

Abstract
Fragments of the mitochondrial genome released into the systemic circulation after mechanical trauma, termed mitochondrial DNA damage-associated molecular patterns (mtDNA DAMPs), are thought to mediate the systemic inflammatory response syndrome. The close association between circulating mtDNA DAMP levels and outcome in sepsis suggests that bacteria also might be a stimulus for mtDNA DAMP release. To test this hypothesis, we measured mtDNA DAMP abundance in medium perfusing isolated rat lungs challenged with an intratracheal instillation of 5 × 107 colony-forming units of Pseudomonas aeruginosa (strain 103; PA103). Intratracheal PA103 caused rapid accumulation of selected 200-bp sequences of the mitochondrial genome in rat lung perfusate accompanied by marked increases in both lung tissue oxidative mtDNA damage and in the vascular filtration coefficient (Kf). Increases in lung tissue mtDNA damage, perfusate mtDNA DAMP abundance, and Kf were blocked by addition to the perfusion medium of a fusion protein targeting the DNA repair enzyme Ogg1 to mitochondria. Intra-arterial injection of mtDNA DAMPs prepared from rat liver mimicked the effect of PA103 on both Kf and lung mtDNA integrity. Effects of mtDNA and PA103 on Kf were also attenuated by an oligodeoxynucleotide inhibitor of Toll-like receptor 9 (TLR-9) by mitochondria-targeted Ogg1 and by addition of DNase1 to the perfusion medium. Collectively, these findings are consistent with a model wherein PA103 causes oxidative mtDNA damage leading to a feed-forward cycle of mtDNA DAMP formation and TLR-9-dependent mtDNA damage that culminates in acute lung injury.
Keywords: mitochondrial DNA, damage-associated molecular patterns, Ogg1, oxidant stress, lung injury
mechanisms linking sepsis to the acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome (MODS) are not completely understood, and pharmacological options to forestall this progression are not available. Several reports suggest a role for damage-associated molecular patterns (DAMPs), which are endogenous signaling molecules that are released to “alarm” the body of cellular damage. One recently described type of DAMP is composed of mitochondrial DNA (mtDNA) fragments (20), possibly associated with mtDNA-binding proteins such as mitochondrial transcription factor A (TFAM) (19). Recent studies show that mtDNA DAMPs are elevated in the plasma of patients that develop the systemic inflammatory response syndrome after trauma (33, 41) as well as in patients with sepsis, ARDS, and MODS (21, 24). Mitochondrial DNA DAMPs prime neutrophils, which activate and recruit additional immune cells to release proinflammatory cytokines, thus eliciting an inflammatory response with the potential to propagate injury to sites distant from the initiating event (20, 34, 41). The molecular trigger for formation and release of mtDNA DAMPs has not been investigated.
A feature common to ARDS and MODS, regardless of the initial insult, is an increase in the production of reactive oxygen species (ROS) (8, 35, 36). Mitochondrial DNA is a target of ROS and is considerably more sensitive to oxidative damage than nuclear DNA (2, 15, 37). Modulation of mtDNA repair exerts coordinate effects on ROS-induced cytotoxicity (7, 11, 20, 28, 29), thus suggesting that mtDNA could function as a molecular sentinel governing cell fate in response to oxidant stress (31). Oxidative damage to the mitochondrial genome leads to a preponderance of strand breaks and abasic sites, which in turn promotes mtDNA degradation (32). In light of these observations, a key goal of the present study was to determine whether bacterial challenge in isolated lungs leads to oxidative mtDNA damage-dependent mtDNA DAMP formation.
MtDNA DAMPs act on Toll-like receptor 9 (TLR-9) found in endosomes or on the plasma membrane of many cell types implicated in the pathogenesis of ARDS, including endothelial cells (13, 30). In this cell population, mtDNA DAMPs cause a TLR-9-dependent decrease in barrier function (10, 34). Accordingly, a second goal of these studies was to determine whether mtDNA DAMPs released in response to bacterial challenge contribute to increases in pulmonary vascular endothelial permeability. Our results point to the intriguing prospect that intratracheal bacterial challenge leads to a regenerative cycle of oxidative mtDNA damage and mtDNA DAMP formation culminating in TLR-9-dependent vascular injury.
MATERIALS AND METHODS
Isolated lung preparation and assessment of the vascular filtration coefficient.
Lungs of adult male Sprague-Dawley rats (250–300 g, Charles River Breeding Laboratories) were isolated and perfused as described previously (7) according to a protocol approved by the IACUC. Briefly, after intraperitoneal (IP) injection of sodium pentobarbital, rats were ventilated using a Harvard rodent ventilator (Harvard Apparatus) with a humidified gas mixture consisting of 21% O2-5% CO2 and N2 at 60 breaths/min, a tidal volume of 2.5 ml, and a positive end-expiratory pressure of 2.5 cmH2O. Following injection of heparin sulfate (100 U), the pulmonary artery and left ventricle were cannulated, and the pulmonary circulation was perfused at a constant flow rate (0.04 ml/g body wt per min) with physiological salt solution (in mM: 3.2 CaCl2, 119, 4.7 KCl, 1.17 MgSO4, 1.18 KH2PO4, 22.6 NaHCO3, and 5.5 d-glucose) containing 4% bovine serum albumin. The heart and lungs were removed en bloc and suspended in a humidified chamber from a force displacement transducer (Grass FT03, Grass Instruments) to record real-time changes in lung weight. In all experiments, zone III conditions were maintained. Pulmonary artery and venous pressures (Pa and Pv, respectively) were continuously monitored with Cobe pressure transducers (Cobe Laboratories) and a Model 7F Grass polygraph recorder. Pulmonary capillary pressure (Pc) was estimated using the double-occlusion method described elsewhere (25). The vascular filtration coefficient, Kf, was calculated as described previously, normalized to 100 g of predicted lung weight, and expressed in ml·min−1·cmH2O−1·100 g−1 lung weight.
Experimental protocols.
After a 30-min equilibration period, the following agents were added directly to the perfusate reservoir and allowed to circulate for 30 min before challenge: N-acetylcysteine (1 mM), mt-targeted Ogg1 fusion protein (10 μg/ml), DNase1 (0.5 U/ml), or an inhibitory oligodeoxynucleotide (Invivogen, 5′ - TCC TGG CGG GGA AGT - 3′) to TLR-9, the receptor for unmethylated CpG DNA (5.3 μg/ml). All pharmacological agents were obtained from Sigma unless specified. Pseudomonas aeruginosa (P. aeruginosa), strain 103 (PA103; 5 × 107 colony-forming units, cfu) instilled intratracheally and exogenous, sonicated mtDNA derived from rat liver (16 μg) were administered as bolus injections into the pulmonary arterial cannula.
