

Abstract
Glucose transporter 1 (GLUT1) is the primary glucose transport protein of the cardiovascular system and astroglia. A recent study proposes that caffeine uncompetitive inhibition of GLUT1 results from interactions at an exofacial GLUT1 site. Intracellular ATP is also an uncompetitive GLUT1 inhibitor and shares structural similarities with caffeine, suggesting that caffeine acts at the previously characterized endofacial GLUT1 nucleotide-binding site. We tested this by confirming that caffeine uncompetitively inhibits GLUT1-mediated 3-O-methylglucose uptake in human erythrocytes [Vmax and Km for transport are reduced fourfold; Ki(app) = 3.5 mM caffeine]. ATP and AMP antagonize caffeine inhibition of 3-O-methylglucose uptake in erythrocyte ghosts by increasing Ki(app) for caffeine inhibition of transport from 0.9 ± 0.3 mM in the absence of intracellular nucleotides to 2.6 ± 0.6 and 2.4 ± 0.5 mM in the presence of 5 mM intracellular ATP or AMP, respectively. Extracellular ATP has no effect on sugar uptake or its inhibition by caffeine. Caffeine and ATP displace the fluorescent ATP derivative, trinitrophenyl-ATP, from the GLUT1 nucleotide-binding site, but d-glucose and the transport inhibitor cytochalasin B do not. Caffeine, but not ATP, inhibits cytochalasin B binding to GLUT1. Like ATP, caffeine renders the GLUT1 carboxy-terminus less accessible to peptide-directed antibodies, but cytochalasin B and d-glucose do not. These results suggest that the caffeine-binding site bridges two nonoverlapping GLUT1 endofacial sites—the regulatory, nucleotide-binding site and the cytochalasin B-binding site. Caffeine binding to GLUT1 mimics the action of ATP but not cytochalasin B on sugar transport. Molecular docking studies support this hypothesis.
Keywords: GLUT1, erythrocyte, ATP, caffeine, glucose transport
the human facilitative glucose transporter, GLUT1, is the prototypic member of the family of carrier proteins responsible for equilibrative, cellular glucose transport (24). GLUT1 is the primary glucose transporter in smooth muscle (47), astrocytes (55), endothelial cells of blood tissue barriers (47, 59), and in primate and cetacean erythrocytes (22, 24). In human red blood cells, GLUT1 constitutes 10% of membrane protein (32). The resulting erythrocyte glucose transport is so rapid that the glucose space of the blood available for exchange with metabolically active tissues includes both the serum and the erythrocyte intracellular water (24). GLUT1 catalyzes trans-cellular glucose transport at the blood-brain barrier to provide the primary metabolic fuel for the central nervous system (CNS) (24, 55). The central role of GLUT1 in cerebral function and development is evinced by GLUT1 deficiency syndrome in which GLUT1 gene mutations produce GLUT1 haploinsufficiency, which in turn gives rise to seizures, microcephaly, and severe developmental delay in infants (27).
GLUT1 activity, sites of expression, and total expression levels are subject to acute and chronic physiologic control (24, 61). In cardiac muscle, fibroblasts, and adipocytes, acute exposure to insulin or hypoxia can increase GLUT1 cell surface expression (7, 19, 30). Acute AMP kinase activation resulting from reduced glucose availability rapidly and reversibly increases cell surface GLUT1 levels and cellular sugar transport in blood-brain barrier endothelial cells (25). Chronic hypoxia, hypoglycemia, and AMP kinase activation increase GLUT1 gene and protein expression (10) thereby increasing blood-brain barrier glucose transport (33, 48) and enhancing glucose transport in some cancers (3, 13, 31).
GLUT1 is allosterically inhibited by ATP, which binds at a single, ATPase-null, GLUT1 nucleotide-binding site (17, 35). Mutagenesis and peptide mapping studies localize the adenosine nucleotide-binding site to the endofacial surface of GLUT1 involving cytoplasmic loop 8-9 and transmembrane helices 8 and 9 (43, 45). ATP binding leads to GLUT1 conformational change involving the cytoplasmic carboxy terminus and large intracellular loop 6-7 (9). These structural changes result in decreased Km and Vmax for zero-trans sugar uptake characteristic of uncompetitive inhibition (17, 43, 44). AMP binds to GLUT1 and acts as a competitive antagonist of ATP inhibition of glucose uptake (17).
GLUT1-mediated sugar transport is also inhibited by families of structurally diverse small molecules, which affect the kinetics of transport in different ways (1, 51). Cytochalasin B (CB) is a micotoxin that binds at the endofacial surface of GLUT1 and functions as a competitive inhibitor of exchange and net sugar efflux and as a noncompetitive inhibitor of net uptake (6). Barbiturates such as phenobarbital, in contrast, appear to act as noncompetitive inhibitors of net sugar uptake and exit but as competitive inhibitors of exchange transport (30). The methylxanthines comprise an additional class of GLUT1 inhibitors (18). Among these, caffeine (1,3,7-trimethylxanthine) is most commonly encountered in a normal diet. Indeed, 80% of the US population consumes caffeine daily, making it the most widely used psychoactive drug in the world (34). Given the widespread use of caffeine and the central role of GLUT1 in cerebral metabolism, an understanding of how caffeine inhibits GLUT1 could be useful in the management of organismal carbohydrate homeostasis in health and disease.
In the present study, we ask whether the uncompetitive inhibition of GLUT1 produced by caffeine (38, 52) and ATP (17) and the structural similarities between caffeine and adenosine reflect a common mechanism of action on GLUT1.1
MATERIALS AND METHODS
Materials.
[3H]3-O-methylglucose ([3H]3-OMG) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Unlabeled 3-OMG, CB, phloretin, ATP, AMP, and d-glucose were purchased from Sigma Aldrich (St. Louis, MO). Trinitrophenyl-ATP (TNP-ATP) was purchased from Life Technologies (Carlsbad, CA). Custom synthesized rabbit anti-GLUT1 COOH-terminal antibody (C-Ab) was produced by New England Peptide and was raised against GLUT1 residues 480-492.
Erythrocyte and ghost preparation.
De-identified whole human blood was purchased from Biological Specialty (Colmar, PA). Erythrocytes were washed three times with cold wash buffer [150 mM KCl, 5 mM HEPES (pH 7.4), and 0.5 mM EDTA] and prepared for transport experiments as previously described (53). Ghosts were prepared from washed erythrocytes as previously described (53) and resealed with erythrocyte wash buffer containing indicated concentrations of ATP or AMP.
Transport measurements.
