

Abstract
Chronic obstructive pulmonary disease (COPD) is a growing cause of morbidity and mortality worldwide. Recent studies have shown that cigarette smoke (CS) induces cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction, which leads to airway-surface liquid (ASL) dehydration. This in turn contributes to the mucus dehydration and impaired mucociliary clearance that are seen in the chronic bronchitis form of COPD. Roflumilast is a phosphodiesterase 4 inhibitor that may improve lung function and reduce the frequency of exacerbations in patients with COPD. Although roflumilast can affect cAMP metabolism, little is known about the downstream pharmacological effects in the airways. We hypothesized that roflumilast would increase ASL rehydration in human bronchial epithelial cultures (HBECs) after chronic CS exposure. cAMP production was measured by Förster resonance energy transfer in HEK293T cells and by ELISA in HBECs. ASL height was measured by xz-confocal microscopy after air exposure or following HBEC exposure to freshly produced CS. Roflumilast had little effect on cAMP or ASL height when applied on its own; however, roflumilast significantly potentiated adenosine-induced increases in cAMP and ASL height in CS-exposed HBECs. Roflumilast increased the rate of ASL height recovery in cultures after CS exposure compared with controls. In contrast, the β2-adrenergic receptor agonists isoproterenol and salmeterol failed to increase ASL height after CS exposure. Our data suggest that roflumilast can increase ASL hydration in CS-exposed HBECs, which is predicted to be beneficial for the treatment of mucus dehydration/mucus stasis in patients with COPD chronic bronchitis.
Keywords: cAMP, cystic fibrosis transmembrane conductance regulator, chronic obstructive pulmonary disease, roflumilast, airway hydration
chronic obstructive pulmonary disease (COPD), the third leading cause of death worldwide (35), is most often caused by chronic tobacco exposure. The chronic bronchitis (CB) form of COPD is characterized by chronic airway inflammation and an increase in mucus production resulting in irreversible airflow obstruction that is not sensitive to bronchodilators (22, 42). Acute exacerbations add to the severity of the disease, and bacterial colonization may contribute to the sustained inflammation and subsequent decline in lung function (25). CB shares some pathological features with cystic fibrosis (CF), a disease caused by defects in the CF transmembrane conductance regulator (CFTR) Cl− channel, including abnormal ion transport, mucus dehydration/mucus accumulation, chronic inflammation, and persistent airway infection (6, 46). Because of dysfunctional CFTR, patients with CF have an impaired ability to secrete anions, leading to airway-surface liquid (ASL) dehydration (27, 36). Defective anion secretion also results in the accumulation of abnormally thick, dehydrated mucus in the airway lumen that prevents mucus clearance and provides a favorable environment for bacterial colonization and proliferation.
Cigarette smoke (CS) exposure is known to reduce CFTR expression, induce CFTR internalization, and disrupt CFTR channel function, all of which lead to ASL dehydration in vitro (10, 15, 48) that is similar to the dehydrated mucus seen in CB airways in vivo (15, 25). For example, Clunes et al. (15) found that mucus from chronic smokers was ∼10% solids (resulting in a dehydrated ASL), whereas mucus from healthy individuals was ∼4%. Additionally, CS-induced CFTR inhibition was not absolute, as ∼45% of normal CFTR function remained intact (15). At present, there is no cure for CB/COPD. Although current anti-inflammatory and bronchodilator therapies are available, they fail to stop the disease progression (44). Moreover, few therapies target the CFTR deficiency itself (3, 8); however, therapies involving the elevation of cAMP, which would serve to activate CFTR, are presently being evaluated for the treatment of COPD (53).
The two main approaches to increase cAMP signaling are 1) activation of an upstream G protein-coupled receptor (GPCR) to stimulate cAMP production or 2) prevention of cAMP degradation by a phosphodiesterase (PDE). Adenylyl cyclase and PDE4 control the formation and degradation of cAMP, respectively, and facilitate spatiotemporal cAMP signaling (16, 26, 37). The adenylyl cyclase activator forskolin is commonly used to increase intracellular cAMP levels (12). Stimulation of GPCRs can also elevate cAMP levels (28). For example, salmeterol, which is used to treat bronchoconstriction in patients with asthma or COPD, activates β2-adrenergic receptors that result in airway smooth muscle relaxation (18, 52). Inhibition of PDE4 augments cAMP signaling and decreases inflammation in epithelial cells (21). Roflumilast is a PDE4 inhibitor that has been approved by the US Food and Drug Administration and the European Commission for the treatment of severe COPD. It has been shown to improve lung function by decreasing inflammation, possibly through the reduction of leukocyte effector responses (49). Roflumilast also activates CFTR-mediated chloride secretion (34). Recently, Lambert et al. (32) demonstrated that roflumilast can partially restore CFTR-dependent Cl− secretion after CS exposure.
In this study, we tested the hypothesis that roflumilast would increase airway hydration in chronically CS-exposed human bronchial epithelial cultures (HBECs). We also tested whether physiologically and pharmacologically relevant GPCR agonists such as adenosine and isoproterenol could elicit ASL rehydration when used alone or in combination with roflumilast.
MATERIALS AND METHODS
Chemicals.
Roflumilast was obtained from Forest Research Institute (Jersey City, NJ), dissolved in DMSO at 10−1 M, and diluted at ≥1,000-fold when added to cultures. Rhodamine-dextran (10 kDa) was obtained from Life Technologies (Carlsbad, CA). The S18 peptide was obtained from AmbioPharm (North Augusta, SC). Unless stated otherwise, all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). For live cell experiments, we used a modified Ringer solution containing (in mM): 120 NaCl, 5.2 KCl, 1.2 MgCl2, 1.2 CaCl2 2H2O, 12 NaHCO3, 24 HEPES, and 10 mM glucose, pH 7.4.
Cell culture.
HEK293T cells were maintained in MEM α media (Life Technologies) and supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 g/ml streptomycin solution. HEK293T cells were typically used 2–3 days after seeding. Cultures were transfected for 4–6 h using Lipofectamine 2000 (Life Technologies) per manufacturer's instructions. After transfecting, cultures were washed and placed in media and allowed to incubate in 5% CO2 at 37°C overnight.
Calu-3 cells were maintained in Eagle's minimal essential medium, supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% nonessential amino acids, and incubated in humidified air containing 5% CO2 at 37°C.
