

Abstract
Familial hypertrophic cardiomyopathy (FHC) is the most common cause of sudden cardiac death in young individuals. Molecular mechanisms underlying this disorder are largely unknown; this study aims at revealing how disruptions in actin-myosin interactions can play a role in this disorder. Cross-bridge (XB) kinetics and the degree of order were examined in contracting myofibrils from the ex vivo left ventricles of transgenic (Tg) mice expressing FHC regulatory light chain (RLC) mutation K104E. Because the degree of order and the kinetics are best studied when an individual XB makes a significant contribution to the overall signal, the number of observed XBs in an ex vivo ventricle was minimized to ∼20. Autofluorescence and photobleaching were minimized by labeling the myosin lever arm with a relatively long-lived red-emitting dye containing a chromophore system encapsulated in a cyclic macromolecule. Mutated XBs were significantly better ordered during steady-state contraction and during rigor, but the mutation had no effect on the degree of order in relaxed myofibrils. The K104E mutation increased the rate of XB binding to thin filaments and the rate of execution of the power stroke. The stopped-flow experiments revealed a significantly faster observed dissociation rate in Tg-K104E vs. Tg-wild-type (WT) myosin and a smaller second-order ATP-binding rate for the K104E compared with WT myosin. Collectively, our data indicate that the mutation-induced changes in the interaction of myosin with actin during the contraction-relaxation cycle may contribute to altered contractility and the development of FHC.
Keywords: left ventricle, mutation of regulatory light chain, polarization of fluorescence
interactions of myosin with actin and associated proteins (sarcomeric proteins) are responsible for muscle contraction (19, 24, 48). These interactions include the mode of attachment of actin to myosin during different physiological states and the kinetics of conformational changes of myosin when interacting with actin. Familial hypertrophic cardiomyopathy (FHC) has been recognized to be caused by mutations in all major sarcomeric proteins of the heart, including the myosin regulatory light chain (RLC) (1). Compared with the myosin heavy chain, mutations in the RLC are rare, but often associated with malignant outcomes (18, 27, 41). It is therefore important to find out whether these FHC-linked mutations result in alterations of the mode of attachment of myosin to actin, and/or whether they lead to alterations of kinetics of acto-myosin interactions. These changes may ultimately contribute to the development of FHC. In this report we have focused on the K104E (lysine to glutamic acid) substitution in the myosin RLC expressed in the heart of transgenic (Tg) mice (25). The mutation was identified in a Danish population by Andersen et al. (2) and was associated with left ventricular and septal hypertrophy and diastolic filling abnormalities.
In the three-dimensional RLC structure, the residue K104 is close to both the Ca2+-Mg2+ binding loop and to the myosin light chain kinase-specific serine-15 phosphorylation site (45). It is therefore possible that this single-point mutation results in alteration of the order with which myosin cross bridges (XBs) interact with actin filaments. Furthermore, because the lever arm and the catalytic domain of a XB are in constant communication (46), the malfunction of the RLC caused by K104E may affect the acto-myosin interaction, most critically the force production and the rates of force development and relaxation. In this report we aimed at measuring the effect of the K104E mutation on the degree of order of XBs in a small observational volume containing fluorescently labeled myofibrils prepared from previously generated Tg-K104E mice expressing the human ventricular RLC-K104E in the heart (25).
The degree of order and XB kinetics are best studied when an individual XB makes a significant contribution to the overall signal, because the mean values of a large number of independent random measurements, each with a well-defined expected value and well-defined variance, are approximately normally distributed (47). Therefore, because the whole ventricle contains at least 1018 molecules of actin or myosin (left ventricle of our B6 mice typically weighs 30 mg), there is no hope of observing differences between spatial distributions of XBs within ventricles. On the other hand, if only a few XBs are observed, the differences in distribution become clear.
An argument that a small number of molecules must be studied when measuring XB kinetics is different: force-generating molecules are unsynchronized, and macroscopic measurements generate only the average values. However, when only a few molecules are studied, the value of a variable is affected by fluctuations around the average (16, 17, 44). The relative size of fluctuations depends on the number of molecules under observation N as . Even if “only” a million XBs were studied, the fluctuation would have been 0.1%. In our experiments, a typical number of molecules was 20, and the corresponding fluctuations were ∼20%. The alternative is to apply rapid transients (11, 26) to synchronize XBs. In the present work we used both approaches.
We report here that the K104E mutation in RLC imposed alterations in the degree of order and kinetics of myosin XBs in contracting myofibrils from the left ventricle of Tg-K104E mice. The mutation resulted in a significantly better order of XBs during steady-state contraction and during rigor, but it had no effect on the degree of order in the relaxed myofibrils. These changes paralleled significantly increased rates of XB binding to the thin filaments and the rate of execution of the power stroke. The stopped-flow experiments revealed significantly faster observed dissociation rates (kobs) in Tg-K104E vs. Tg-wild-type (WT) myosin at low MgATP concentrations, and the linear regression analysis of the kobs-MgATP concentration ([MgATP]) dependence showed a smaller second-order ATP-binding rate for the K104E compared with WT myosin.
MATERIALS AND METHODS
Chemicals and solutions.
All chemicals were from Sigma-Aldrich (St. Louis, MO). The dye SeTau-647-monomaleimide was from SETA BioMedicals (Urbana, IL). Glycerinating solution contained 50% glycerol, 150 mM KCl, 10 mM Tris·HCl, pH 7.5, 5 mM MgCl2, 5 mM EGTA, 5 mM ATP, 1 mM DTT, 2 mM PMSF, and 0.1% β-mercaptoethanol. Rigor solution contained 50 mM KCl, 10 mM Tris·HCl, pH 7.5, and 2 mM MgCl2. EDTA-rigor contained 50 mM KCl, 10 mM Tris·HCl, pH 7.5, and 5 mM EDTA. Ca-rigor contained 50 mM KCl, 10 mM Tris·HCl, pH 7.5, 2 mM MgCl2, and 0.1 mM CaCl2. Contracting solution contained 50 mM KCl, 10 mM Tris·HCl, pH 7.5, 5 mM MgCl2, 0.1 mM CaCl2, 5 mM ATP, 20 mM creatine phosphate, and 10 U/ml of 1 mg/ml creatine kinase. Relaxing solution contained 50 mM KCl, 10 mM Tris·HCl, pH 7.5, 5 mM MgCl2, 5 mM ATP, and 2 mM EGTA.
Preparation of mouse cardiac myosin and glycerinated left ventricular muscle strips.
