

Abstract
The present study was carried out to investigate if hepatic nuclear factor (HNF)4α contributed to the high glucose-induced increase in stromal interacting molecule (STIM)1 protein abundance in glomerular mesangial cells (MCs). Western blot and immunofluorescence experiments showed HNF4α expression in MCs. Knockdown of HNF4α using a small interfering RNA approach significantly increased mRNA expression levels of both STIM1 and Orai1 and protein expression levels of STIM1 in cultured human MCs. Consistently, overexpression of HNF4α reduced expressed STIM1 protein expression in human embryonic kidney-293 cells. Furthermore, high glucose treatment did not significantly change the abundance of HNF4α protein in MCs but significantly attenuated HNF4α binding activity to the Stim1 promoter. Moreover, knockdown of HNF4α significantly augmented store-operated Ca2+ entry, which is known to be gated by STIM1 and has recently been found to be antifibrotic in MCs. In agreement with those results, knockdown of HNF4α significantly attenuated the fibrotic response of high glucose. These results suggest that HNF4α negatively regulates STIM1 transcription in MCs. High glucose increases STIM1 expression levels by impairing HNF4α binding activity to the Stim1 promoter, which subsequently releases Stim1 transcription from HNF4α repression. Since the STIM1-gated store-operated Ca2+ entry pathway in MCs has an antifibrotic effect, inhibition of HNF4α in MCs might be a potential therapeutic option for diabetic kidney disease.
Keywords: hepatic nuclear factor 4α, stromal interacting molecule 1, store-operated Ca2+ entry, mesangial cells, high glucose, diabetic nephropathy
store-operated Ca2+ entry (SOCE) via store-operated Ca2+ channels (SOCs) is a ubiquitous signaling mechanism in both nonexcitable and excitable cells. This Ca2+ signaling regulates diverse cellular functions ranging from cell proliferation and gene expression to cell contraction and secretion (35). Although SOCE was originally described over two decades ago, its molecular mediators were unknown until recently. By high-throughput RNA inhibition screening, two protein families, stromal interacting molecule (STIM) (27, 36) and Orai (13, 48, 59), were identified as required components of SOCE. STIM1 is a single-pass transmembrane protein located primarily in the endoplasmic reticulum (ER) membrane and functions as an ER Ca2+ sensor to sense the ER luminal Ca2+ concentration. Orai1 is a small plasma membrane protein and is believed to be the pore-forming unit of SOCs. Upon depletion of ER Ca2+, STIM1 aggregates and translocates to ER-plasma membrane junctions, where it physically associates and subsequently activates Orai1 and causes Ca2+ entry into the cytosol (8, 50). Over the past several years, intensive attention has been focused on the gating mechanism of SOC and STIM1/Orai1 interactions (21, 33, 43, 44, 57). Studies on the regulation of SOCE and its molecular components at transcriptional level are extremely scarce. Recently, we found that prolonged high glucose (HG) treatment enhanced SOCE in glomerular mesangial cells (MCs) by increasing STIM1/Orai1 protein expression (1). We further demonstrated that HG upregulated STIM1 expression by promoting its transcription. However, the mechanism for HG-stimulated STIM1 transcription has not been defined.
Hepatocyte nuclear factor (HNF)4α is a tissue-specific transcription factor expressed mainly in the liver, kidney, and intestine and activates the transcription of many genes involved in glucose, fatty acid, and lipid metabolism (18, 20, 31, 41, 45). Mutations in the HNF4α gene in humans give rise to maturity-onset diabetes of the young (37). However, very little is known about the role of HNF4α in diabetes. A previous study by Xie et al. (54) reported that levels of HNF4α protein and mRNA were decreased in the liver of db/db mice (type II diabetes model) but increased in streptozotocin-treated mice (type I diabetes model). A recent study by Niehof and Borlak (32) showed that HNF4α protein expression was repressed in kidneys of both diabetic rats and humans and that the reduced HNF4α might contribute to diabetic nephropathy by inhibiting canonical transient receptor potential (TRP)C1 protein in the kidney (32). Since TRPC1 is a Ca2+-permeable channel expressed in kidney cells (10), the results of Niehof and Borlak's study suggest that HNF4α may affect diabetic renal changes by regulating renal cell Ca2+ signaling. STIM1-gated Ca2+ channels participate in the Ca2+ response in glomerular MCs (39, 42), and dysfunction of MCs contributes to the progression of diabetic kidney disease. Furthermore, the promoter region of Stim1 has a HNF4α-binding consensus, which was identified by the F-MATCH program (http://www.gene-regulation.com/cgi-bin/pub/programs/fmatch/ffa2.cgi). Therefore, the present study was carried out to investigate if STIM1 is a target of HNF4α and whether impairment of HNF4α-regulated STIM1 transcription is a mechanism for the HG-induced increase in STIM1 expression in MCs.
MATERIALS AND METHODS
Cell culture and transient transfection.
Human MCs were purchased from Lonza (Walkersville, MD) and cultured as described in our previous studies (1, 51). Cells were growth arrested with 0.5% FBS medium during treatments. Only subpassages of less than nine generations were used in this study. For experiments with HG treatment, MCs were incubated with culture medium containing 25 mM glucose for the time periods indicated. As a control, MCs were cultured in medium containing 5.6 mM glucose plus 20 mM mannitol [normal glucose (NG)] for the same time periods.
Human embryonic kidney (HEK)-293 cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured as previously described (16). HEK-293 cells were subcultured to no more than 50 generations.
All transfections were transient transfection. Small interfering (si)RNA oligonucleotides were transfected into MCs using DharmaFECT 2 transfection reagent (Thermo Scientific, Rockford, IL) following protocols provided by the manufacturer. Expression plasmids were transfected into HEK-293 cells using LipofectAmine and Plus reagent (Invitrogen-BRL, Carlsbad, CA) following protocols provided by the manufacturer. Experiments were conducted 48–72 h after transfection.
Plasmids.
pcDNA-HA-hHNF4α2, the expression plasmid of human HNF4α, was a generous gift from Dr. Fukamizu (University of Tsukuba, Tsukuba, Japan) (19, 20). Yellow fluorescent protein (YFP)-STIM expression plasmid (pDS_XB_YFP-STIM1-II) was a kind gift from Dr. Tobias Meyer (Stanford University School of Medicine) (27).
Quantitative real-time RT-PCR.
Total RNA was isolated from cultured human MCs using a PerfectPure RNA cultured cell kit (5 Prime, Hamburg, Germany) following the manufacturer's protocol. Human HNF4α2 primers (forward: 5′-GAGTGACATCCAGGAGGAATAAG-3′ and reverse: 5′-GGGAGGAAGGGAGGATTAAATG-3′), human STIM1 primers (forward: 5′-ACAGGGACTGTGCTGAAGATGACA-3′ and reverse: 5′-ACCAGCATGAAGTCCTTGAGGTGA-3′), and β-actin primers (forward: 5′-ACTGTGTGGATTACATGGGCCAGA-3′ and reverse: 5′-AGGATTGCCTCCACAATCCGTACA-3′) were synthesized by IDT (Coralville, IA). An iScript cDNA synthesis kit (Bio-Rad) was used for reverse transcription reactions with 1.0 μg total RNA in a final volume of 20 μl following the manufacturer's reaction protocol. Real-time PCR used 0.2 μg of reverse transcription product and 100 nM primer and was performed using iQ SYBR Green supermix (Bio-Rad) in a final volume of 20 μl. The PCR mix was denatured at 95°C for 10 min followed by 40 cycles of melting at 95°C for 15 s and 60°C for 1 min. After amplification, a melting curve analysis from 60 to 95°C with a heating rate of 0.02°C/s with a continuous fluorescence acquisition was made. The assay was run on a C1000TM Thermal Cycler (Bio-Rad). The average threshold cycle (Ct) of fluorescence units was used to analyze mRNA levels. STIM1 and HNF4α mRNA levels were normalized by β-actin mRNA. Quantification was calculated as follows: mRNA levels = 2ΔCt, where ΔCt = Ct,STIM1 or Ct,HNF4α − Ct,actin.