Human donor lungs were obtained by The University of North Carolina at Chapel Hill CF Center Tissue Core under protocols approved by the UNC Institutional Committee for the Protection of the Rights of Human Subjects. Bronchial epithelial cells were isolated as described and plated on 12-mm Transwell clear culture inserts (Corning, Corning, NY) that were coated with collagen from human placenta. Cells were used after 3–5 wk of culture (45).
Förster resonance energy transfer.
To measure Förster resonance energy transfer (FRET), cells expressing cyan fluorescent protein (CFP)/yellow fluorescent protein-tagged constructs were placed on an inverted Ti-U microscope (Nikon Instruments, Melville, NY) with a 60 × 1.4 NA oil objective lens, switchable filter wheels (Ludl, Hawthorne, NY), and either an Orca CCD or Orca Flash 4.0 CMOS camera (Hamamatsu, Bridgewater, NJ). CFP (excitation, 430 nm; emission, 470 nm) and FRET (excitation, 430 nm; emission, 575 nm) images were obtained over time as described (57). After background subtraction, the FRET ratio was determined by dividing the 430/470 image intensity by the 430/575 image intensity. All images were analyzed using Image J (NIH Freeware, Bethesda, MD).
cAMP measurements.
To measure intracellular cAMP, medium was aspirated after vehicle or agonist addition, HBECs were lysed with 0.1 N HCl, the lysate was centrifuged, and the supernatants were assayed using an enzyme immunoassay kit (Enzo Life Science, Farmingdale, NY). Pellets were assayed for protein content using the BCA method (Pierce Thermo Scientific, Rockford, IL). Absorbance was read by a Tecan infinite multi-plate reader, and cellular cAMP was normalized to total protein as described previously (57).
ASL height measurements.
To measure ASL height, PBS (20 μl) containing 2 mg/ml rhodamine-dextran was added to cultures at the start of the experiment. ASL secretion was measured by aspirating excess fluid with a Pasteur pipette to decrease ASL height to ∼7 μm, as described (51). After CS exposure, 100 μM roflumilast, isoproterenol, or salmeterol was added basolaterally. Adenosine was suspended in perfluorocarbon (PFC) and added apically to activate CFTR via A2B adenosine receptors (A2BR) (17). In all cases, five predetermined points (1 central and 4 2 mm from the edge of the culture) were xz scanned using a confocal microscope (Leica SP5; glycerol ×63 immersion lens) as previously described (17). Cultures were returned to the incubator between time points. For all studies, PFC was added mucosally during imaging to prevent evaporation of the ASL.
CS exposure.
HBECs were placed in a specially built smoke-exposure chamber that exposed apical but not basolateral surfaces to CS (14, 15). CS was then generated according to the International Organization of Standardization (ISO) standards (35-ml draw over 2 s) using an LC1 smoke engine (Borgwaldt, Richmond, VA) and applied to the cultures at a rate of 1 puff every 30 s until the cigarette was exhausted (∼12 puffs over ∼5 min). For ASL experiments, cells were treated with CS for ∼5 min and placed in an incubator for 30 min before addition of roflumilast and other compounds (∼1 h). This protocol has previously been shown to drive removal of CFTR from the plasma membrane without inducing gross cellular toxicity (14, 15).
Statistical analyses.
All data were checked for homogeneity of variance and analyzed using ANOVA followed by Tukey's post hoc test. All values are expressed as means ± SE, and α was set to 0.05. Airway cultures derived from three or more donors were used per experiment, and experiments using cell lines were repeated on at least three separate occasions; n refers to the number of cultures used per experiment. All analyses were conducted using Instat or Prism 5 (GraphPad, La Jolla, CA) software.
RESULTS
Roflumilast potentiates adenosine-stimulated cAMP signaling.
A FRET-based approach was used initially to study the effects of roflumilast on intracellular cAMP. HEK293T cells transfected with the cytoplasmic cAMP FRET sensor indicator of cAMP using Epac2 (ICUE2) (56) were stimulated with adenosine and measured by epifluorescence microscopy to determine the FRET ratio. Roflumilast alone had no effect on intracellular cAMP levels (n = 4; data not shown). However, adenosine is a purine nucleoside that is present in the ASL and acts as a physiological regulator of CFTR by stimulating A2BRs (13, 33). At a dose of 1 μM, which is lower than the EC50 for the A2BR (∼2 μM), adenosine caused a small but rapid increase in FRET ratio (Fig. 1, A and B). To investigate the effect of increasing doses of roflumilast on intracellular cAMP signaling, a dose response was generated using FRET ratio as the readout. A 30-min pretreatment with roflumilast significantly potentiated the adenosine response with an EC50 of 1.48 μM (Fig. 1, B and C). We next examined the effect of varying doses of adenosine and forskolin with or without roflumilast on cAMP. Roflumilast significantly potentiated the 1 and 10 μM adenosine-induced increases in cAMP levels compared with untreated cells. A maximum increase in cAMP was obtained with either 10 μM adenosine or forskolin (Fig. 1D).
Fig. 1.

Roflumilast potentiates adenosine-stimulated cAMP signaling. A: overlay images of cyan fluorescent protein and yellow fluorescent protein fluorescence from HEK293T cells expressing indicator of cAMP using Epac2 (ICUE2) before and after adenosine (ADO) exposure. B: typical changes in Förster resonance energy transfer (FRET) ratio in HEK293T cells transfected with the ICUE2 FRET sensor with 1 μM ADO and varying amounts of roflumilast. Red, cells alone; green, 0.01 μM roflumilast + ADO; blue, 1 μM + ADO; gray, 10 μM roflumilast + ADO; black, 1,000 μM roflumilast + ADO. C: roflumilast dose-response curve generated using FRET ratio in HEK293T cells transfected with the ICUE2 FRET sensor and stimulated with 1 μM ADO and varying amounts of roflumilast. D: mean change in ICUE2 FRET ratio in HEK293T cells exposed to varying amounts of ADO or forskolin and roflumilast. Black, 1 μM ADO + 10 μM forskolin; gray, roflumilast + 1 μM ADO + 10 μM forskolin; red, 10 μM ADO + 10 μM forskolin; orange, roflumilast + 10 μM ADO + 10 μM forskolin. E: intracellular cAMP levels (% above basal) measured by ELISA in Calu-3 cells following increased amounts of serosal roflumilast ± 0.5 μM mucosal ADO; △, roflumilast; ▲, roflumilast + ADO. F: dose response to roflumilast generated by measuring the rate of airway-surface liquid (ASL) secretion in human bronchial epithelial cultures (HBECs) following serosal exposure to roflumilast (30 min) and apical exposure to ADO; n = 9–12.