Exogenous mtDNA DAMPs were prepared from liver of adult male Sprague-Dawley rats weighing 250–350 g. Animals were given an IP injection of sodium pentobarbital, after which one lobe of the liver was dissected and immersed in a tube containing 0.9% sodium chloride and placed on ice. Liver mitochondria were isolated using the Mitochondria Isolation Kit for Tissue (ThermoScientific) according to the manufacturer's protocol. DNA was immediately isolated from mitochondria using the DNeasy Blood and Tissue Kit (Qiagen), again following the manufacturer's instructions. Eluted DNA was immediately sonicated to produce mtDNA fragments using a Vibra-Cell sonicator (Sonics and Materials) at 100% amplitude, 10 times for 30 s, with 30-s intervals between sonications according to methods described by Zhang et al. (41). The lengths of mtDNA fragments prepared as such were determined by gel electrophoresis to be 50–150 base pairs. Aliquots were stored at −20°C.
Ogg1 fusion protein.
A codon-optimized construct of the fusion protein (containing the base excision repair enzyme, Ogg1) attached to a TAT sequence for cellular uptake, a mitochondrial-targeting sequence from manganese superoxide dismutase, a hemagglutinin tag for immunological localization, and a histidine tail were placed in a plasmid for expression in Escherichia coli. Liquid cultures of the transfected bacteria containing the construct were grown to an OD60 = 0.6 and induced with isopropylthiogalactoside for 3 h. After centrifugation, bacteria were resuspended in Buffer A [20 mM Tris·HCl pH 8.0, 500 mM NaCl, 1× protease inhibitor cocktail EDTA-free (Calbiochem), 100 mM PMSF, and 5 mM imidazole]. Bacteria were lysed with sonication using a Branson Sonifier 250 (ThermoFisher Scientific). Following sonication, bacterial lysates were spun in a Beckman ultracentrifuge for 20 min at 105 g. Cleared lysates were then incubated with Ni-NTA-agarose, which was then placed in a column and washed with several volumes of wash buffer (Buffer A with 30 mM imidazole). Elution buffer (Buffer A with 500 mM imidazole) was used to elute bound protein. Purity was assessed using SDS-PAGE analysis. All reagents used in the production of the Ogg1 fusion protein were purchased from Sigma Aldrich, unless otherwise noted. In all experiments, 10 μg/ml of the Ogg1 fusion protein was used as determined by previous studies (7). In addition, our prior work demonstrated in isolated, buffer-perfused rat lungs that the fusion protein distributed almost exclusively into the mitochondrial fraction with little or no nuclear deposition (7).
Quantitative Southern blot analysis of mtDNA damage.
At termination of perfusion, lungs were snap-frozen in liquid nitrogen and stored at −80°C. Lungs were then powdered with a mortar and pestle, and oxidative mtDNA damage was assessed using previously described methods (7, 15). In brief, purified DNA samples were digested with the restriction enzymes PpuMI and AhdI (New England Biolaboratories). Digested samples were precipitated, dissolved in Tris-EDTA buffer, and quantified using a VersaFluor fluorometer (Bio-Rad) with a Quant-IT Picogreen dsDNA Assay kit (Molecular Probe). To reveal oxidative base damage, the DNA sample was divided into two aliquot parts; one was treated with formamidopyrimidine glycosylase (Fpg), a bacterial DNA repair enzyme that recognizes and cleaves oxidized purines to create single-strand breaks (New England Biolaboratories), whereas the other was untreated. Samples with and without Fpg treatment were then incubated with 0.1 N NaOH for 15 min at 37°C to cleave the strand at abasic sites or sites of damage to the deoxyribose backbone, mixed with loading dye, and resolved in a 0.6% agarose alkaline gel. Following electrophoresis, DNA was vacuum-transferred to a nylon membrane (Roche Diagnostics) and hybridized with a PCR-generated probe to a specific region of mtDNA. This probe was generated using rat mtDNA sequence as a template and the following primers: 5′-CCCTACTTACTGGCTTCAATCTAC-3′ for the sense strand and 5′-CATACCATACCTATATATCCGAAGG-3′ for the anti-sense strand and subsequently labeled with a DIG-labeled kit (Roche Diagnostics). The 1,016-bp product was then hybridized with a 13.6-kb fragment of rat mtDNA obtained after PpuMI and AhdI digestion. Hybridization bands were detected with Amersham Hyperfilm Enhanced Chemiluminescence (GE Healthcare) and a Gel Logic 1500 Imaging System (Kodak). Changes in equilibrium lesion density were calculated as negative ln of the quotient of hybridization intensities in Fpg-treated and untreated (alkali only) control bands and normalized to 10 kb, as described previously (7).
Isolation of DNA from perfusion medium and determination of mtDNA abundance.
Approximately 50 ml of perfusate was collected after each isolated perfused rat lung experiment and stored at −80°C until further processing. After samples were thawed, 10 ml was centrifuged at 18,000 g at 4°C for 20 min to pellet cells and organelles. The upper 7 ml of the supernatant was then transferred to a 50-ml tube and centrifuged at 100,000 g at 4°C for 30 min to remove debris, leaving any free DNA suspended in the supernatant. Following this final centrifugation step, DNA was isolated from 1 ml of the supernatant using the DNeasy Blood and Tissue Kit (Qiagen) following manufacturer's instructions. Isolated DNA was stored at −80°C.
Quantitative real-time PCR was used to detect selected 200-bp sequences of the mitochondrial genome, a sequence of the nuclear genome encoding 28S rRNA, and a bacterial DNA sequence encoding 16S rRNA. Primers, developed using Beacon Designer software, are shown in Table 1. Mitochondrial DNA sequences quantified included the displacement loop (D-loop), which is the transcriptional origin of the genome and is particularly sensitive to oxidative injury (27) as well as regions encoding cytochrome C oxidase subunit II (COX2), ATP synthase subunit 6 (ATP6), and NADH dehydrogenase subunit 4 (ND4). ATP6 and ND4 lie within the mtDNA common deletion sequence, a 4,977-bp region of the mitochondrial genome whose deletion is frequently associated with various pathologies, whereas COX2 and D-loop flank either side of the deleted sequence. Quantitative RT-PCR was done using the HotStart-IT SYBR Green One-Step qRT-PCR kit (USB) following manufacturer's instructions. An iCycler with the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad) was used to analyze each sample. The amplification efficiency of each primer set was determined by calculating the slope of the calibration curve, which was constructed by performing PCR with serial dilutions of a known concentration of template DNA.