All transport reactions were performed in triplicate at ice temperature using 20 μl of a 50% suspension of either erythrocytes or ghosts as previously described (53). Uptake was initiated by the addition of 100 μl of uptake solution containing 2.5 μCi/ml [3H]3-OMG and the indicated concentrations of unlabeled 3-OMG and caffeine. After 30 s, reactions were stopped by addition of 1 ml ice-cold stop solution containing 10 μM CB and 100 μM phloretin in PBS. Cells were pelleted and washed once with 1 ml stop solution, then lysed with 0.5 ml of 3% perchloric acid. Clarified lysates were assayed in duplicate for the radiolabel using liquid scintillation counting.
Kinetic analysis.
Nonlinear regression analysis of transport data was performed using GraphPad Prism software (version 6; GraphPad, La Jolla, CA). The concentration dependence of 3-OMG uptake was fit to Eq. 1
| (1) |
where v is the rate of 3-OMG uptake, [S] is the 3-OMG concentration, and Vmax and Km are the velocity and Michaelis constants, respectively, for transport. Caffeine dose-response data were fit to Eq. 2
| (2) |
where v0 is the amount of sugar uptake in the absence of caffeine, [I] is the caffeine concentration, v0 − vi is the sugar uptake remaining at saturating caffeine concentration, and Ki(app) (apparent inhibition constant) is that concentration of caffeine reducing transport by vi/2.
TNP-ATP binding competition assay.
GLUT1 protein was purified to ≥90% purity from washed erythrocyte membranes as previously described (36). Binding reactions were assayed at room temperature in 50 mM Tris·HCl, pH 7.4, 5 mM MgCl2 by adding 100 nM purified GLUT1 to 5 μM TNP-ATP. After a 2 min incubation to permit equilibrium binding (21), interfering ligands were added and the suspension was assayed for fluorescence at 500–600 nm with excitation at 408 nm (Photon Technologies International, Edison, NJ). Data were plotted using GraphPad Prism software (version 6) and a locally weighted scatterplot-smoothing (LOWESS) curve fit.
[3H]cytochalasin B binding.
[3H]cytochalasin B ([3H]CB; 100 nM) binding to purified human GLUT1 (1 μg GLUT1 in 40 μl reaction medium) was measured as described previously (36).
COOH-terminal antibody accessibility assay.
ELISA determination of GLUT1 COOH-terminal antibody binding was performed as previously described (9). Briefly, 200 ng of purified GLUT1 in PBS was bound to 96-well microtiter dishes at 37°C for 2 h. Plates were blocked with 3% bovine serum albumin in PBS with the indicated concentrations of ligand at 37°C for 2 h. Bound protein was probed with COOH-terminal antibody solutions (0.3 μg/ml) containing indicated concentrations of ligand at 37°C for 2 h followed by five washes with PBS. Each well was then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (0.5 μg/ml) and ligand for a further 2 h. After washing five times with PBS, wells were developed with ABTS (Pierce). Absorbance was measured at 415 nm using an iMark microplate reader (Bio-Rad).
Docking studies.
The GLUT1 crystal structure was obtained from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) using the pdb code 4PYP (28). The β-nonylglucoside molecule was removed from the pdb structure. Structures for ATP, caffeine, and CB were obtained from ZINC (http://zinc.docking.org) (40). Computational docking was performed with AutoDock Vina (58). The ligands and protein were prepared for docking using Autodock Tools version 1.5.6 (50, 54). Docking was constrained to a cube with dimensions of 44 × 28 × 28 Å volume encompassing loop 6 and the endofacial transport cavity of 4PYP.
RESULTS
Caffeine is an uncompetitive inhibitor of 3-OMG uptake by human erythrocytes.
The reported uncompetitive inhibition of erythrocyte sugar uptake by caffeine (38, 52) represents a relatively unusual form of transport inhibition. To confirm the inhibitory nature of caffeine on GLUT1, the uptake of 100 μM 3-OMG into sugar-free erythrocytes was measured over an interval of 30 s at ice temperature. Under these conditions, transport allows ∼33% equilibration of cell water space with substrate, ensuring the measurement of close to initial rates of transport (53).
Exposure to caffeine produces a robust and dose-dependent inhibition of 3-OMG transport with an apparent Ki of 3.5 ± 0.2 mM (Fig. 1A). Analysis of the concentration dependence of 3-OMG uptake in the absence and presence of 5 mM caffeine indicates that caffeine reduces both Vmax and Km for sugar uptake (Fig. 1B). Vmax for transport falls from 151 ± 12 μmol·l−1·min−1 to 32 ± 1 μmol·l−1·min−1. Km falls from 1.4 ± 0.3 mM to 0.4 ± 0.1 mM (Fig. 1B). This result is characteristic of uncompetitive inhibition and is consistent with previous studies of caffeine-mediated GLUT1 inhibition (38, 52). The decreases in Vmax and Km induced by caffeine are of the same magnitude (4.7 ± 0.6 and 3.5 ± 2.2-fold, respectively).
Fig. 1.
