagic flux and lysosomal function are essential for autophagy-mediated cardioprotection. We have provided evidence that sequestration of mitochondria in autophagosomes may mediate the cardioprotective effects of chloramphenicol in the absence of lysosomal function. Chloramphenicol-induced infarct size reduction was preserved when the lysosomal inhibitor chloroquine was administered in isolated ischemia/reperfused rat hearts (19). Although autophagic sequestration of nutrient sources without lysosomal degradation during an episode of metabolic stress seems counterintuitive, it is also a process by which regulated lysosomal function participates in protective energy management.
In contrast, lysosomal function appears to play an important role in cardioprotection in other contexts. Enhanced lysosome biogenesis was shown to attenuate apoptosis mediated by overexpression of BNIP3 in neonatal rat cardiomyocytes (53). Increased lysosome biogenesis (via cobalt protoporphyrin treatment) contributes to autophagy-mediated cardioprotection in sepsis (90). It is noteworthy that mTOR is associated with the lysosome, and during starvation, mTOR is inactivated until amino acids derived from the lysosome restore mTORC1 signaling (106), enabling new protein synthesis required for cellular repair processes. It may be that sequestration of mitochondria (or other cellular components) into autophagosomes is protective as a short-term strategy to minimize cellular injury in ischemia-reperfusion, and delayed/inhibited lysosomal degradation slows the negative feedback on autophagy via mTOR.
The serine/threonine kinase GSK3β has been extensively investigated in the past decade with respect to its role in cardioprotection. Ser9 phosphorylation of GSK3β is considered to exert its cardioprotective effects through inhibition of mitochondrial permeability transition pore (mPTP) opening. The precise nature of the GSK3β-mPTP interaction is unclear; however, several mechanisms have been proposed including preservation of hexokinase II in the mPTP complex (72), suppression of the interaction between adenine nucleotide translocase and cyclophilin D (67), and inhibition of p53 (97). GSK3β inhibition has been implicated in the cardioprotection mediated by a multitude of agents and therapeutic strategies (13, 20, 70, 92); however, little attention has been given to the original process from which it derives its name - in regulating glycogen turnover. GSK3β inhibition during ischemia should favor glycogen synthesis and decreased degradation, which in turn may reduce the availability of glycolytic substrate, the oxidation of which promotes ischemic acidosis and worsened reperfusion injury. Although a role for GSK3β in regulating glycophagy is speculative at this time, two separate reports have recently demonstrated that GSK3β-inhibition promotes lysosome biogenesis (56, 71). If GSK3β inhibition in preconditioned hearts promotes lysosomal biogenesis, this may hypothetically prime the cell for enhanced autophagic glycogen disposal. Similarly GSK3β has been shown to regulate autophagic activity in both tumor and nontumor cell lines (18). Thus it should be noted that pharmacologic agents that target GSK3β (some of which have been proposed as cardioprotective drugs) may at least partially act through a mechanism involving glycogen action.
Glycogen turnover in the preconditioned heart.
Glycogenolysis contributes significantly to glycolytic substrate supply during ischemia, and a number of studies have sought to determine a relationship between ischemic glycogen consumption and infarct severity (5, 15, 80). Ischemic glycogenolysis may have beneficial effect through increased glycolytic ATP supply or detrimental effect due to enhanced cellular lactate and proton production. Interventions that increase pre-ischemic glycogen content have been shown to be both beneficial and detrimental to the ischemic heart (80). One study demonstrated that separate cardioprotective interventions, which deplete or enhance pre-ischemic glycogen, can confer a similar level of cardioprotection (7). Because any intervention intended to manipulate glycogen levels before ischemia will likely involve parallel activation of signaling pathways related to cardioprotection, delineating the precise contribution of glycogen depletion to ischemic injury is problematic. Indeed, it has recently been reported that ischemic preconditioning and insulin administration independently protected against ischemia/reperfusion injury while having differential impacts on glycogen turnover (17). When both ischemic preconditioning and insulin administration were combined, cardioprotection was abolished.