Preparation of nuclear extracts.
Preparation of nuclear extracts from human MCs was performed using a Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific) following the manufacturer's protocol. Extracts were stored at −80°C until use.
Isolation of glomeruli and extracting glomerular and liver proteins.
The study protocol was approved by the Institutional Animal Care and Use Committee of the University of North Texas Health Science Center. Male Sprague-Dawley rats at an age of 8–10 wk were purchased from Harlan (Indianapolis, IN). Kidneys and liver were removed after rats were anesthetized. Glomeruli were isolated as described in our previous publication (17). The isolated glomeruli and a piece of homogenized liver tissue (∼100 mg) were solubilized in lysis buffer. Samples were sonicated and centrifuged at 100,000 g for 45 min at 4°C. Supernatants were collected for Western blot analysis.
Western blot analysis.
Western blot analysis was performed as described in our previous publications (1, 51). Briefly, whole cell lysates or nuclear extracts were fractionated by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed with primary STIM1 or actin or lamin A/C or HNF4α antibody. Bound antibodies were visualized with Super Signal West Femto or Pico Luminol/Enhancer Solution (Thermo Scientific). The specific protein bands were visualized and captured using an AlphaEase FC Imaging System (Alpha Innotech, San Leandro, CA). The integrated density value of each band was measured by drawing a rectangle outlining the band using Alpha-Ease FC software with auto background subtraction. Expression levels of STIM1 and HNF4α proteins were quantified by normalization of the integrated density values of those protein bands to that of actin or lamin A/C bands on the same blot.
Immunofluorescent staining.
Kidney sections of 5 μm thickness from a normal rat from our previously published study (1) were fixed with 4% paraformaldehyde for 15 min at room temperature. After being washed with PBS, sections were incubated with ice-cold acetone at −20°C for 10 min. After 30 min of incubation with blocking buffer, sections were incubated with HNF4α and CD90/Thy 1.1 (OX-7) antibodies at 1:50 and 1:100, respectively, in PBS plus 10% donkey serum and 0.2% Triton X-100 at 4°C overnight. OX-7 is an antibody to Thy1.1, a MC-specific antigen, and was used to label MCs (9, 40, 46, 47). After three washes with PBS, sections were then incubated with donkey anti-goat secondary antibodies conjugated with Alexa fluor 568 (for HNF4α) or with donkey anti-rabbit fluor 488 (for OX-7) (Invitrogen) at a concentration of 1:500 for 1 h at room temperature. Sections were examined using an Olympus microscope (BX41) equipped for epifluorescence and an Olympus DP70 digital camera with DP manager software (version 2.2.1). Images were converted to 16-bit format and uniformly adjusted for brightness and contrast using ImageJ [version 1.47, National Institutes of Health (NIH)].
Fluorescent immunocytochemistry.
Human MCs were plated on 22 × 22.1-mm glass coverslips. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature. After being washed with PBS, cells were then incubated with ice-cold acetone at −20°C for 10 min. After 30 min of incubation with blocking buffer, cells were incubated with HNF4α primary antibody at 1:50 in PBS plus 10% donkey serum and 0.2% Triton X-100 at 4°C overnight. After three washes with PBS, cells were incubated with goat anti-rabbit secondary antibodies conjugated with Alexa fluor 568 (Invitrogen) at a concentration of 1:500 for 1 h at room temperature. 4′,6-Diamidino-2-phenylindole (Invitrogen) was used to stain nuclei. For a control, an equal amount of rabbit IgG was used instead of primary antibodies. Fluorescent staining was examined using an Olympus microscope (BX41) equipped for epifluorescence and an Olympus DP70 digital camera with DP manager software (version 2.2.1). Images were converted to 16-bit format and uniformly adjusted for brightness and contrast using ImageJ (version 1.47, NIH).
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assay was performed using a double-stranded oligonucleotide containing a consensus binding sequence for HNF4α (5′-CTCAGCTTGTACTTTGGTACAACTA-3′), where the underlined sequence represents the HNF4α-binding consensus. The nucleotides were end labeled with biotin using a biotin 3′-end DNA labeling kit (Thermo Scientific). Nuclear extracts (5 μg protein) were incubated in 1× binding buffer containing 2.5% glycerol, 50 ng poly(dI-dC), 5 mM MgCl2, 0.05% Nonidet P-40, and 4 pmol biotin-labeled oligonucleotide in a total volume of 20 μl at room temperature for 20 min. The reaction mixture was then subjected to electrophoresis in a 6% polyacrylamide gel using 0.5 Tris-borate-EDTA as the running buffer. For competition experiments, nuclear extracts were incubated with a 50-fold molar excess of unlabeled oligonucleotide. Transfer to a nylon membrane was conducted at 100 V for 30 min at 4°C. The membrane was then cross linked for 60 s in an ultraviolet cross-linker. The binding complexes were then developed using a Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific) following the manufacturer's protocol. HNF4α-specific bands were visualized and captured using an AlphaEase FC Imaging System (Alpha Innotech). The integrated density value of each band was measured by drawing a rectangle outlining the band using Alpha-Ease FC software with auto background subtraction.
Chromatin immunoprecipitation assay.