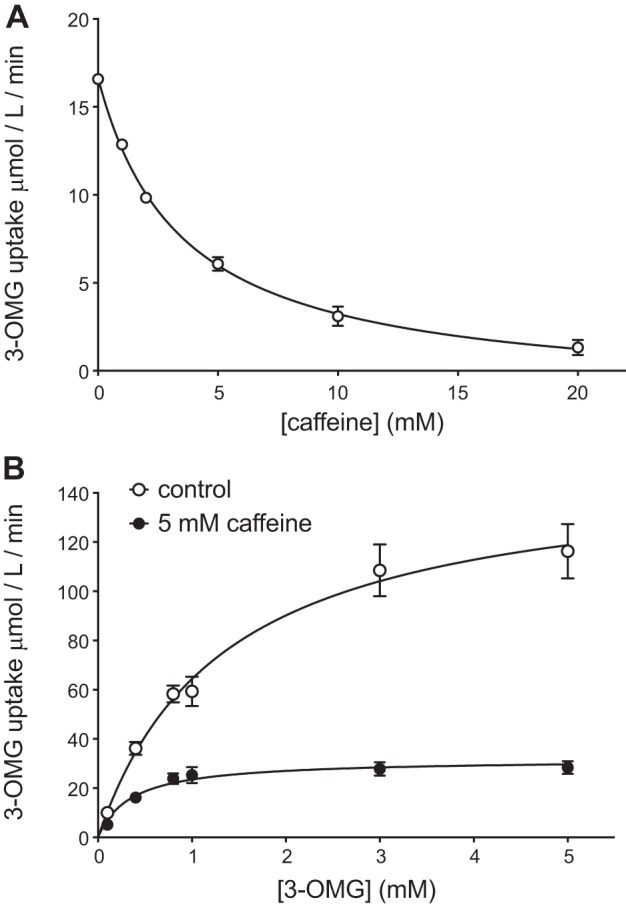
A: concentration dependence of caffeine inhibition of 100 μM 3-O-methylglucose (3-OMG) uptake. Ordinate: rate of sugar uptake in μmol·l cell water−1·min−1; abscissa: caffeine concentration in the uptake medium in mM. Results are shown as means ± SE for at least 4 measurements made in duplicate. The curve drawn through the points was computed by nonlinear regression assuming that saturable inhibition of transport is described by Eq. 2. The results are: v0 = 16.7 ± 0.2 μmol·l−1·min−1; Vi = 18.1 ± 0.3 μmol·l−1·min−1; apparent inhibition constant Ki(app) = 3.5 ± 0.2 mM, R2 = 0.99. B: caffeine is an uncompetitive inhibitor of 3-OMG uptake by human red blood cells (RBCs). Ordinate: rate of sugar uptake in μmol·l cell water−1·min−1; abscissa: extracellular 3-OMG concentration in mM. Results are shown as means ± SE for at least 4 measurements made in duplicate with control cells (○) and for cells exposed to 5 mM caffeine (●). Control experiments indicate that results obtained by preincubating cells with caffeine for 15 min are indistinguishable from the those obtained by simply adding caffeine during the 30 s of sugar uptake measurement. The curves drawn through the points were computed by nonlinear regression assuming that transport is described by Michaelis-Menten kinetics (Eq. 1). The results are: control, Vmax = 151 ± 12 μmol·l−1·min−1; Km = 1.3 ± 0.3 mM, R2 = 0.87; Caffeine, Vmax = 32 ± 2 μmol·l−1·min−1; Km = 0.4 ± 0.1 mM, R2 = 0.80.
ATP antagonizes caffeine-mediated transport inhibition.
Uncompetitive inhibition of GLUT1 by ATP has previously been described by our laboratory (17, 43, 44). Cytoplasmic (but not extracellular) ATP causes a dose-dependent decrease in both Vmax and Km for zero-trans sugar uptake (8- and 18-fold, respectively). This unbalanced effect of ATP on Vmax and Km contrasts with the more balanced reduction in both parameters induced by caffeine and results in an apparent stimulation of net sugar transport under subsaturating [3-OMG] conditions. Reduction of Vmax and Km by ATP and caffeine suggests a common mechanism of action, a hypothesis further supported by the structural similarities between adenosine and caffeine.
To explore this possibility, we investigated the effects of ATP on caffeine-mediated GLUT1 inhibition. As with erythrocytes, resealed erythrocyte ghosts are inhibited by caffeine in a dose-dependent manner with an apparent Ki of 0.91 ± 0.34 mM (Fig. 2, no ATP). When ghosts are resealed with either 5 mM ATP or AMP, Ki increases to 2.6 ± 0.6 mM and 2.4 ± 0.5 mM, respectively (Fig. 2, ATP or AMP inside). The effect is specific to the cytoplasmic side of the membrane, as including ATP in the uptake solutions and not inside the ghosts resulted in an apparent Ki of 0.96 ± 0.63 mM (Fig. 2, ATP outside). The antagonism of caffeine-mediated GLUT1 inhibition by only intracellular adenosine nucleotide (Fig. 2) is consistent with existing evidence indicating that ATP binds to GLUT1 at a cytoplasmic nucleotide-binding site (44) and suggests that caffeine also functions at the cytosolic surface of the protein. Ki(app) values for ATP and AMP antagonism of caffeine inhibition of 3-OMG transport (assuming simple, competitive antagonism) are 2.7 ± 0.8 and 3.1 ± 0.7 mM, respectively. Recent studies suggest that Kd(app) (apparent dissociation constant) values for ATP and AMP binding to GLUT1 are 0.6–2 mM and 2.2 mM, respectively (9).
Fig. 2.
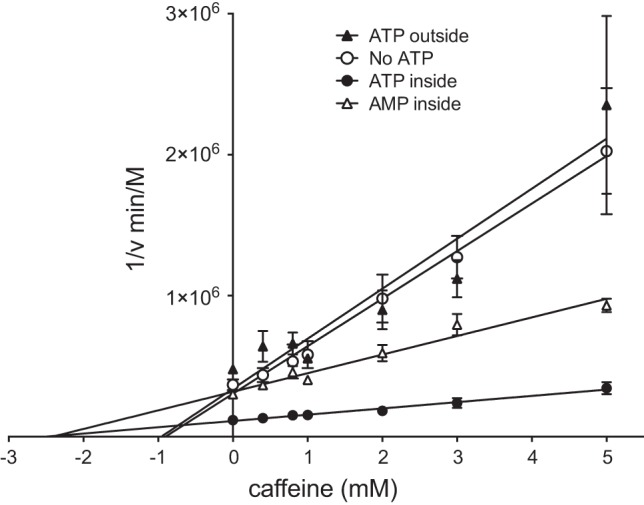
Dixon plot of caffeine inhibition of 3-OMG uptake in RBC ghosts. Ordinate: 1/sugar uptake (min·mol−1·l−1); Abscissa: caffeine concentration in mM. Results are shown as means ± SE for at least 3 measurements made in duplicate and are shown for control ghosts (○), ghosts containing 4 mM ATP (●), ghosts containing 4 mM AMP (△), and nucleotide-free, resealed ghosts exposed to 4 mM extracellular ATP (▲). The lines drawn through the points were computed by linear regression and have the following constants: control slope = 337.0 ± 44.9 min, x-intercept = −0.91 ± 0.34 mM, y-intercept = 305,226 ± 111,768 min/M, R2 = 0.72; 4 mM intracellular ATP slope = 44.5 ± 4.6 min, x-intercept = −2.58 ± 0.55 mM, y-intercept = 111,949 ± 10,641 min/M, R2 = 0.83; 4 mM intracellular AMP slope = 131.8 ± 11.1 min, x-intercept = −2.41 ± 0.45 mM, y-intercept = 318,946 ± 27,465 min/M, R2 = 0.85; 4 mM extracellular ATP slope = 354.6 ± 56.7 min, x-intercept = −0.96 ± 0.63 mM, y-intercept = 340,677 ± 135,181 min/M, R2 = 0.67. In all instances the slope is significantly different from zero (P < 0.0001).