Myosin was isolated from the left (LV) and right (RV) ventricles of Tg-K104E and Tg-WT mice as described in Kazmierczak et al. (28). Briefly, after death, whole hearts were isolated, and the atria were removed. LV and RV were flash-frozen and stored at −80°C until processed. The ventricular tissue was later thawed in an ice-cold Guba Straub-type buffer (pH 6.5) consisting of 300 mM NaCl, 100 mM NaH2PO4, 50 mM Na2HPO4, 1 mM MgCl2, 10 mM EDTA, 0.1% NaN3, 10 mM Na4P2O7, 1 mM DTT, and 1 μl/ml protease inhibitor cocktail (Sigma-Aldrich) in a volume of 0.75 ml buffer/0.2 g tissue. Ventricles kept on ice were first minced by hand and then homogenized for 2 min at a frequency of 30 Hz in a Mixer-Mill MM301 (Retsch). The homogenate was then incubated on ice for 40 min before centrifugation for 1 h at 200,000 g. The supernatant was then diluted 60-fold (by volume) with 2 mM DTT and incubated on ice for 1 h. The samples were centrifuged again for 10 min at 8,000 g, and the pellets were then resuspended in a minimal volume of buffer containing 0.4 M KCl, 10 mM MOPS (pH 7.0), 5 mM DTT, and 1 μl/ml protease inhibitor cocktail. Samples were then diluted 1:1 with glycerol, mixed gently, and stored at −20°C for up to ∼10 days. On average, one myosin preparation was obtained from a pool of approximately five hearts. All animal studies were conducted in accordance with the University of Miami Miller School of Medicine institutional guidelines, and we have received required Institutional Animal Care and Use Committee approval. Muscle strips were prepared from flash-frozen hearts of Tg-WT mice (52) and Tg-K104E mice (25). LV were isolated and dissected into small muscle bundles in a cold room in ice-cold pCa 8 solution containing 30 mM butanedione monoxime and 15% glycerol (42). They were then chemically skinned in 50% glycerol and 50% pCa 8 solution {10−8 M Ca2+ concentration, 1 mM free Mg2+ concentration [total MgPr (propionate) = 3.88 mM], 7 mM EGTA, 2.5 mM [MgATP], 20 mM MOPS, pH 7.0, 15 mM creatine phosphate, and 15 U/ml of phosphocreatine kinase, ionic strength = 150 mM adjusted with KPr} containing 1% Triton X-100 for 24 h at 4°C. Mouse LV muscle bundles were then transferred to the same solution without Triton X-100 and shipped to the laboratory in Fort Worth, TX (Borejdo).
Preparation and cross-linking of myofibrils.
To observe a few XBs, it is preferable to observe myofibril preparations rather than isolated myocytes. Myofibrils are much thinner (∼0.5 μm) than the depth-of-view of a confocal microscope (∼3 μm) with the result that the volume from which fluorescence is detected (hence the number of XBs) is smaller than if the fluorescence was collected from the entire confocal volume (This procedure reduces the number of observed XBs from thousands to 20–40.). In addition, because they are much thinner than myocytes, they are easier to penetrate by fluorescent peptide and thus prevent the problems associated with the nonuniformity of labeling of sarcomeres. To prepare myofibrils, LV was washed three times with ice-cold EDTA-rigor solution for 30 min to wash out ATP present in the glycerinating solution without causing contraction. It was then washed thoroughly with rigor solution and homogenized by the Cole-Palmer LabGen 125 homogenizer for 10 s followed by further 10-s homogenization after a cool down period of 30 s. Contracting myofibrils were immobilized without affecting their ATPase by cross-linking with the water-soluble cross-linker 1-ethyl-3-[3-(dimethylamino)-propyl]carbodiimide (EDC) (4, 5, 22, 50). Myofibrils (1 mg/ml) were incubated for 20 min at room temperature with 20 mM EDC in Ca-rigor solution. The reaction was stopped by adding 20 mM DTT. The pH of the solution (7.5) remained unchanged throughout the 20-min reaction. The absence of shortening was verified by labeling the myofibrils with 10 nM rhodamine-phalloidin and observing contraction in a TIRF microscope.
Myosin light chain 1 labeling and exchange into myofibrils.
Recombinant myosin light chain 1 (LC1) was purchased from Prospec (ProSpec-Tany TechnoGene, Rehovot, Israel). It was labeled at position 182 with five molar excess of dye (SeTau-647-maleimide) overnight in ice. Labeled LC1 was purified by passing through a Sephadex G50 LP column to eliminate the free dye. LC1 (and not RLC) was chosen as the site of labeling to ensure that the mutation site was located on a different polypeptide chain so as not to cause any disturbance to the mutation. Freshly prepared myofibrils (1 mg/ml) were exchanged with an exchange solution as described previously (32) with 10 nM SeTau-LC1 under mild conditions (20 min at 30°C; see Ref. 51). The exchange was deliberately made highly inefficient so that only a small fraction of native LC1s were fluorescent. Myosin exchanged with fluorescent LC1 and bound to myofibrils has high anisotropy, in contrast to free LC1, i.e., it is well immobilized by myofibril even though it is bound at one site only.
Choice of the dye.
The fluorophore of choice was SeTau-647. It is excited in the red and hence bypasses most contributions of autofluorescence (30). SeTau is also very resistant to photobleaching (the initial rate of photobleaching was 2.4/s) because of nanoencapsulation of the squaraine moiety of the dye chromophore system in a mixed aliphatic-aromatic macrocycle. SeTau has a large Stokes shift (44 nm), high extinction coefficient, and quantum yield (0.65). Its overall fluorescence intensity was 4.2 times larger than fluorescence intensity of Alexa647.
Data collection and statistical analysis.
The fluorescence was measured by a PicoQuant MT 200 inverse time-resolved fluorescence instrument coupled to an Olympus IX 71 microscope. Before each experiment, fluorescence of an isotropic solution of a long fluorescence lifetime dye (i.e., with 0 anisotropy, such as rhodamine 700) was measured to make sure that the emitted light was split equally by a birefringent prism into the parallel and perpendicular The 640-nm excitation beam was focused by an Olympus ×60, 1.2 numerical aperture water immersion objective to the diffraction limit on the overlap band of a myofibril. For each overlap band, the fluorescence intensity was measured 20,000 times over 20 s. The normalized ratio of the difference between parallel and perpendicular channels is known as polarization of fluorescence (PF). PF is known to be a sensitive indicator of the orientation of fluorophore's transition dipole moment (12, 23, 24, 40, 43, 48, 49). A histogram of PFs, i.e., a plot of PF vs. frequency with which it occurs over 20 s, was constructed from these 20,000 measurements. Myofibrils (∼20–30) were examined for each physiological state (i.e., rigor, contraction, or relaxation), location (i.e., LV or RV), and state of mutation (i.e., WT or MUT) of a ventricle.
The differences between full width at half-maximum (FWHM) values of PF of different populations of ventricles are statistically interpretable only when the signal from each half-sarcomere is similar (38). This is because the relative value of FWHM of a strong random signal is small (relative to a signal), and FWHM of a weak signal is relatively large. To make sure that a signal was equal, the power was adjusted within the 1- to 2-μW range. Comparisons between groups were performed using an unpaired Student's t-test (Sigma Plot 11; Systat Software, San Jose, CA) or Origin version 8.6 (Northampton, MA). The significance was defined as P < 0.05. SigmaPlot 11 was used to compute histograms. Origin was used to compute autocorrelation functions (ACFs).
The number of observed myosin molecules.
