

Abstract
Inflammasomes are cytosolic protein complexes that promote the cleavage of caspase-1, which leads to the maturation and secretion of proinflammatory cytokines, including interleukin-1β (IL-1β) and IL-18. Among the known inflammasomes, the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3)-dependent inflammasome is critically involved in the pathogenesis of various acute or chronic inflammatory diseases. Carbon monoxide (CO), a gaseous molecule physiologically produced in cells and tissues during heme catabolism, can act as an anti-inflammatory molecule and a potent negative regulator of Toll-like receptor signaling pathways. To date, the role of CO in inflammasome-mediated immune responses has not been fully investigated. Here, we demonstrated that CO inhibited caspase-1 activation and the secretion of IL-1β and IL-18 in response to lipopolysaccharide (LPS) and ATP treatment in bone marrow-derived macrophages. CO also inhibited IL-18 secretion in response to LPS and nigericin treatment, another NLRP3 inflammasome activation model. In contrast, CO did not suppress IL-18 secretion in response to LPS and poly(dA:dT), an absent in melanoma 2 (AIM2)-mediated inflammasome model. LPS and ATP stimulation induced the formation of complexes between NLRP3 and apoptosis-associated speck-like protein, or NLRP3 and caspase-1. CO treatment inhibited these molecular interactions that were induced by LPS and ATP. Furthermore, CO inhibited mitochondrial ROS generation and the decrease of mitochondrial membrane potential induced by LPS and ATP in macrophages. We also observed that the inhibitory effect of CO on the translocation of mitochondrial DNA into the cytosol was associated with suppression of cytokine secretion. Our results suggest that CO negatively regulates NLRP3 inflammasome activation by preventing mitochondrial dysfunction.
Keywords: interleukin-18; mitochondria; nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3
inflammasomes are multiprotein complexes responsible for the activation of caspase-1 and downstream immune responses that include the maturation and secretion of interleukin (IL)-1β and IL-18 (12, 51). The cytoplasmic NOD-like receptors (NLRs), critical components of the inflammasome, interact with apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), which in turn recruits procaspase-1. Among the identified NLRs, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3, also known as a cryopirin or NALP3) is expressed in myeloid cells and upregulated by stimulation with pathogen-associated molecular patterns (PAMPs) (12, 56). The NLRP3 inflammasome activates caspase-1 after forming an oligomerized structure. The NLRP3 inflammasome is activated by a diverse series of agonists. Foreign activators of the NLRP3 inflammasome include pathogens (e.g., bacteria, fungi, and viruses) and environment-derived sterile activators such as asbestos, silica, and ultraviolet radiation. Self-activators or damage-associated molecular patterns (DAMPs) of NLRP3 include ATP, monosodium urate crystals, glucose, and cholesterol (12, 51). Thus, the NLRP3 inflammasome is not only linked primarily to infectious diseases but also to a wide range of human diseases such as type II diabetes, cancer, and autoimmune diseases (12). Of note, it has been shown that circulating levels of IL-18 are significantly associated with disease severity in various human diseases (5, 8, 13, 59). The mechanisms by which the NLRP3 inflammasome is activated include several key factors such as reactive oxygen species (ROS), ion flux, and lysosomal destabilization (12, 51). Moreover, several autophagy or apoptosis-related molecules (e.g., Beclin 1, Atg16L1, LC3B, and Bcl-2) have been identified as negative regulators of caspase-1-dependent cytokine secretion by preventing mitochondrial dysfunction (15, 20, 37, 50, 65).
Carbon monoxide (CO), a byproduct of heme catabolism by the heme oxygenase-1 (HO-1) enzyme system, has been shown to exert various physiological effects, such as the modulation of inflammation, apoptosis, and cell proliferation in vitro and in vivo (49). Notably, the anti-inflammatory effects of CO have been extensively studied in various animal models of tissue injury (2, 38, 41). The pathways by which CO inhibits the inflammatory response include the inhibition of Toll-like receptor 4 (TLR4) trafficking to lipid rafts and the activation of p38 MAPK (38, 41). Although recent studies suggest that CO modulates inflammasome-mediated IL-1β production in response to bacteria (62), it remains unclear whether CO can regulate the NLRP3 inflammasome pathway, including IL-18 secretion in vivo and in vitro. Here, we examined the effect of CO on NLRP3 inflammasome activation. We also investigated the mechanism by which CO regulates inflammasome-mediated immune responses in macrophages.
MATERIALS AND METHODS
Animals.
Male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were allowed to acclimate for 1 wk with rodent chow and water ad libitum. Animal care and use for all experiments were approved by the Harvard Medical Area Standing Committee on Animals of Harvard Medical School.
Cell culture and treatments.
Bone marrow-derived macrophages (BMDMs) were prepared as previously described (37, 56). Bone marrow collected from mouse femurs and tibias was plated on sterile petri dishes and then incubated for 7 days in DMEM containing 10% (vol/vol) heat-inactivated fetal calf serum (FCS), penicillin and streptomycin, and 25% (vol/vol) conditioned medium from L929 mouse fibroblasts (37). Cells were incubated for 6 h with lipopolysaccharide (LPS, 10 ng/ml) and then treated with ATP (2 mM) for 15–60 min depending on the assays (15 min for flow cytometry, 15 and 30 min for detecting caspase-1 activation and IL-1β expression, and 60 min for measuring cytokine secretion) (37, 56). J774A.1 macrophages were grown in DMEM supplemented with 10% (vol/vol) FCS. For activation of caspase-1, cells were incubated for 4 h with LPS (200 ng/ml), and then cells were stimulated with ATP (37, 44).
CO exposure.