Effects of caffeine on nucleotide and cytochalasin B binding to GLUT1.
ATP antagonism of caffeine inhibition of glucose transport suggests that ATP and caffeine compete for binding to GLUT1. Competition for binding could result if ATP and caffeine bind at a common site or if ATP- and caffeine-binding sites are physically distinct but mutually exclusive. To test for competitive binding, we evaluated the ability of caffeine to interfere with the binding of the fluorescent ATP analog TNP-ATP to GLUT1 protein purified from human erythrocytes. TNP-ATP mimics the effect of ATP on GLUT1-mediated 3-OMG transport kinetics (21). When bound to purified GLUT1 in unsealed proteoliposomes, the probe exhibits an enhanced and blue-shifted fluorescence (Fig. 3A “control”). This bound fluorescence is unaffected by either the presence of 5 mM d-glucose or the well-characterized GLUT1 inhibitor, CB (10 μM) (Fig. 3A). The addition of 5 mM ATP, however, displaces prebound TNP-ATP, reducing fluorescence and restoring red-shifted fluorescence. This effect is mimicked by the addition of 5 mM caffeine (Fig. 3A). Control reactions lacking GLUT1 reveal that TNP-ATP autofluorescence in the absence of protein is modulated by some ligands (Fig. 3A, dashed lines). Following correction of fluorescence maxima for these changes, we observe that ATP and caffeine produce 28% and 27% reductions in TNP-ATP fluorescence, respectively, while d-glucose and CB are without effect (Fig. 3B). These results demonstrate that ATP and caffeine compete with TNP-ATP for binding to GLUT1. Caffeine inhibits [3H]CB binding to human GLUT1 but ATP does not (Table 1). d-Glucose competitively displaces CB from GLUT1 (62).
Fig. 3.
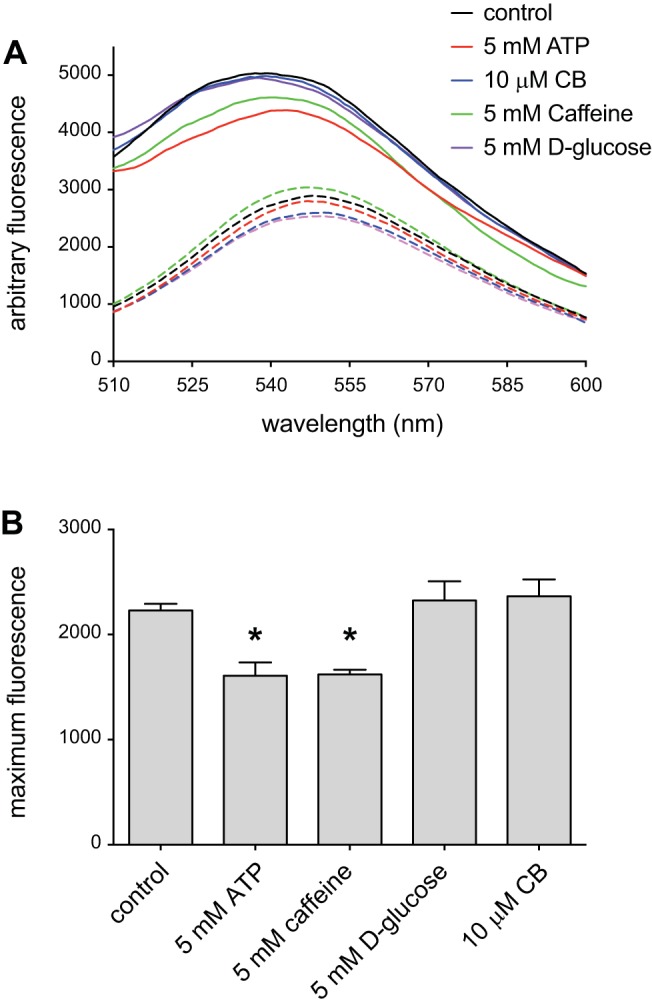
Caffeine displaces ATP from glucose transporter 1 (GLUT1). A: emission spectra of 5 μM trinitrophenyl-ATP (TNP-ATP) in the absence (dashed lines) or presence (continuous lines) of 100 nM GLUT1. Suspensions were excited at 408 nm, and the resulting emission (ordinate) was measured over 500–600 nm (abscissa). Results are shown for TNP-ATP emission in the absence of added ligands (black lines) and for emission in the presence of 5 mM d-glucose (purple lines), 10 μM cytochalasin B (CB; blue lines), 5 mM ATP (red lines), or 5 mM caffeine (green lines). B: GLUT1-specific TNP-ATP maximum fluorescence shown in the absence or presence of ligands. Ordinate: fluorescence; abscissa: ligand additions. Results are shown as means ± SE for 3 separate measurements. *Measured emission is significantly less than that observed in the absence of added ligand (P ≤ 0.037, 1-tailed, paired t-test).
Table 1.
Effect of adenine nucleotides on Ki(app) for caffeine inhibition of 3-OMG uptake
| Experimental Condition | Caffeine Ki(app), mM | Nucleotide Ki(app), mM |
|---|---|---|
| 0 ATPi, 0 ATPo | 0.9 ± 0.3 | NA |
| 4 mM ATPi, 0 ATPo | 2.6 ± 0.6 | 2.2 |
| 0 ATPi, 4 mM ATPo | 1.0 ± 0.6 | NA |
| 4 mM AMPi, 0 ATPo | 2.4 ± 0.5 | 2.4 |
Values are means ± SE. Effects of ATP and AMP on apparent inhibition constant Ki(app) for caffeine inhibition of 3-OMG uptake were obtained by analysis of data in Fig. 2. ATP and/or AMP (4 mM) were either absent or present on the inside (e.g., ATPi) or outside (e.g., AMPo) of red cell ghosts. Nucleotides were added to the interior of ghosts during resealing of hypotonically lysed red cells. Ki(app) for caffeine inhibition of 3-OMG uptake was computed as −x-intercept of the Dixon plot of Fig. 2. Ki(app) for nucleotide-dependent competition with caffeine for inhibition of uptake was computed assuming that caffeine and intracellular nucleotide compete for binding at the same site. 3-OMG, 3-O-methylglucose; NA, not applicable.
Caffeine and ATP induce similar structural changes in GLUT1.