After MCs were treated with HG for 6 days, cells were cross linked by 1% formaldehyde for 10 min at room temperature. Glycine was then added at a final concentration of 0.125 M to neutralize formaldehyde. After two washes with PBS, cells were scraped and collected by centrifugation (750 g). Cells were then resuspended in lysis buffer 1 [50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5 Nonidet P-40, 0.25% Triton X-100, and a proteinase inhibitor mixture] and incubated at 4°C for 10 min. Nuclei were then isolated by centrifugation (1,350 g) and lysed in lysis buffer 2 [10 mM Tris·HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and a protease inhibitor mixture]. The pelleted chromatin was then resuspended in lysis buffer 3 [10 mM Tris·HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, and proteinase inhibitor]. After sonication, a 1/10th volume of 10% Triton X-100 was added to the lysates followed by centrifugation at 20,000 g at 4°C for 10 min, and the supernatants were incubated with 2 μg HNF4α antibody (sc-8987, Santa Cruz Biotechnology) or control rabbit IgG (Cell Signaling Technology, Danvers, MA) overnight at 4°C. After immunoprecipitation, samples were incubated with 25 μl magnetic protein A/G beads (Thermo Scientific) for 2 h at room temperature. The immune complexes were collected by Magnetic Stand and washed four times with radioimmune precipitation assay buffer (10 mM Tris·HCl, 0.25 M LiCl, 0.5% Nonidet P-40, and 0.5% sodium deoxycholate, pH 7.5) followed by two washes with Tris-EDTA buffer with 50 mM NaCl. Then, 50 μl of 10% Chelex 100 were added to the washed bead pellets, and samples were boiled for 10 min at 100°C. Pellets were then incubated with RNase for 1 h and proteinase K for 30 min at 55°C. Supernatants were collected after centrifugation at 20,000 g at room temperature and subjected to quantitative real-time PCR or regular PCR. A volume of 2 μl of immunoprecipitated DNA was analyzed by real-time PCR (25-μl reaction mixture) using iQ SYBR Green Supermix (Bio-Rad). The specificity of the assay was tested by amplification of the GAPDH gene not bound by HNF4α (negative control). Primers flanking HNF4α sites at the STIM1 promoter were 5′-CTGGGAGGCTAACGTCGTGT-3′ and 5′-TCCCTGACCCGCAGTTACTCA-3′, designed to amplify the 151-bp product. The primers for the negative control were 5′-ATGGTTGCCACTGGGGATCT-3′ and 5′-TGCCAAAGCCTAGGGGAAGA-3′, designed to amplify the 174-bp product. The negative control primers flank a region of genomic DNA between the GAPDH gene and chromosome condensation-related smooth muscle cell-associated protein (CNAP)1 gene (55). A volume of 2 μl of the immunoprecipitated chromatin from the cells with different treatments was analyzed by real-time PCR. The PCR was performed in duplicate using iQ SYBR Green Supermix reagents (Bio-Rad). Real-time PCR conditions for GAPDH were 95°C for 3 min and 40 cycles with 95°C for 15 s, 55°C for 30 s, and 72°C for 30 s, and real-time PCR conditions for STIM1 were 95°C for 15 s, 57°C for 30 s, and 72°C for 30 s. Melting curves were measured in the PCR machine between 60 and 95°C with a resolution of 0.5°C.
Fluorescence measurements of intracellular Ca2+ concentration.
Measurements of intracellular Ca2+ concentration ([Ca2+]i) in MCs using fura-2 were performed using dual-excitation wavelength fluorescence microscopy. MCs grown on a coverslip (22 × 22 mm) were loaded with 2 μM fura-2 AM plus 0.018 g/dl Pluronic F-127 (Invitrogen) for 50 min at room temperature followed by an additional 20 min of incubation in fura-2-free physiological saline solution. The coverslip was then placed in a perfusion chamber (model RC-2OH, Warner) mounted on the stage of a Nikon Diaphot inverted microscope. Fura-2 fluorescence was monitored at 340- and 380-nm excitation wavelengths and at 510-nm emission wavelength using NIS Elements AR software (Nikon Instruments) at room temperature. [Ca2+]i was calculated using the software following the manufacturer's instructions. Calibrations were performed at the end of each experiment, and conditions of high [Ca2+]i were achieved by the addition of 5 μM ionomycin, whereas conditions of low [Ca2+]i were obtained by the addition of 5 mM EGTA.
Materials and reagents.
siRNA oligonucleotides against human HNF4α and control scrambled siRNA were obtained from Origene (Rockville, MD). Cyclopiazonic acid was purchased from Alomone labs (Har Hotzvim Hi-Tech Park). Rabbit polyclonal anti-STIM1 antibody was purchased from ProteinTech (Chicago, IL). Rabbit polyclonal anti-HNF4α antibody (sc-8987) was used for immunohistochemistry, immunocytochemistry, and chromatin immunoprecipitation assay, and a goat polyclonal anti-HNF4α antibody (sc-6556) was used for Western blot analysis. Both antibodies were purchased from Santa Cruz Biotechnology. Rabbit anti-lamin A/C antibody was purchased from Biolegend (San Diego, CA). All other chemicals and antibodies were purchased from Sigma-Aldrich (St. Louis, MO) unless indicated in other places.
Statistical analysis.
Data are reported as means ± SE. One-way ANOVA plus Student-Newman-Keuls post hoc analysis and Student's unpaired t-test were used to analyze differences among multiple groups and between two groups, respectively. P values of <0.05 were considered statistically significant. Statistical analysis was performed using SigmaStat (version 3.0, Jandel Scientific, San Rafael, CA).
RESULTS
Expression of HNF4α in glomerular MCs.
HNF4α expression is tissue and cell type specific (18, 19, 31, 41, 45). To determine if HNF4α was expressed in MCs, we performed immunocytochemistry and Western blot analysis using cultured human MCs. As shown in Fig. 1A, HNF4α was localized in the nucleus of MCs. Western blot analysis of MC lysates showed a HNF4α-specific band comparable with the HNF4α band in extracts of the rat liver, an organ with high HNF4α expression (Fig. 1B, left) (18, 19, 23). Furthermore, immunoblot analysis of rat glomerular extracts (MCs are located in the glomerulus) also showed a band that had the same molecular weight as HNF4α in MCs (Fig. 1B, right). Immunofluorescent staining revealed a broad distribution of HNF4α in the kidney, in both the glomerulus and tubule regions (Fig. 1C). Further examination showed a good colocalization of HNF4α signals with staining of OX-7, which is an antibody recognizing Thy1.1, a MC-specific antigen (Fig. 1, C and D) (9, 40, 46, 47), suggesting MC localization of HNF4α. These data indicate that HNF4α is present in glomerular MCs.
Fig. 1.

Expression of hepatic nuclear factor (HNF)4α in glomerular mesangial cells (MCs). A: immunocytochemistry showing nuclear localization of HNF4α (red signals) in cultured human MCs. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue signals). B: Western blots showing HNF4α protein expression in human MCs and rat glomeruli. Mouse liver tissue extracts were used as a positive control. Actin served as a loading control. C–F: immuofluorescence staining showing HNF4α expression (C; red signals) in both glomerular MCs and tubule regions in a rat kidney. Glomeruli are indicated by arrows. MCs were labeled with OX-7 antibody (D; green signals). E: overlapping HNF4α and OX-7 signals are indicated by yellow signals. F: enlarged image inside the dashed box in E. The yellow signals indicate colocalization of HNF4α with OX-7. Original magnification: ×100.
Knockdown of HNF4α increased STIM1, Orai1 mRNA, and STIM1 protein expressions in cultured human MCs.
Like HNF4α, STIM1 and Orai1 are also expressed in MCs, as demonstrated by our previous studies (1, 39) using both in vitro and in vivo assays. To determine if HNF4α regulated STIM1 and Orai1 expression, we knocked down HNF4α expression using siRNA against human HNF4α (siHNF4α) in human MCs. Compared with MCs without siRNA treatment and with scrambled siRNA treatment, cells treated with siHNF4α showed a significantly low level of HNF4α mRNA (Fig. 2A). To verify the knocking down effect of siHNF4α on HNF4α protein, we overexpressed HNF4α protein by transfection of HNF4α expression plasmid (pcDNA3-HA-hHNF4α2) to HEK-293 cells. As shown in Fig. 2, B and C, siHNF4α significantly reduced both transfected and endogenous HNF4α protein in HEK-293 cells.