Mice or macrophages were exposed to compressed air or CO [250 parts/million (ppm)] as previously described (35, 41). For cell culture experiments, 5% CO2 was also present for buffering requirements. CO at a concentration of 1% (10,000 ppm) in compressed air was mixed with compressed air with or without CO2 in a stainless steel mixing cylinder before being delivered into the exposure chamber. Flow into the 3.70-ft2 Plexiglas animal chamber was maintained at a rate of 12 l/min and into the 1.2-ft2 cell culture chamber at a rate of 2 l/min. The cell culture chamber was humidified and maintained at 37°C. A CO analyzer (Interscan, Chatsworth, CA) was used to measure CO levels continuously in the chambers. Gas samples were introduced to the analyzer through a port in the top of the chambers at a rate of 1 l/min and were analyzed by electrochemical detection, with a sensitivity of 10–600 ppm. Concentration levels were measured hourly, and there were no fluctuations in the CO concentrations after the chamber had equilibrated (∼5 min).
Reagents.
The following antibodies were used for immunoprecipitation or immunoblotting: antibody to NLRP3 (ALX-804-881; Enzo Life Science, Farmingdale, NY), mouse antibody to ASC (04–147; Millipore), rabbit antibody to mouse caspase-1 (sc-514; Santa Cruz Biotechnology, Dallas, TX), and rabbit antibody to mouse IL-1β (5129-100; Biovision, Milpitas, CA). Mito-TEMPO was from Enzo Life Sciences. LPS (Escherichia coli) was from Invivogen. ATP, nigericin, poly(dA:dT), rotenone, and cyclosporine A were from Sigma (St. Louis, MO).
Flow cytometry.
Mitochondrial ROS were measured in cells by MitoSOX (Life Technologies, Grand Island, NY) staining (5 μM for 15 min at 37°C), followed by 15 min of ATP treatment (37). To assess mitochondrial membrane potential (ΔΨm), LPS-primed BMDMs were stained for 20 min at 37°C with 100 nM tetramethylrhodamine, ethyl ester (TMRE; Abcam, Cambridge, MA), followed by incubation with ATP for 15 min (33). For measurement of mitochondrial mass, cells were stained for 15 min at 37°C with 25 nM MitoTracker Green FM and MitoTracker Deep Red FM (Life Technologies), followed by 15 min of ATP treatment (37, 57, 65). Data were acquired with a FACS Canto II (BD Biosciences, San Jose, CA) and were analyzed with FlowJo analytical software (FlowJo, Ashland, OR).
Immunoblot analysis.
Proteins were separated by electrophoresis as described (32, 37). Lysates were boiled 5 min in NuPAGE sample-loading buffer (Life Technologies). Proteins were separated by electrophoresis through NuPAGE 4–12% Bis-Tris gels and were transferred to polyvinylidene difluoride or nitrocellulose membranes by electroblotting.
ELISA.
IL-1β and IL-18 levels in culture supernatants, serum, or lung homogenates were measured with ELISA kits (R&D Systems, Minneapolis, MN).
Coimmunoprecipitation.
To assess the protein composition and association of proteins in the inflammasome, BMDM cultures (2 × 106 cells) were lysed in 600 μl of RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) with protease inhibitor mixture (Sigma-Aldrich, St. Louis, MO) as described (61). Approximately 500 μg of cell lysates were immunoprecipitated with anti-ASC or anti-NLRP3 antibodies using protein A/G beads (SC-2003; Santa Cruz). Cell lysates were precleared by adding 20 μl of protein A/G beads to 500 μg of lysate in a microcentrifuge tube. The mixture was incubated for 1 h at 4°C, and beads were pelleted by centrifugation at 6,000 g for 60 s (61). The supernatant was recovered and immunoprecipitated with 5 μg of anti-ASC or anti-NLRP3 and incubated at 4°C overnight. Protein A/G beads (40 μl) were added to the mixture, incubated for 2 h, and then centrifuged at 6,000 g for 3 min. The pelleted beads were washed five times in RIPA lysis buffer, resuspended in Nu-PAGE loading buffer, and heated at 95°C for 5 min before analysis by immunoblotting using antibodies against ASC, caspase-1, and NLRP3.
Quantitative real-time PCR.
Quantitative PCR (2 independent reactions) was used for measurement of mitochondrial DNA (mtDNA) with SYBR Green PCR Master mix (Life Technologies) and established primers for a mitochondrial gene. For measurement of mtDNA in cytosol, 1 × 107 cells were homogenized with a Dounce homogenizer in 100 mM Tricine-NaOH solution, pH 7.4, containing 0.25 M sucrose, 1 mM EDTA, and protease inhibitor and then were centrifuged at 700 g for 10 min at 4°C (37). Protein concentration and volume of the supernatant were normalized, followed by centrifugation at 10,000 g for 30 min at 4°C for the production of a supernatant corresponding to the cytosolic fraction. DNA was isolated from 200 μl of the cytosolic fraction with a DNeasy Blood and Tissue kit (Qiagen, Valencia, CA) as described (37, 64). The copy number of DNA encoding cytochrome c oxidase 1 was measured by quantitative real-time PCR with the same volume of the DNA solution. The following primers were used: mouse cytochrome c oxidase I forward 5′-GCCCCAGATATAGCATTCCC-3′ and reverse 5′-GTTCATCCTGTTCCTGCTCC-3′ (37, 57).
Protein transfection.
BMDM prepared at 70–80% confluency in 24 wells were washed with warmed DMEM without FBS two times and were then transfected with 3 μg of DNase I or lactate dehydrogenase by using PULSin Reagent (Polyplus Transfection, New York, NY) for 4 h at 37°C according to the instruction manual (37). After the media were removed, cells were incubated in full growth media for 2 h. Next, cells were primed with LPS for 4 h, followed by stimulation with ATP.
In vivo experiments.
Male mice (8–10 wk old) were injected intraperitoneally with E. coli LPS at a dose of 2.5 mg/kg body wt (12.5 × 106 endotoxin U/kg).
Statistical analysis.
Data are presented as means ± SE. Differences in measured variables between the experimental and control groups were assessed using an unpaired, two-tailed Student's t-test and between multiple groups and conditions using 2-way ANOVA and Bonferroni's posttests. P values were calculated, and minimum statistical significance was accepted at P < 0.05.