To validate the FRET assay, we tested the effect of roflumilast on adenosine-stimulated cAMP production in Calu-3 airway epithelial cells using an ELISA-based approach. Roflumilast was added serosally to mimic oral delivery in patients. This significantly potentiated the adenosine-induced cAMP increase in CS compared with air controls. A 30-min treatment with a range of roflumilast doses had no detectable effect on cAMP levels (Fig. 1E). Again, a low dose of adenosine (0.5 μM) significantly increased cAMP levels in the presence of roflumilast with an EC50 of 0.017 μM (Fig. 1E). We have previously shown that adenosine, A2BR, and cAMP stimulate CFTR to increase ASL height (33). When using the ASL height assay as an indirect measure of CFTR function and while keeping adenosine constant at 1 μM, roflumilast increased ASL height in a dose-dependent manner with an EC50 of 4.3 μM (Fig. 1F).
Roflumilast enhances cAMP production in airway epithelia exposed to CS.
To determine whether roflumilast would be effective in airway epithelia after CS exposure, cAMP levels were measured in air- or CS-exposed polarized airway cultures. HBECs pretreated serosally with vehicle (PBS and 0.1% DMSO) or roflumilast were exposed to air or the smoke from one cigarette, followed by a second vehicle (PBS) or adenosine in PBS. After PBS/adenosine exposure, cells were lysed, and the intracellular cAMP was measured by ELISA. Whereas adenosine alone had no significant effect on cAMP (Fig. 2), roflumilast pretreatment significantly enhanced the effects of adenosine. Surprisingly, the roflumilast/adenosine-induced cAMP levels were significantly greater in airway epithelia exposed to CS than to air (control; Fig. 2).
Fig. 2.

Roflumilast (Rof) enhances intracellular cAMP production in airway epithelia exposed to cigarette smoke (CS). Intracellular cAMP levels were measured by ELISA in HBECs pretreated with roflumilast, exposed to air or CS, and then stimulated with 0.5 μM ADO mucosally; n = 12. *P < 0.05 vs. control. ***P < 0.001 vs. control.
Prophylactic roflumilast prevents ASL dehydration caused by CS exposure.
Normal airway epithelia modulate ion transport, including CFTR-mediated anion secretion, to maintain ASL height at ∼7 μm (33). CS inhibits CFTR and causes an acute but persistent reduction in ASL volume that cannot be rescued by the addition of high levels of adenosine (100 μM) (15). To determine whether roflumilast could help increase ASL hydration in CS-exposed airway epithelia by increasing cAMP/CFTR-mediated anion/ASL secretion, HBECs were pretreated for 30 min with roflumilast or vehicle. Cells were then exposed to air (control) or CS, and adenosine was added mucosally to stimulate cAMP/CFTR-mediated ASL secretion. As predicted, adenosine alone increased ASL height in air-exposed but not in CS-exposed HBECs (Fig. 3, A and B). With roflumilast pretreatment, adenosine significantly increased ASL height in air-exposed cultures and restored ASL height to the normal range after CS exposure (Fig. 3, A and B).
Fig. 3.

Roflumilast prevents ASL dehydration caused by CS exposure. A: images of ASL (red) after exposure to ADO and either air or 1 cigarette ± serosal roflumilast. B: mean data taken from A. HBECs were pretreated with vehicle or roflumilast, and ASL was measured immediately after air or CS exposure. Open bar, control before CS exposure; shaded bar, CS exposure; solid bar, ADO before and after CS exposure; n = 12. *P < 0.05 vs. control; †P < 0.05 vs. ± CS in the presence of roflumilast.
To test whether the increase in ASL height induced by roflumilast/adenosine was indeed attributable to cAMP/CFTR-mediated ASL secretion, we performed the following experiments. First, we added bumetanide to inhibit Cl− secretion via the basolateral Na+/K+/2 Cl− cotransporter (51). This maneuver significantly abolished roflumilast/adenosine-induced ASL secretion (Fig. 4A). In contrast, addition of the epithelial sodium channel (ENaC) antagonist S18 (24) significantly enhanced the ASL secretory response post-CS exposure, suggesting that roflumilast/adenosine did not act by inhibiting ENaC. Finally, roflumilast and adenosine were unable to elicit ASL secretion in CF HBECs derived from ΔF508 homozygous patients, suggesting that this is a CFTR-mediated response (Fig. 4B).
Fig. 4.

The roflumilast/ADO response is dependent on CFTR conductance regulator-mediated Cl−/ASL secretion. A: all HBECs were pretreated with 10 μM serosal roflumilast 1 h before measurements were taken. Where indicated, 10 μM bumetanide was added serosally at this time. ASL was then measured before and 30 min after CS exposure. In all cases, ADO was then added apically as a dry powder in perfluorocarbon after CS exposure. Where indicated, S18 peptide was added along with the ADO. Open bar, control before CS exposure; shaded bar, CS exposure; closed bar, ADO after CS exposure; n = 6. B: CF HBECs (ΔF508 homozygote) do not increase ASL height upon exposure to roflumilast/ADO; n = 6. Open bar, control. Solid bar, roflumilast/ADO. *P < 0.05 vs. control or CS as appropriate; †P < 0.05 vs. ± CS/ADO.
Roflumilast aids in ASL height recovery after acute CS exposure.
Because prophylactic roflumilast treatment prevented CS-induced ASL dehydration, we next tested whether roflumilast could also aid in ASL height recovery when added to HBECs after CS exposure. We washed HBEC apical surfaces immediately before loading to shift the balance toward Na+ absorption and inhibit Cl− secretion (51). Neither air nor adenosine (0.5 mg/ml) exposure had any significant effect on ASL height, which remained at ∼7 μm under baseline conditions (Fig. 5A). Next, HBECs were exposed to CS from one cigarette and treated with serosal roflumilast or vehicle, followed by mucosal adenosine (Fig. 5B). After 20 min, the ASL height of cultures exposed to CS was significantly reduced compared with air controls (Fig. 5, A and B). Following 20 min of roflumilast treatment after CS exposure, ASL height was partially restored to normal levels. Again, a combination of adenosine and roflumilast potentiated the response compared with either compound alone and returned ASL height to the normal range (Fig. 5B). We next determined the rate of ASL rehydration after CS exposure by measuring the slope of the ASL height recovery (i.e., by comparing data points immediately and 20 min after CS exposure). Treatment with adenosine and roflumilast significantly increased the rate at which HBEC mucosal surfaces regained fluid (Fig. 5C).