The detection volume (DV) was estimated by measuring the width of axial and lateral dimensions of an image of 20-nm fluorescent beads. It was 1.7 μm3 and contained ∼3 × 105 XBs. The number of observed (fluorescent) XBs in this volume was reduced by four orders of magnitude (see below). Data are expressed as means of n experiments ± SD. A calibration curve that plotted the number of SeTau molecules in the DV vs. fluorescence intensity was constructed. The number of molecules in the DV was estimated using fluorescence correlation spectroscopy. The ACF of fluctuations of fluorescence (because of free SeTau entering and leaving the DV) at delay time 0 is equal to the inverse of the number of molecules contributing to the fluctuations (14, 15, 35). Six correlation functions were obtained from solutions of the fluorophore in the range 1.29–77.3 nM. Extrapolating the plot to one molecule revealed that a single molecule of SeTau illuminated with 0.2 μW of laser power gave 225 counts/s (38). We used this fact to estimate the number of molecules contributing to the signal as 20–40. However, it should be emphasized that, as long as the number of XBs is mesoscopic (i.e., when the number of observed molecules is so small that fluorescence is affected by fluctuations around the average; see Ref. 44), the exact number does not matter, i.e., 20 molecules should give the same result as 40 molecules, etc. The precision of measurement is approximately equal to 1/. To measure with 1% precision we must analyze 10,000 fluctuations. The characteristic lifetime of one fluctuation is of the order of 1 ms (26), and 10,000 fluctuations occur in 10 s. We collected the data over 20 s.
Stopped-flow measurements.
Three Tg-WT and seven Tg-K104E mouse hearts were used for mouse cardiac myosin preparation (see above). Myosins at 0.25 μM were mixed with 0.25 μM pyrene-labeled F-actin (stabilized by 0.25 μM phalloidin) in rigor buffer containing 0.4 M KCl, 1 mM DTT, and 10 mM MOPS, pH 7.0. The complexes were mixed in a 1:1 ratio (vol/vol) with varying concentrations of MgATP (10–80 μM) dissolved in the same buffer, and the dissociation of Tg-K104E vs. Tg-WT myosin from F-actin was observed by monitoring the time course of the change in pyrene fluorescence. Measurements were performed at 21°C using a BioLogic (Claix, France) model SFM-20 stopped-flow instrument outfitted with a Berger ball mixer and an FC-20 observation cuvette. The data were collected and digitized using a JASCO 6500 Fluorometer. The estimated dead time was 8.2 ms. The pyrene-F-actin was excited at 347 nm, and emission was monitored at 404 nm using monochromators set to 20-nm bandwidths. Typically, 8–14 stopped-flow records were averaged and fit to a single exponential equation to obtain the rate of a given MgATP concentration. A plot of the observed myosin dissociation rates as a function of [MgATP] was linear, and the slope corresponded to the rate constant expressed in molar per second.
RESULTS
The degree of order (distribution of XB orientations).
An informative way to characterize spatial distribution of XBs is to construct a histogram of polarization values vs. the number of times that a given polarization (i.e., orientation) occurs during a 20-s experiment. Figure 1 is a typical example of histograms of contracting Tg-WT (left) and contracting MUT myofibrils (right). The graphs are Gaussian. The regular residuals are shown (Fig. 1). Goodness of fit to an ideal Gaussian was assessed by adjusted coefficient of determination, AR2. It is a better measure of goodness of fit than χ2 (typically employed to assess nonlinear curve fitting) because it takes into account scaling of the dependent variable. The closer AR2 is to the value of one, the better the fit. Table 1 lists average AR2 of 25 experiments of contracting WT and 31 of contracting MUT muscles.
Fig. 1.

Random examples of the probability distribution of orientations of cross-bridges (XBs) of cardiac myofibrils of contracting wild-type (WT) and mutant (MUT) ventricles. Gray line is the best fit to a 3-parameter Gaussian. Graphs on the bottom show regular residuals of the counts. Left: WT muscle, FWHM = 0.464, adjusted coefficient of determination (AR2) = 0.953, mean intensity = 6.3 photons/ms, mean polarization = −0.444. Right: MUT muscle, FWHM = 0.380, AR2 = 0.947, mean intensity = 6 photons/ms, mean polarization = −0.156.
Table 1.
Comparison of FWHM's of 25 probability distributions of Tg-WT and 31 experiments of Tg-K104E of contracting myofibrils
| Contracting Muscle | FWHM | Intensity, counts/ms | Mean Polarization | AR2 |
|---|---|---|---|---|
| Tg-WT | 0.506 ± 0.129* | 5.83 ± 2.011 | −0.352 ± 0.153* | 0.897 ± 0.138 |
| Tg-K104E | 0.449 ± 0.046* | 4.76 ± 1.112 | 0.148 ± 0.022* | 0.886 ± 0.040 |
Errors are SDs of 28 experiments.
FWHM, full width at half-maximum; Tg, transgenic; WT, wild type; AR2, adjusted coefficient of determination.
Statistically significant difference.
Table 1 compares FWHMs of probability distributions of contracting Tg-WT and Tg-K104E (MUT) myofibrils. Detailed results of 25 experiments of contracting Tg-WT are shown in Table 2. Detailed results of 31 experiments of Tg-K104E are shown in Table 3.
Table 2.
Detailed results of probability distributions of contracting Tg-WT myofibrils
| Experiment No. | FWHM | TI | Mean Orientation | Adjusted R2 |
|---|---|---|---|---|
| 1 | 0.464 | 6.39 | −0.44 | 0.953 |
| 2 | 0.35 | 6.98 | −0.627 | 0.978 |
| 3 | 0.378 | 8.834 | −0.652 | 0.971 |
| 4 | 0.48 | 5.918 | −0.393 | 0.972 |
| 5 | 0.821 | 6.017 | −0.361 | 0.902 |
| 6 | 0.356 | 10.98 | −0.369 | 0.985 |
| 7 | 0.584 | 5.86 | −0.183 | 0.953 |
| 8 | 0.76 | 1.92 | −0.319 | 0.536 |
| 9 | 0.635 | 4.08 | −0.3445 | 0.909 |
| 10 | 0.511 | 4.87 | −0.444 | 0.968 |
| 11 | 0.489 | 6.164 | −0.4006 | 0.955 |
| 12 | 0.463 | 5.47 | −0.291 | 0.951 |
| 13 | 0.597 | 3.11 | −0.375 | 0.761 |
| 14 | 0.4312 | 6.631 | −0.4407 | 0.951 |
| 15 | 0.52038 | 4.233 | −0.291 | 0.916 |
| 16 | 0.448 | 5.56 | −0.3044 | 0.946 |
| 17 | 0.4719 | 7.6 | −0.3203 | 0.965 |
| 18 | 0.491 | 4.42 | −0.397 | 0.935 |
| 19 | 0.525 | 4.138 | −0.2306 | 0.874 |
| 20 | 0.46061 | 4.337 | −0.0175 | 0.851 |
| 21 | 0.656 | 5.12 | −0.012 | 0.855 |
| 22 | 0.613 | 4.582 | −0.6209 | 0.411 |
| 23 | 0.489 | 7.47 | −0.4001 | 0.965 |
| 24 | 0.449 | 9.66 | −0.3359 | 0.975 |
| 25 | 0.2088 | 5.58 | −0.232 | 0.976 |
| Average | 0.50608 | 5.837 | −0.35206 | 0.89656 |
| SD | 0.12932 | 2.0114 | 0.153920258 | 0.138636 |
TI, total intensity.
Table 3.