RESULTS
CO inhibits caspase-1 activation by regulating the NLRP3 inflammasome.
To examine the role of CO in the inflammasome response, we first evaluated caspase-1-dependent cytokine secretion in primarily macrophages. BMDMs were incubated with LPS, followed by stimulation with ATP, an NLRP3 inflammasome activation model in macrophages. We observed that CO exposure significantly inhibited the secretion of IL-1β and IL-18 in response to LPS and ATP (Fig. 1A). The inhibitory effect of CO on IL-1β secretion was comparable between cells pretreated with CO and cells cotreated with CO and LPS (Fig. 1B). In addition, IL-1β release was significantly suppressed in response to LPS and ATP when the cells were posttreated with CO beginning 3 h after LPS treatment (Fig. 1B). In a manner similar to that observed in BMDMs, CO treatment significantly inhibited the secretion of IL-1β and IL-18 in J774A.1 macrophages in response to LPS and ATP (Fig. 1C). We also observed that CO suppressed IL-18 secretion in macrophages stimulated with LPS and nigericin, another NLRP3 inflammasome activation model (Fig. 1D) (12). In contrast, CO did not significantly reduce the secretion of IL-18 in BMDMs in response to LPS and poly(dA:dT) treatment, which activates the absent in melanoma 2 (AIM2) inflammasome pathway (Fig. 1E) (12, 47). These results suggest that CO selectively regulated NLRP3 inflammasome activation in macrophages. Next, we examined the effect of CO on cytokine secretion in serum from mice treated with LPS, an animal model of NLRP3-dependent acute inflammation (56). Consistent with previous reports, IL-1β production was significantly suppressed by CO exposure (250 ppm) (Fig. 1F) (35). We also observed that CO exposure significantly reduced the levels of IL-18 in serum and lung homogenates of mice treated with LPS (Fig. 1F). Because secretion and maturation of IL-1β and IL-18 is tightly regulated by caspase-1, we examined the effect of CO on caspase-1 activation [expression of the cleaved form of caspase-1 (p10)]. Caspase-1 activation was significantly inhibited by CO in BMDMs treated with LPS and ATP (Fig. 2). CO had no effect on expression levels of the P2X7 receptor, ASC, or NLRP3 under basal or LPS-induced conditions, critical cofactors for caspase-1 activation induced by LPS and ATP (Fig. 2). While CO moderately inhibited pro-IL-1β synthesis by LPS, the cleaved form of IL-1β (mature IL-1β) was substantially inhibited by CO (Fig. 2). To investigate the upstream mechanism by which CO regulates caspase-1 activation, we examined the effect of CO on inflammasome activation. By performing coimmunoprecipitation assays, we observed that complex formation of NLRP3 and ASC was increased after ATP treatment in LPS-stimulated cells (Fig. 3, A and B). In addition, the interaction of NLRP3 and procaspase-1 was also increased by LPS and ATP stimulation (Fig. 3B). Importantly, CO treatment inhibited the formation of both complexes in the stimulated cells (Fig. 3, A and B). These data suggest that CO negatively regulated caspase-1 activation by inhibiting NLRP3 inflammasome formation.
Fig. 1.

Carbon monoxide (CO) suppressed interleukin (IL)-1β and IL-18 secretion in vitro and in vivo. A: bone marrow-derived macrophages (BMDMs) were stimulated with lipopolysaccharide (LPS, 10 ng/ml) for 6 h and then stimulated with ATP (2 mM) for an additional 1 h in the presence or absence of CO [250 parts/million (ppm)]. IL-1β and IL-18 in the supernatants were measured by ELISA. Data are representative of 3 experiments. B: CO exposure was initiated at different time points (−2 h, 2 h before LPS treatment; 0 h, cotreatment with LPS; +3, 3 h after LPS treatment; +5, 5 h after LPS treatment). IL-1β secretion was analyzed by ELISA. Data are representative of 3 experiments. C: J774A.1 macrophages were stimulated with LPS (200 ng/ml) for 4 h, followed by ATP (3 mM) for 1 h. IL-1β and IL-18 secretion was analyzed by ELISA. Data are representative of 3 experiments. D: LPS-primed BMDMs were stimulated with nigericin (3.3 μM). IL-18 in the supernatant was analyzed by ELISA. E: LPS-primed BMDMs were transfected with poly(dA:dT) (1 μg/ml). IL-18 in the supernatant was analyzed by ELISA. Data are representative of 3 experiments. All data represent means ± SE. **P < 0.01 by unpaired, 2-tailed Student's t-test (A–D). F: mice were exposed to 250 ppm CO or ambient room air (RA) for 1 h before receiving ip injections of LPS (2.5 mg/kg) or PBS; n = 6/group. Blood and lungs were collected 24 h after LPS treatment. The levels of IL-1β and IL-18 in serum and lung homogenates were analyzed by ELISA. Data are representative of 2 experiments. All data represent means ± SE. *P < 0.05 vs. RA + PBS, #P < 0.05 vs. CO + PBS, and †P < 0.05 vs. RA + LPS by 2-way ANOVA with Bonferroni's posttests.
Fig. 2.
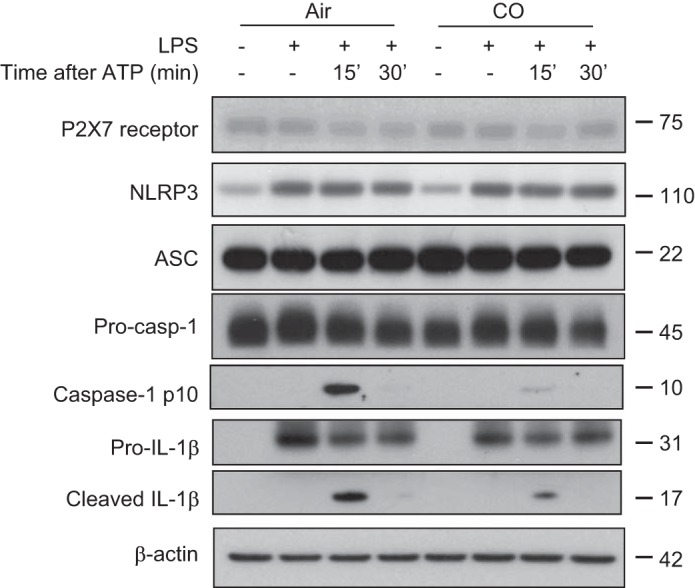
CO inhibited caspase-1 activation in macrophages. BMDMs were incubated with LPS for 6 h, followed by stimulation with ATP for the indicated time in the presence or absence of CO. Cell lysates were analyzed by immunoblotting for caspase-1, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3), IL-1β, P2X7 receptor, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), and β-actin. Data are representative of 3 experiments.