Fig. 2.

Effect of knocking down HNF-4α on stromal interacting molecule (STIM)1 and Orai1 expression levels in human MCs. A, D, and E: quantitative real-time RT-PCR showing mRNA expression levels of HNF4α (A), STIM1 (D), and Orai1 (E) in cultured human MCs without transfection (Untran) and transfected with scrambled small interfering (si)RNA (Scr) or siRNA against human HNF4α (siHNF4α). Cells were harvested 3 days after transfection. Expression levels of HNF4α and STIM1 mRNA in Scr and siHNF4α cells were normalized to Untran cells. †P < 0.05 compared with both Untran and Scr. n is the number of independent experiments. B and C: Western blots in human embryonic kidney (HEK)-293 cells showing HNF4α protein expression in HEK-293 cells without transfection (Untran), transfected with HNF4α expression plasmid (HNF4α), and cotransfected with HNF4α expression plasmid with siHNF4α (HNF4α + siHNF4α). B: representative blot. Actin was used as a loading control. Both transfected HNF4α (Ex-HNF4α) and endogenous HNF4α (En-HNF4α) are indicated by arrows. C: summary data. HNF4α expression levels in HNF4α and HNF4α + siHNF4α cells were normalized to Untran cells. †P < 0.05 compared with both Untran and HNF4α + siHNF4α cells. n is the number of independent experiments. F–H: Western blot analysis showing STIM1 (F and G) and Orai1 (H) protein expression in human MCs without transfection (Untran) or transfected with scrambled siRNA (Scr) or siRNA against human HNF4α (siHNF4α). F: representative Western blot of STIM1. Actin was used as a loading control. G and H: summary data. STIM1 and Orai1 expression levels in Scr and siHNF4α cells were normalized to Untran cells. †P < 0.05 compared with both Untran and Scr cells. n is the number of independent experiments.
Corresponding to a decrease in HNF4α mRNA, the abundance of both STIM1 and Orai1 mRNA increased significantly in siHNF4α-treated MCs (Fig. 2, D and E). Furthermore, knocking down HNF4α significantly increased abundance of STIM1 protein (Fig. 2, F and G). The Orai1 protein expression level had tendency to increase in cells treated with HNF4α siRNA, but this response did not reach a statistically significant level (Fig. 2H). In summary, these data suggest that HNF4α negatively regulates STIM1 and Orai1 expressions at the transcriptional level. Because HG treatment significantly promoted STIM1 but not Orai1 mRNA expression (1), we decided to focus on the regulation of HNF4α on STIM1 transcription in the following experiments to explore the mechanism for HG-stimulated STIM1 transcription.
Overexpression of HNF4α decreased STIM1 protein expression in HEK-293 cells.
If HNF4α represses STIM1 expression, then overexpression of HNF4α would be expected to reduce STIM1 protein expression. This hypothesis was tested in HEK-293 cells cotransfected with the human STIM1 expression plasmid (pDS_XB_YFP-STIM1) and the human HNF4α expression plasmid (pcDNA3-HA-hHNF4α2). As shown in Fig. 3, the expression level of transfected STIM1 (YFP tagged) was markedly increased in MCs transfected with pDS_XB_YFP-STIM1. However, cotransfection of HNF4α significantly reduced the exogenous STIM1 compared with cells cotransfected with pcDNA3, a control vector. These data are consistent with the results of knocking down HNF4α and further support a negative regulation of STIM expression by HNF4α.
Fig. 3.
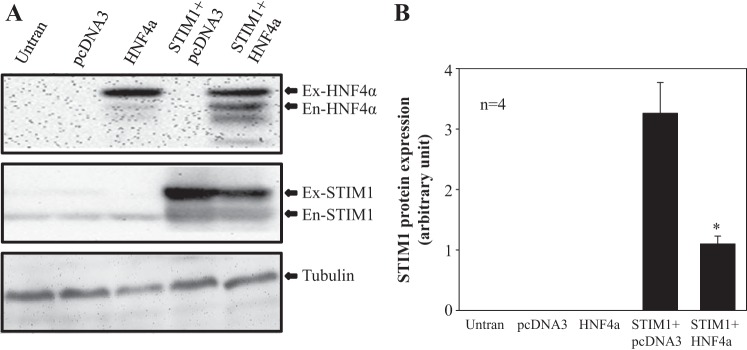
Effect of overexpression of HNF-4α on STIM1 expression in HEK-293 cells. A: representative experiment showing STIM1 expression (middle) in HEK-293 cells without transfection (Untran) or transfected with vector pcDNA3 (pcDNA3), HNF4α expression plasmid (HNF4α), STIM1 expression plasmid plus pcDNA3 vector (STIM1 + pcDNA3), and STIM1 expression plasmid plus HNF4α expression plasmid (STIM1 + HNF4α). HNF4α expression in the five groups of cells is shown in the top. Tubulin (TB; bottom) was used as a loading control. Ex-HNF4α, exogenous HNF4α; En-HNF4α, endogenous HNF4α; Ex-STIM1, exogenous STIM1; En-STIM1, endogenous STIM1. B: summary data averaged from four independent experiments shown in A (n = 4). Ex-STIM1 protein expression levels were normalized to TB. *P < 0.05 compared with STIM1 + pcDNA3.
HG did not alter HNF4α protein expression in human MCs.
We have previously demonstrated that HG treatment increased STIM1 expression at the transcriptional level in human MCs (1). To determine if HNF4α played a role in the HG response, we conducted Western blot analysis and examined time course- and dose-dependent effects of HG on HNF4α expression. MCs treated with different concentrations of glucose for 3–4 days did not significantly change the expression level of HNF4α (Fig. 4, A and B). We chose this time period because the HG-induced increase of STIM1 protein required 6–7 days. If the HG effect was mediated by altering expression levels of HNF4α protein, HG treatment should change the abundance of HNF4α in a shorter time period (i.e., <6–7 days). Furthermore, there was no significant difference in HNF4α expression levels between NG (5.6 mM) and HG (25 mM) treatments for time periods from 1 to 7 days (Fig. 4, C and D). However, the STIM1 protein expression level was significantly increased by 7-day HG treatment, which is consistent with our previous report (1).
Fig. 4.

High glucose (HG) effect on HNF4 and STIM1 protein expression in cultured human MCs. A–D: Western blot analysis showing HNF4α protein expression in response to different concentrations of glucose (Glu; A and B) and different time periods of HG treatment (C and D). In A and B, whole cell lysates were used. MCs were cultured in 0.5% FBS medium containing different concentration of glucose for 3–4 days. Appropriate concentrations of α-mannitol (Man) were used as an osmotic control. In C and D, nuclear extracts were used. MCs were incubated in 0.5% FBS containing normal glucose (NG; 5.6 mM glucose + 20 mM mannitol) and HG (25 mM glucose) for 1 day (1D), 3 days (3D), and 7 days (7D). A and C: representative immunoblots. TB and lamin A/C were used as loading controls. B and D: summary data from experiments shown in A and C, respectively. HNF4α protein expression levels were normalized to TB (B) or lamin A/C (D) and then further normalized to either group of 5 mM glucose with 0 mM mannitol (B) or the NG group of each HG treatment time period (D). n is the number of independent experiments. E and F: time course effect of HG treatment on STIM1 protein expression (whole cell lysates). MCs were incubated in 0.5% FBS containing NG (5.6 mM glucose + 20 mM mannitol) and HG (25 mM glucose) for 1, 3, and 7 days. Actin served as a loading control. STIM1 protein expression levels were normalized to actin and then further normalized to the NG group of each HG treatment time period. *P < 0.05 compared with the NG group with 7-day treatment. n is the number of independent experiments.