ATP binding to GLUT1 promotes GLUT1 conformational changes involving the cytosolic COOH terminus and the large intracellular loop connecting trans-membrane helices 6 and 7 (9). One consequence of these changes is reduced GLUT1 COOH-terminal peptide-directed antibody (C-Ab) binding to GLUT1 in an ELISA-based assay (9, 17). We explored the possibility that caffeine may induce comparable structural changes in GLUT1. Purified GLUT1 was adsorbed to microtiter ELISA wells and probed with C-Ab in media containing increasing concentrations of either ATP or caffeine. Similar to previous findings, we observed that ATP induces a dose-dependent decrease in antibody binding to the COOH terminus (9, 17). Interestingly, this effect is also seen with caffeine, suggesting that caffeine binding to GLUT1 promotes similar GLUT1 conformational changes in (Fig. 4). Neither the inclusion of 5–20 mM d-glucose nor 5–20 μM CB resulted in significant perturbation of C-Ab binding (Fig. 4). Caffeine and ATP have no effect on COOH-terminal antibody recognition by HRP-conjugated secondary antibody over the concentrations of ligand used (data not shown).
Fig. 4.

Caffeine and ATP promote conformational change in the GLUT1 COOH terminus. Ordinate: COOH-terminal antibody (C-Ab) binding to GLUT1 measured by ELISA (relative antibody binding, %); abscissa: C-Ab binding conditions. C-Ab binding to unsealed GLUT1 proteoliposomes was measured in the presence and absence of ATP (5–20 mM, gray bars), caffeine (5–20 mM, diagonal bars), d-glucose (d-Glc, 5–20 mM, black bars), or CB (5–20 μM, hatched bars). The numbers below the chart indicate the concentration of ligand present during the binding assay. Results are shown as means ± SE of 3–16 measurements made in duplicate. *Unpaired, 1-tailed t-test analysis indicates that the results are significantly less than that observed in control antibody binding (P ≤ 0.0027).
Molecular docking analysis.
We undertook a docking analysis of caffeine, ATP, and CB binding to the recently published structure of human GLUT1 (28). Several putative binding sites are obtained for all three ligands. Figure 5 summarizes ATP, caffeine, and CB binding at their highest affinity sites in GLUT1. While these studies are in silico and require biochemical verification, a number of points are worthy of comment. 1) The ATP- and caffeine-binding sites persist in both the e1 conformation of GLUT1 and in an e2 conformation obtained by homology modeling GLUT1 on the XylE e2 conformation but the CB-binding site is lost in the e2 conformation (data not shown). This is consistent with the observation that caffeine and ATP are uncompetitive with respect to sugar uptake but CB is a noncompetitive inhibitor of sugar uptake and a competitive inhibitor of sugar exit (6, 16). 2) ATP has previously been demonstrated to interact with GLUT1 transmembrane helices (TMs) 8 and 9 (44). 3) These highest affinity docking sites suggest that ATP- and caffeine-binding sites overlap, that caffeine- and CB-binding sites overlap but that ATP- and CB-binding sites do not overlap. These predictions are consistent with the ligand-binding studies reported here (Fig. 3 and Table 1).
Fig. 5.

Docking analysis of caffeine, ATP, and CB binding to GLUT1. A: GLUT1 crystal structure (28) showing ATP, caffeine, and CB located in their respective, highest affinity docking sites. The images were generated in PyMol and show GLUT1 in cartoon representation. ATP (red), caffeine (blue), and CB (yellow) are shown as space-filling representations. GLUT1 transmembrane helix (TM) 5 is eliminated to reveal the putative ligand binding sites. TMs 8 and 9, which have previously been demonstrated to be the site of azidoATP photo-incorporation (44), are indicated in red. TMs 1, 3, 6, 8, 9, and 10 are numbered. The approximate margins of the lipid bilayer are indicated by the horizontal lines. Residues coordinating ATP binding (and their TM locations) are: F26 (TM1), Q161 (TM5), Q282 and Q283 (TM7), E380 and P385 (TM10), N411 and N415 (TM11). The computed ΔG for ATP binding is −8.5 kcal/mol. Residues coordinating caffeine binding (and their TM locations) are: Q161 (TM5) and P385 (TM10). The computed ΔG for ATP binding is −5.7 kcal/mol. Residues coordinating CB binding (and their TM and loop locations) are: V83 (TM2), M142 (TM4), W388, F389 and A392 (TM10 and loop 10–11), I404 and G408 (TM 11). The computed ΔG for CB binding is −10.3 kcal/mol. B: space-filling models of ATP (red) and caffeine (blue) and their corresponding stick models docked in their respective GLUT1-binding sites. C: space-filling models of caffeine (blue) and CB (yellow) and their corresponding stick models docked in their respective GLUT1-binding sites. D: space-filling models of ATP (red) and CB (yellow) and their corresponding stick models docked in their respective GLUT1-binding sites.
DISCUSSION
GLUT1 caffeine-binding site.
This study demonstrates that caffeine and TNP-ATP compete for binding to isolated, human GLUT1. Several mechanisms could explain this. 1) Caffeine and ATP share identical or overlapping binding sites. 2) Caffeine binds at an endo- or exofacial site that is physically distinct from the ATP-binding site but where ATP or caffeine binding promotes a conformational change in the competing ligand's binding site resulting in an indirect competition. Caffeine is also a competitive inhibitor of cytochalasin B binding to GLUT1 (52). We too observe caffeine inhibition of GLUT1 CB binding but find that cytochalasin B and ATP binding are not mutually exclusive. If caffeine inhibition of ATP and cytochalasin B binding results from steric overlap, this suggests that the caffeine-binding site must bridge the otherwise independent endofacial ATP- and cytochalasin B-binding sites.
Ojeda and colleagues (52) have previously demonstrated that caffeine is an uncompetitive inhibitor of GLUT1-mediated sugar uptake, a noncompetitive inhibitor of sugar exit into sugar-free medium, and a noncompetitive inhibitor of GLUT1-mediated equilibrium exchange sugar transport (52). The absence of competitive inhibition establishes that caffeine does not bind at the endo- or exofacial sugar-binding sites of GLUT1 (16). One curious observation made by Ojeda and colleagues is that the methylxanthines caffeine and theophylline decrease Vmax for infinite-cis sugar exit (efflux of sugar from cells containing saturating [sugar] into media containing varying [sugar]) without affecting the affinity of the external sugar-binding site for sugar. However, pentoxifylline (a methylxanthine containing a 5-oxohexyl group in place of a methyl group at position 1 of the purine) reduces Vmax for infinite-cis exit but increases Km(app) for sugar interaction with the exofacial sugar-binding site (52). These results were interpreted to be consistent with an exofacial methylxanthine-binding site independent from the substrate-binding site. Kinetic experiments of this type, however, do not permit unambiguous interpretation of site or sidedness of action (16, 29, 56). While definitive characterization of the GLUT1 methylxanthine-binding site(s) awaits either direct labeling studies or mutagenesis of putative interaction domains, molecular docking studies can provide some insights.