Detailed results of probability distributions of contracting Tg-K104E myofibrils
| Experiment No. | FWHM | TI | Mean Orientation | Adjusted R2 |
|---|---|---|---|---|
| 1 | 0.456 | 4.56 | −0.131 | 0.887 |
| 2 | 0.47001 | 4.15 | −0.167 | 0.881 |
| 3 | 0.443 | 5.24 | −0.117 | 0.887 |
| 4 | 0.4332 | 4.94 | −0.1455 | 0.909 |
| 5 | 0.403 | 5.321 | −0.1553 | 0.91 |
| 6 | 0.453 | 4.684 | −0.1466 | 0.905 |
| 7 | 0.392 | 6.15 | −0.123 | 0.943 |
| 8 | 0.394 | 5.97 | −0.1502 | 0.924 |
| 9 | 0.38 | 6.225 | −0.156 | 0.947 |
| 10 | 0.473 | 4.06 | −0.1118 | 0.853 |
| 11 | 0.423 | 5.29 | −0.1133 | 0.899 |
| 12 | 0.403 | 5.388 | −0.156 | 0.921 |
| 13 | 0.497 | 3.834 | −0.1042 | 0.851 |
| 14 | 0.47 | 4.21 | −0.1255 | 0.874 |
| 15 | 0.466 | 4.008 | −0.1139 | 0.846 |
| 16 | 0.48859 | 4.136 | −0.157 | 0.854 |
| 17 | 0.498 | 4.051 | −0.222 | 0.897 |
| 18 | 0.554 | 2.95 | −0.177 | 0.786 |
| 19 | 0.4418 | 5.108 | −0.1859 | 0.899 |
| 20 | 0.336 | 8.309 | −0.173 | 0.974 |
| 21 | 0.455 | 4.54 | −0.1849 | 0.899 |
| 22 | 0.5511 | 3.28 | −0.173 | 0.807 |
| 23 | 0.476 | 4.29 | −0.1008 | 0.85 |
| 24 | 0.479 | 3.81 | −0.1618 | 0.862 |
| 25 | 0.444 | 4.618 | −0.157 | 0.891 |
| 26 | 0.428 | 5.232 | −0.172 | 0.931 |
| 27 | 0.412 | 5.231 | −0.184 | 0.907 |
| 28 | 0.473 | 4.09 | −0.127 | 0.873 |
| 29 | 0.487 | 3.77 | −0.114 | 0.833 |
| 30 | 0.456 | 4.34 | −0.1609 | 0.874 |
| 31 | 0.408 | 5.139 | −0.137 | 0.893 |
| Average | 0.4498 | 4.765 | −0.148348 | 0.8860323 |
| SD | 0.04684 | 1.112 | 0.0299128 | 0.0401501 |
The t-test revealed that the 0.057 difference of FWHM was statistically significant (t = 2.436, P = 0.018) with 54 degrees of freedom. The 95% confidence interval of this difference is from 0.010 to 0.103. The 1.073 difference of intensities was statistically insignificant (t = 2.532, P = 0.014) with 54 degrees of freedom. The 95% confidence interval of this difference is from 0.223 to 1.922.
The differences in FWHM and polarization values were significant, but the differences in intensities were insignificant. This is important because, as already mentioned in materials and methods, the stochastic nature of a Gaussian signal causes a relative FWHM to depend on the strength of the signal. Weak signal results in a large FWHM, and strong signal results in small FWHM. Thus, when comparing FWHMs of two populations, it is crucial that the difference in photon counts between the two populations be statistically insignificant (38). The matter is more fully discussed in the discussion. The −0.498 difference of mean polarization was statistically significant (t = 17.744, P < 0.001) with 54 degrees of freedom. The 95% confidence interval of this difference is from −0.554 to −0.441.
Figure 2 is a typical example of histograms of rigor Tg-WT (left) and rigor MUT myofibrils (right). As before, the graphs are Gaussian. The regular residuals are shown in Fig. 2.
Fig. 2.

Random examples of the probability distribution of orientations of XBs of cardiac myofibrils of rigor transgenic (Tg)-WT and MUT ventricles. Gray line is the best fit to a 3-parameter Gaussian. Graphs on the bottom show regular residuals of the counts. Left: WT muscle, FWHM = 0.469, AR2 = 0.932, mean intensity = 4.7 photons/ms, mean polarization = −0.269. Right: mutated muscle, FWHM = 0.563, AR2 = 0.942, mean intensity = 6.817 photons/ms, mean polarization = −0.223.
Table 4 shows a comparison of FWHMs of 25 probability distributions of rigor Tg-WT and 34 experiments of Tg-K104E.
Table 4.
Comparison of FWHM's of 28 probability distributions of Tg-WT and Tg-K104E of rigor myofibrils
| Rigor Muscle | FWHM | Intensity, counts/min | Mean Polarization | AR2 |
|---|---|---|---|---|
| Tg-WT | 0.579 ± 0.223* | 5.68 ± 1.72 | −0.226 ± 0.290 | 0.885 ± 0.101 |
| Tg-K104E | 0.484 ± 0.083* | 5.53 ± 0.93 | −0.229 ± 0.085 | 0.922 ± 0.032 |
Values are means ± SD. Errors are SDs.
Statistically significant difference.
The t-test revealed that the 0.094 difference of FWHM was statistically significant (t = 2.253, P = 0.028) with 55 degrees of freedom. The 95% confidence interval of this difference is from 0.010 to 0.178. The 0.158 difference of intensities is statistically insignificant (t = 0.4477, P = 0.656) with 55 degrees of freedom. The 95% confidence interval of this difference is from −0.549 to 0.865. The importance of this difference being insignificant is addressed in the discussion. The 0.003 difference of mean polarization is statistically insignificant (t = 0.057, P = 0.954) with 55 degrees of freedom. The 95% confidence interval of this difference is from −0.10244 to 0.10844. Statistically significant differences are shown.
A relevant negative control experiment is to measure distribution in relaxation because here neither WT nor K104E XBs interact with the thin filaments. We therefore expect no differences. Figure 3 is a typical example of histograms of relaxed Tg-WT (left) and relaxed MUT myofibrils (right). As before, the graphs are Gaussian. The regular residuals are shown in Fig. 3.
Fig. 3.

Random examples of the probability distribution of orientations of XBs of relaxed cardiac myofibrils of rigor Tg-WT and MUT ventricles. Gray line is the best fit to a 3-parameter Gaussian. Graphs on the bottom show regular residuals of the counts. Left: WT muscle, FWHM = 0.429, AR2 = 0.927, mean intensity = 5.0 photons/ms, mean polarization = −0.074. Right: mutated muscle, FWHM = 0.467, mean intensity = 4.1 photons/ms, mean polarization = −0.100.
Table 5 is a comparison of FWHMs of 28 probability distributions of relaxed Tg-WT and Tg-K104E.
Table 5.
Comparison of FWHM's of 28 probability distributions of Tg-WT and Tg-K104E of relaxed myofibrils
| Relaxed Muscle | FWHM | Intensity, counts/min | Mean Polarization | AR2 |
|---|---|---|---|---|
| Tg-WT | 0.438 ± 0.037 | 4.99 ± 0.770 | −0.144 ± 0.085* | 0.910 ± 0.054 |
| Tg-K104E | 0.440 ± 0.043 | 4.72 ± 0.76 | −0.096 ± 0.022* | 0.901 ± 0.056 |
Errors are SDs of 28 experiments.
Statistically significant difference.