Table 1.
Primers used for RT-PCR
| Direction | Primer Sequence | |
|---|---|---|
| COX2 | Forward | GCTGTCATTCTTATTCTAA |
| Reverse | GGATTATGTAGGAGTCAA | |
| ATP6 | Forward | CGAAACTATCAGCCTATT |
| Reverse | AGTAGAAGTAGAATAATAAATGTAA | |
| ND4 | Forward | CTCCGCAACAGAACTAAT |
| Reverse | GTTGAGTGTTCCTATTGAGT | |
| D-loop | Forward | ATTTATCCTCATAGACAAAG |
| Reverse | TTTACCAATGCTAAGATTT | |
| Bacterial 16S | Forward | TTCGGACCTCACGCTATCAG |
| Reverse | ATCATCCTCTCAGACCAGTT | |
| Nuclear 28S | Forward | GATTCCCACTGTCCCTACC |
| Reverse | ACCTCTCATGTCTCTTCACC |
Analysis of the indicated mitochondrial, nuclear and bacterial DNA sequences. COX2, cytochrome C oxidase subunit II; ND4, NADH dehydrogenase subunit 4; D-loop, displacement loop.
Statistical analyses.
Data are expressed as means ± SE. Determination of significant differences between all groups was done with a one-way ANOVA and a Newman-Keuls post hoc test. Significance was defined as P < 0.05.
RESULTS
P. aeruginosa causes time-dependent increases in vascular permeability accompanied by mtDNA DAMP release into the perfusion medium.
As shown in Fig. 1A, intratracheal instillation of PA103 caused time-dependent increases in Kf. Within 15 min of PA103 administration, Kf was significantly increased above baseline and at 60 min post-PA103 challenge was higher than both baseline and 15 min.
Fig. 1.
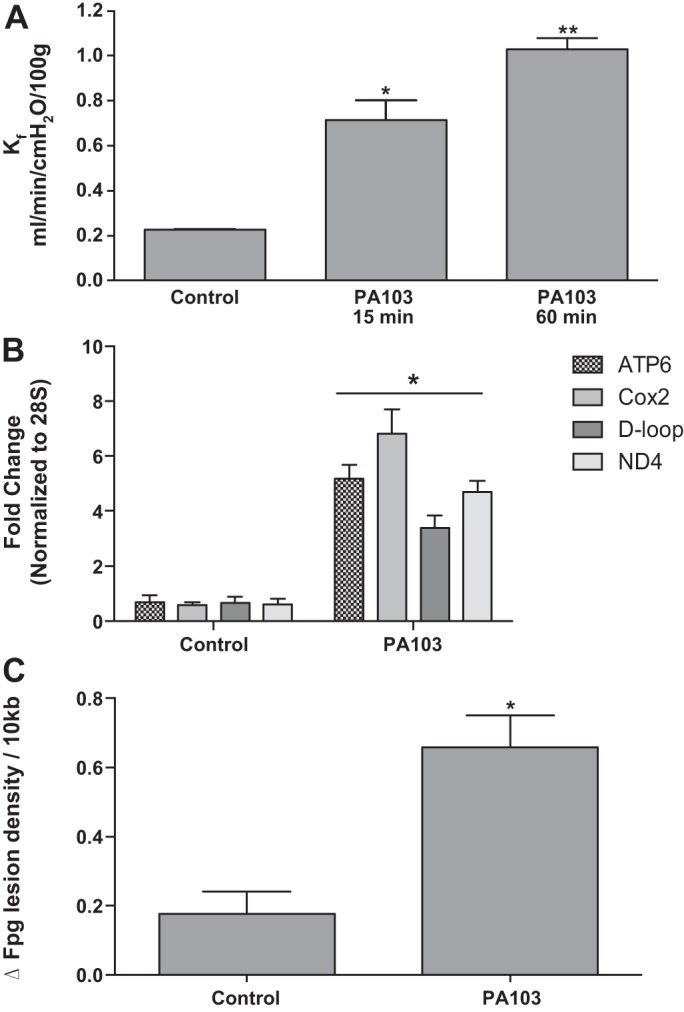
Pseudomonas aeruginosa (P. aeruginosa) causes time-dependent increases in vascular permeability accompanied by mitochondrial DNA (mtDNA) damage-associated molecular pattern (DAMP) release into the perfusion medium. Rat lungs were isolated, mechanically ventilated, and perfused at a constant flow rate with physiological salt solution containing albumin as a colloid. P. aeruginosa, strain 103 [PA103; 5 × 107 colony-forming units (CFU) in 100 μl physiological salt solution] was instilled as a bolus into the trachea. As shown in A, relative to control preparations not challenged with bacteria (n = 12), instillation of PA103 (n = 9) caused time-dependent increases in the vascular filtration coefficient (Kf). *Different from control at P < 0.05; **different from both control and 15 min post-PA103 at P < 0.05. In results of companion experiments shown in B, perfusate contents of 200-bp sequences of the indicated mtDNA regions measured by qRT-PCR were increased at 15 min after PA103 instillation relative to controls although not all sequences were increased to the same extent (n = 4 for both groups). *Different from control at P < 0.05. Finally, in results of separate experiments shown in C, formamidopyrimidine DNA glycosylase (Fpg)-detectable oxidative base damage in the mtDNA genome, determined by quantitative Southern blot analysis, was increased in lung tissue challenged 15 min previously with PA103 relative to damage levels in control perfused lung tissue (n = 4 for both groups). *Different from control at P < 0.05. Cox2, cytochrome C oxidase subunit II; ND4, NADH dehydrogenase subunit 4; D-loop, displacement loop.