Molecular docking studies suggest that the so-called “e1” conformation of GLUT1 (28) binds cytochalasin B, ATP, and caffeine in decreasing order of affinity. Although several permutations of binding sites were computed for each ligand, the highest affinity binding sites are consistent with the transport and ligand-binding studies presented here and with previous biochemical analyses of ATP and CB binding to GLUT1. ATP is predicted to interact with TMs 8 and 9, a GLUT1 region previously demonstrated to become covalently labeled by azido-ATP (44). CB is predicted to interact with the cytoplasmic loops of GLUT1 and especially with those extending between TM10 and 11, a region previously demonstrated to be essential for CB binding (39). The predicted ATP- and caffeine-binding sites persist in a GLUT1 “e2” conformation obtained by homology modeling GLUT1 on the XylE e2 conformation (57), but the CB-binding site is lost in the e2 conformation (data not shown). These docking studies suggest that sites occupied by the purine groups of ATP and caffeine overlap, that the methyl group at position 1 of caffeine sterically hinders binding of the benzene ring of CB but that ATP and CB-binding sites do not overlap—predictions consistent with the ligand-binding studies reported here (Fig. 3 and Table 2). Docking studies also suggest that the purine group of pentoxifylline overlays the space occupied by the ribose moiety of GLUT1-liganded ATP (not the purine-binding site occupied by the adenine moiety of ATP nor the purine of caffeine) and extends its 5-oxohexyl group into space occupied by the triphosphate group of ATP or into a hydrophobic pocket between TMs 7, 8, and 10 (data not shown). This supports the hypothesis that pentoxifylline binding to GLUT1 may not involve the GLUT1 purine-binding site and thus provides a rationale for the different effects of caffeine and pentoxyfilline on GLUT1-mediated sugar transport observed by Ojeda and colleagues (52).
Table 2.
Effects of caffeine and ATP on [3H]cytochalasin B binding to GLUT1 proteoliposomes
| Binding Condition | CBbound/CBfree | P |
|---|---|---|
| Control | 0.71 ± 0.03 | |
| Caffeine 1 mM | 0.60 ± 0.01 | 0.0488 |
| Caffeine 5 mM | 0.54 ± 0.01 | 0.0015 |
| Caffeine 20 mM | 0.39 ± 0.02 | 0.000035 |
| ATP 5 mM | 0.76 ± 0.03 | 0.1749 |
| Cytochalasin B 20 μM | 0.24 ± 0.04 | 0.000003 |
Values are means ± SE for a minimum of 3 measurements made in duplicate. Equilibrium binding of 100 nM [3H]cytochalasin B ([3H]CB) to unsealed glucose transporter 1 (GLUT1) proteoliposomes was measured in the absence (control) or presence of caffeine (1 to 20 mM), ATP (5 mM), or unlabeled cytochalasin B (20 μM). Binding was measured as the ratio of bound to free [3H]CB. The significance of the binding assay result was computed by a one-tailed, unpaired t-test and is shown as the probability that binding in the presence of the ligand is identical to binding in the absence of ligand (control).
GLUT1 structural changes induced by caffeine binding.
ATP binding promotes GLUT1 conformational changes involving the cytoplasmic COOH terminus and loop 6–7 (9, 17). The structural similarities between adenosine and caffeine, and the observation that caffeine binding also occludes the GLUT1 COOH terminus in a dose-dependent manner, suggests that caffeine and ATP binding to GLUT1 promote similar structural changes.
ATP binding drives transport inhibition by bringing the COOH terminus of GLUT1 in close proximity to the COOH-terminal half of intracellular loop 6–7 (9). Quench flow experiments in erythrocytes indicate that rapid translocation of sugar through the transport pore is unaffected by ATP, however, the release of sugar into the cytosol is significantly slowed (8). It has been suggested, therefore, that the GLUT1 COOH terminus and loop 6–7 form a cytosolic “cage,” trapping the substrate in the transport channel thereby preventing its release. A similar, “ball and chain,” mechanism mediates Na and K channel inactivation (2, 49). The trapped sugar has a greater probability of reentering the translocation pore thereby promoting its return to the extracellular environment and thus causing the apparent saturation of sugar uptake at lower extracellular sugar concentrations (15). The net effect (reduced Vmax and Km) resembles uncompetitive inhibition in which the inhibitor stabilizes the enzyme-substrate complex preventing catalysis and product release (transport).
Given these considerations, we propose that caffeine inhibition of GLUT1-mediated sugar transport mirrors the mechanism of action of ATP on transport. In this model, caffeine binding at a cytoplasmic domain of the transporter leads to a stabilized interaction between the GLUT1 COOH terminus and loop 6–7. This interaction results in a physical barrier preventing sugar release into the cell and, therefore, decreased net sugar uptake. It should be emphasized that this explanation of transport inhibition by caffeine and ATP applies equally well to the simple (alternating conformer) carrier model for sugar transport, which presents only one sugar-binding site (exofacial or endofacial) at any instant (41, 60), and to the multisite carrier model in which the transporter presents exofacial, endofacial, and cavity (occluded) sugar-binding sites simultaneously (4, 5, 15, 23). The multisite carrier model is consistent with experimental observations showing that glucose becomes physically occluded within GLUT1 only in the presence of cytochalasin B (8) and that occupancy of an internal ligand-binding site can exert a negative or positive allosteric effect on ligand binding to an external site and vice versa depending on the nature of the ligands that bind (16, 20, 37). In sum, the effects of caffeine on sugar transport support previous demonstrations that sugar binding at exofacial domains may be subject to modification by occupancy of the carrier by endofacial ligands.
Implications for GLUT1 inhibition by caffeine.
Our studies and those of previous groups indicate that membrane resident GLUT1 is profoundly inhibited by caffeine. This effect is observed at high concentrations with half-maximal inhibition seen at 2.5–3.5 mM caffeine. Serum caffeine levels typically measured in humans are ∼6 μM (14), suggesting that the sugar transport inhibition resulting from normal consumption of caffeine is <1%.