The t-test revealed that the −0.0025 difference of FWHM was statistically insignificant (t = 0.251, P = 0.802) with 65 degrees of freedom. The 95% confidence interval of this difference is from −0.0223712 to 0.0173712. The −0.271 difference of intensities was statistically insignificant (t = −1.406, P = 0.165 with 63 degrees of freedom). The 95% confidence interval of this difference is from −0.655 to 0.114. The −0.0478000 difference of mean polarization was statistically significant (t = 2.893, P = 0.052 with 55 degrees of freedom). The 95% confidence interval of this difference is from −0.0807955 to −0.0148045.
XB kinetics.
Because the number of observed molecules was small, we were able to measure fluctuations in equilibrium values of PF. These fluctuations contain information about the rate constants characterizing the enzymatic cycle of actin-myosin interaction. An ACF of PF fluctuations characterizes these rate constants. The shape of the ACF depends on the model assumed for the chemical reaction that drives the cycling of XBs. We use combination of models of Lymn and Taylor (34) and of Coureux et al. (10) that includes transitions between three fundamental states as illustrated in Fig. 4 where a XB, actin, ATP, and ADP are denoted by M, A, T, and D, respectively.
Fig. 4.

Conventional model of muscle contraction. In vitro measurements (see materials and methods) have shown that steady-state anisotropy associated with different cross-bridge states are 0 for MDP and MT (dissociated XBs) states, intermediate for the AMDP state, and maximum for AM and AM* states (data not shown). M, XB; A, actin; T, ATP; D, ADP.
The enzymatic (and the mechanical cycle coupled to it) begins with a state MT where M has already dissociated from thin filament by binding T and T is hydrolyzed to D and P to assume the MDP state (34). The products of hydrolysis remain bound to M. In this state cleft between upper and lower subdomains of a 50-kDa domain is partially closed, and the lever arm is in the up position (10). M and MT are free to rotate in the myofilament space and have the lowest steady-state anisotropy (and PF). MDP binds actin with a rate constant k1 forming a weakly bound, prepower stroke state, AMDP. It is partially immobilized. In this state the cleft between the upper and lower subdomains of a 50-kDa domain is completely closed, and the lever arm is less up than previously (10). Next, P dissociates from AMDP forming a strongly bound rigor complex AMD. It is assumed that this transition is very rapid. The transition from AMD to AM state is a power stroke that occurs with the rate constant k2. In AM state the cleft is completely closed and the lever arm is in the down position. The anisotropy is high. Finally, AM isomerizes to a “rigor-like” state with a rate constant k3 (10). In this state the cleft is opened and the lever arm becomes uncoupled from actin. Anisotropy is rigor-like. The true relaxation is the transition from AM* to MT state characterized by the rate kobs.
A representative experimental ACF is plotted in Fig. 5. The red line shows the best nonlinear fit to theoretically predicted expression of the ACF of a system of in Fig. 4 (an analytical expression of ACF is shown in Ref. 36). The kinetic constants were reported earlier by Huang et al. (25) and are reproduced as follows: k1 = 205 ± 93.7/s, k2 = 0.617 ± 0.496/s, and k3 = 0.689 ± 0.499/s for Tg-WT myofibrils and k1 = 779 ± 572.1/s, k2 = 1.162 ± 1.198/s, and k3 = 0.300 ± 0.242/s for Tg-K104E myofibrils.
Fig. 5.
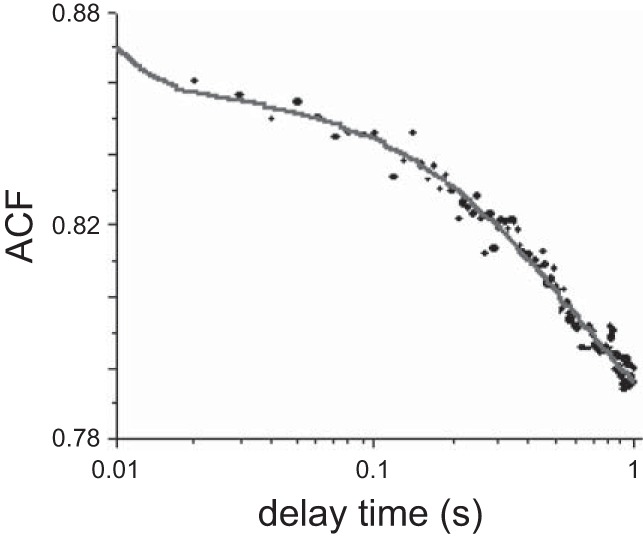
A representative trace of a normalized autocorrelation function (ACF) of polarization of fluorescence of contracting myofibril prepared from the left ventricle of Tg-WT mouse. Symbols are experimental data, and a gray line is the fit to equation shown in Ref. 36. The results of each experiment are presented in Table 4. The fact that ACF decays to a value >0 is due to the fact that mean polarization was nonzero.
The t-test revealed that the −574 difference of k1 was statistically significant (t = −4.764, P < 0.001) with 50 degrees of freedom. The −0.545 difference of k2 was statistically significant (t = −2.091, P = 0.041) with 53 degrees of freedom. The +0.389 difference of k3 was statistically significant (t = 3.809,P = 0.001) with 53 degrees of freedom.
The detailed results of kinetics experiments on WT and mutated ventricles are shown in Table 6.
Table 6.
Detailed results of 23 kinetics experiments on WT ventricles and 29 experiments on mutated ventricles
| k1 WT | k1 MUT | k2 WT | k2 MUT | k3 WT | k3 MUT |
|---|---|---|---|---|---|
| 244.2160 | 529.4700 | 0.7680 | 0.3310 | 0.8530 | 0.1590 |
| 189.4840 | 471.8200 | 0.1140 | 0.0450 | 0.1200 | 0.0281 |
| 2.1650 | 343.5200 | 0.2850 | 3.4400 | 0.8770 | 0.2700 |
| 1.8450 | 347.6000 | 0.4020 | 0.0440 | 0.5240 | 0.0330 |
| 138.5100 | 368.3400 | 0.6970 | 0.5300 | 1.8400 | 0.3100 |
| 319.8440 | 1,535.0060 | 0.4850 | 0.3330 | 0.9630 | 0.1810 |
| 218.8400 | 376.1900 | 1.5280 | 3.3350 | 1.3590 | 0.9800 |
| 359.5600 | 1,644.4000 | 0.4440 | 1.0400 | 0.2500 | 0.4850 |
| 226.4500 | 1,442.2400 | 0.9170 | 1.2680 | 0.5600 | 0.5970 |
| 284.5820 | 416.8900 | 0.7800 | 0.6290 | 1.2700 | 0.0580 |
| 219.4140 | 337.1720 | 0.0530 | 0.0320 | 0.0390 | 0.0150 |
| 208.1330 | 1,477.1570 | 0.6300 | 2.0100 | 0.6690 | 0.6200 |
| x | 355.6100 | 0.7370 | 0.1670 | 0.5590 | 0.0400 |
| 200.7300 | 422.7700 | 0.2500 | 2.1570 | 0.3140 | 0.3320 |
| 238.0140 | 308.4370 | 0.0370 | 0.0710 | 0.0290 | 0.0230 |
| 263.8200 | 1,519.7680 | 0.3700 | 0.9530 | 0.3410 | 0.4600 |
| 213.2600 | 306.0660 | 0.1660 | 0.4080 | 0.1730 | 0.2340 |
| 302.3300 | 1,444.8200 | 0.8620 | 4.7900 | 1.2080 | 0.5600 |
| 226.8060 | 340.6200 | 0.5610 | 0.2700 | 0.4030 | 0.1620 |
| −0.1250 | 402.2800 | 0.3160 | 0.1246 | 0.2180 | |
| 247.4680 | x | 2.3700 | 2.2700 | x | 0.7900 |
| x | 450.8900 | 0.6400 | 2.3900 | 0.5170 | 0.3930 |
| 224.1660 | 419.7400 | 0.6730 | 2.3000 | 1.3670 | 0.3120 |
| 232.5190 | 304.6900 | 0.5211 | 0.0548 | 0.9610 | 0.0230 |
| 174.0300 | 1,690.4300 | 0.5260 | 0.8750 | 1.2250 | 0.3900 |
| 1,036.8661 | 0.9740 | 0.5240 | |||
| 1,839.0600 | 1.4200 | 0.3330 | |||
| 397.8100 | 0.0700 | 0.0540 | |||
| x | 2.1760 | 0.1219 | |||
| 1,596.7300 | 0.7430 | 0.3590 | |||
| 367.5150 | 0.6020 | 0.2360 |
Values are means ± SD. x, Extreme values of constants that were removed from the original data. Extremes were defined as differing by more than a factor of 5 from the mean.