HG treatment inhibited DNA binding activity of HNF4α.
We next examined if HG affected the DNA binding activity of HNF4α. Electrophoretic mobility shift assay was performed using a commercial probe specific for HNF4α binding. In this line of experiments, MCs were incubated with NG and HG medium for 6 days. This time period was selected because HG required 6–7 days to increase STIM1 protein expression (1). As shown in Fig. 5, nuclear extracts from NG-treated human MCs produced an intense HNF4α-DNA complex (Fig. 5A, lane 2). When MCs were treated with HG for 6 days, the formation of the complex was significantly inhibited (Fig. 5, A and B, lane 4). The HNF4α-DNA complex was dramatically reduced by a 50-fold excess of the same unlabeled HNF4α oligonucleotide in nuclear extracts from both NG- and HG-treated MCs (Fig. 5A, lanes 3 and 5), indicating that the complex formation was specific. These results in combination with the data shown in Fig. 4 suggest that HG inhibited HNF4α-DNA binding activity but not the expression level of HNF4α.
Fig. 5.

HG effect on DNA-binding activity of HNF4α in human MCs (electrophoretic mobility shift assay). A: representative experiment. Nuclear extracts were extracted from MCs treated with NG or HG for 6 days. Unlabeled oligonucleotides were 50-fold greater than labeled oligonucleotides. B: summary data from five independent experiments (n = 5) showing HNF4α expression levels in cells treated with NG and HG plus labeled probes (lanes 2 and 5 in A). The HNF4α expression level in HG + labeled probe-treated MCs was normalized to that in NG + labeled probe-treated MCs. *P < 0.05 vs. NG + labeled probe.
HG treatment inhibited HNF4α binding to the Stim1 promoter in human MCs.
Consistent with results from electrophoretic mobility shift assays, chromatin immunoprecipitation assays showed that HG treatment for 6 days inhibited HNF4α but not control IgG binding to the Stim1 promoter in human MCs (Fig. 6, A and B). As a negative control, HG treatment did not stimulate HNF4α binding to a region of genomic DNA between the GAPDH gene and CNAP1 gene, in which there is no transcription factor-binding site (Fig. 6, A and C). These results suggest that HG specifically inhibited HNF4α-Stim1 binding.
Fig. 6.
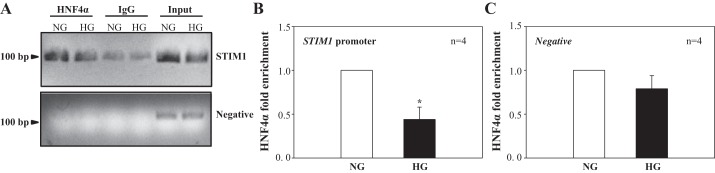
HG effect on HNF-4α binding to the STIM1 promoter region in human MCs. Cells were treated with NG and HG for 6 days, and chromatin immunoprecipitation analyses were performed using anti-HNF4 antibody. Promoter copy number was quantified by quantitative real-time PCR in duplicate using a specific primer that amplifies an HNF4-binding site in the STIM1 promoter. Normal rabbit IgG was used as the negative control for the specificity of immunoprecipitation. A 174-bp genomic region flanking the GAPDH gene and chromosome condensation-related smooth muscle cell-associated protein 1 gene, which does not have an HNF4α-binding site, was used as a negative control (Negative). As a positive control, aliquots (1/10th of immunoprecipitates) of chromatin fragments obtained before immunoprecipitation were also subjected to PCR analysis (Input). A: representative experiment showing HNF4-bound DNA fragments in 2% agarose gel. B and C: summary data from four independent experiments (n = 4) shown in A. Data are expressed as HNF4α enrichment by normalization of HNF4-STIM1 binding (B) or HNF4-negative control binding (C) to their corresponding input chromatins. *P < 0.05 compared with NG treatment.
Knockdown of HNF4α augmented SOCE in human MCs.
STIM1 is an essential component of the SOCE pathway by gating SOCs (27, 36). If HNF4α reduced abundance of STIM1 protein, then a decrease in the HNF4α level would be expected to increase SOCE due to release of STIM1 from inhibition of HNF4α. Thus, we carried out Ca2+ imaging experiments to examine the functional consequence of HNF4α inhibition on STIM1 protein expression. Cyclopiazonic acid was used to activate SOCs in MCs with and without knockdown of HNF4α. The Ca2+ entry response was evaluated using a classical Ca2+ omission-addition protocol (1). As shown in Fig. 7, the cyclopiazonic acid-stimulated SOCE response was significantly greater in MCs transfected with siHNF4α compared with the response in MCs without transfection or transfected with scrambled control siRNA. These fura-2 data provided functional evidence supporting a negative regulation of STIM1 by HNF4α.
Fig. 7.

Effect of knocking down HNF-4α on store-operated Ca2+ entry (SOCE) in human MCs. Fura-2 fluorescence ratiometry was used to assess the intracellular Ca2+ concentration ([Ca2+]i). SOCE was evaluated using a Ca2+-readmission protocol. A–C: representative traces showing cyclopiazonic acid (CPA; 25 μM)-evoked Ca2+ responses in human MCs without transfection (Untran), transfected with scrambled siRNA (Scr), or transfected with siRNA against human HNF4α (siHNF4α). Traces were smoothed using SigmaPlot program (version 11.0). [Ca2+]B is the Ca2+ concentration in the bathing solution. SOCE was the increase in [Ca2+]i upon switch of the bathing solution from Ca2+-free to 2 mM Ca2+ solution. The numbers inside the horizontal bar on the top of each graph indicate the Ca2+ concentration in the bathing solution. D: summary data showing SOCE in Untran, Scr, and siHNF4α groups of human MCs. *P < 0.05 compared with both Untran and Scr groups. n is the number of cells analyzed in each group.
Knockdown of HNF4α reduced fibronectin and collagen type IV protein expression in human MCs.
Our previous in vitro and in vivo study (53) demonstrated that activation of SOCs suppressed extracellular matrix protein expression. Since HNF4α repressed STIM1 expression and reduced SOCE, we speculated that knockdown of HNF4α should decrease extracellular matrix protein expression by enhancing SOCE. This hypothesis was tested by Western blot analysis in MCs with and without knockdown of HNF4α. We found that expression levels of fibronectin (Fig. 8, A and B) and collagen type IV (Fig. 8, C and D) proteins were significantly reduced in MCs treated with siHNF4α compared with that in cells without treatment or treated with scrambled siRNA. These results suggest that the negative regulation of STIM1 by HNF4α may have physiological and pathological relevance by regulation of the renal fibrosis pathway.
Fig. 8.