Fig. 5.
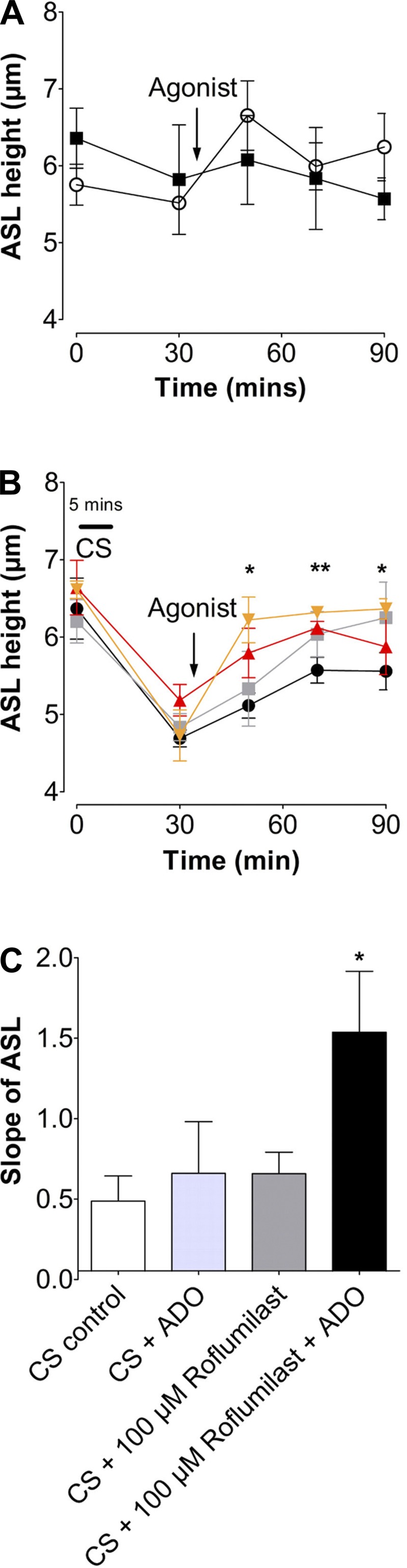
Roflumilast increases the rate of ASL height recovery after acute CS exposure. A: graph showing mean ASL height over time, as measured by xz confocal microscopy in HBECs exposed to air and ADO. ■, air; ○, air + adenosine; n = 12. B: mean ASL height over time in HBECs exposed to CS followed by roflumilast ± ADO or vehicle. ■, CS; ●, CS + ADO; ▲, roflumilast; ▼, roflumilast + ADO; n = 12. C: slope of ASL secretion taken from B. Slope shows ASL height recovery after treatment. *P < 0.05; **P < 0.01 vs. CS control.
Roflumilast, but not β2-agonists, initiate ASL height recovery after chronic CS exposure.
Thus far, we have shown that roflumilast is effective after exposure to CS from one cigarette. To determine whether roflumilast could help restore ASL to normal levels after chronic CS exposure, HBECs were exposed to the smoke of one cigarette per day for 5 days. This maneuver had no significant effect on the transepithelial resistance (air, 620; CS, 590 Ω/cm2), and the cultures remained viable as indicated by the maintenance of a thin film of ASL and the exclusion of rhodamine-dextran from the cellular and paracellular compartments (data not shown). On day 5 of chronic CS exposure, HBECs were exposed to CS and left untreated (CS control) or were treated with serosal roflumilast with or without mucosal GPCR agonists (Fig. 6A). ASL height was lower in chronic CS-exposed cultures than in air-exposed cultures (Fig. 6B). After 20 min of roflumilast and adenosine treatment, ASL height was restored toward normal ASL levels, whereas the ASL height of the CS/adenosine controls did not recover. Furthermore, adenosine significantly potentiated the roflumilast-induced recovery (Fig. 6B). Analysis of ASL height-recovery rates (Fig. 6C) revealed that chronic CS exposure significantly impeded recovery compared with acute CS exposure (Figs. 5C and 6C). Despite this, roflumilast or adenosine alone produced a similar increase in ASL rehydration after chronic smoke exposure, whereas the combination of roflumilast and adenosine induced a highly significant increase in ASL recovery (P < 0.001; Fig. 6C).
Fig. 6.
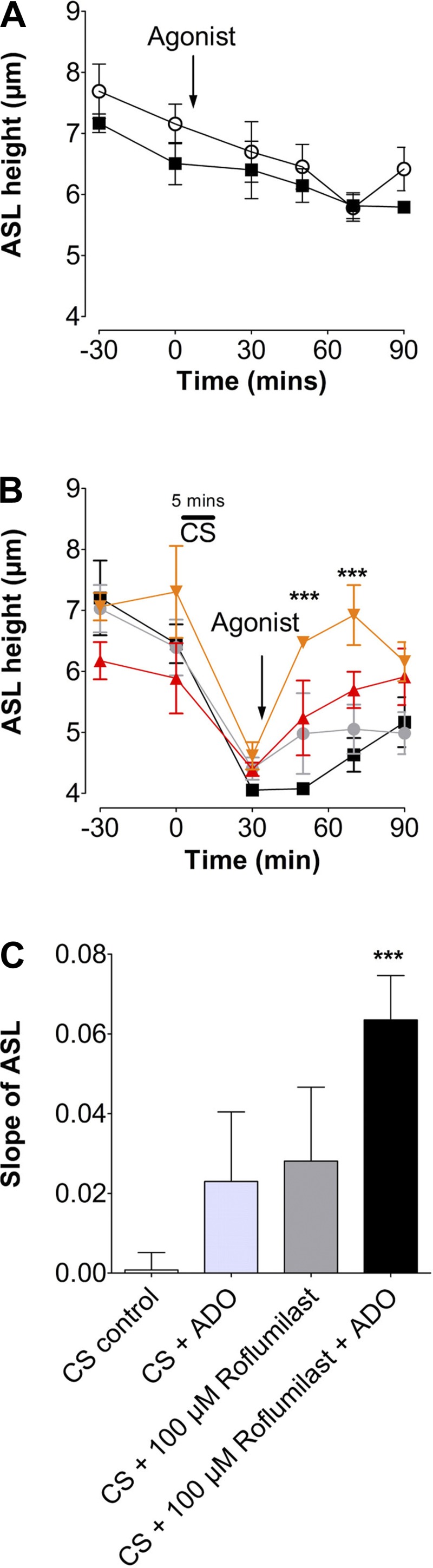
Roflumilast increases the rate of ASL height recovery after chronic CS exposure. HBECs were exposed daily to CS or air, and ASL height was measured before and after the last exposure. A: graph showing mean ASL height over time, as measured by xz confocal microscopy in HBECs exposed to air ± ADO. ■, air; ○, air + ADO; n = 12. B: mean ASL height over time in HBECs exposed to CS for 5 days followed by roflumilast ± ADO or vehicle after the 5th CS exposure. ■, CS; ●, CS + ADO; ▲, roflumilast; ▼, roflumilast + ADO; n = 12. C: slope of ASL secretion taken from B. Slope shows ASL height recovery after agonist treatment. ***P < 0.001 vs. control.