To determine whether mitochondrial or nuclear DNA sequences accumulated in perfusion medium in a manner temporally associated with the bacteria-induced rise in Kf, perfusion medium was reserved at 15 min after PA103, and quantitative RT-PCR was used to measure the perfusate concentration of 200-bp DNA sequences of ATP6, COX2, ND4, and D-loop mtDNA regions. A 200-bp sequence of the nuclear genome encoding 28S rRNA also was determined. Low levels of this nuclear DNA sequence detected in control lungs did not change in lungs instilled with the bacteria (vide infra). This sequence was, accordingly, used to normalize changes in the abundance of mtDNA sequences evoked by PA103. Figure 1B shows that circulating abundances of mtDNA sequences were increased in the perfusion medium within 15 min of PA103 administration relative to lungs not challenged with PA103. The released sequences did not accumulate to the same extent; for example, COX2 was most prominently elevated compared with the other three sequences (significant at P < 0.05). To test for the presence of bacterial DNA, primers for a 200-bp sequence of the bacterial 16S rRNA gene were also generated. This sequence, however, remained undetectable (data not shown).
We next determined whether mtDNA release is associated with lung tissue oxidative mtDNA damage. DNA was isolated from lungs harvested 15 min after PA103 challenge, and quantitative Southern blot analysis was used to detect oxidative base damage in a 10.8-kb sequence of the mitochondrial genome harboring most of the coding region and the common deletion sequence. As shown in Fig. 1C, within 15 min of intratracheal administration of PA103, there was an approximately fourfold increase in oxidative mtDNA damage in intact lung tissue.
Mitochondria-targeted Ogg1 reduces PA103-induced increases in Kf and perfusate mtDNA sequences and decreases oxidative mtDNA damage.
As shown in Fig. 2A, whereas addition of 10 μg/ml mt-targeted Ogg1 fusion protein to the perfusate reservoir failed to influence Kf in control lungs, 15-min treatment with the DNA repair enzyme before intratracheal instillation of PA103 prevented increases in Kf normally associated with bacterial challenge. Similarly, Fig. 2B shows that mt-targeted Ogg1, although not influencing baseline levels of circulating mtDNA sequences, prevented elevation-evoked PA103. In addition, Fig. 2B displays the perfusion medium abundances of 200-bp sequences of nuclear DNA encoding 28S rRNA, which was used to normalize the abundances of mtDNA sequences. Importantly, no changes in this nuclear sequence from control were noted in perfusate from lungs treated with PA103 or mt-targeted Ogg1 given either alone or in combination. Thus changes in perfusion medium contents of mtDNA fragments do not seem to be linked to release of total cell DNA. Finally, as shown in Fig. 2C, lung tissue oxidative mtDNA base damage caused by intratracheal PA103 also was blocked by pretreatment with mt-targeted Ogg1.
Fig. 2.

Mitochondria-targeted 8-oxoguanine DNA glycosylase (Ogg1) reduces PA103-induced increases in Kf and perfusate mtDNA sequences and decreases oxidative mtDNA damage. Rat lungs were isolated, mechanically ventilated, and perfused at a constant flow rate with physiological salt solution containing albumin as a colloid. In some preparations, a fusion protein targeting the DNA repair enzyme, Ogg1, was added to the perfusate to achieve a final concentration of 10 μg/ml and allowed to recirculate for 30 min before intratracheal instillation of P. aeruginosa (PA103; 5 × 107 CFU in 100 μl physiological salt solution). As shown in A, relative to control and PA103-challenged preparations (n = 12 and 9), treatment with mt-targeted Ogg1 alone (n = 4) failed to alter baseline Kf but prevented the increase normally evoked by PA103 (n = 4). B demonstrates that, whereas Ogg1 alone (n = 4) failed to alter baseline perfusate mtDNA DAMP levels, the fusion protein inhibited the increase normally evoked by PA103. In addition, neither PA103 nor Ogg1, given alone or in concert, altered accumulation of a ≈200-bp nuclear DNA sequence encoding 28S rRNA (n = 4). C shows that, although Ogg1 alone (n = 4) failed to alter the baseline level of Fpg-detectable oxidative mtDNA damage, the fusion protein inhibited the increase normally evoked by PA103 (n = 4). Note that, in all three panels, bars depicting control and PA103 responses in the absence of mt-targeted Ogg1 are identical to those displayed in Fig. 1. *Significantly different from all other groups at P < 0.05.
Evidence that mtDNA DAMPs mediate PA103 effects on lung endothelial permeability.
Because PA103 increased mtDNA concentration in the perfusion medium and because mtDNA DAMPs activate inflammatory (20, 41) and endothelial cells (10, 34), we tested the idea that mtDNA DAMPs contributed to PA103-induced increase in Kf. First, we determined whether mtDNA DAMPs prepared from rat liver and administered as a bolus injection into the pulmonary artery mimicked the effect of PA103 on Kf. The outcome of these studies, displayed in Fig. 3A, shows that, 15 min after intrapulmonary arterial injection of 16 μg exogenous mtDNA DAMPs, Kf increased to a level similar to the response evoked by intratracheal instillation of PA103 (5 × 107 cfu/100 μl).
Fig. 3.