The mesoscopic method was unable to measure the rate of XB dissociation of Tg-K104E and Tg-WT myosin from thin filaments (kobs). To measure this rate actin was labeled with pyrene (31). The time course of the change in pyrene fluorescence was monitored as a function of ATP concentrations. Myosins were stoichiometrically mixed with pyrene-F-actin, and the complexes were mixed in a 1:1 volume ratio with increasing concentrations of MgATP (10–80 μM) in a stopped-flow apparatus. An increase in the fluorescence intensity was monitored as a function of time as the myosin heads dissociated from pyrene-F-actin on the addition of MgATP. The representative time-dependent dissociation profiles for Tg-K104E myosins are presented in Fig. 6A. The kobs, derived from the averaged fluorescence traces and fitted with a single exponential, are presented in Table 7. The results revealed significantly faster kobs in Tg-K104E vs. Tg-WT myosin for 10 and 25 μM MgATP and no differences between K104E and WT at higher ATP concentrations. Linear regression analysis of the plot of kobs vs. increasing [MgATP] yielded the effective second-order ATP-binding rate for the K104E equal to 0.68 ± 0.01 μM−1/s compared with 0.74 ± 0.02 μM−1s for Tg-WT (Fig. 6B) showing a small but significant (P < 0.05) change in the slope indicating a smaller MgATP binding rate for the mutant.
Fig. 6.

Fast kinetics of interaction between Tg-K104E and Tg-WT myosins with pyrene-labeled F-actin. The myosin-pyrene-actin complex was mixed with various MgATP concentrations (range from 10 to 80 μM), and the recovery of pyrene fluorescence was monitored on the dissociation of actomyosin complex. A: representative traces of a real-time increase of pyrene fluorescence within 1 s after MgATP was added to the Tg-WT-F-actin system. B: observed dissociation rates (kobs)-[MgATP] dependence and the effective second-order MgATP-binding rates for Tg-K104E vs. Tg-WT myosins. The average values of kobs ± SE for each MgATP concentration are presented in Table 7.
Table 7.
Observed dissociation rates (s−1) for Tg-WT and Tg-K104E myosins
| Myosin/[ATP], μM |
|||||
|---|---|---|---|---|---|
| 10 | 25 | 40 | 60 | 80 | |
| Tg-WT | 3.95 ± 0.081 | 11.75 ± 0.69 | 26.31 ± 0.41 | 36.26 ± 1.24 | 60.56 ± 2.84 |
| n | 11 | 11 | 11 | 11 | 11 |
| Tg-K104E | 6.71 ± 0.49* | 18.08 ± 0.45* | 27.26 ± 0.41 | 39.08 ± 0.89 | 55.81 ± 1.44 |
| n | 22 | 22 | 22 | 22 | 22 |
Errors are SEs of n sets of experiments with each set including at least 5 independent measurements. Concentrations refer to the amount of [ATP].
P < 0.01.
DISCUSSION
We studied the effect of the K104E mutation in RLC on the degree of order of ventricular myosin during steady-state contraction, relaxation and rigor, and on the XB kinetics during contraction of left ventricular myofibrils. Autofluorescence and photobleaching were minimized by labeling the myosin lever arm with a relatively long-lived red-emitting dye. Inhomogenities were minimized by focusing the exciting laser light on a single half-sarcomere. The dye was immobilized during critical transient states of the contraction cycle, in spite of the fact that it was attached to LC1 at one point only, rather than two points, which is more stable (9). By using the mesoscopic approach, we were able to measure the average kinetics of ∼20 XBs.
The degree of order.
As mentioned before, comparison of the probability distributions of orientations of Tg-WT and Tg-K104E XBs can be misleading unless the means of distributions are the same (38). The relative FWHM of distribution [for normal distribution FWHM = 2 = 2.355 SD] is sharply dependent on the strength of the signal. A weak Gaussian signal is associated with a broad distribution, and a strong Gaussian signal is associated with a narrow distribution. The same is true for any ratio of two Gaussian signals (like PF) (38). Therefore, meaningful comparison of FWHMs (or SDs) of the polarization of the two Gaussian signals requires that they have nearly equal mean. In practice, we ensure this by keeping the average photon count in each series of experiments similar (by adjusting the power of the illuminating laser). Because the difference in the intensities was statistically insignificant, we could conclude that, during contraction and rigor, the Tg-WT XBs were distributed more widely than mutated Tg-XBs. The fact that the spatial distribution of contracting XBs was significantly narrower in the Tg-K104E myofibrils was not surprising since mutated muscle was found to develop less tension than WT muscle (25), implying that K104E-XBs cannot reach as many actin-binding sites as WT-XBs. The observation of XB order in contraction is consistent with earlier work (7). The difference in polarization values of XBs of relaxed K104E and WT myofibrils was significant, perhaps because some myosin-actin interaction occurs at relatively low ionic strength used here (8). It was surprising to find that FWHM values for both types of myofibrils were not wider in relaxation compared with rigor.
Kinetics of contracting myofibrils.
The analysis of the ACF leads to two immediate conclusions about the mode of action of XBs. First, the action of XBs is not synchronized. This is because the ACF obtained from XBs within a single half-sarcomere were not periodic. If they were periodic, the XBs would have to be synchronized because ACF of a periodic signal is periodic (6). Second, although cross-linking prevented shortening, it did not prevent XBs from executing the power stroke. The present work shows that XBs rotate and are able to reach the neighboring target zones. Not only enzymatic but also mechanical cycles go on, in spite of the cross-linking (22).
Kinetic results show that the K104E mutation exerts significant effects on the interaction between actin and myosin. It increases the rates of XB binding, execution of the power stroke, and XB dissociation from the thin filaments. In particular, the rate of XB dissociation from the thin filaments is increased. This suggests that a mutated ventricle is prone to relaxation abnormalities, ultimately leading to diastolic dysfunction in patients carrying the mutation. The molecular effects of the mutation are shown schematically in Fig. 7. The blue cones (that indicate the cone of angles within which the transition dipole can rotate) suggest that the degree of disorder of XBs in WT ventricle (top) is larger than in K104E XBs (bottom). This implies that K104E-XBs cannot reach as many actin-binding sites as WT-XBs, i.e., we could predict that, in vivo, the mutated ventricles would develop less tension compared with WT. This prediction was confirmed by Huang et al. (25).