Isoproterenol and salmeterol do not induce ASL height recovery after CS exposure.
β2-Agonists are frequently administered to patients with COPD and are used to relax airway smooth muscle (11). β2-Adrenergic agonists work by elevating intracellular cAMP. However, because they act via GPCRs, the subsequent increase in cAMP may have a different spatiotemporal pattern than that elicited by PDE inhibitors such as roflumilast (41), and the duration of CFTR-mediated ASL secretion may be different. To investigate whether β2-agonists could help restore ASL levels after CS exposure, HBECs were acutely exposed to the smoke from one cigarette and treated with the short-acting β2-agonist isoproterenol, the long-acting β-agonist salmeterol, or left untreated (CS control). Neither air exposure nor adenosine affected ASL height (Fig. 7A). CS-exposed cultures had reduced ASL height that did not recover to normal ranges after receiving isoproterenol, salmeterol, or vehicle alone (Fig. 7B). When adenosine was added, the response from isoproterenol and salmeterol was potentiated (Fig. 7C). As with the roflumilast-treated cultures, the slope of ASL recovery indicated that neither isoproterenol nor salmeterol was capable of restoring ASL height when added alone (Fig. 7D). To confirm that isoproterenol and salmeterol were effective in stimulating cAMP in airway epithelia after air or CS exposure, intracellular cAMP levels were measured by ELISA after acute CS exposure. Adenosine had no significant effect on cAMP (Fig. 7E); however, isoproterenol and salmeterol pretreatment significantly enhanced the effects of adenosine. Similar results were seen in CS-exposed HBECs. Consistent with our previous observations, the isoproterenol/adenosine-induced increase in cAMP levels was greater in CS-exposed than in air-exposed airway epithelia (Fig. 7E).
Fig. 7.

Isoproterenol and salmeterol alone do not alter ASL recovery rates after acute CS exposure. A: graph showing mean ASL height over time, as measured by xz confocal microscopy in HBECs exposed to air ± ADO. ■, air; ○, adenosine; n = 12. B: mean ASL height over time in HBECs exposed to CS followed by salmeterol, isoproterenol, or vehicle. ■, CS; ○, isoproterenol; ▲, salmeterol. C: mean ASL height over time in HBECs exposed to CS followed by salmeterol, isoproterenol, or vehicle with adenosine. ■, CS; ○, isoproterenol; ▲, salmeterol; n = 12. D: slope of ASL secretion taken from B. Slope shows ASL height recovery after treatment. E: intracellular cAMP levels were measured by ELISA in HBECs pretreated with roflumilast (Rof), isoproterenol (Iso), and salmeterol (Sal), exposed to air or CS, and then stimulated with 0.5 μM ADO mucosally; n = 12. *P < 0.05; **P < 0.01.
Salmeterol inhibits the ability of roflumilast to aid ASL height recovery after acute CS exposure.
We next investigated whether the combination of salmeterol and roflumilast could further potentiate ASL height recovery after CS exposure. Air exposure alone had no effect on ASL height (Fig. 8A), whereas CS exposure caused a rapid diminution of ASL height that was not reversed following adenosine exposure (Fig. 8, B and C). Similarly, salmeterol alone did not restore ASL height after CS exposure. Surprisingly, the combination of roflumilast and salmeterol decreased ASL recovery compared with the roflumilast and adenosine addition (Fig. 8, B and C).
Fig. 8.

Salmeterol inhibits the ability of roflumilast to induce ASL secretion after acute CS exposure. A: graph showing mean ASL height over time, as measured by xz confocal microscopy in HBECs exposed to air ± ADO. ■, air; ○ air + ADO; n = 12. B: mean ASL height over time in HBECs exposed to CS ± agonist. ■, CS; ●, salmeterol + ADO; ▲, roflumilast + ADO; ▼, roflumilast + salmeterol + ADO; n = 12. C: slope of ASL secretion taken from B. Slope shows ASL height recovery after treatment. *P < 0.05 vs. control; **P < 0.01.
DISCUSSION
Chronic inflammation is a hallmark of COPD (1), and roflumilast is thought to reduce inflammation in patients with COPD by favorably altering gene expression (5). Indeed, there is considerable evidence that gene expression in the lung is partially under the control of cAMP-sensitive promoters (29) and that cAMP is anti-inflammatory (5, 47). In addition to increasing inflammation, CS exposure also triggers the internalization of CFTR, leading to ASL/mucus dehydration that likely contributes to decreased mucus clearance (15, 48). Accordingly, a new therapeutic focus on restoring CFTR function in patients with COPD has emerged. As roflumilast has been shown to activate CFTR in normal airway epithelia (32, 34), we tested whether this compound can affect ASL height after CS exposure.