Effect of knocking down HNF-4α on fibronectin (FN) and collagen type IV (Col IV) protein expression in human MCs. A: representative Western blots showing FN expression in MCs without transfection or transfected with siRNA against human HNF4α (siHNF4α) or scrambled siRNA (Scr). Actin was used as a loading control. B: summary data from the experiments shown in A. FN expression levels were normalized to actin. *P < 0.05 compared with both Untran and Scr groups. n is the number of independent experiments. C: representative Western blots (duplicate) showing Col IV expression in MCs without transfection or transfected with siRNA against human HNF4α (siHNF4α) or scrambled siRNA (Scr). Actin was used as a loading control. D: summary data from the experiments shown in C. Col IV expression levels were normalized to actin. *P < 0.05 compared with both Untran and Scr groups. n is the number of independent experiments.
DISCUSSION
STIM1 has been known to gate SOCs by interacting with the channel pore-forming unit Orai1 (8, 50). Over past 10 yr, intensive studies have been carried out to investigate the mechanisms for controlling the channel function, and much attention was focused on regulation of STIM1-Orai1 interactions (21, 33, 43, 44, 57). Little is known about the regulation of the abundance of STIM1, which could also affect channel function. In this regard, the transcription factor NF-κB has been recently reported to promote STIM1 protein expression in mast cells and in vascular endothelial cells (7, 12). In the present study, we showed that HNF4α, a nuclear transcription factor, regulated STIM1 expression by functioning as a repressor in MCs. The consequence of this repression is attenuation of SOCE. Thus, the STIM1 expression level is under the control of multiple transcription factors. Because HNF4α expression is tissue specific (11, 23, 41), our results suggest that transcription factor-regulated STIM1 expression might be cell context dependent. Indeed, we failed to observe an NF-κB effect on STIM1 expression in MCs (data not shown), as previously described in mast cell and pulmonary vascular endothelial cells (7, 12).
HNF4α is generally thought as a transcriptional activator and is known to activate a wide variety of genes involved in glucose, fatty acid, cholesterol, and amino acid metabolism (18, 20, 31, 45). However, a recent study (38) revealed that HNF4α directly inhibits transcription of the epithelial-to-mesenchymal transition master regulatory genes Snail, Slug, and high-mobility group AT-hook 2 in hepatocytes. Thus, HNF4α can function as both a transcriptional activator and repressor. The present study supports a repression role of HNF4α by providing evidence that HNF4α inhibits STIM1 transcription by interacting with the promoter of Stim1.
One main finding of this study was that HNF4α mediated HG-stimulated STIM1 expression. Presumably, a decrease in the abundance of HNF4α and/or function of HNF4α (i.e., DNA-binding activity) would promote STIM1 expression because of release of STIM1 transcription from repression by HNF4α. Our study suggests that suppression of HNF4α activity is the mechanism for the HG effect on STIM1 protein expression because HG did not alter the amount of HNF4α protein but inhibited the binding activity of HNF4α to the Stim1 promoter. The mechanism for this inhibitory effect of HG is unknown in the present study. However, several HG-associated factors might be involved. For instance, transforming growth factor (TGF)-β1 has been reported to impair HNF4α DNA-binding activity on target gene promoters through glycogen synthase kinase-3β and Smad3 pathways in hepatocytes (2, 26). It is known that TGF-β1 and its signaling pathway are activated by HG in MCs (22, 49, 52). Histone deacetylase (HDAC) is another potential candidate. Histone deacetylation mediated by HDAC generally leads to transcriptional repression. In hepatocytes, recruitment of HDAC to cytochrome P-450 (CYP)7A1 chromatin is one mechanism for TGF-β1-reduced HNF4α binding to CYP7A1 chromatin (26). HDAC is expressed in MCs and regulates MC function by regulating transcription of several genes (14, 25, 56). We have previously demonstrated that HG stimulates the NF-κB p65 subunit to recruit HDAC into the promoter region of Trpc6 to repress Trpc6 transcription in human MCs (51). Thus, it is possible that HG treatment activates HDAC in MCs and that the activated HDAC is recruited to the Stim1 gene and impairs HNF4α binding to the Stim1 promoter.
HNF4α is mainly expressed in the liver, kidney, pancreas, and intestine (5, 11, 18, 20, 23, 24, 29, 54). In the kidney, HNF4α is abundant in proximal tubules. Jiang et al. (23) previously reported that HNF4α protein was detected in proximal tubular epithelial cells by immunohistochemistry using their own HNF4α antibody but was absent in the glomerulus as well as in distal and collecting tubular epithelial cells. It should be noted that the antibody they generated only recognizes P1 promoter-driven HNF4α isoforms (HNF4α1/α2/α3) but does not recognize HNF4α isoforms originated from P2 promoter (HNF4α7/α8/α9). Indeed, using this antibody, they could not detect HNF4α expression in pancreas and mucosal epithelial cells of the stomach, two organs that are known to have high expression of HNF4α (6, 11). This is probably because the HNF4α isoforms expressed in the pancreas and stomach are derived from the P2 promoter. Thus, the negative staining in the glomerular region in Jiang et al.'s study might indicate the P2 promoter origin of HNF4α in the glomerulus. We noted that not all glomeruli showed HNF4α staining in the present study. Although we do not have a good explanation on the different behaviors of different anti-HNF4α antibodies at present, it does suggest a possible influence of HNF4α antibody properties on immunofluorescence studies.
In the kidney, only little is known about the physiological and pathological role of HNF4α. Previous studies revealed that HNF4α regulates “drug” transporters during development of the kidney proximal tubule (15, 29), stimulates the mesenchymal-epithelial transition in early nephrogenesis (24), and suppresses renal cell carcinoma (28). Recently, Niehof and Borlak (32) reported that HNF4α protein was significantly reduced in kidneys from diabetic rats and humans. The decrease in HNF4α in the diabetic kidney downregulated expression of TRPC1 protein, a Ca2+ channel that regulates Ca2+ signals in kidney cells (10). In the present study, we provide evidence that HNF4α can regulate another type of Ca2+ channel, i.e., SOCs, by repressing STIM1 transcription.
Diabetes/HG effects on STIM1 expression/SOC function are cell context dependent (1, 3, 4, 30, 34, 58). Like in platelets and vascular endothelial cells (4, 58), prolonged HG treatment in MCs increased STIM1/Orai1 protein expression levels and augmented SOCE. In a recent study (53), we further showed that activation of STIM1/Orai1-mediated SOCs reduced fibronectin and collagen type IV expression, suggesting a renoprotective effect of SOCE. Thus, the increase in STIM1/Orai1-mediated SOCE in response to diabetes/HG might be a compensatory mechanism in the kidney to counteract kidney damages induced by diabetes/HG. Our findings suggest that the compensatory increase in STIM1 protein is mediated by dysfunction of HNF4α under HG conditions. In MCs, HG impairs the repression of HNF4α on STIM1 transcription, resulting in increased abundance of STIM1 protein and consequently enhanced SOCE. Therefore, HNF4α in MCs might be a therapeutic target for patients with diabetic kidney disease.
In summary, our study indicates that HNF4α acts as a novel repressor of STIM1 in MCs. HG increased STIM1 protein expression in MCs by impairing HNF4α-binding activity to the Stim1 promoter, thus releasing Stim1 transcription from HNF4α repression (Fig. 9). Because STIM1-gated SOCs in MCs are renoprotective, inhibition of HNF4α in MCs might be a therapeutic option for patients with diabetic kidney disease.