Fig. 7.
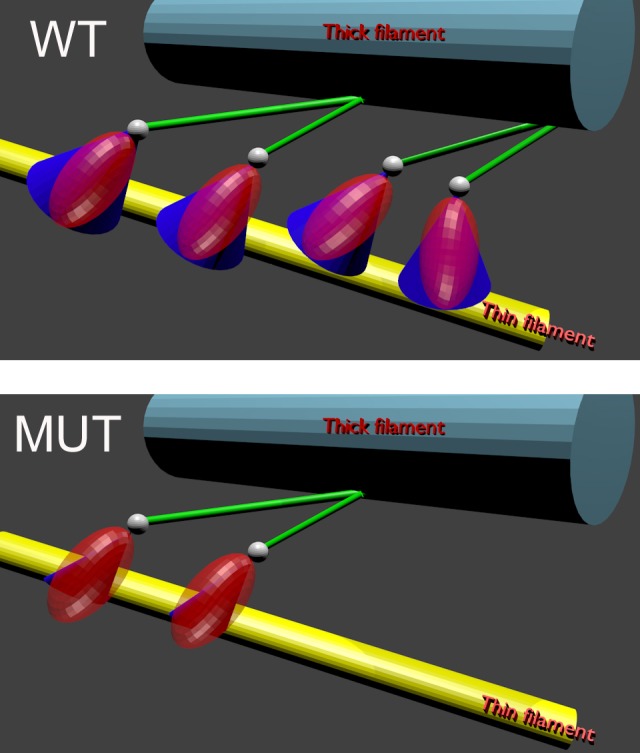
Schematic representation of XBs in contracting WT (top) and K104E (bottom) ventricles. WT XBs are more disorganized than K104E XBs [they rotate within larger cone of angles (blue)]. K104E XBs dissociate faster than WT (there are less XBs on bottom). Gray spheres are the fluorescent LC1s. Even though they can interact with actin (3), for simplicity it is drawn here separate from actin. Red ellipsoid represents the 50 + 20-kDa NH2-terminal domains of the myosin head. Myosin thick filament is shown in light blue, actin filament in yellow, and the COOH-terminal domain of XB (lever arm) in green.
The difference in XB distribution and kinetics between K104E and WT may constitute a molecular hallmark of FHC. Ventricles carrying familiar hypertrophy mutations at different sites also show obvious differences in the kinetics compared with WT muscles (13, 20, 21, 29, 36, 37, 39). Differences are also present between the α- and β-cardiac myosins (33).
In conclusion, we report here that the K104E mutation significantly alters the degree of order and the kinetics of single myosin XBs carrying the mutated RLC. The mutation-imposed molecular changes monitored in Tg-K104E myofibrils are followed by physiological changes in heart contractility of transgenic mice. We conclude that the differences in XB distribution and kinetics between the K104E mutant and WT muscles are sufficient to trigger pathological heart remodeling and FHC.
GRANTS
This work was supported by National Institutes of Health Grants R01-AR-048622 (J. Borejdo), R01-HL-108343 (D. Szczesna-Cordary), and R01-HL-090786 (J. Borejdo and D. Szczesna-Cordary) and by American Heart Association Grant 12PRE12030412 (W. Huang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.D., J.N., R.R., W.H., K.M., R.F., and D.S.-C. performed experiments; D.D., J.N., R.R., K.M., and J.B. analyzed data; D.D. and J.N. prepared figures; R.R. and J.B. interpreted results of experiments; R.R., I.G., and J.B. approved final version of manuscript; R.F. edited and revised manuscript; I.G., D.S.-C., and J.B. conception and design of research; J.B. drafted manuscript.
Glossary
- ACF
Autocorrelation function
- DV
Detection volume
- FHC
Familial hypertrophic cardiomyopathy
- FCS
Fluorescence correlation spectroscopy
- FWHM
Full width at half-maximum
- LC1
Myosin light chain 1
- MF
Cardiac myofibrils
- PF
Polarization of fluorescence
- RLC
Regulatory light chain of myosin
- WT
Wild type
- XB
Cross bridge
REFERENCES
- 1.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19: 104–110, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Andersen PS, Havndrup O, Bundgaard H, Moolman-Smook JC, Larsen LA, Mogensen J, Brink PA, Borglum AD, Corfield VA, Kjeldsen K, Vuust J, Christiansen M. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations (Abstract). J Med Genet 38: E43, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreev OA, Borejdo J. Binding of heavy-chain and essential light-chain 1 of S1 to actin depends on the degree of saturation of F-actin filaments with S1. Biochemistry 34: 14829–14833, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Barman T, Brune M, Lionne C, Piroddi N, Poggesi C, Stehle R, Tesi C, Travers F, Webb MR. ATPase and shortening rates in frog fast skeletal myofibrils by time-resolved measurements of protein-bound and free Pi. Biophys J 74: 3120–3130, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bershitsky SY, Tsaturyan AK, Bershitskaya ON, Mashanov GI, Brown P, Burns R, Ferenczi MA. Muscle force is generated by myosin heads stereospecifically attached to actin. Nature 388: 186–190, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Bracewell R. The Fourier Transform and Its Applications. New York, NY: McGraw-Hill, 1965. [Google Scholar]
- 7.Burghardt TP, Ando T, Borejdo J. Evidence for cross-bridge order in contraction of glycerinated skeletal muscle. Proc Natl Acad Sci USA 80: 7515–7519, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalovich JM, Stein LA, Greene LE, Eisenberg E. Interaction of isozymes of myosin subfragment 1 with actin: effect of ionic strength and nucleotide. Biochemistry 23: 4885–4889, 1984. [DOI] [PubMed] [Google Scholar]
- 9.Corrie JE, Craik JS, Munasinghe VR. A homobifunctional rhodamine for labeling proteins with defined orientations of a fluorophore. Bioconjug Chem 9: 160–167, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Coureux PD, Sweeney HL, Houdusse A. Three myosin V structures delineate essential features of chemo-mechanical transduction. Embo J 23: 4527–4537, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dantzig JA, Higuchi H, Goldman YE. Studies of molecular motors using caged compounds. Methods Enzymol 291: 307–348, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Dos Remedios CG, Millikan RG, Morales MF. Polarization of tryptophan fluorescence from single striated muscle fibers. A molecular probe of contractile state. J Gen Physiol 59: 103–120, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumka D, Talent J, Akopova I, Guzman G, Szczesna-Cordary D, Borejdo J. E22K mutation of RLC that causes familial hypertrophic cardiomyopathy in heterozygous mouse myocardium: effect on cross-bridge kinetics. Am J Physiol Heart Circ Physiol 291: H2098–H2106, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Elson EL. Fluorescence correlataion spectroscopy and photobleaching recovery. Annu Rev Phys Chem 36: 379–406, 1985. [Google Scholar]
- 15.Elson EL. Introduction to FCS. Fort Worth, TX: UNT, 2007. [Google Scholar]
- 16.Elson EL, Magde D. Fluorescence correlation spectroscopy: coceptual basis and theory. Biopolymers 13: 1–28, 1974. [DOI] [PubMed] [Google Scholar]
- 17.Elson EL, Webb WW. Concentration correlation spectroscopy: a new biophysical probe based on occupation number fluctuations. Annu Rev Biophys Bioeng 4: 311–334, 1975. [DOI] [PubMed] [Google Scholar]
- 18.Flavigny J, Richard P, Isnard R, Carrier L, Charron P, Bonne G, Forissier JF, Desnos M, Dubourg O, Komajda M, Schwartz K, Hainque B. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med 76: 208–214, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Geeves MA, Holmes KC. The molecular mechanism of muscle contraction. Adv Protein Chem 71: 161–193, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg MJ, Watt JD, Jones M, Kazmierczak K, Szczesna-Cordary D, Moore JR. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol 46: 108–115, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, Wang Y, Kerrick WG, Potter JD. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem 280: 37183–37194, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann C, Lionne C, Travers F, Barman T. Correlation of ActoS1, myofibrillar, and muscle fiber ATPases. Biochemistry 33: 4148–4154, 1994. [DOI] [PubMed] [Google Scholar]