The role of myocardial glycogen breakdown in the cardioprotection observed with preconditioning is controversial: one study reported that ischemic preconditioning attenuated glycogenolysis in rat hearts (99), whereas another study showed that 24-h in vivo fasting resulted in increased glycogen content and greater glycogen utilization during ischemia/reperfusion in a Langendorff model (80), attributed to activation of phosphorylase a (94). Interestingly, it was also observed that insulin pretreatment increased glycogen content to a similar extent but suppressed glycogen utilization during ischemia/reperfusion, resulting in increased injury and function (80). Fasting upregulates autophagy, whereas insulin treatment suppresses both autophagy and glycogen utilization, which might explain the differential effects. The adenosine A1 agonist CCPA (2-chloro-N6-cyclopentyladenosine) has been shown to increase pre-ischemic glycogen content and suppress glycogen utilization in the first 2–5 min of ischemia, but total glycogen utilization over the ischemic period was increased relative to the ischemic preconditioning group (7). These findings must also be considered in the context of studies showing that adenosine or adenosine A1 agonists inhibited glycolysis and glycolytic proton production yet had no effect on glucose oxidation, resulting in tighter coupling of glycolysis to glucose oxidation (14). In a related study it was observed that A1 agonists promoted glycogen synthesis without affecting glycogen degradation; yet glycogen turnover was accelerated (16). Little information is available to address the question of whether stimulated glycogenolysis occurred in the cytosol or the lysosome. We have demonstrated that cardioprotection by CCPA is dependent on autophagy (105), which would be consistent with lysosomal glycogen breakdown. Although no clear answers have emerged, it may be speculated that autophagic engulfment of glycogen particles (i.e., glycophagy) might sequester glycogen and prevent its utilization during acute ischemic stress, thereby limiting lactic acidosis and risk of Ca2+ overload.
Glycophagy in the ischemic heart.
An issue of major importance relating to ischemic glycogen turnover that has been largely overlooked is the contribution of autophagic (i.e., glycophagic) sequestration of glycogen. Cytosolic glycogen degradation is normally regulated via the actions of the cytosolic phosphorylase and debranching enzymes, and it could be also assumed that some glycogen is constitutively degraded via lysosomes. The advantage of sequestering glycogen into autophagosomes for subsequent delivery to the lysosomes for degradation is not yet understood. Cytosolic glycogen is localized within distinct myocyte subcellular regions, including intermyofibrillar, intramyofibrillar, and subsarcolemmal compartments (65). In stimulated skeletal muscle fibers, rates of glycogen depletion differ depending upon subcellular localization (64). It is conceivable, therefore, that in the setting of local high rates of glycogenolysis, glycophagy may function to supplement the cytosolic machinery and that localized glycophagic hotspots provide important metabolic support under ischemic stress. In cardiomyocytes we have been able to observe sequestered glycogen particles positioned adjacent to free glycogen particles between myofibrillar bundles (Fig. 5). This suggests the possibility of a differential functional role for glycogen stored intra- and extraphagosomally.
Fig. 5.

Electron micrograph of glycogen and a glycophagosome in an adult rat ventricular cardiomyocyte. Free glycogen particles (short arrowhead) are aligned between myofibrils. Some glycogen particles are seen sequestered in a glycophagosome (arrow). m, Mitochondria. Bar indicates 500 nm.
A second possibility is that glycogen destined for autophagic degradation is qualitatively different from normal glycogen and therefore requires a separate means of degradation. Glycogen molecules are heterogeneous in size; an ordered distribution of branch points contributes to the formation of a sphere whereby the nonreducing chain ends are exposed to the cytosol, rendering the molecule soluble (69). In Lafora disease, loss of function of the phosphatase laforin or the E3 ubiquitin ligase malin results in disordered glycogen structure. It is characterized by the accumulation of abnormally branched, hyperphosphorylated glycogen, leading to the formation of insoluble aggregates within the cell (88). Glycogen phosphorylation may compromise glycogen solubility indirectly by interfering with intermolecular bonds between adjacent branches, or directly by preventing branching due to phosphorylation at C6 of the glucose residues. Understanding of how excess glycogen phosphorylation confers macromolecular solubility is limited; however, recent evidence suggests that this phosphorylation is a regulated process (9, 69). Overexpression of laforin has been shown to augment autophagy (1). Whether glycophagy serves to deliver abnormally branched glycogen to the more amenable lysosomal acidic environment to achieve more complete molecular breakdown is a pressing question, and whether this route of degradation assumes more importance in an ischemic context is not yet established.