Fig. 3.
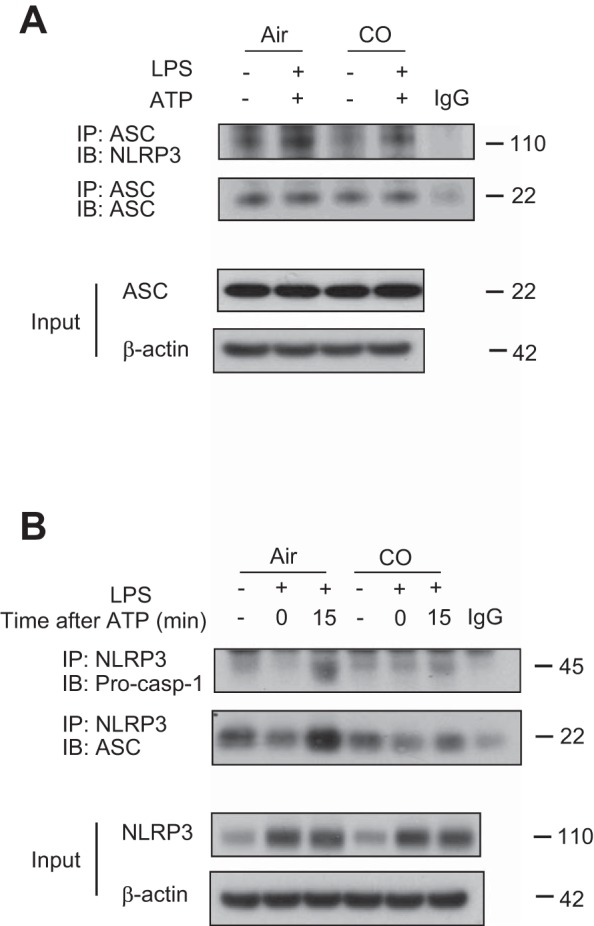
Effect of CO on NLRP3 inflammasome complex formation. A: BMDMs were incubated with LPS for 6 h, followed by stimulation with ATP for 15 min in the presence or absence of CO. Interaction between ASC and NLRP3 was analyzed by immunoprecipitation and immunoblotting. Expression of ASC and β-actin was analyzed by immunoblotting using the identical total cell lysates. Data are representative of 3 experiments. B: BMDMs were incubated with LPS for 6 h, followed by stimulation with ATP for 15 min in the presence or absence of CO. Interaction between NLRP3 and caspase-1 or NLRP3 and ASC was analyzed by immunoprecipitation and immunoblotting. Expression of NLRP3 and β-actin was analyzed by immunoblotting using the identical total cell lysates. Data are representative of 3 experiments.
CO inhibits mitochondrial ROS generation.
Mitochondria have been shown to be critically involved in caspase-1 activation, especially through NLRP3 inflammasome activation (18, 37, 65). Our group and others have previously shown that LPS and ATP stimulation in macrophages enhances mitochondrial ROS generation, which promotes caspase-1 activation and the secretion of IL-1β and IL-18 (36, 37, 63). To investigate the upstream mechanism by which CO regulates the NLRP3 inflammasome, we assessed the effect of CO on mitochondrial ROS generation in LPS- and ATP-stimulated BMDMs. We observed that mitochondrial ROS generation induced by LPS and ATP was inhibited by CO treatment (Fig. 4A). Neither LPS alone nor CO alone had any significant effect on mitochondrial ROS generation in macrophages (Fig. 4A, data not shown). To examine whether the inhibitory effect of CO on mitochondrial ROS generation was associated with the effect of CO on the NLRP3 inflammasome, we scavenged mitochondrial ROS by Mito-TEMPO, a derivative of the antioxidant TEMPO that concentrates in the mitochondrial matrix and acts as an O2− scavenger (22, 58). Consistent with our previous reports, Mito-TEMPO treatment significantly inhibited IL-18 secretion in BMDMs stimulated with LPS and ATP (Fig. 4B) (37, 63). We observed that the inhibitory effect of CO on IL-18 secretion was impaired in cells treated with Mito-TEMPO (Fig. 4B). These data, taken together, suggest that CO inhibits caspase-1 activation in part by inhibition of mitochondrial ROS generation in macrophages.
Fig. 4.

Effect of CO on mitochondrial reactive oxygen species (ROS) production in response to LPS and ATP. A: LPS-primed BMDMs were stained with MitoSOX for 15 min before stimulation with ATP for 15 min in the absence or presence of CO, and then analyzed by flow cytometry. The x-axis shows the fluorescent signal intensity, and the y-axis represents the cell number normalized as a percentage of the maximum (%max). Data are representative of 3 experiments. B: LPS-primed macrophages were treated with Mito-TEMPO (100 μM) for 30 min before incubation with ATP for 1 h in the presence or absence of CO. IL-18 secretion in the supernatants was analyzed by ELISA. Data are representative of 3 experiments. All data represent means ± SE. **P < 0.01 by unpaired, 2-tailed Student's t-test (A and B).
CO suppressed IL-1β and IL-18 secretion by preserving mitochondrial membrane potential.