- 23.Hopkins SC, Sabido-David C, Corrie JE, Irving M, Goldman YE. Fluorescence polarization transients from rhodamine isomers on the myosin regulatory light chain in skeletal muscle fibers. Biophys J 74: 3093–3110, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins SC, Sabido-David C, van der Heide UA, Ferguson RE, Brandmeier BD, Dale RE, Kendrick-Jones J, Corrie JE, Trentham DR, Irving M, Goldman YE. Orientation changes of the myosin light chain domain during filament sliding in active and rigor muscle. J Mol Biol 318: 1275–1291, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Huang W, Liang J, Kazmierczak K, Muthu P, Duggal D, Farman G, Sorensen L, Pozios I, Abraham T, Moore J, Borejdo J, Szczesna-Cordary D. Hypertrophic cardiomyopathy associated Lys104Glu mutation in the myosin regulatory light chain causes diastolic disturbance in mice. J Mol Cell Card 74: 318–329, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huxley AF, Simmons RM. Proposed mechanism of force generation in striated muscle. Nature 233: 533–538, 1971. [DOI] [PubMed] [Google Scholar]
- 27.Kabaeva ZT, Perrot A, Wolter B, Dietz R, Cardim N, Correia JM, Schulte HD, Aldashev AA, Mirrakhimov MM, Osterziel KJ. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet 10: 741–748, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Kazmierczak K, Muthu P, Huang W, Jones M, Wang Y, Szczesna-Cordary D. Myosin regulatory light chain mutation found in hypertrophic cardiomyopathy patients increases isometric force production in transgenic mice. Biochem J 442: 95–103, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerrick WG, Kazmierczak K, Xu Y, Wang Y, Szczesna-Cordary D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J 23: 855–865, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakowicz JR. Principles of Fluorescence Spectroscopy. New York, NY: Springer-Verlag, 2006. [Google Scholar]
- 31.Lehrer SS, & Ishii Y. Fluorescence properties of acrylodan-labeled tropomyosin and tropomyhosin actin: evidence for myosin subfragment-1 induced changes in geometry between tropomyosin and actin. Biochemistry 27 5899–5906, 1988. [DOI] [PubMed] [Google Scholar]
- 32.Ling N, Shrimpton C, Sleep J, Kendrick-Jones J, Irving M. Fluorescent probes of the orientation of myosin regulatory light chains in relaxed, rigor, and contracting muscle. Biophys J 70: 1836–1846, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM. Transgenic mouse alpha-and beta-cardiac myosins containing the R403Q mutation show isoform dependent transient kinetic differences. J Biol Chem 288: 14780–14787, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lymn RW, Taylor EW. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10: 4617–4624, 1971. [DOI] [PubMed] [Google Scholar]
- 35.Magde D, Elson EL, Webb WW. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13: 29–61, 1974. [DOI] [PubMed] [Google Scholar]
- 36.Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, Zhao J, Szczesna-Cordary D, Borejdo J. Cross-bridge kinetics in myofibrils containing familial hypertrophic cardiomyopathy R58Q mutation in the regulatory light chain of myosin. J Theor Biol 284: 71–81, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mettikolla P, Luchowski R, Gryczynski I, Gryczynski Z, Szczesna-Cordary D, Borejdo J. Fluorescence lifetime of actin in the FHC transgenic heart. Biochemistry 48: 1264–1271, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Midde K, Rich R, Marandos P, Fudala R, Li A, Gryczynski I, Borejdo J. Comaprison or orientation and rotational motion of cross-bridges containing phosphorylated and dephosphorylated myosin regulatory light chain. J Biol Chem 288: 7012–7023, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang Y, Xu Y, Kerrick WG, Potter JD. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem 276: 3743–3755, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Morales MF. Calculation of the polarized fluorescence from a labeled muscle fiber. Proc Nat Acad Sci USA 81: 145–149, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, Waldenstrom A. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 35: 841–849, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Muthu P, Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski I, Szczesna-Cordary D, Borejdo J. Single molecule kinetics in the familial hypertrophic cardiomyopathy D166V mutant mouse heart. J Mol Cell Cardiol 48: 989–998, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nihei T, Mendelson RA, Botts J. Use of fluorescence polarization to observe changes in attitude of S1 moieties in muscle fibers. Biophys J 14: 236–242, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian H, Saffarian S, Elson EL. Concentration fluctuations in a mesoscopic oscillating chemical reaction system. Proc Natl Acad Sci USA 99: 10376–10381, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan R. Structure of the actin-myosin complex and its implications for muscle contraction. Science 261: 58–65, 1993. [DOI] [PubMed] [Google Scholar]
- 46.Rayment I, Rypniewski W, Schmidt-Base K, Smith R, Tomchik DR, Benning MM, Winkelman DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261: 50–58, 1993. [DOI] [PubMed] [Google Scholar]
- 47.Rice J. Mathematical Statistics and Data Analysis (2nd ed). N. Scituate, MA: Duxbury, 1995. [Google Scholar]
- 48.Sabido-David C, Brandmeier B, Craik JS, Corrie JE, Trentham DR, Irving M. Steady-state fluorescence polarization studies of the orientation of myosin regulatory light chains in single skeletal muscle fibers using pure isomers of iodoacetamidotetramethylrhodamine. Biophys J 74: 3083–3092, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tregear RT, Mendelson RA. Polarization from a helix of fluorophores and its relation to that obtained from muscle. Biophys J 15: 455–467, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsaturyan AK, Bershitsky SY, Burns R, Ferenczi MA. Structural changes in the actin-myosin cross-bridges associated with force generation induced by temperature jump in permeabilized frog muscle fibers. Biophys J 77: 354–372, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ushakov DS, Caorsi V, Ibanez-Garcia D, Manning HB, Konitsiotis AD, West TG, Dunsby C, French PM, Ferenczi MA. Response of rigor cross-bridges to stretch detected by fluorescence lifetime imaging microscopy of myosin essential light chain in skeletal muscle fibers. J Biol Chem 286: 842–850, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Xu Y, Kerrick WGL, Wang Y, Guzman G, Diaz-Perez Z, Szczesna-Cordary D. Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol 361: 286–299, 2006. [DOI] [PubMed] [Google Scholar]