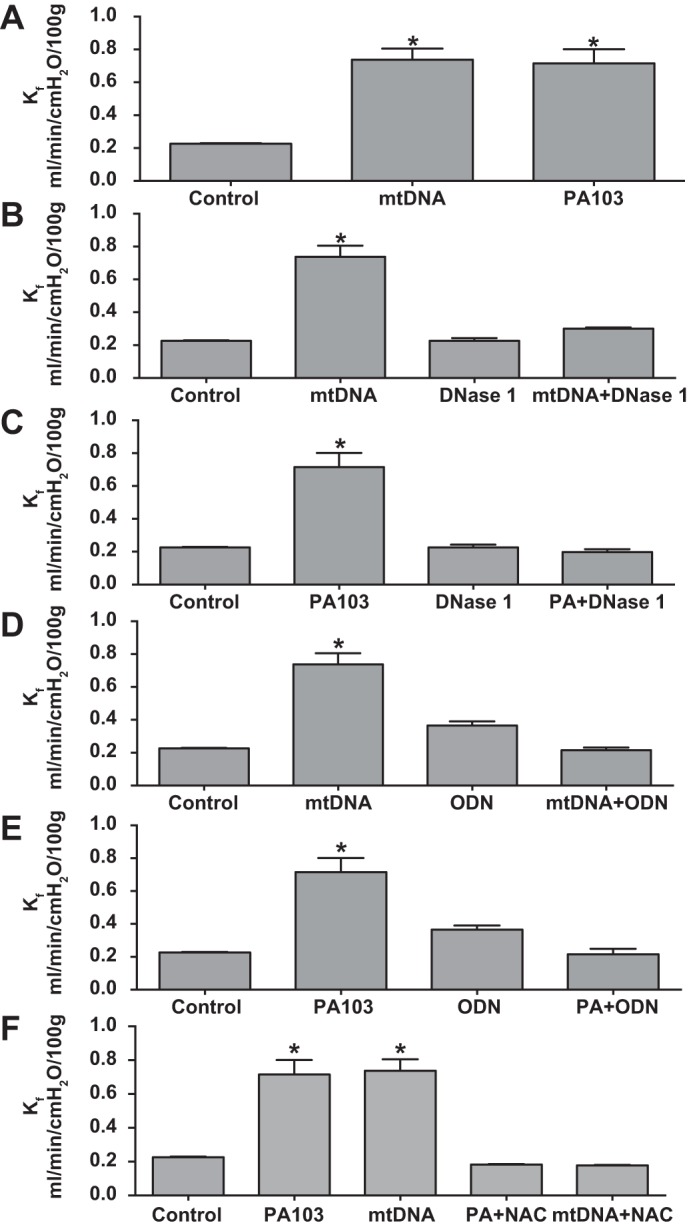
Evidence that mtDNA DAMPs mediate PA103 effects on lung endothelial permeability. Results shown in A–F were generated from experiments in isolated, mechanically ventilated rat lungs perfused at a constant flow rate with physiological salt solution containing albumin as a colloid. A shows that 16 μg exogenous mtDNA DAMPs (n = 4) increased Kf to a level similar to that evoked by intratracheal administration of 5 × 107 CFU PA103. B and C show that enhanced DNA degradation with DNase 1 (0.5 U/ml) added to the perfusion medium 15 before mtDNA or PA103 challenge, respectively, inhibited the increase in Kf evoked by both stimuli. Similarly, D and E show that the effects of mtDNA DAMPs and PA103 on Kf are both suppressed by a Toll-like receptor 9 inhibitory oligodeoxynucleotide (ODN; added to the perfusate reservoir to achieve a final concentration of 5.3 μg/ml; n = 4 for all groups, except control PA103, for which n = 12 and 9, respectively). F shows that the antioxidant N-acetylcysteine (NAC) added to the perfusate reservoir to attain a final concentration of 1 mM also inhibited increases in Kf evoked by PA103 and mtDNA. *Significantly different from all other groups at P < 0.05.
We next determined whether DNase1, which degrades DNA, exerted similar inhibitory effects on mtDNA- and PA103-induced increases in Kf. Results of these studies, displayed in Fig. 3, B and C, respectively, show that DNase1 added to the perfusion medium inhibits increases in Kf evoked by both stimuli. Finally, because mtDNA DAMPs activate neutrophils and endothelial cells at least in part through a TLR-9-dependent mechanism (10, 20, 34, 41), we evaluated the effect of an oligodeoxynucleotide antagonist of TLR-9 on increases in Kf initiated by rat liver-derived mtDNA DAMPs and PA103. Figure 3, D and E, shows that, as expected, the oligodeoxynucleotide TLR-9 antagonist inhibited the increase in Kf evoked by exogenous mtDNA DAMPs as well as PA103.
Lastly, we determined whether addition of N-acetylcysteine to the perfusion medium blocked PA103- and mtDNA-induced increases in Kf. On the basis of earlier reports (5, 9, 17), we expected that the antioxidant would suppress the response to PA103, thus implicating ROS as a link between bacterial challenge and increases in Kf. Whether the antioxidant would exert similar actions on the response to mtDNA was less certain. As shown in Fig. 3F, we found that, as anticipated, N-acetylcysteine suppressed the response to PA103. Perhaps more interestingly though, the antioxidant also blocked the mtDNA DAMP-induced decrements in rat lung endothelial function.
Exogenous mtDNA DAMPs cause oxidative mtDNA damage-dependent increases in Kf.
In light of the similarities in the actions of PA103 and exogenous mtDNA DAMPs on endothelial barrier function in the isolated rat lung, experiments were conducted to determine whether the effects of mtDNA DAMPs, like PA103, were mediated by oxidative mtDNA damage. First, we determined whether exogenous mtDNA DAMPs caused oxidative DNA damage after intra-arterial administration to perfused rat lungs. As shown in Fig. 4A, quantitative Southern blot analysis revealed that, like PA103, exogenous mtDNA caused an approximate sevenfold increase in the density of oxidative mtDNA damage in intact lung tissue. The increase in lesion density was abolished by addition of mt-targeted Ogg1 to the perfusion medium 15 min before mtDNA challenge. Figure 4B shows that, similar to its actions on mtDNA damage, the Ogg1 fusion protein prevented increases in Kf normally provoked by exogenous mtDNA DAMPs
Fig. 4.
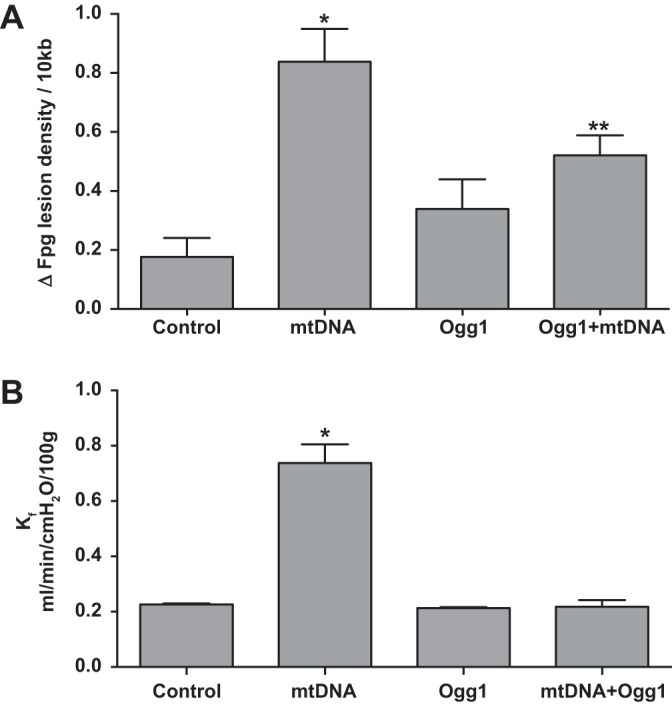
Exogenous mtDNA DAMPs cause oxidative mtDNA damage-dependent increases in Kf. Results shown in A indicate that mtDNA isolated from lungs perfused for 15 min after challenge with 16 μg intra-arterial bolus injections of exogenous mtDNA DAMPs displayed more oxidative damage relative to mtDNA isolated from control lungs. Recirculating perfusion with 10 μg/ml mitochondrially targeted Ogg1 for 15 min failed to alter baseline oxidative mtDNA damage but attenuated the increase normally evoked by mtDNA DAMPs, n = 4/group. *Significantly increased from control at P < 0.05; **significantly different from control and mtDNA DAMPs alone at P < 0.05. B displays results of similarly designed studies, except Kf was measured instead of oxidative mtDNA damage. Here, too, treatment with mitochondrially targeted Ogg1, while not impacting baseline Kf, inhibited the increase evoked by exogenous mtDNA DAMPs; n = 4/group. *Significantly different from all other groups at P < 0.05.