Fig. 9.
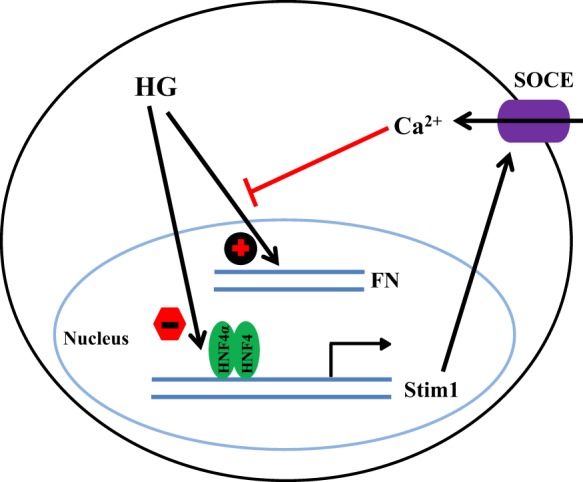
Schematic illustration of the negative regulation of STIM1 by HNF4α in human MCs. In MCs, HNF4α represses Stim1 transcription. Under the condition of diabetes, HG stimulates FN expression. Simultaneously, HG impairs HNF4α binding to the Stim1 promoter region, which releases Stim1 transcription from repression by HNF4α. An increase in STIM1 protein, in combination with other factors, results in the augmentation of SOCE. The Ca2+ signals of SOCE inhibit the fibrotic pathway of HG.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant 5-RO1-DK-079968 (to R. Ma).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.W. and R.M. conception and design of research; Y.W., S.C., and Y.R. performed experiments; Y.W., S.C., Y.R., and R.M. analyzed data; Y.W., S.C., and R.M. interpreted results of experiments; Y.W. and R.M. prepared figures; Y.W. and R.M. drafted manuscript; Y.W., S.C., Y.R., and R.M. approved final version of manuscript; R.M. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Akiyoshi Fukamizu (University of Tsukuba, Tsukuba, Japan) for providing the pcDNA-HA-hHNF4α2 plasmid and Dr. Tobias Meyer (Stanford University School of Medicine) for the pDS_XB_YFP-STIM1-II plasmid.
REFERENCES
- 1.Chaudhari S, Wu P, Wang Y, Ding Y, Yuan J, Begg M, Ma R. High glucose and diabetes enhanced store-operated Ca2+ entry and increased expression of its signaling proteins in mesangial cells. Am J Physiol Renal Physiol 306: F1069–F1080, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cozzolino AM, Alonzi T, Santangelo L, Mancone C, Conti B, Steindle C, Musone M, Cicchini C, Tripodi M, Marchetti A. TGFβ overrides HNF4α tumor suppressing activity through GSK3β inactivation: implication for hepatocellular carcinoma gene therapy. J Hepatol 58: 65–72, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Curtis TM, Major EH, Trimble ER, Scholfield CN. Diabetes-induced activation of protein kinase C inhibits store-operated Ca2+ uptake in rat retinal microvascular smooth muscle. Diabetologia 46: 1252–1259, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Daskoulidou N, Zeng B, Berglund LM, Jiang H, Chen GL, Kotova O, Bhandari S, Ayoola J, Griffin S, Atkin SL, Gomez MF, Xu SZ. High glucose enhances store-operated calcium entry by upregulating Ora/STIM via calcineurin-NFAT signaling. J Mol Med. In press. [DOI] [PubMed] [Google Scholar]
- 5.David-Silva A, Freitas HS, Okamoto MM, Sabino-Silva R, Schaan BD, Machado UF. Hepatocyte nuclear factor 1α/4α and forkhead box A2 regulate the solute carrier 2A2 (Slc2a2) gene expression in the liver and kidney of diabetic rats. Life Sci 93: 805–813, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Dean S, Tang JI, Seckl JR, Nyirenda MJ. Development and tissue-specific regulation of hepatocyte nuclear factor 4-α (HNF4-α) isoforms in rodents. Gene Expr 14: 337–344, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DebRoy A, Vogel SM, Soni D, Sundivakkam PC, Malik AB, Tiruppathi C. Cooperative signaling via transcription factors NF-κB and AP1/c-Fos mediates endothelial cell STIM1 expression and hyperpermeability in response to endotoxin. J Biol Chem 289: 24188–24201, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng X, Wang Y, Zhou Y, Soboloff J, Gill DL. STIM and Orai: dynamic intermembrane coupling to control cellular calcium signals. J Biol Chem 284: 22501–22505, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du J, Ding M, Sours-Brothers S, Graham S, Ma R. Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am J Physiol Renal Physiol 294: F909–F918, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Du J, Sours-Brothers S, Coleman R, Ding M, Graham S, Kong D, Ma R. Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. J Am Soc Nephrol 18: 1437–1445, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Erdmann S, Senkel S, Arndt T, Lucas B, Lausen J, Klein-Hitpass L, Ryffel GU, Thomas H. Tissue-specific transcription factor HNF4α inhibits cell proliferation and induces apoptosis in the pancreatic INS-1 β-cell line. Biol Chem 388: 91–106, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Eylenstein A, Schmidt S, Gu S, Yang W, Schmid E, Schmidt EM, Alesutan I, Szteyn K, Regel I, Shumilina E, Lang F. Transcription factor NF-κB regulates the expressions of a pore-forming unit, Orai1, and its activator, STIM1, to control Ca2+ entry and affect cellular functions. J Biol Chem 287: 2719–2730, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441: 179–185, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Freidkin I, Herman M, Tobar A, Chagnac A, Ori Y, Korzets A, Gafter U. Effects of histone deacetylase inhibitors on rat mesangial cells. Am J Physiol Renal Physiol 298: F426–F434, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Gallegos T, Martovetsky G, Kouznetsova V, Bush KT, Nigam SK. Organic anion and cation SLC22 “drug” transporter (Oat1, Oat3, and Oct1) regulation during development and maturation of the kidney proximal tubule. PLoS One 7: e40796, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, Ma R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J Biol Chem 285: 23466–23476, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham S, Ding M, Sours-Brothers S, Yorio T, Ma JX, Ma R. Downregulation of TRPC6 protein expression by high glucose, a possible mechanism for the impaired Ca2+ signaling in glomerular mesangial cells. Am J Physiol Renal Physiol 293: F1381–F1390, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 21: 1393–1403, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirota K, Daitoku H, Matsuzaki H, Araya N, Yamaqata K, Asada S, Suqaya T, Fukamizu A. Hepatocyte nuclear factor-4 is a novel downstream target of insulin via FKHR as a signal-regulated transcriptional inhibitor. J Biol Chem 278: 13056–13060, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Hirota K, Sakamaki JI, Ishida J, Shimamoto Y, Nishihara S, Kodama N, Ohta K, Yamamoto M, Tanimoto K, Fukamizu A. A combination of HNF-4 and Foxo1 is required for reciprocal transcriptional regulation of glucokinase and glucose-6-phosphatase genes in response to fasting and feeding. J Biol Chem 283: 32432–32441, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, ICRAC and TRPC1 channels. Nat Cell Biol 8: 1003–1010, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Inoki K, Haneda M, Maeda S, Koya D, Kikkawa R. TGF-β1 stimulates glucose uptake by enhancing GLUT1 expression in mesangial cells. Kidney Int 55: 1704–1712, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Jiang S, Tanaka T, Iwanari H, Hotta H, Yamashita H, Kumakura J, Watanabe Y, Uchiyama Y, Aburatani H, Hamakubo T, Kodama T, Naito M. Expression and localization of P1 promoter-driven hepatocyte nuclear factor-4α (HNF4α) isoforms in human and rats. Nucl Recept 1: 5, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanazawa T, Ichii O, Otsuka S, Namiki Y, Hashimoto Y, Kon Y. Hepatocyte nuclear factor 4 alpha is associated with mesenchymal-epithelial