Excess production of mitochondrial ROS is associated with a decrease of ΔΨm, which induces mitochondrial membrane permeability (28, 29). Because mitochondrial ROS generation was increased during NLRP3 inflammasome activation (Fig. 4A), we first assessed whether ΔΨm is decreased by LPS and ATP by using TMRE, a fluorescent probe sensitive to mitochondrial membrane potential (33). Treatment with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), a representative uncoupling agent, decreased ΔΨm (Fig. 5A) (46). We also observed that stimulation with LPS and ATP decreased ΔΨm in BMDMs compared with control cells (53), implying that the proinflammatory stimuli increased the occurrence of depolarized or inactive mitochondria (Fig. 5A). Because scavenging mitochondrial ROS with Mito-TEMPO inhibited IL-18 secretion in response to NLRP3 inflammasome stimuli, we tested whether Mito-TEMPO can preserve ΔΨm in the stimulated macrophages. As expected, cotreatment with Mito-TEMPO inhibited the reduction of ΔΨm induced by LPS and ATP (Fig. 5B). Mito-TEMPO treatment alone did not regulate ΔΨm (Fig. 5B). Because CO inhibited mitochondrial ROS generation (Fig. 4A), we next assessed the effect of CO on ΔΨm in cells stimulated with LPS and ATP. The decrease of ΔΨm induced by LPS and ATP was suppressed by CO treatment (Fig. 5C). We also assessed the effect of CO on ΔΨm in cells stimulated with LPS and ATP using MitoTracker Deep Red, a fluorescent probe sensitive to the mitochondrial inner transmembrane potential (37, 57, 65). To measure the total mitochondrial pool, BMDMs were counterstained with MitoTracker Green, a probe that stains mitochondrial membrane lipids independently of membrane potential. Similar to the results obtained by using TMRE, LPS and ATP treatment increased the percentage of mitochondria exhibiting loss of membrane potential (MitoTracker Deep Red negative-MitoTracker Green positive) from 1.43 to 35.8% of the total population. CO treatment reduced the number of depolarized mitochondria as the result of LPS and ATP treatment (19.8% of the total cell population relative to 0.33% in the untreated control) (Fig. 5D). We further investigated whether preservation of ΔΨm by CO was associated with the caspase-1-mediated immune response. To examine this, macrophages were cotreated with the mitochondrial complex I inhibitor rotenone, a mitochondrial membrane depolarization inducer (21, 34), during stimulation with LPS and ATP in the absence or presence of CO. Consistent with our previous report (37), we observed that rotenone treatment strongly enhanced caspase-1 activation and secretion of IL-1β and IL-18 in BMDMs stimulated with LPS and ATP (Fig. 5E) (37). Importantly, the potent enhancement of LPS- and ATP-induced cytokine secretion by rotenone was significantly suppressed in CO-exposed macrophages (Fig. 5E). We also examined whether the inhibitory effect of CO on the decrease in ΔΨm is associated with the effect of CO on the NLRP3 inflammasome by using cyclosporine A, a potent mitochondrial permeability transition inhibitor (6). Cyclosporine A treatment dose-dependently inhibited IL-18 secretion in LPS- and ATP-stimulated BMDMs (37), and the inhibitory effect of CO on IL-18 secretion was impaired in the cells treated with cyclosporine A (Fig. 5F). These data imply that preservation of mitochondrial membrane potential by CO is associated with downregulation of caspase-1-mediated immune response by CO in macrophages.
Fig. 5.

CO inhibited decrease of mitochondrial membrane potential in response to LPS and ATP. A: LPS-primed BMDMs were stained with tetramethylrhodamine, ethyl ester (TMRE) for 15 min, followed by stimulation with ATP for 15 min. BMDMs treated with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 20 μM) for 15 min were used as a positive control. MFI, mean fluorescence intensity. The x-axis shows the fluorescent signal intensity, and the y-axis represents the cell number normalized as a percentage of the maximum. Data are representative of 3 experiments. B: LPS-primed BMDMs were stained with TMRE for 15 min, followed by stimulation with ATP for 15 min in the presence or absence of Mito-TEMPO (100 μM). The x-axis shows the fluorescent signal intensity, and the y-axis represents the cell number normalized as a percentage of the maximum. Data are representative of 3 experiments. C: LPS-primed BMDMs were stained with TMRE for 15 min, followed by stimulation with ATP for 15 min in the presence or absence of CO. The x-axis shows the fluorescent signal intensity, and the y-axis represents the cell number normalized as a percentage of the maximum. Data are representative of 3 experiments. D: LPS-primed BMDMs were stained with MitoTracker Deep Red and MitoTracker Green for 15 min, followed by stimulation with ATP for 15 min in the presence or absence of CO. Data are representative of 3 experiments. E: LPS-primed macrophages were incubated with rotenone (5 μM) or DMSO for 45 min before stimulation with ATP for 1 h in the presence or absence of CO. Cytokine secretion was analyzed by ELISA. Data are representative of 3 experiments. F: LPS-primed macrophages were incubated with 5 or 10 μM of cyclosporine A or DMSO for 1 h before stimulation with ATP for 1 h. IL-18 secretion was measured by ELISA. Data are representative of 3 experiments. All data represent means ± SE. **P < 0.01 or *P < 0.05 by unpaired, 2-tailed Student's t-test (A–C, E, and F).
CO inhibited mtDNA translocation into the cytosol.
Mitochondrial membrane permeability is associated with release of mtDNA in the cytosol in irradiated mice (42, 43). Recent studies have demonstrated that mtDNA translocates to the cytosol and regulates NLRP3 inflammasome-mediated cytokine secretion in response to stimuli that cause mitochondrial dysfunction, including decrease of ΔΨm (36, 37, 53). To further investigate the effects of CO on caspase-1 activation, we examined the effect of CO on mtDNA release in BMDMs. We observed that the copy number of mtDNA in the cytosol was increased by LPS and ATP treatment (36, 37, 53), which was significantly inhibited by cotreatment with CO (Fig. 6A). Finally, we transfected DNase I into the cytosol to degrade cytosolic DNA (37) and then stimulated the cells with LPS and ATP to examine the immunological role of cytosolic mtDNA as well as the effect of CO on this process. CO inhibited IL-1β secretion (∼50%) in response to LPS and ATP treatment, whereas CO had a minor inhibitory effect on IL-1β secretion after transfection with DNase I (Fig. 6B). Similarly, inhibition of IL-18 secretion by CO was not significant in the cell transfected with DNase I (Fig. 6B). These data suggest that CO inhibited IL-1β and IL-18 secretion by preventing mtDNA translocation into cytosol.