DISCUSSION
Multiple lines of evidence from studies in human subjects suggest that mtDNA DAMPs play important roles in trauma and severe illness leading to ARDS and MODS. For example, mtDNA fragments are elevated in plasma from patients with severe trauma or illness; in both instances, increases in plasma mtDNA are associated with adverse outcomes (21, 24, 33, 41). The potential contribution of mtDNA DAMPs to tissue injury is also supported by persuasive evidence from cell culture and animal models where administration of exogenous mtDNA fragments or prevention of their accumulation leads to concordant effects on cytotoxicity, cellular dysfunction, and tissue inflammation (10, 13, 30, 34, 38, 41). Although these intriguing observations suggest an important pathophysiological role for mtDNA DAMPs in injury propagation and tissue damage, little is known about mechanisms governing mtDNA DAMP formation and release. In eosinophils, mtDNA DAMP release is strikingly rapid, described as “catapult-like,” and requires NADPH-dependent ROS production as an early signaling event (39, 40). Studies in isolated mitochondria also show that formation of mtDNA fragments can be initiated by oxidant stress, with export from the organelle occurring through the permeability transition pore in a size-dependent manner (14). Mechanisms of mtDNA DAMP release have not been yet been examined at the whole organ level in pathologically relevant contexts.
The initial aim of the present study was to determine whether bacterial challenge provoked mtDNA DAMP release from isolated lungs. Validation of this hypothesis would extend the stimuli initiating mtDNA DAMP release from mechanical trauma to an infectious stress, both of which are known to have high levels of ROS and mitochondrial bioenergetic dysfunction. We employed isolated, buffer-perfused lung preparations to eliminate and reduce, respectively, the potentially obfuscating contribution of other organs or circulating blood cells, and we used PA103 as an infectious stimulus because, in an intact rat model, this opportunistic pathogen causes MODS (1). The results demonstrate that an intratracheal dose of PA103 causing prominent decrements in pulmonary endothelial barrier function initiates substantial accumulation of mtDNA in the perfusion medium. The contention that DNA detected in perfusate originates from the mammalian mitochondrial genome and not from bacteria is derived from the inability to detect a unique bacterial DNA encoding 16S rRNA in perfusate over the same time frame. Our confidence that the DNA detected is mitochondrial in origin and not derived from nuclear pseudogenes (23) relates to two considerations, first, that nuclear DNA encoding 28S rRNA did not change with PA103 administration, and, second, that the primers selected for amplification of the mtDNA sequences were specific for rat mtDNA and had no homology to nuclear sequences. The idea that mtDNA was released in fragments rather than intact mitochondrial genome is derived from the fact that the sequences were detected in different abundances; if the sequences detected represented one contiguous segment, it would be expected that their abundances would be similar. Finally, it seems unlikely that the sequences detected originated from intact cells or mitochondria shed or released (22) into the perfusion medium because low- and high-speed centrifugation steps were applied before PCR analysis specifically to remove cells and cellular debris.
Results of the present study support the idea that oxidative damage to the mitochondrial genome initiated formation and release of mtDNA DAMPs into the perfusion medium. First, evidence that an oxidant stress was important for release of mtDNA DAMPs was derived from the finding that, as expected, the antioxidant N-acetylcysteine suppressed the increase in Kf evoked by PA103. We also found that, within 15 min of PA103 instillation into the trachea, the levels of oxidative base damage in lung tissue rose sharply. In this instance, involvement of an oxidant stress is indicated by the specificity of Fpg to recognize and excise 8-oxoguanine and closely related oxidized base products (26). Treatment of perfused lungs with a fusion protein construct targeting the DNA repair glycosylase, Ogg1, to mitochondria (7) also prevented both lung tissue oxidative base damage and mtDNA DAMP accumulation in perfusion medium. In this latter context, whereas enhancing mtDNA repair using targeted Ogg1 did indeed prevent oxidative mtDNA damage, it might be noted that damage repair per se might not be the only mechanism whereby the fusion protein suppresses mtDNA DAMP release (7, 31); it is possible that by binding to oxidatively damaged mtDNA, a prerequisite to initiation of base excision repair, the mt-targeted Ogg1 fusion protein stabilizes the molecule to prevent its fragmentation into mtDNA DAMPs. Completion of repair could thus be a downstream step not directly related to prevention of mtDNA DAMP formation. Additional studies will be required to explore this possibility.
The prospect that oxidative mtDNA damage triggers mtDNA DAMP formation and release raises many interesting questions. First, specific sequences in the mitochondrial genome are known to be prone to oxidative mtDNA damage (12, 15, 16, 27). Because the repair intermediate or degradation product of oxidative base damage, abasic sites, leads to strand instability and DNA degradation (32), it is interesting to speculate that site-specific oxidative mtDNA damage could be a determinant of the break sites in mtDNA and therefore the sequence characteristics of mtDNA DAMPs. This, along with the size of the base damaged-induced fragment, could explain why PA103 engendered differential accumulation of specific mtDNA sequences in perfusion medium; there could be preferential formation of certain fragments as determined by the site-specific pattern of oxidative base damage and attendant differences in the ability of the individual sequences to be exported.