We first measured the effect of roflumilast on intracellular cAMP in HEK293T cells using the ICUE2 FRET sensor (56) (Fig. 1, A and B). The basal cAMP concentration is low in HEK293T cells, and roflumilast alone had no observable effect on intracellular cAMP levels (data not shown). These results were consistent with the primary mode of action of roflumilast as an inhibitor of cAMP degradation rather than a stimulator of cAMP production. Adenosine was used in these studies to elevate cAMP, as it is present in the ASL (54) and activates CFTR in normal airways (33). Adenosine is also thought to play a role in COPD pathogenesis, and smokers are known to have significantly increased concentrations of adenosine compared with nonsmokers (19, 55). Our results show that, in contrast to roflumilast alone, a submaximal dose of adenosine (1 μM) acutely elevated cAMP levels, likely by stimulation of the A2BR (Fig. 1, B–D). Coaddition of roflumilast and adenosine resulted in a further increase in intracellular cAMP. Keeping the adenosine level constant (1 μM), we found that increases in roflumilast potentiated the FRET response with an EC50 of 1.5 μM. This potentiation was not seen with 10 μM adenosine, indicating that the response had likely become saturated. A2BR is Gs-linked and uses adenylate cyclase to increase cAMP (39). Therefore, we bypassed A2BR and directly activated adenylate cyclase with forskolin, which resulted in a similar increase in FRET. This effect was similarly potentiated by roflumilast, indicating that the use of 1 μM adenosine instead of forskolin was valid for this study.
We next tested whether roflumilast was efficacious in airway epithelia (Fig. 1, E and F). Under our experimental conditions, there is very little basal cAMP production (Fig. 1). Roflumilast alone had no effect on cAMP levels, but the effect of roflumilast was significantly potentiated by the addition of adenosine, with an EC50 of 0.02 μM (Fig. 1E). Our results also show that roflumilast induced ASL secretion in a dose-dependent manner (Fig. 1F). Interestingly, the EC50 was shifted significantly to the right of that observed for cAMP production in airway epithelia (4.2 μM). These differences could be attributable to a number of factors. It has previously been noted that efficacy in airway epithelia under thin film conditions is usually significantly worse than in more reductionist systems because the epithelia can remove or metabolize xenobiotics (23, 50). For example, when amiloride is added mucosally, it has a half-life of ∼9 min in the ASL. This is because it is a substrate for organic cation transporters and ATP, which is degraded by ecto-enzymes, has a half-life of ∼30 s (23, 50). Although roflumilast is known to be metabolized to its active form by cytochrome P450 (31), to date there is no evidence suggesting that roflumilast is a substrate for multidrug resistance protein or any of the xenobiotic transporters expressed in the lung. Thus, although we added roflumilast serosally at relatively high concentrations, we do not know the terminal concentration of roflumilast or whether it may indeed be a substrate for these types of transporters at certain concentrations.
Surprisingly, we found that roflumilast/adenosine-induced cAMP levels were greater in CS- than air-exposed airway epithelia (Fig. 2). Sloane et al. (48) previously reported that the forskolin-induced cAMP concentration was elevated in airway epithelia 24 h after CS extract exposure (48). An increase in cGMP, but not cAMP, has been observed when rats were exposed to CS (30). We do not know the mechanism underlying the CS-induced increase in cAMP; however, CS has previously been shown to affect the actin cytoskeleton (43). Furthermore, in polarized airway epithelia, actin helps locate PDEs to a subapical location (2). Thus CS exposure may disrupt cAMP signaling via cytoskeleton alterations in the actin cytoskeleton. Changes in CFTR surface densities have previously been linked to altered cAMP signaling, via changes in GPCRs (57). Because CS exposure alters CFTR levels, maybe this also impacts on cAMP production. Whether enhanced cAMP production extends to other pathological insults remains to be determined.
Clunes et al. (15) previously demonstrated that HBECs were unable to rehydrate their mucosal surfaces after whole CS exposure when stimulated with either adenosine or isoproterenol alone, suggesting that GPCR-mediated increases in cAMP were unable to sufficiently activate CFTR. Our studies also demonstrate that adenosine alone does not elicit a response after CS exposure, most likely attributable to the decrease in plasma membrane CFTR levels (Fig. 3). Because PDE4 inhibitors can increase intracellular levels of cAMP and activate CFTR (2, 38), we hypothesized that roflumilast could increase ASL after CS exposure. With a 30-min roflumilast pretreatment, adenosine was capable of bringing ASL height into the normal range after CS exposure in HBECs (Fig. 3). These results indicated that roflumilast enhanced intracellular cAMP levels to a greater extent than adenosine and maximally activated the remaining CFTR. This activation may have increased anion secretion and prevented ASL dehydration caused by CS exposure. That this response was due to CFTR was confirmed by our observations that 1) roflumilast/adenosine failed to induce ASL secretion in ΔF508 CF HBECs and 2) bumetanide inhibited this response (Fig. 4, A and B). Interestingly, when the short palate and nasal epithelial clone 1 (SPLUNC1)-derived peptide S18, a known ENaC antagonist (24), was added after CS exposure along with the adenosine, ASL height was significantly increased, suggesting that the observed effect with the roflumilast/adenosine combination was not due itself to an inhibition of ENaC and that subsequent ENaC inhibition with S18 would enhance surface rehydration (Fig. 4B).
Lambert et al. (32) recently demonstrated that a 24-h pretreatment with roflumilast could mitigate the effects caused by CS on ASL height (32). However, patients with COPD are more likely to take roflumilast after ASL dehydration has already occurred. Therefore, we performed additional studies with roflumilast administered in combination with adenosine after acute CS exposure. These studies demonstrated that the combination treatment resulted in an increased rate of ASL height recovery (Fig. 5). CB, however, is often the result of long-term smoking, so the ability of roflumilast to restore ASL after chronic smoke exposure is crucial. The typical life of these primary, nonimmortalized cultures is ∼6 wk, and, because it is not feasible to expose HBECs to CS over the time frame that it would take to develop COPD (i.e., years), we exposed them to CS every day for 5 days, which induced a persistent inhibition of ASL height without adversely affecting HBEC integrity. After this time, a single dose of roflumilast with adenosine (i.e., 1 h of exposure) was also effective at rehydrating chronic CS-exposed HBECs (Fig. 6). This further highlighted the potential role of roflumilast in ASL rehydration for patients with COPD. It is likely that the residual CFTR activity seen with chronic smoke is sufficient to restore ASL hydration, assuming that this CFTR is fully activated.