Fig. 6.
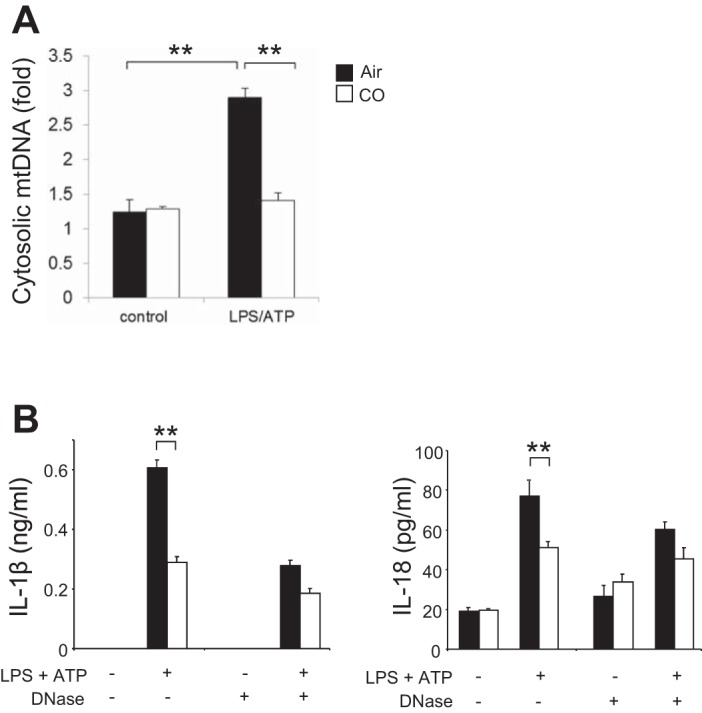
CO decreased release of mitochondrial DNA (mtDNA) into cytosol. A: LPS-primed macrophages were incubated with ATP for 15 min in the presence or absence of CO. Cytosolic mtDNA copy number was measured by quantitative PCR. Data are representative of 3 experiments. B: BMDMs were transfected with 3 μg of DNase I for 4 h and then stimulated with LPS, followed by ATP stimulation for 1 h in the absence or presence of CO. Cytokine secretion into the supernatants was analyzed by ELISA. Data are representative of 3 experiments. All data represent means ± SE. **P < 0.01 by unpaired, 2-tailed Student's t-test (A and B).
DISCUSSION
In this study, we demonstrated that CO can act as a negative regulator of NLRP3 inflammasome activation and secretion of IL-1β and IL-18 in vitro and in vivo. The mechanism by which CO inhibited the caspase-1-mediated immune response involved an inhibitory effect of CO on mitochondrial dysfunction, including mitochondrial ROS generation, decrease of ΔΨm, and mtDNA translocation, which represent core mechanisms for NLRP3 inflammasome activation (37, 65). We observed that CO inhibited mitochondrial ROS generation in response to LPS and ATP in macrophages. In addition, decrease of ΔΨm and release of mtDNA into the cytosol induced by LPS and ATP were also prevented by CO. Our data have demonstrated a novel mechanism by which CO regulates proinflammatory signaling pathways and secretion of IL-18.
Previously, CO has been shown to inhibit TLR4 signaling pathways, an upstream step of NF-κB activation (38). Although it has been reported that CO inhibits IL-1β secretion in RAW 264.7 cells (7, 41, 49), the role of CO on inflammasome activation has not been fully investigated (62). Because deficiency of Casp1, Asc, and Nlrp3 causes defects in IL-1β and IL-18 maturation and secretion both in primary macrophages and mice in the context of endotoxin models (30, 56), it is important to investigate the role of CO on the inflammasome, a central signaling platform for IL-1β and IL-18 activation. Here, we show that CO regulated this critical posttranscriptional inflammatory signaling pathway, based on the following observations. First, we observed that CO strongly inhibited the cleavage of caspase-1 (expression of p10), whereas LPS-induced NLRP3 expression was comparable between air-treated cells and CO-treated cells. Second, although CO exposure mildly suppressed pro-IL-1β synthesis induced by LPS (2), the degree of conversion from pro-IL-1β to cleaved IL-1β in CO-treated cells was smaller than that observed in air-treated cells. Third, complex formation of ASC and NLRP3, as well as of NLRP3 and caspase-1, was initiated after ATP treatment in LPS-primed cells and inhibited by CO exposure. In addition, the differential regulation of CO on inflammasome (i.e., NLRP3 and AIM2) suggests that the inhibitory effect of CO on NLRP3 inflammasome activation is not mainly dependent on regulation of the TLR4 signaling pathway but rather on regulation of second signals (e.g., ATP). These results suggest that CO is a potent negative regulator of the NLRP3 inflammasome pathway.