transition in developing kidneys of C57BL/6 mice. J Vet Med Sci 73: 601–607, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki SI, Isshiki K, Isono M, Uzu T, Guarente L, Kashiwagi A, Koya D. SIRT1 inhibits transforming growth factor β-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J Biol Chem 282: 151–158, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Li T, Chiang JYL. A novel role of transforming growth factor β1 in transcriptional repression of human cholesterol 7α-hydroxylase gene. Gastroenterology 133: 1660–1669, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Meyer T. STIM is a Ca2+ store sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Current Biol 15: 1235–1241, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas B, Grigo K, Erdmann S, Lausen J, Klein-Hitpan L, Ryffel GU. HNF4α reduces proliferation of kidney cels and affects genes deregulated in renal cell carcinoma. Oncogene 24: 6418–6431, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Martovetsky G, Tee JB, Nigam SK. Hepatocyte nuclear factor 4α and 1α regulate kidney developmental expression of drug-metabolizing enzymes and drug transporters. Mol Pharmacol 84: 808–823, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mita M, Ito K, Taira K, Nakagawa J, Walsh MP, Shoji M. Attenuation of store-operated Ca2+ entry and enhanced expression of TRPC channels in caudal artery smooth muscle from type 2 diabetic Goto-Kakizaki rats. Clin Exp Pharmacol Physiol 37: 670–678, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Mogilenko DA, Dizhe EB, Shavva VS, Lapikov IA, Orlov SV, Perevozchikov AP. Role of the nuclear receptors HNF4α, PPARα, and LXRs in the TNFα-mediated inhibition of human apolipoprotein A-I gene expression in HepG2 cells. Biochem 48: 11950–11960, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Niehof M, Borlak J. Hepatic nuclear factor 4 alpha and the Ca-channel TRPC1 are novel disease candidate genes in diabetic nephropathy. Diabetes 57: 1069–1077, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Ong E, Nesin V, Long CL, Bai CX, Guz JL, Ivanov IP, Abramowitz J, Birnbaumer L, Humphrey MB, Tsiokas L. A TRPC1 protein-dependent pathway regulates osteoclast formation and function. J Biol Chem 288: 22219–22232, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang Y, Hunton DL, Bounelis P, Marchase RB. Hyperglycemia inhibits capacitative calcium entry and hypertrophy in neonatal cardiomyocytes. Diabetes 51: 3461–3467, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev 85: 757–810, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435–445, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryffel GU. Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol 27: 11–29, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, Bonzo JA, Gonzalez FJ, Alonzi T, Amicone L, Tripodi M. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4α. Hepatology 53: 2063–2074, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen B, Zhu J, Zhang J, Jiang F, Wang Z, Zhang Y, Li J, Huang D, Ke D, Ma R, Du J. Attenuated mesangial cell proliferation related to store-operated Ca2+ entry in aged rat: the role of STIM1 and Orai1. AGE 35: 2093–2202, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimizu H, Hori Y, Kaname S, Yamada K, Nishiyama N, Matsumoto S, Miyata K, Oba M, Yamada A, Kataoka K, Fujita T. siRNA-Based therapy ameliorates glomerulonephritis. J Am Soc Nephrol 21: 622–633, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sladek FM, Zhong W, Lai E, Darnell JE. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev 4: 2353–2365, 1990. [DOI] [PubMed] [Google Scholar]
- 42.Sours-brothers S, Ding M, Graham S, Ma R. Interaction between TRPC1/TRPC4 assembly and STIM1 contributes to store-operated Ca2+ entry in mesangial cells. Exp Biol Med 234: 673–682, 2009. [DOI] [PubMed] [Google Scholar]
- 43.Srikanth S, Jew M, Kim KD, Yee MK, Abramson J, Gwack Y. Junctate is a Ca2+-sensing structural component of orai1 and stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 109: 8682–8687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol 12: 436–446, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4α regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci USA 94: 13209–13214, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takabatake Y, Isaka Y, Mizui M, Kawachi H, Shimizu F, Ito T, Hori M, Imai E. Exploring RNA interference as a therapeutic strategy for renal disease. Gene Ther 12: 965–973, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Tsujie M, Isaka Y, Nakamura H, Imai E, Hori M. Electroporation-mediated gene transfer that targets glomeruli. J Am Soc Nephrol 12: 949–954, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312: 1220–1223, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB. Inhibition of the JAK/STAT signaling pathway prevents the high glucose-induced in TGF-β and fibronectin synthesis in mesangial cells. Diabetes 51: 3505–3509, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal 3: pe42-1–pe42-4, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Ding M, Chaudhari S, Ding Y, Yuan J, Stankowska D, He S, Krishnamorthy R, Cunningham JT, Ma R. Nuclear factor κB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells. J Biol Chem 288: 12852–12865, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weigert C, Sauer U, Brodbeck K, Pfeiffer A, Häring HU, Schleicher ED. AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-β1 promoter in mesangial cells. J Am Soc Nephrol 11: 2007–2016, 2000. [DOI] [PubMed] [Google Scholar]
- 53.Wu P, Wang Y, Davis ME, Zuckerman JE, Chaudhari S, Begg M, Ma R. Store-operated Ca2+ channel in mesangial cells inhibits matrix protein expression. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie X, Liao H, Dang H, Pang W, Guan Y, Wang X, Shyy JYJ, Zhu Y, Sladek FM. Down-regulation of hepatic HNF4α gene expression during hyperinsulinemia via SREBPs. Mol Endocrinol 23: 434–443, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu G, Kanezaki R, Toki T, Watanabe S, Takahashi Y, Terui K, Kitabayashi I, Ito E. Physical association of the patient-specific GATA1 mutants with RUNX1 in acute megakaryoblastic leukemia accompanying Down syndrome. Leukemia 20: 1002–1008, 2006. [DOI] [PubMed] [Google Scholar]
- 56.Yu Z, Kone BC. Targeted histone H4 acetylation via phosphoinositide 3-kinase- and p70s6-kinase-dependent pathways inhibits iNOS induction in mesangial cells. Am J Physiol Renal Physiol 290: F496–F502, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11: 337–343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zbidi H, Lopez JJ, Amor NB, Bartegi A, Salido GM, Rosado JA. Enhanced expression of STIM1/Orai1 and TRPC3 in platelets from patients with type 2 diabetes mellitus. Blood Cells Mol Dis 43: 211–213, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Zhang SL, Yeromin AV, Zhang XHF, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA 103: 9357–9362, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]