Bronchodilators such as isoproterenol and salmeterol relax the airway smooth muscles (11) and are one of the main therapies used to treat decreased airflow in COPD. These bronchodilators stimulate Gs-linked β2-adrenergic receptors and increase the concentration of intracellular cAMP. Because β2-adrenergic agonists also activate CFTR (7), the ability of salmeterol and isoproterenol to restore ASL height after CS exposure was investigated and compared with adenosine with or without roflumilast (Fig. 6). In contrast to adenosine, which had a small effect, neither salmeterol nor isoproterenol had any significant effect on ASL height when added apically (data not shown) or basolaterally. To confirm that salmeterol and isoproterenol were effective in airway epithelia after air and CS exposure, intracellular cAMP levels were measured by ELISA in air- and CS-exposed HBECs. As observed previously with roflumilast, salmeterol and isoproterenol pretreatment significantly enhanced the effects of adenosine to increase intracellular cAMP (Fig. 6E). Similar results were seen in CS-exposed HBECs. Additionally, the isoproterenol/adenosine-induced cAMP levels were significantly greater in the CS- than the air-exposed airway epithelia (Fig. 6E). β2-Adrenergic receptors associate with CFTR in the apical plasma membrane in a similar fashion to A2BR (40, 57). Furthermore, CFTR increases plasma membrane A2BR protein levels, and, subsequently, adenosine-induced cAMP production is enhanced in the presence of CFTR, whereas isoproterenol-induced cAMP production is not (57). These data suggest that the interaction between GPCRs, CFTR, and adenylate cyclase is very different. In our studies, coadministration of adenosine and roflumilast after CS exposure resulted in a higher ASL compared with roflumilast treatment alone. We do not presently know why β2-agonists were significantly less effective at increasing ASL height than adenosine; studies on these interactions are ongoing.
We next hypothesized that the clinically relevant combination of salmeterol and roflumilast would lead to an increase in ASL height that was greater than that seen with roflumilast alone. However, roflumilast, but not salmeterol, led to a significant increase in ASL height (Fig. 8, A and B). Interestingly, when the two compounds were combined, there was an inhibition of the roflumilast effect (Fig. 8B). Although there are no previous reports of salmeterol affecting the ability of roflumilast to inhibit PDE4, the data from this study support this possibility and may explain the loss of ASL recovery following coadministration. It has recently been reported that patients taking roflumilast had improved FEV1 (both before and after bronchodilator application) that was similar to results seen with patients taking corticosteroids (9). Previous clinical studies on patients already using other therapies before receiving roflumilast indicate that it may be beneficial. For example, in two 6-mo studies, patients who had moderate to severe COPD and were already receiving salmeterol showed significant additional improvements in lung function with the addition of roflumilast (4, 20). Results from our study indicate that roflumilast may help ASL rehydration in patients with moderate to severe COPD and that this beneficial effect may be reduced when administered along with a LABA.
In summary, roflumilast is capable of elevating intracellular cAMP in human airway epithelia. Pretreatment with roflumilast elevates intracellular cAMP after CS exposure and prevents ASL dehydration in HBECs exposed to CS. Roflumilast also aids ASL recovery after chronic smoke exposure, restoring the ASL to within normal ranges (∼7 μm). Treating ASL dehydration associated with CB would be beneficial to the wellbeing of patients with COPD. Our results suggest that roflumilast could be used as a therapy for patients with COPD who have dehydrated ASL attributable to acquired CFTR dysfunction caused by CS and other possible inhaled toxins.
GRANTS
This work was funded by NIH Grants HL108927, HL034322, DK065988, CF Foundation R026-CR11, and by a Forest Research International Grant.
DISCLOSURES
R. Tarran has equity in Spyryx Biosciences. X. Qian and J. Freire are employees of Forest Research Institute. The authors declare no other conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
Author contributions: J.T., X.Q., J.F., and R.T. conception and design of research; J.T. and R.T. performed experiments; J.T. and R.T. analyzed data; J.T., X.Q., J.F., and R.T. interpreted results of experiments; J.T. and R.T. drafted manuscript; J.T., X.Q., J.F., and R.T. edited and revised manuscript; J.T., X.Q., J.F., and R.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Mr. Michael Watson for technical assistance and The University of North Carolina at Chapel Hill Cystic Fibrosis Center Tissue Core for isolating and providing cells from donor lungs. ICUE2 was kindly donated by Dr. John Violin (Duke University).
REFERENCES
- 1.Angelis N, Porpodis K, Zarogoulidis P, Spyratos D, Kioumis I, Papaiwannou A, Pitsiou G, Tsakiridis K, Mpakas A, Arikas S, Tsiouda T, Katsikogiannis N, Kougioumtzi I, Machairiotis N, Argyriou M, Kessisis G, Zarogoulidis K. Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis 6: S167–S172, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes AP, Livera G, Huang P, Sun C, O'Neal WK, Conti M, Stutts MJ, Milgram SL. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem 280: 7997–8003, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 56: 515–548, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Bateman ED, Rabe KF, Calverley PM, Goehring UM, Brose M, Bredenbroker D, Fabbri LM. Roflumilast with long-acting beta2-agonists for COPD: influence of exacerbation history. Eur Respir J 38: 553–560, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Boswell-Smith V, Page CP. Roflumilast: a phosphodiesterase-4 inhibitor for the treatment of respiratory disease. Expert Opin Investig Drugs 15: 1105–1113, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58: 157–170, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest 78: 1245–1252, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calverley PM, Anderson JA, Celli B, Ferguson GT, Jenkins C, Jones PW, Yates JC, Vestbo J. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 356: 775–789, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176: 154–161, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, Zielenski J, Durie P. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med 173: 1139–1144, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Cazzola M, Page CP, Calzetta L, Matera MG. Pharmacology and therapeutics of bronchodilators. Pharmacol Rev 64: 450–504, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci 14: 310–317, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clancy JP, Ruiz FE, Sorscher EJ. Adenosine and its nucleotides activate wild-type and R117H CFTR through an A2B receptor-coupled pathway. Am J Physiol Cell Physiol 276: C361–C369, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Clunes LA, Bridges A, Alexis N, Tarran R. In vivo versus in vitro airway surface liquid nicotine levels following cigarette smoke exposure. J Anal Toxicol 32: 201–207, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clunes LA, Davies CM, Coakley RD, Aleksandrov AA, Henderson AG, Zeman KL, Worthington EN, Gentzsch M, Kreda SM, Cholon D, Bennett WD, Riordan JR, Boucher RC, Tarran R. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J 26: 533–545, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481–511, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 354: 241–250, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Donohue JF. Therapeutic responses in asthma and COPD. Bronchodilators. Chest 126: 125S–137S; discussion 159S–161S, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis 148: 91–97, 1993. [DOI] [PubMed] [Google Scholar]