The effect of CO on the caspase-1-mediated immune response is likely to be mediated by suppressing mitochondrial ROS generation induced by LPS and ATP, since CO had no effect on the caspase-1-mediated immune response in cells treated with Mito-TEMPO. Previously, we showed that CO suppresses LPS-induced NADPH oxidase-dependent ROS generation and that the anti-inflammatory effect of CO was abolished in gp91phox-deficient macrophages (38). It is possible that the inhibitory effect of CO on IL-1β secretion may be partially mediated through regulation of NADPH oxidase-mediated ROS generation, since CO moderately inhibited pro-IL-1β synthesis induced by TLR4 signaling pathway activation. We have recently observed that CO increases mitochondrial ROS generation in epithelial cell lines, where CO exerts anti-apoptotic properties that are mediated by the enhancement of mitochondrial ROS generation (27). Because mitochondrial ROS are involved in various cellular functions, including proliferation, metabolism, cell death, or immune responses (16, 18, 39), the differential role of CO on mitochondrial ROS may be dependent on cell type or stimulation. In our models, inhibition of mitochondrial membrane permeability by CO was associated with the negative regulation of the NLRP3 inflammasome by CO, since the inhibitory effect on cytokine secretion by CO was abolished when cells were pretreated with DNase I. Previously, we have shown that mitochondrial ROS generation is situated upstream of NLRP3, whereas decrease of ΔΨm and mtDNA translocation is dependent on NLRP3 in LPS and ATP models (37). It is likely that CO inhibits loss of ΔΨm and mtDNA leak by suppressing mitochondrial ROS generation, which results in the negative regulatory effect of CO on the NLRP3 inflammasome-mediated immune response. We have recently reported that Mito-TEMPO does not inhibit AIM2 inflammasome-mediated cytokine secretion (37). Thus, CO and Mito-TEMPO are likely to regulate inflammasome in a similar fashion, since CO and Mito-TEMPO selectively regulate NLRP3 inflammasome. HO-1 catalyzes the rate-limiting step in the metabolic conversion of heme to the bile pigments (i.e., biliverdin and bilirubin) and thus constitutes a major intracellular source of CO (49). Previous studies show that induction and translocation of HO-1 to mitochondria prevents mitochondrial oxidative stress and tissue injury (3, 4, 10), suggesting that mitochondria are key targets of CO for regulation of the inflammasome. It is important to note that our exogenous treatment of CO may exert additional effects on mitochondria because of higher intracellular concentration of CO compared with endogenous CO derived from heme catabolism (49).
Although our current study reveals a novel role of CO on inflammasome-mediated immune responses by preventing mitochondrial dysfunction, the precise mechanism by which CO regulates mitochondrial function remains unclear. Proinflammatory cytokines such as tumor necrosis factor (TNF) have been shown to induce mitochondrial dysfunction, including mitochondrial ROS production (23, 48). Because CO negatively regulates TLR4 signaling pathways, including NF-κB (38), it is also possible that the mechanism by which CO negatively regulates mitochondrial ROS generation may include inhibition of TLR4-mediated proinflammatory cytokine production, such as TNF. Recently, Wegiel et al. reported that CO (250 ppm) enhances caspase-1 activation (e.g., increased expression of the cleaved form of caspase-1 and secretion of IL-1β) in macrophages infected with live bacteria such as E. coli or E. faecalis (62). This report demonstrated that CO increased bacteria-derived ATP production and that the secreted extracellular ATP in turn activates NLRP3 inflammasome through the P2X7 receptor in macrophages. Because our current model did not use live bacteria, it is unlikely that CO exposure increased the levels of extracellular ATP for activation of the P2X7 receptor-mediated NLRP3 inflammasome in macrophages. The effect of CO on inflammasome may be dependent on distinct pathogens, PAMPs or DAMPs. Although further studies will be needed to clarify the precise mechanism by which CO regulates the inflammasome, our results demonstrate that CO is a novel candidate as a negative regulator of the NLRP3 inflammasome and secretion of IL-1β and IL-18.
For decades, mitochondria have been known as dynamic cellular organelles for ATP production, ion homeostasis, or apoptosis. The role of mitochondria in innate immunity was first reported in the context of viral infection models in 2005 (52). Subsequently, mitochondrial dysfunction has been shown to underlie viral-mediated immune responses, including type 1 interferon production (57). In addition, recent reports link mitochondrial ROS and NLRP3 inflammasome activation (37, 65). Deregulation of mitochondria is associated with various human diseases, such as neurodegeneration, cancer, or metabolic disorders (24, 31). Moreover, mitochondrial dysfunction is also involved in the pathogenesis of liver, brain, or muscle in critically ill patients (11, 17, 60). Accumulation of mitochondrial damage in cells or tissues may promote inflammasome-mediated immune responses. It is speculated that caspase-1-mediated immune responses are associated with disease severity in patients with inflammatory diseases, and blockage of this pathway could have an impact on the pathogenesis and prognosis of the patients (8, 25, 55). Whereas IL-1β contributes to the pathogenesis of various human diseases such as type II diabetes, gout, or rheumatoid arthritis (1, 9, 26, 54), IL-18 also has been shown as a significant predictor of cardiovascular events and hospitalization of hemodialysis patients (8, 59). Moreover, IL-18 has been shown to be significantly increased in plasma from patients with critical illnesses such as acute respiratory distress syndrome, and sepsis, which represent leading causes of death in the intensive care unit (13, 19, 37, 40). Importantly, patients with elevated IL-18 levels at the time of medical intensive care unit hospitalization have increased mortality (13), suggesting that inflammasome-mediated immune responses could be critically involved in the pathogenesis of severe inflammatory diseases. Given the potent negative regulation of the NLRP3 inflammasome by CO treatment, CO might be exploited as a therapeutic to inhibit excess inflammasome activation through the preservation of mitochondrial function.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants T32-HL-007633 to K. Nakahira, R01-HL-060234 to S. W. Ryter, and P01-HL-108801, R01-HL-060234, R01-HL-55330, R01-HL-079904 to A. M. K. Choi, who was also supported by a FAMRI Clinical Innovator Award.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.-S.J., J.-S.M., J.X., E.I., and K.N. performed experiments; S.-S.J., J.X., and K.N. analyzed data; S.-S.J., J.X., S.W.R., and K.N. interpreted results of experiments; S.-S.J. and K.N. prepared figures; S.-S.J. and K.N. drafted manuscript; S.W.R., A.M.C., and K.N. edited and revised manuscript; A.M.C. and K.N. conception and design of research; A.M.C. and K.N. approved final version of manuscript.