Conclusions and Directions
An understanding of the role of autophagic processes in the management of cardiac metabolic stress responses is advancing rapidly and progressing beyond a conceptualization of the autophagosome as a simple cell recycling depot. The importance of autophagy dysregulation in diabetic cardiomyopathy and in ischemic heart disease - both conditions comprising the majority of cardiac disease burden - has now become apparent. New findings have revealed that specific autophagic processes may operate in the cardiomyocyte, specialized for selective recognition and management of mitochondria and glycogen particles in addition to protein macromolecular structures. Thus mitophagy, glycophagy, and macroautophagy regulatory pathways have become the focus of intensive experimental effort, and delineating the signaling pathways involved in these processes offers potential for targeted therapeutic intervention. A body of evidence is emerging that suggests that cytosolic autophagosomal sequestering of cellular structures has a role in acute energy management and cell viability preservation. Subsequent metabolic access to sequestered stores appears to rely on more complex lysosomal-autophagosomal regulatory mechanisms yet to be delineated. Chronically elevated macroautophagic activity in the diabetic myocardium is generally observed in association with structural and functional cardiomyopathy; yet there are also numerous reports of detrimental effect of autophagy suppression in diabetes. Autophagy induction has been identified as a key component of protective mechanisms that can be recruited to support the ischemic heart, but in this setting benefit may be mitigated by adverse downstream autophagic consequences. New studies are required in both these disease contexts to undertake detailed mapping of convergent AMPK and PI3K/Akt signaling pathways to identify metabolic leverage points to optimize macroautophagic responses. Recent report of glycophagy upregulation in diabetic cardiomyopathy opens up a new area of investigation. Similarly, a role for glycogen management in ischemia protection through glycophagy initiation is an exciting prospect under investigation.
GRANTS
L. M. D. Delbridge acknowledges research funding from the National Health and Medical Research Council of Australia and Diabetes Australia Research Trust. R. A. Gottlieb is supported by National Heart, Lung, and Blood Institute Grant NIH P01HL-112730 and the Dorothy and E. Phillip Lyon Chair in Molecular Cardiology. K. M. Mellor is supported by the Rutherford Foundation Postdoctoral Fellowship, New Zealand. Figure 5 has been contributed by Maryam Ghochani.3
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.M.D., K.M.M., D.J.T., and R.A.G. prepared figures; L.M.D., K.M.M., D.J.T., and R.A.G. drafted manuscript; L.M.D., K.M.M., D.J.T., and R.A.G. edited and revised manuscript; L.M.D., K.M.M., D.J.T., and R.A.G. approved final version of manuscript.
REFERENCES
- 1.Aguado C, Sarkar S, Korolchuk VI, Criado O, Vernia S, Boya P, Sanz P, de Cordoba SR, Knecht E, Rubinsztein DC. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet 19: 2867–2876, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alfarano C, Suffredini S, Fantappie O, Mugelli A, Cerbai E, Manni ME, Raimondi L. The effect of losartan treatment on the response of diabetic cardiomyocytes to ATP depletion. Pharmacol Res 63: 225–232, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 6: 462–472, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou IP, Chiu YP, Ding ST, Liu BH, Lin YY, Chen CY. Adiponectin receptor 1 overexpression reduces lipid accumulation and hypertrophy in the heart of diet-induced obese mice—possible involvement of oxidative stress and autophagy. Endocr Res 39: 173–179, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res 78: 482–491, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Cui M, Yu H, Wang J, Gao J, Li J. Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J Diabetes Res 2013: 852754, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Jonge R, de Jong JW, Giacometti D, Bradamante S. Role of adenosine and glycogen in ischemic preconditioning of rat hearts. Eur J Pharmacol 414: 55–62, 2001. [DOI] [PubMed] [Google Scholar]
- 8.de Waal EJ, Vreeling-Sindelarova H, Schellens JP, James J. Starvation-induced microautophagic vacuoles in rat myocardial cells. Cell Biol Int Rep 10: 527–533, 1986. [DOI] [PubMed] [Google Scholar]
- 9.DePaoli-Roach AA, Contreras CJ, Segvich DM, Heiss C, Ishihara M, Azadi P, Roach PJ. Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J Biol Chem 290: 841–850, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K. Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res 61: 288–296, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7: 643–644, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eguchi M, Kim YH, Kang KW, Shim CY, Jang Y, Dorval T, Kim KJ, Sweeney G. Ischemia-reperfusion injury leads to distinct temporal cardiac remodeling in normal versus diabetic mice. PLoS One 7: e30450, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology 103: 987–995, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Finegan BA, Lopaschuk GD, Gandhi M, Clanachan AS. Ischemic preconditioning inhibits glycolysis and proton production in isolated working rat hearts. Am J Physiol Heart Circ Physiol 269: H1767–H1775, 1995. [DOI] [PubMed] [Google Scholar]