- 20.Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, Rabe KF. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long-acting bronchodilators: two randomised clinical trials. Lancet 374: 695–703, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Hatzelmann A, Morcillo EJ, Lungarella G, Adnot S, Sanjar S, Beume R, Schudt C, Tenor H. The preclinical pharmacology of roflumilast—a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther 23: 235–256, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Hirota N, Martin JG. Mechanisms of airway remodeling. Chest 144: 1026–1032, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Hirsh AJ, Sabater JR, Zamurs A, Smith RT, Paradiso AM, Hopkins S, Abraham WM, Boucher RC. Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J Pharmacol Exp Ther 311: 929–938, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Hobbs CA, Blanchard MG, Alijevic O, Tan CD, Kellenberger S, Bencharit S, Cao R, Kesimer M, Walton WG, Henderson AG, Redinbo MR, Stutts MJ, Tarran R. Identification of SPLUNC1′s ENaC-inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airway cultures. Am J Physiol Lung Cell Mol Physiol 305: L990–L1001, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol 4: 435–459, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 35: 91–100, 2009. [DOI] [PubMed] [Google Scholar]
- 27.Jiang C, Finkbeiner WE, Widdicombe JH, McCray PB Jr, Miller SS. Altered fluid transport across airway epithelium in cystic fibrosis. Science 262: 424–427, 1993. [DOI] [PubMed] [Google Scholar]
- 28.Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol 117: 18–24; quiz 25, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Kaur M, Holden NS, Wilson SM, Sukkar MB, Chung KF, Barnes PJ, Newton R, Giembycz MA. Effect of β2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am J Physiol Lung Cell Mol Physiol 295: L505–L514, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Klass DJ. Cigarette smoke exposure in vivo increases cyclic GMP in rat lung. Arch Environ Health 35: 347–350, 1980. [DOI] [PubMed] [Google Scholar]
- 31.Lahu G, Nassr N, Hunnemeyer A. Pharmacokinetic evaluation of roflumilast. Expert Opin Drug Metab Toxicol 7: 1577–1591, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Lambert JA, Raju SV, Tang LP, McNicholas CM, Li Y, Courville CA, Farris RF, Coricor GE, Smoot LH, Mazur MM, Dransfield MT, Bolger GB, Rowe SM. CFTR activation by roflumilast contributes to therapeutic benefit in chronic bronchitis. Am J Respir Cell Mol Biol 50: 549–558, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S, Boucher RC. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J Biol Chem 279: 36855–36864, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Veilleux A, Zhang L, Young A, Kwok E, Laliberte F, Chung C, Tota MR, Dube D, Friesen RW, Huang Z. Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J Pharmacol Exp Ther 314: 846–854, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance—United States, 1971–2000. MMWR Surveill Summ 51: 1–16, 2002. [PubMed] [Google Scholar]
- 36.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95: 1005–1015, 1998. [DOI] [PubMed] [Google Scholar]
- 37.McCahill AC, Huston E, Li X, Houslay MD. PDE4 associates with different scaffolding proteins: modulating interactions as treatment for certain diseases. Hand Exp Pharmacol 186: 125–166, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Milara J, Armengot M, Banuls P, Tenor H, Beume R, Artigues E, Cortijo J. Roflumilast N-oxide, a PDE4 inhibitor, improves cilia motility and ciliated human bronchial epithelial cells compromised by cigarette smoke in vitro. Br J Pharmacol 166: 2243–2262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mundell SJ, Olah ME, Panettieri RA, Benovic JL, Penn RB. Regulation of G protein-coupled receptor-adenylyl cyclase responsiveness in human airway smooth muscle by exogenous and autocrine adenosine. Am J Respir Cell Mol Biol 24: 155–163, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci USA 100: 342–346, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oldenburger A, Maarsingh H, Schmidt M. Multiple facets of cAMP signalling and physiological impact: cAMP compartmentalization in the lung. Pharmaceuticals (Basel) 5: 1291–1331, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pappas K, Papaioannou AI, Kostikas K, Tzanakis N. The role of macrophages in obstructive airways disease: Chronic obstructive pulmonary disease and asthma. Cytokine 64: 613–625, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Poggi P, Rota MT, Boratto R. The volatile fraction of cigarette smoke induces alterations in the human gingival fibroblast cytoskeleton. J Periodontal Res 37: 230–235, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Rab A, Rowe SM, Raju SV, Bebok Z, Matalon S, Collawn JF. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. Am J Physiol Lung Cell Mol Physiol 305: L530–L541, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Randell SH, Fulcher ML, O'Neal W, Olsen JC. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol 742: 285–310, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 136: 1554–1560, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol 39: 127–132, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloane PA, Shastry S, Wilhelm A, Courville C, Tang LP, Backer K, Levin E, Raju SV, Li Y, Mazur M, Byan-Parker S, Grizzle W, Sorscher EJ, Dransfield MT, Rowe SM. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One 7: e39809, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taegtmeyer AB, Leuppi JD, Kullak-Ublick GA. Roflumilast—a phosphodiesterase-4 inhibitor licensed for add-on therapy in severe COPD. Swiss Med Wkly 142: w13628, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell 8: 149–158, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J Gen Physiol 127: 591–604, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 11: 149, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ulrik CS. Clinical benefit of fixed-dose dual bronchodilation with glycopyrronium and indacaterol once daily in patients with chronic obstructive pulmonary disease: a systematic review. Int J Chron Obstruct Pulmon Dis 9: 331–338, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van den Berge M, Hylkema MN, Versluis M, Postma DS. Role of adenosine receptors in the treatment of asthma and chronic obstructive pulmonary disease: recent developments. Drugs R D 8: 13–23, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Versluis M, ten Hacken N, Postma D, Barroso B, Rutgers B, Geerlings M, Willemse B, Timens W, Hylkema M. Adenosine receptors in COPD and asymptomatic smokers: effects of smoking cessation. Virchows Arch 454: 273–281, 2009. [DOI] [PubMed] [Google Scholar]
- 56.Violin JD, DiPilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ. Beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem 283: 2949–2961, 2008. [DOI] [PubMed] [Google Scholar]
- 57.Watson MJ, Worthington EN, Clunes LA, Rasmussen JE, Jones L, Tarran R. Defective adenosine-stimulated cAMP production in cystic fibrosis airway epithelia: a novel role for CFTR in cell signaling. FASEB J 25: 2996–3003, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]