REFERENCES
- 1.Abramson SB, Amin A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology (Oxford) 41: 972–980, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Bilban M, Bach FH, Otterbein SL, Ifedigbo E, d'Avila JC, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O, Otterbein LE. Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 24: 601–610, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, Bandyopadhyay U. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem 286: 39387–39402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolisetty S, Traylor A, Zarjou A, Johnson MS, Benavides GA, Ricart K, Boddu R, Moore RD, Landar A, Barnes S, Darley-Usmar V, Agarwal A. Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells. Am J Physiol Renal Physiol 305: F255–F264, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boraschi D, Dinarello CA. IL18 in autoimmunity: review. Eur Cytokine Network 17: 224–252, 2006. [PubMed] [Google Scholar]
- 6.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem 264: 7826–7830, 1989. [PubMed] [Google Scholar]
- 7.Chhikara M, Wang S, Kern SJ, Ferreyra GA, Barb JJ, Munson PJ, Danner RL. Carbon monoxide blocks lipopolysaccharide-induced gene expression by interfering with proximal TLR4 to NF-kappaB signal transduction in human monocytes. PloS one 4: e8139, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang CK, Hsu SP, Pai MF, Peng YS, Ho TI, Liu SH, Hung KY, Tsai TJ. Interleukin-18 is a strong predictor of hospitalization in haemodialysis patients. Nephrol Dial Transplant 19: 2810–2815, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol 4: 34–42, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Converso DP, Taille C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J 20: 1236–1238, 2006. [DOI] [PubMed] [Google Scholar]
- 11.d'Avila JC, Santiago AP, Amancio RT, Galina A, Oliveira MF, Bozza FA. Sepsis induces brain mitochondrial dysfunction. Crit Care Med 36: 1925–1932, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Ann Rev Immunol 29: 707–735, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L, Nakahira K, Haspel JA, Landazury R, Eppanapally S, Christie JD, Meyer NJ, Ware LB, Christiani DC, Ryter SW, Baron RM, Choi AM. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 185: 1225–1234, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faustin B, Chen Y, Zhai D, Le Negrate G, Lartigue L, Satterthwait A, Reed JC. Mechanism of Bcl-2 and Bcl-X(L) inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proc Natl Acad Sci USA 106: 3935–3940, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 287: 4434–4440, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fredriksson K, Tjader I, Keller P, Petrovic N, Ahlman B, Scheele C, Wernerman J, Timmons JA, Rooyackers O. Dysregulation of mitochondrial dynamics and the muscle transcriptome in ICU patients suffering from sepsis induced multiple organ failure. PLoS One 3: e3686, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333: 1109–1112, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grobmyer SR, Lin E, Lowry SF, Rivadeneira DE, Potter S, Barie PS, Nathan CF. Elevation of IL-18 in human sepsis. J Clin Immunol 20: 212–215, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286: 9587–9597, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isenberg JS, Klaunig JE. Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells. Toxicol Sci 53: 340–351, 2000. [DOI] [PubMed] [Google Scholar]
- 22.Jiang J, Stoyanovsky DA, Belikova NA, Tyurina YY, Zhao Q, Tungekar MA, Kapralova V, Huang Z, Mintz AH, Greenberger JS, Kagan VE. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat Res 172: 706–717, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kastl L, Sauer SW, Ruppert T, Beissbarth T, Becker MS, Suss D, Krammer PH, Gulow K. TNF-alpha mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-kappaB activation in liver cells. FEBS Lett 588: 175–183, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 87: 99–163, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Ann Rev Cell Dev Biol 28: 137–161, 2012. [DOI] [PubMed] [Google Scholar]
- 26.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, Kim YS. Carbon Monoxide Activates Autophagy via Mitochondrial Reactive Oxygen Species Formation. Am J Respir Cell Mol Biol 45: 867–873, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516–8525, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80: 401–411, 1995. [DOI] [PubMed] [Google Scholar]
- 31.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol 9: 866–872, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J Neurochem 93: 1199–1208, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, Otterbein SL, Otterbein LE, Choi AM. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem 278: 36993–36998, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 109: 11282–11287, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112: 481–490, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Oberholzer A, Steckholzer U, Kurimoto M, Trentz O, Ertel W. Interleukin-18 plasma levels are increased in patients with sepsis compared to severely injured patients. Shock 16: 411–414, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61: 3100–3103, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev AI. Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion 6: 43–47, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol 180: 7147–7157, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci 125: 5745–5757, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11: 395–402, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153: 521–534, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456: 264–268, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Schroder K, Tschopp J. The inflammasomes. Cell 140: 821–832, 2010. [DOI] [PubMed] [Google Scholar]
- 52.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669–682, 2005. [DOI] [PubMed] [Google Scholar]
- 53.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout (Abstract). Arthritis Res Ther 9: R28, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature 481: 278–286, 2012. [DOI] [PubMed] [Google Scholar]
- 56.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24: 317–327, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 106: 2770–2775, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trnka J, Blaikie FH, Logan A, Smith RA, Murphy MP. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic Res 43: 4–12, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Troseid M, Seljeflot I, Hjerkinn EM, Arnesen H. Interleukin-18 is a strong predictor of cardiovascular events in elderly men with the metabolic syndrome: synergistic effect of inflammation and hyperglycemia. Diabetes Care 32: 486–492, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 365: 53–59, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol 182: 3809–3818, 2009. [DOI] [PubMed] [Google Scholar]
- 62.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP, Otterbein LE. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest 124: 4926–4940, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu JF, Washko GR, Nakahira K, Hatabu H, Patel AS, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, Ross JC, Estepar RS, Diaz AA, Li HP, Qu JM, Himes BE, Come CE, D'Aco K, Martinez FJ, Han MK, Lynch DA, Crapo JD, Morse D, Ryter SW, Silverman EK, Rosas IO, Choi AM, Hunninghake GM, Investigators CO. Statins and pulmonary fibrosis: the potential role of NLRP3 inflammasome activation. Am J Respir Crit Care Med 185: 547–556, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011. [DOI] [PubMed] [Google Scholar]