- 15.Fraser H, Lopaschuk GD, Clanachan AS. Assessment of glycogen turnover in aerobic, ischemic, and reperfused working rat hearts. Am J Physiol Heart Circ Physiol 275: H1533–H1541, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Fraser H, Lopaschuk GD, Clanachan AS. Alteration of glycogen and glucose metabolism in ischaemic and post-ischaemic working rat hearts by adenosine A1 receptor stimulation. Br J Pharmacol 128: 197–205, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fullmer TM, Pei S, Zhu Y, Sloan C, Manzanares R, Henrie B, Pires KM, Cox JE, Abel ED, Boudina S. Insulin suppresses ischemic preconditioning-mediated cardioprotection through Akt-dependent mechanisms. J Mol Cell Cardiol 64: 20–29, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavilan E, Sanchez-Aguayo I, Daza P, Ruano D. GSK-3[beta] signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis 4: e572, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giricz Z, Mentzer RM Jr, Gottlieb RA. Cardioprotective effects of chloramphenicol are mediated by autophagy. J Am Coll Cardiol 57: E1015, 2011. [Google Scholar]
- 20.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 117: 2761–2768, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: all fluxed up. Circ Res 116: 504–514, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottlieb RA, Mentzer RM Jr, Linton PJ. Impaired mitophagy at the heart of injury. Autophagy 7: 1573–1574, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta 1832: 1136–1148, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res 104: 150–158, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han JC, Goo S, Barrett CJ, Mellor KM, Taberner AJ, Loiselle DS. The afterload-dependent peak efficiency of the isolated working rat heart is unaffected by streptozotocin-induced diabetes. Cardiovasc Diabetol 13: 4, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardie DG. AMPK and autophagy get connected. EMBO J 30: 2511, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 62: 1270–1281, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He J, Kelley DE. Muscle glycogen content in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 287: E1002–E1007, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Hill JA. Autophagy in cardiac plasticity and disease. Pediatr Cardiol 32: 282–289, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hobson-Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late-onset Pompe disease: a case report and systematic review of the literature. Mol Genet Metab 106: 462–469, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6: e20975, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM Jr, Gottlieb RA. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol 298: H570–H579, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 3: 365–373, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Qi Q, Hua X, Li X, Zhang W, Sun H, Li S, Wang X, Li B. Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol Rep 31: 1761–1767, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Ingwall JS, DeLuca M, Sybers HD, Wildenthal K. Fetal mouse hearts: a model for studying ischemia. Proc Natl Acad Sci USA 72: 2809–2813, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli-Roach AA, Wells CD, Skurat AV, Roach PJ. Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem 285: 34960–34971, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang S, Wells CD, Roach PJ. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun 413: 420–425, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 584: 1287–1295, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalamidas SA, Kotoulas OB, Hann AC. Studies on glycogen autophagy: effects of phorbol myristate acetate, ionophore A23187, or phentolamine. Microsc Res Tech 57: 507–511, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, Nagashima K, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol 174: 1705–1714, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy. Microsc Res Tech 64: 10–20, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Lajoie C, Calderone A, Trudeau F, Lavoie N, Massicotte G, Gagnon S, Beliveau L. Exercise training attenuated the PKB and GSK-3 dephosphorylation in the myocardium of ZDF rats. J Appl Physiol 96: 1606–1612, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Lancel S, Montaigne D, Marechal X, Marciniak C, Hassoun SM, Decoster B, Ballot C, Blazejewski C, Corseaux D, Lescure B, Motterlini R, Neviere R. Carbon monoxide improves cardiac function and mitochondrial population quality in a mouse model of metabolic syndrome. PLoS One 7: e41836, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Lee JH, Jin M, Han SD, Chon GR, Kim IH, Kim S, Kim SY, Choi SB, Noh YH. Diet control to achieve euglycemia induces significant loss of heart and liver weight via increased autophagy compared with ad libitum diet in diabetic rats. Exp Mol Med 46: e111, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee Y, Hong Y, Lee SR, Chang KT, Hong Y. Autophagy contributes to retardation of cardiac growth in diabetic rats. Lab Anim Res 28: 99–107, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Tang Y, Feng Z, Liu J, Liu J, and Long J. (-)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic Res 48: 898–906, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, Welty TK, Lee ET, Devereux RB. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol 37: 1943–1949, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Lizaso A, Tan KT, Lee YH. Beta-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy 9: 1228–1243, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8: 297–309, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia-reperfusion injury. Circulation 125: 3170–3181, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mancini GM, Beerens CE, Verheijen FW. Glucose transport in lysosomal membrane vesicles. Kinetic demonstration of a carrier for neutral hexoses. J Biol Chem 265: 12380–12387, 1990. [PubMed] [Google Scholar]
- 56.Marchand B, Arsenault D, Raymond-Fleury A, Boisvert FM, Boucher MJ. Glycogen synthase kinase-3 (GSK3) inhibition induces pro-survival autophagic signals in human pancreatic cancer cells. J Biol Chem 290: 5592–5605, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsh SA, Powell PC, Dell'italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92: 648–656, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol 50: 1035–1043, 2011. [DOI] [PubMed] [Google Scholar]
- 60.Mellor KM, Reichelt ME, Delbridge LM. Autophagy anomalies in the diabetic myocardium. Autophagy 7: 1263–1267, 2011. [DOI] [PubMed] [Google Scholar]
- 61.Mellor KM, Varma U, Stapleton D, Delbridge LMD. Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. Am J Physiol Heart Circ Physiol 306: H1240–H1245, 2014. [DOI] [PubMed] [Google Scholar]
- 62.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130: 554–564, 2014. [DOI] [PubMed] [Google Scholar]
- 63.Murphy RT, Mogensen J, McGarry K, Bahl A, Evans A, Osman E, Syrris P, Gorman G, Farrell M, Holton JL, Hanna MG, Hughes S, Elliott PM, Macrae CA, McKenna WJ. Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol 45: 922–930, 2005. [DOI] [PubMed] [Google Scholar]
- 64.Nielsen J, Cheng AJ, Ortenblad N, Westerblad H. Subcellular distribution of glycogen and decreased tetanic Ca2+ in fatigued single intact mouse muscle fibres. J Physiol 592: 2003–2012, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nielsen J, Ortenblad N. Physiological aspects of the subcellular localization of glycogen in skeletal muscle. Appl Physiol Nutr Metab 38: 91–99, 2013. [DOI] [PubMed] [Google Scholar]
- 66.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ 16: 31–38, 2009. [DOI] [PubMed] [Google Scholar]
- 67.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol 43: 564–570, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep 3: 64–69, 2003. [DOI] [PubMed] [Google Scholar]
- 69.Nitschke F, Wang P, Schmieder P, Girard JM, Awrey DE, Wang T, Israelian J, Zhao X, Turnbull J, Heydenreich M, Kleinpeter E, Steup M, Minassian BA. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab 17: 756–767, 2013. [DOI] [PubMed] [Google Scholar]
- 70.Park SS, Zhao H, Mueller RA, Xu Z. Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3beta. J Mol Cell Cardiol 40: 708–716, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol Cell Biol 32: 4410–4418, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65: 10545–10554, 2005. [DOI] [PubMed] [Google Scholar]
- 73.Pei XM, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Siu PM. Protective effects of desacyl ghrelin on diabetic cardiomyopathy. Acta Diabetol. In press. [DOI] [PubMed] [Google Scholar]
- 74.Pham T, Loiselle D, Power A, Hickey AJ. Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart. Am J Physiol Cell Physiol 307: C499–C507, 2014. [DOI] [PubMed] [Google Scholar]
- 75.Raben N, Roberts A, Plotz PH. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol 26: 45–48, 2007. [PMC free article] [PubMed] [Google Scholar]
- 76.Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy—a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol 65: 67–75, 2013. [DOI] [PubMed] [Google Scholar]
- 77.Riehle C, Abel ED. Insulin regulation of myocardial autophagy. Circ J 78: 2569–2576, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest 122: 3919–3930, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sandhu R, Thomas U, Diaz RJ, Wilson GJ. Effect of ischemic preconditioning of the myocardium on cAMP. Circ Res 78: 137–147, 1996. [DOI] [PubMed] [Google Scholar]
- 80.Schaefer S, Ramasamy R. Glycogen utilization and ischemic injury in the isolated rat heart. Cardiovasc Res 35: 90–98, 1997. [DOI] [PubMed] [Google Scholar]
- 81.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol 32: 275–281, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125: 1134–1146, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 284: 28319–28331, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shearer J, Ross KD, Hughey CC, Johnsen VL, Hittel DS, Severson DL. Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab 301: E31–E39, 2011. [DOI] [PubMed] [Google Scholar]