Glucose transport across the endothelial cells of the blood-brain barrier into the brain is also catalyzed by GLUT1 (55). GLUT1 mutation can cause impaired glucose transport across the blood-brain barrier, resulting in severe neurological disorders (38). Currently, the clinical recommendation for such individuals is to avoid the consumption of caffeine due to a possible exacerbation of glucose transport deficiency (11). In light of the results presented here and the absence of clear clinical evidence, it seems unlikely that normal caffeine consumption (≤300 mg/day; Ref. 46) could give rise to significant inhibition of GLUT1-mediated glucose transport unless caffeine or its metabolites are concentrated in GLUT1-expressing membranes. Caffeine also enhances Ca release via the sarcoplasmic reticulum ryanodine receptor (63) and inhibits cAMP-phosphodiesterase (12). Thus it is possible that blood-brain barrier glucose transport could be impacted via elevated cytoplasmic Ca or cAMP levels. However, the IC50 for caffeine modulation of these off-target effects is >1 mM, suggesting again that normal caffeine consumption is unlikely to inhibit sugar transport via these pathways. The arousal effects of caffeine appear to be mediated by high affinity antagonism at adenosine A2A receptors present in specific areas of the CNS (26, 42). It is possible, therefore, that increased neuronal activity resulting from CNS arousal could exacerbate demand for an already limited glucose supply in GLUT1 deficiency.
Conclusions.
ATP and caffeine compete for binding to the endofacial, GLUT1 nucleotide-binding site and act as uncompetitive inhibitors of GLUT1. The action of caffeine on glucose transport is half-maximal at concentrations at least 2 orders of magnitude greater than those produced by normal consumption of caffeine.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-44888 and DK-36018.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.S., A.J.C., K.P.L., and A.C. conception and design of research; J.M.S., A.J.C., and K.P.L. performed experiments; J.M.S., K.P.L., and A.C. analyzed data; J.M.S., K.P.L., and A.C. interpreted results of experiments; J.M.S., K.P.L., and A.C. prepared figures; J.M.S., A.J.C., and K.P.L. drafted manuscript; J.M.S., A.J.C., K.P.L., and A.C. edited and revised manuscript; J.M.S., A.J.C., K.P.L., and A.C. approved final version of manuscript.
Footnotes
This article is the topic of an Editorial Focus by Richard J. Naftalin (51a).
REFERENCES
- 1.Afzal I, Cunningham P, Naftalin RJ. Interactions of ATP, oestradiol, genistein and the anti-oestrogens, faslodex (ICI 182780) and tamoxifen, with the human erythrocyte glucose transporter, GLUT1. Biochem J 365: 707–719, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol 70: 567–590, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayala FR, Rocha RM, Carvalho KC, Carvalho AL, da Cunha IW, Lourenço SV, Soares FA. GLUT1 and GLUT3 as potential prognostic markers for Oral Squamous Cell Carcinoma. Molecules 15: 2374–2387, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker GF, Widdas WF. The asymmetry of the facilitated transfer system for hexoses in human red cells and the simple kinetics of a two component model. J Physiol 231: 143–165, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker GF, Naftalin RJ. Evidence of multiple operational affinities for d-glucose inside the human erythrocyte membrane. Biochim Biophys Acta 550: 474–484, 1979. [DOI] [PubMed] [Google Scholar]
- 6.Basketter DA, Widdas WF. Asymmetry of the hexose transfer system in human erythrocytes. Comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J Physiol 278: 389–401, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker C, Sevilla L, Tomas E, Palacin M, Zorzano A, Fischer Y. The endosomal compartment is an insulin-sensitive recruitment site for GLUT4 and GLUT1 glucose transporters in cardiac myocytes. Endocrinology 142: 5267–5276, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Blodgett DM, Carruthers A. Quench-flow analysis reveals multiple phases of GluT1-mediated sugar transport. Biochemistry 44: 2650–2660, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Blodgett DM, De Zutter JK, Levine KB, Karim P, Carruthers A. Structural basis of GLUT1 inhibition by cytoplasmic ATP. J Gen Physiol 130: 157–168, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boado RJ, Pardridge WM. Glucose deprivation and hypoxia increase the expression of the GLUT1 glucose transporter via a specific mRNA cis-acting regulatory element. J Neurochem 80: 552–554, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Brockmann K. Towards a more palatable treatment for Glut1 deficiency syndrome. Dev Med Child Neurol 53: 580–581, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Butcher RW, Sutherland EW. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J Biol Chem 237: 1244–1250, 1962. [PubMed] [Google Scholar]
- 13.Cantuaria G, Fagotti A, Ferrandina G, Magalhaes A, Nadji M, Angioli R, Penalver M, Mancuso S, Scambia G. GLUT-1 expression in ovarian carcinoma. Cancer 92: 1144–1150, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Cao C, Loewenstein DA, Lin X, Zhang C, Wang L, Duara R, Wu Y, Giannini A, Bai G, Cai J, Greig M, Schofield E, Ashok R, Small B, Potter H, Arendash GW. High blood caffeine levels in MCI linked to lack of progression to dementia. J Alzheimers Dis 30: 559–572, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carruthers A. Mechanisms for the facilitated diffusion of substrates across cell membranes. Biochemistry 30: 3898–3906, 1991. [DOI] [PubMed] [Google Scholar]
- 16.Carruthers A, Helgerson A. Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane. Evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry 30: 3907–3915, 1991. [DOI] [PubMed] [Google Scholar]
- 17.Carruthers A, Helgerson AL. The human erythrocyte sugar transporter is also a nucleotide binding protein. Biochemistry 28: 8337–8346, 1989. [DOI] [PubMed] [Google Scholar]
- 18.Challiss JR, Taylor LP, Holman GD. Sugar transport asymmetry in human erythrocytes–the effect of bulk haemoglobin removal and the addition of methylxanthines. Biochim Biophys Acta 602: 155–166, 1980. [DOI] [PubMed] [Google Scholar]
- 19.Clark AE, Holman GD, Kozka IJ. Determination of the rates of appearance and loss of glucose transporters at the cell surface of rat adipose cells. Biochem J 278: 235–241, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cloherty EK, Levine KB, Carruthers A. The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry 40: 15549–15561, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Cloherty EK, Levine KB, Graybill C, Carruthers A. Cooperative nucleotide binding to the human erythrocyte sugar transporter. Biochemistry 41: 12639–12651, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Craik JD, Young JD, Cheeseman CI. GLUT-1 mediation of rapid glucose transport in dolphin (Tursiops truncatus) red blood cells. Am J Physiol Regul Integr Comp Physiol 274: R112–R119, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham P, Naftalin RJ. Reptation-induced coalescence of tunnels and cavities in Escherichia Coli XylE transporter conformers accounts for facilitated diffusion. J Membr Biol 247: 1161–1179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cura AJ, Carruthers A. Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr Physiol 2: 863–91439, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cura AJ, Carruthers A. AMP kinase regulation of sugar transport in brain capillary endothelial cells during acute metabolic stress. Am J Physiol Cell Physiol 303: C808–C814, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daly JW, Butts-Lamb P, Padgett W. Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell Mol Neurobiol 3: 69–80, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Vivo DC, Leaiy L, Wang D. Glucose transporter 1 deficiency syndrome and other glydolytic defects. J Child Neurol 17, Suppl 3: 3S15–3S23, 2002. [PubMed] [Google Scholar]