MtDNA DAMPs seem to play a key role in the PA103-mediated erosion of pulmonary vascular endothelial barrier integrity. The contention is based on two general lines of evidence. First, the ability of PA103 to increase Kf in perfused rat lungs was mimicked by exogenous mtDNA. Second, blockade of TLR-9, known to mediate the effects of mtDNA DAMPs (20, 41), and degradation of DNA with DNase1 both suppressed PA103-induced and exogenous mtDNA DAMP-induced increases in Kf.
The most surprising and potentially significant observation of the present study was that exogenous mtDNA caused lung tissue oxidative mtDNA damage-accompanied increases in pulmonary vascular endothelial permeability. The causal role of oxidative mtDNA damage in triggering increased pulmonary vascular endothelial permeability was demonstrated several years ago, also in isolated rat lungs, where it was shown that glucose oxidase-generated H2O2 increased Kf and caused mtDNA damage by a mechanism that was inhibited by mt-targeted Ogg1 (7). In the present study, we found that exogenous mtDNA DAMPs prepared by sonicating rat liver mtDNA also increased Kf and caused oxidative base damage in the mitochondrial genome. Both events were abrogated by mt-targeted Ogg1. It thus appears that mtDNA DAMPs, like other oxidant stressors, cause mtDNA damage-dependent endothelial barrier disruption in isolated, buffer-perfused rat lungs. It should be noted that the intra-arterial dose of exogenous mtDNA used to increase Kf in the present study was quite high, 16 μg, and cannot be considered physiological. However, the idea that mtDNA is released from oxidatively stressed cells in pure form, that is, without bound proteins like TFAM or in the absence of other mitochondrial DAMPs like formylated peptides (20), seems unlikely. If mtDNA is released in concert and acts synergistically with other mitochondria-derived DAMPs as shown by previous studies (18, 19), then it seems reasonable to speculate that mtDNA DAMPs accumulating in pathophysiological contexts could promote deleterious effects in substantially lower concentrations than those used here. Whether the synergy between mtDNA and other mt-derived DAMPs extends to the effect of mtDNA on integrity of the mitochondrial genome remains to be determined.
The finding that PA103 causes mtDNA damage-sensitive and TLR-9-sensitive increases in Kf viewed in concert with observations that exogenous mtDNA DAMPs caused oxidative mtDNA damage-dependent increases in Kf points to the intriguing prospect that mtDNA DAMPs generated in response to PA103-induced oxidant stress participate in a feed-forward process culminating in acute lung injury (Fig. 5). In this model, PA103 causes an oxidant stress, which leads to fragmentation of the mitochondrial genome into DAMPs, their export from the organelle, and subsequent activation of TLR-9, which in turn causes additional mtDNA damage and more mtDNA DAMP formation culminating in loss of endothelial barrier integrity and pulmonary edema. If this feed-forward pathway is validated in subsequent studies, it could provide an explanation for long-standing observations that neither early eradication of bacterial infection with antibiotics nor effective repair and resuscitation after severe injury are uniformly effective in preventing progression of ARDS and MODS. In such instances, it is possible that the initial event, whether it be sepsis or injury, activates the feed-forward pathway of mtDNA damage and DAMP release, which proceeds unabated regardless of the effectiveness of therapy directed against the initiating event. As an extension of this idea, it is interesting to speculate that suppression of mtDNA damage or degradation of released mtDNA DAMPs could emerge as novel pharmacological targets in ARDS and MODS.
Fig. 5.
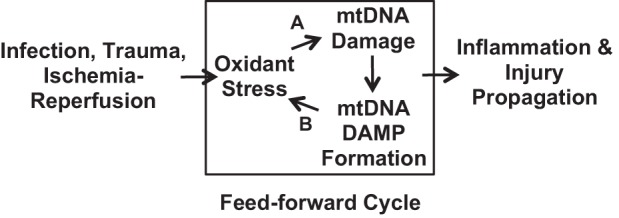
Proposed feed-forward cycle linking oxidative mtDNA damage and DAMP formation to injury propagation. Proposed model linking oxidant stress induced by bacteria, trauma, or ischemia-reperfusion injury to oxidative mtDNA damage, mtDNA DAMP formation, and regenerative mtDNA damage in a feed-forward cycle culminating in inflammation and tissue damage. The model, if valid, explains why treatment of the initial insult, regardless of its specific etiology, often fails to prevent propagation of the insult to distant organs and the occurrence of delayed organ dysfunction. The model also points to two new isolated targets for pharmacological intervention. Both inhibition (A) of oxidative mtDNA damage and enhanced degradation (B) of mtDNA DAMPs would be expected to forestall injury progression. Note that the model fails to consider the potentially complex interactions between alveolar macrophages, resident neutrophils, and vascular endothelial cells in driving the postulated feed-forward pathway. See text for additional details.
The present study also asks a number of interesting and related questions related to the mechanisms of action and sources of mtDNA DAMPs in PA103-challenged lungs. First, what are the specific sequences of the mitochondrial genome released, and why are these sequences released preferentially? Second, do the released sequences exert identical or disparate biological activities? And third, what is the cellular basis of mtDNA DAMP release? In the present study, PA103 was administered to the airways, but mtDNA DAMP accumulation was detected in the perfusion medium. The cellular signaling pathways involved in this response are unknown. It is tempting to speculate that an alveolar or distal airway cell, like alveolar macrophages, or resident neutrophils are important in initiating or transducing the response, but whether and how multiple cell types contribute to the increase in circulating mtDNA DAMPs is presently unclear. Finally, Piantadosi and colleagues (3, 4, 6) have shown that mtDNA repair associated with accrual of Ogg1 and mitochondrial biogenesis are important determinants of survival in the setting of sepsis. These observations further underscore the need for studies on how pathways promoting mtDNA fragmentation and mtDNA DAMP release interact with those regulating mtDNA repair and biogenesis to govern the overall response to sepsis or severe injury.
GRANTS
This work was supported in part by grants from the National Institutes of Health (HL058234, HL073244, and Project #3 in PO1 HL66299).