- 85.Srinivasan S, Sathish G, Jayanthi M, Muthukumaran J, Muruganathan U, Ramachandran V. Ameliorating effect of eugenol on hyperglycemia by attenuating the key enzymes of glucose metabolism in streptozotocin-induced diabetic rats. Mol Cell Biochem 385: 159–168, 2014. [DOI] [PubMed] [Google Scholar]
- 86.Stapleton D, Nelson C, Parsawar K, McClain D, Gilbert-Wilson R, Barker E, Rudd B, Brown K, Hendrix W, O'Donnell P, Parker G. Analysis of hepatic glycogen-associated proteins. Proteomics 10: 2320–2329, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thorburn AW, Gumbiner B, Bulacan F, Brechtel G, Henry RR. Multiple defects in muscle glycogen synthase activity contribute to reduced glycogen synthesis in non-insulin dependent diabetes mellitus. J Clin Invest 87: 489–495, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, Kameka AP, Pencea N, Zhao X, Ackerley CA, Minassian BA. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol 68: 925–933, 2010. [DOI] [PubMed] [Google Scholar]
- 89.Tuttle DL, Lewin AS, Dunn WA Jr. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur J Cell Biol 60: 283–290, 1993. [PubMed] [Google Scholar]
- 90.Unuma K, Aki T, Funakoshi T, Yoshida K, Uemura K. Cobalt protoporphyrin accelerates TFEB activation and lysosome reformation during LPS-induced septic insults in the rat heart. PLoS One 8: e56526, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M, Borbely A, van der Velden J, Stienen GJ, Laarman GJ, Niessen HW, Paulus WJ. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117: 43–51, 2008. [DOI] [PubMed] [Google Scholar]
- 92.Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, Duplaa C, Jaspard-Vinassa B. GSK-3beta at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res 90: 49–56, 2011. [DOI] [PubMed] [Google Scholar]
- 93.Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, Bai WW, Liu XQ, Zhao YX. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 18: 1599–1611, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang LF, Ramasamy R, Schaefer S. Regulation of glycogen utilization in ischemic hearts after 24 hours of fasting. Cardiovasc Res 42: 644–650, 1999. [DOI] [PubMed] [Google Scholar]
- 95.Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 288: H2102–H2110, 2005. [DOI] [PubMed] [Google Scholar]
- 96.Warren S. The effect of insulin on pathologic glycogen deposits in diabetes mellitus. American Journal of Medical Science 179: 482–488, 1930. [Google Scholar]
- 97.Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. J Biol Chem 278: 48872–48879, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wei C, Li H, Han L, Zhang L, Yang X. Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J Cardiovasc Pharmacol 61: 416–422, 2013. [DOI] [PubMed] [Google Scholar]
- 99.Weiss RG, de Albuquerque CP, Vandegaer K, Chacko VP, Gerstenblith G. Attenuated glycogenolysis reduces glycolytic catabolite accumulation during ischemia in preconditioned rat hearts. Circ Res 79: 435–446, 1996. [DOI] [PubMed] [Google Scholar]
- 100.Xie Z, He C, Zou MH. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 7: 1254–1255, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60: 1770–1778, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol 5: 61–63, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288: 18077–18092, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu X, Ren J. Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes (Lond) 39: 387–396, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM Jr, Gottlieb RA. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol 104: 157–167, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu X, Long YC. Autophagy modulates amino acid signaling network in myotubes: differential effects on mTORC1 pathway and the integrated stress response. FASEB J 29: 394–407, 2014. [DOI] [PubMed] [Google Scholar]
- 107.Zhang YL, Yao YT, Fang NX, Zhou CH, Gong JS, Li LH. Restoration of autophagic flux in myocardial tissues is required for cardioprotection of sevoflurane postconditioning in rats. Acta Pharmacol Sin 35: 758–769, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao M, Klionsky DJ. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab 13: 119–120, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang H, Ren J, Zang WJ. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol Biochem 32: 601–613, 2013. [DOI] [PubMed] [Google Scholar]
- 110.Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, Peng Y, Dong X, Huang H, Si L, Zhang X, Zhang L, Li J, Wang W, Zhou L, Gao X. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One 8: e75927, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhong Y, Wang QJ, Yue Z. Atg14L and Rubicon: yin and yang of Beclin 1-mediated autophagy control. Autophagy 5: 890–891, 2009. [DOI] [PubMed] [Google Scholar]
- 112.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117: 1782–1793, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]