- 28.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Crystal structure of the human glucose transporter GLUT1. Nature 510: 121–125, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Deves R, Krupka RM. Testing transport systems for competition between pairs of reversible inhibitors. J Biol Chem 255: 11870–11874, 1980. [PubMed] [Google Scholar]
- 30.el-Barbary A, Fenstermacher JD, Haspel HC. Barbiturate inhibition of GLUT-1 mediated hexose transport in human erythrocytes exhibits substrate dependence for equilibrium exchange but not unidirectional sugar flux. Biochemistry 35: 15222–15227, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta 1805: 141–152, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorga FR, Lienhard GE. Changes in the intrinsic fluorescence of the human erythrocyte monosaccharide transporter upon ligand binding. Biochemistry 21: 1905–1908, 1982. [DOI] [PubMed] [Google Scholar]
- 33.Harik SI, Behmand RA, LaManna JC. Hypoxia increases glucose transport at blood-brain barrier in rats. J Appl Physiol 77: 896–901, 1994. [DOI] [PubMed] [Google Scholar]
- 34.Harland BF. Caffeine and nutrition. Nutrition 16: 522–526, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Heard KS, Fidyk N, Carruthers A. ATP-dependent substrate occlusion by the human erythrocyte sugar transporter. Biochemistry 39: 3005–3014, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Hebert DN, Carruthers A. Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J Biol Chem 267: 23829–23838, 1992. [PubMed] [Google Scholar]
- 37.Helgerson AL, Carruthers A. Equilibrium ligand binding to the human erythrocyte sugar transporter. Evidence for two sugar-binding sites per carrier. J Biol Chem 262: 5464–5475, 1987. [PubMed] [Google Scholar]
- 38.Ho YY, Yang H, Klepper J, Fischbarg J, Wang D, De Vivo DC. Glucose transporter type 1 deficiency syndrome (Glut1DS): methylxanthines potentiate GLUT1 haploinsufficiency in vitro. Pediatr Res 50: 254–260, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Inukai K, Asano T, Katagiri H, Anai M, Funaki M, Ishihara H, Tsukuda K, Kikuchi M, Yazaki Y, Oka Y. Replacement of both tryptophan residues at 388 and 412 completely abolished cytochalasin B photolabelling of the GLUT1 glucose transporter. Biochem J 302: 355–361, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52: 1757–1768, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jardetzky O. Simple allosteric model for membrane pumps. Nature 211: 969–970, 1966. [DOI] [PubMed] [Google Scholar]
- 42.Lazarus M, Shen HY, Cherasse Y, Qu WM, Huang ZL, Bass CE, Winsky-Sommerer R, Semba K, Fredholm BB, Boison D, Hayaishi O, Urade Y, Chen JF. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci 31: 10067–10075, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine KB, Cloherty EK, Fidyk NJ, Carruthers A. Structural and physiologic determinants of human erythrocyte sugar transport regulation by adenosine triphosphate. Biochemistry 37: 12221–12232, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Levine KB, Cloherty EK, Hamill S, Carruthers A. Molecular determinants of sugar transport regulation by ATP. Biochemistry 41: 12629–12638, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Levine KB, Hamill S, Cloherty EK, Carruthers AL. Alanine scanning mutagenesis of the human erythrocyte glucose transporter putative ATP binding domain. Blood Cells Mol Dis 27: 139–142, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Mandel HG. Update on caffeine consumption, disposition and action. Food Chem Toxicol 40: 1231–1234, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Mann GE, Yudilevich DL, Sobrevia L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol Rev 83: 183–252, 2003. [DOI] [PubMed] [Google Scholar]
- 48.McCall AL, Fixman LB, Fleming N, Tornheim K, Chick W, Ruderman NB. Chronic hypoglycemia increases brain glucose transport. Am J Physiol Endocrinol Metab 251: E442–E447, 1986. [DOI] [PubMed] [Google Scholar]
- 49.Miller C. 1990: annus mirabilis of potassium channels. Science 252: 1092–1096, 1991. [DOI] [PubMed] [Google Scholar]
- 50.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naftalin RJ, Afzal I, Cunningham P, Halai M, Ross C, Salleh N, Milligan SR. Interactions of androgens, green tea catechins and the antiandrogen flutamide with the external glucose-binding site of the human erythrocyte glucose transporter GLUT1. Br J Pharmacol 140: 487–499, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51a.Naftalin RJ. Definitively, my cup of tea. Focus on “Caffeine inhibits glucose transport by binding at the GLUT1 nucleotide-binding site.” Am J Physiol Cell Physiol. doi: 10.1152/ajpcell.00083.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ojeda P, Perez A, Ojeda L, Vargas-Uribe M, Rivas CI, Salas M, Vera JC, Reyes AM. Noncompetitive blocking of human GLUT1 hexose transporter by methylxanthines reveals an exofacial regulatory binding site. Am J Physiol Cell Physiol 303: C530–C539, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Sage JM, Carruthers A. Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am J Physiol Cell Physiol 306: C910–C917, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model 17: 57–61, 1999. [PubMed] [Google Scholar]
- 55.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27: 1766–1791, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein WD. Transport, and Diffusion Across Cell Membranes. New York: Academic, 1986. [DOI] [PubMed] [Google Scholar]
- 57.Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 490: 361–366, 2012. [DOI] [PubMed] [Google Scholar]
- 58.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vannucci SJ, Reinhart R, Maher F, Bondy CA, Lee WH, Vannucci RC, Simpson IA. Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res 107: 255–264, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Widdas WF. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J Physiol 118: 23–39, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18: 1437–1446, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zoccoli MA, Baldwin SA, Lienhard GE. The monosaccharide transport system of the human erythrocyte. Solubilization and characterization on the basis of cytochalasin B binding. J Biol Chem 253: 6923–6930, 1978. [PubMed] [Google Scholar]
- 63.Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 49: 1–51, 1997. [PubMed] [Google Scholar]