DISCLOSURES
Mark N. Gillespie is co-owner of a start-up company, Exscien Corporation, which has licensed rights to the fusion protein construct targeting Ogg1 to mitochondria and used as a pharmacological agent in some of the research presented in this report. The authors declare no other conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
Author contributions: J.L.K., V.M.P., J.D.S., and M.N.G. conception and design of research; J.L.K., B.O.O., O.M.G., V.M.P., and J.K. performed experiments; J.L.K., B.O.O., O.M.G., V.M.P., J.K., J.D.S., and M.N.G. analyzed data; J.L.K., O.M.G., V.M.P., J.K., J.D.S., and M.N.G. interpreted results of experiments; J.L.K., B.O.O., and M.N.G. prepared figures; J.L.K. and M.N.G. drafted manuscript; J.L.K., V.M.P., J.D.S., and M.N.G. edited and revised manuscript; J.L.K., V.M.P., J.D.S., and M.N.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Mykhaylo V. Ruchko, Y.-L. Lee, and Glenn L. Wilson for helpful and constructive discussions concerning this work.
REFERENCES
- 1.Audia JP, Lindsey AS, Housley NA, Ochoa CR, Zhou C, Toba M, Oka M, Annamdevula NS, Fitzgerald MS, Frank DW, Alvarez DF. In the absence of effector proteins, the Pseudomonas aeruginosa type three secretion system needle tip complex contributes to lung injury and systemic inflammatory responses. PLoS One 8: e81792, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballinger SW, Patterson C, Yan CN, Doan R, Burow DL, Young CG, Yakes FM, Van Houten B, Ballinger CA, Freeman BA, Runge MS. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res 86: 960–966, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Bartz RR, Fu P, Suliman HB, Crowley SD, MacGarvey NC, Welty-Wolf K, Piantadosi CA. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS One 9: e100912, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartz RR, Suliman HB, Fu P, Welty-Wolf K, Carraway MS, MacGarvey NC, Withers CM, Sweeney TE, Piantadosi CA. Staphylococcus aureus sepsis and mitochondrial accrual of the 8-oxoguanine DNA glycosylase DNA repair enzyme in mice. Am J Respir Crit Care Med 183: 226–233, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernard GR, Lucht WD, Niedermeyer ME, Snapper JR, Ogletree ML, Brigham KL. Effect of N-acetylcysteine on the pulmonary response to endotoxin in the awake sheep and upon in vitro granulocyte function. J Clin Invest 73: 1772–1784, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med 78: 179–189, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chouteau JM, Obiako B, Gorodnya OM, Pastukh VM, Ruchko MV, Wright AJ, Wilson GL, Gillespie MN. Mitochondrial DNA integrity may be a determinant of endothelial barrier properties in oxidant-challenged rat lungs. Am J Physiol Gastrointest Liver Physiol 301: G892–G898, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christofidou-Solomidou M, Muzykantov VR. Antioxidant strategies in respiratory medicine. Treat Respir Med 5: 47–78, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Cuzzocrea S, Costantino G, Mazzon E, Caputi AP. Protective effect of N-acetylcysteine on multiple organ failure induced by zymosan in the rat. Crit Care Med 27: 1524–1532, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Diebel LN, Liberati DM, Ledgerwood AM, Lucas CE. Changes in lymph proteome induced by hemorrhagic shock: the appearance of damage-associated molecular patterns. J Trauma Acute Care Surg 73: 41–50; discussion 51, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am J Physiol Lung Cell Mol Physiol 283: L205–L210, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Driggers WJ, Holmquist GP, LeDoux SP, Wilson GL. Mapping frequencies of endogenous oxidative damage and the kinetic response to oxidative stress in a region of rat mtDNA. Nucleic Acids Res 25: 4362–4369, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El Kebir D, Jozsef L, Pan W, Wang L, Filep JG. Bacterial DNA activates endothelial cells and promotes neutrophil adherence through TLR9 signaling. J Immunol 182: 4386–4394, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Garcia N, Chavez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 81: 1160–1166, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Grishko V, Solomon M, Wilson GL, LeDoux SP, Gillespie MN. Oxygen radical-induced mitochondrial DNA damage and repair in pulmonary vascular endothelial cell phenotypes. Am J Physiol Lung Cell Mol Physiol 280: L1300–L1308, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Grishko VI, Druzhyna N, LeDoux SP, Wilson GL. Nitric oxide-induced damage to mtDNA and its subsequent repair. Nucleic Acids Res 27: 4510–4516, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groeneveld AB. Vascular pharmacology of acute lung injury and acute respiratory distress syndrome. Vascul Pharmacol 39: 247–256, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, Crouser ED. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol 189: 433–443, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One 8: e72354, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32: 157–164, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Kung CT, Hsiao SY, Tsai TC, Su CM, Chang WN, Huang CR, Wang HC, Lin WC, Chang HW, Lin YJ, Cheng BC, Su BY, Tsai NW, Lu CH. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J Transl Med 10: 130, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis 5: e1312, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 13: 481–492, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, Lawler LA, Christie JD, Meyer NJ, Mc Causland FR, Waikar SS, Waxman AB, Chung RT, Bueno R, Rosas IO, Fredenburgh LE, Baron RM, Christiani DC, Hunninghake GM, Choi AM. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10: e1001577; discussion e1001577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker JC, Townsley MI. Evaluation of lung injury in rats and mice. Am J Physiol Lung Cell Mol Physiol 286: L231–L246, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Prakash A, Doublie S, Wallace SS. The Fpg/Nei family of DNA glycosylases: substrates, structures, and search for damage. Prog Mol Biol Transl Sci 110: 71–91, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothfuss O, Gasser T, Patenge N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res 38: e24, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi AB, Gillespie MN. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am J Physiol Lung Cell Mol Physiol 288: L530–L535, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radic Biol Med 50: 1107–1113, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneberger D, Caldwell S, Kanthan R, Singh B. Expression of Toll-like receptor 9 in mouse and human lungs. J Anat 222: 495–503, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306: L962–L974, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37: 2539–2548, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, Gillespie MN, Richards WO. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg 258: 591–596; discussion 596–598, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 8: e59989, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal 10: 739–753, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Wilson JN, Pierce JD, Clancy RL. Reactive oxygen species in acute respiratory distress syndrome. Heart Lung 30: 370–375, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 94: 514–519, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao X, Wigginton JG, Maass DL, Ma L, Carlson D, Wolf SE, Minei JP, Zang QS. Estrogen-provided cardiac protection following burn trauma is mediated through a reduction in mitochondria-derived DAMPs. Am J Physiol Heart Circ Physiol 306: H882–H894, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14: 949–953, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Yousefi S, Simon D, Simon HU. Eosinophil extracellular DNA traps: molecular mechanisms and potential roles in disease. Curr Opin Immunol 24: 736–739, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]