

Abstract
This paper is focused on unique insights provided by the preterm lamb physiological model of bronchopulmonary dysplasia (BPD). Connections are also made to insights provided by the former preterm baboon model of BPD, as well as to rodent models of lung injury to the immature, postnatal lung. The preterm lamb and baboon models recapitulate the clinical setting of preterm birth and respiratory failure that require prolonged ventilation support for days or weeks with oxygen-rich gas. An advantage of the preterm lamb model is the large size of preterm lambs, which facilitates physiological studies for days or weeks during the evolution of neonatal chronic lung disease (CLD). To this advantage is linked an integrated array of morphological, biochemical, and molecular analyses that are identifying the role of individual genes in the pathogenesis of neonatal CLD. Results indicate that the mode of ventilation, invasive mechanical ventilation vs. less invasive high-frequency nasal ventilation, is related to outcomes. Our approach also includes pharmacological interventions that test causality of specific molecular players, such as vitamin A supplementation in the pathogenesis of neonatal CLD. The new insights that are being gained from our preterm lamb model may have important translational implications about the pathogenesis and treatment of BPD in preterm human infants.
Keywords: neonatal chronic lung disease, alveolarization, alveolar simplification, pulmonary hypertension, airway expiratory resistance, nitric oxide, vitamin A, endothelial nitric oxide synthase, nasal CPAP, insulin-like growth factor-1, epigenetics
acute respiratory failure of preterm human infants reflects immaturity of the lung following preterm birth, disrupted developmental processes, and dysregulated repair responses. This reflection occurs in the neonatal intensive care setting of continuous invasive mechanical ventilation support with oxygen-rich gas that is necessary to keep many preterm human infants alive. Among survivors of preterm birth and initial days of ventilation support with oxygen-rich gas, human infants who require prolonged invasive ventilation support with oxygen-rich gas frequently develop bronchopulmonary dysplasia (BPD). BPD is a significant pediatric health problem because, almost four decades after its initial description (134), this disease of preterm infants remains the most common cause of long-term pour outcomes, including postnatal growth failure, recurrent hospitalization for respiratory tract infections, and intraventricular hemorrhage (53, 178). In the United States, since 2006, the overall rate of prematurity is 1 in 8 births, or ∼12% (74, 118). However, the underlying pathogenic mechanisms are incompletely understood.
Gaps in knowledge about the underlying pathogenic mechanisms that contribute to BPD are related, in part, to the challenge of establishing suitable animal models that include preterm birth and respiratory failure requiring prolonged invasive mechanical ventilation with oxygen-rich gas for days, weeks, or months. Two types of animal models have been developed: large-animal models and small-animal models. Large-animal models provide unique opportunity for physiological studies of evolving disease pathogenesis in preterm neonates that are supported in a neonatal intensive care setting for days or weeks, akin to preterm human infants. Small-animal models provide elegant opportunities to manipulate the genome or molecular pathways to reveal mechanisms that impact the immature lung of postnatal pups (113). Insights provided by both types of models are the focus of this paper.
Species-Specific Lung Anatomy
Species variations are evident in lung anatomy. For context for this review, comparison of lung anatomy is made among humans, monkeys, sheep, rats, and mice. Focus on baboons and sheep is because they are the large-animal models of BPD, rats and mice because they are the small-animal models. Key features of lung comparative anatomy are summarized in Table 1. This table exposes developmental and anatomic similarities and differences among the five species that may be helpful to keep in mind.
Table 1.
Comparative anatomy of human, monkey, sheep, rat, and mouse lungs
| Lung Anatomical Feature | Humans | Monkeys | Sheep | Rats | Mice |
|---|---|---|---|---|---|
| Developmental stage at full term | Alveolar | Alveolar | Alveolar | Saccular | Saccular |
| Lobes | 3 right; 2 left | 4 right; 3 left | 4 right; 2 left | 4 right; 1 left | 4 right; 1 left |
| Airway generations | 17–21 | 13–17 | 13–17 | 13–17 | 13–17 |
| Airway branching pattern | Symmetric | Symmetric | Symmetric | Asymmetric | Asymmetric |
| Trachea | |||||
| Epithelial thickness | 50–100 μm | 20–30 μm | 65–70 μm | ND | 11–14 μm |
| Submucosal glands | Present | Present | Present | Present | Proximal 1/3rd |
| Goblet cells | 9% | 17% | 5% | 1% | <1% |
| Club cells | None | <1% | None | 45% | ∼50% |
| Main bronchus diameter | ∼10–15 mm | ND | ND | 2–3 mm | ∼1 mm |
| Intrapulmonary airways | |||||
| Cartilage | Yes | Yes | Yes | Yes | No |
| Epithelial thickness | 40–50 μm | ∼30 μm | 50–60 μm | 10–16 μm | 8–16 μm |
| Submucosal glands | Present | Present | Present | Absent | Absent |
| Goblet cells | 10% | 15% | Present | Rare | <1% |
| Club cells | 3% | 5% | Rare | 45% | ∼60% |
| Terminal bronchioles | |||||
| Diameter | ∼600 μm | ∼500 μm | ND | ND | ∼10 μm |
| Epithelial thickness | ND | ND | ND | ∼11 μm | ∼8 μm |
| Submucosal glands | Absent | Absent | Absent | Absent | Absent |
| Goblet cells | 2% | 20% | Absent | Absent | Absent |
| Club cells | 11–41% | Absent | ∼60% | ∼75% | 60–80% |
| Respiratory bronchioles | |||||
| Number | Many (∼150,000) | Many | Many | Absent (or one) | Absent (or one) |
| Club cells | ∼20% | >90% | ∼60% | >50% | >50% |
| Ratio of lung parenchyma/total lung volume | ∼12% | ND | ND | ND | ∼18% |
| Alveolar diameter | 100–200 μm | 100–200 μm | 100–200 μm | 50–90 μm | 40–80 μm |
| Blood-air barrier thickness | ∼0.70 μm | ∼0.50 μm | ∼0.50 μm | ∼0.40 μm | ∼0.30 μm |
| Pulmonary venule location | Interlobular septa | Interlobular septa | Interlobular septa | Next to bronchioles | Next to bronchioles |
ND, not determined. Compiled from Refs. 27, 51, 55, 67, 70, 79, 86, 88, 93, 97, 116, 121, 126, 142–145, 149, 169, 170, 172, 184, 185, 190–192 (Humans and Monkeys); Refs. 8, 117, 146 (Sheep); Refs. 32, 34, 40, 71, 80, 92, 98, 112, 125, 127, 128, 137, 138, 141, 144, 158, 180, 187, 194, 198, 201 (Rats and Mice).
At term gestation, the developmental stage is different among the five species (Table 1) (191). The developmental stage at term gestation of the lung of humans, monkeys, and sheep is at the beginning of the alveolar stage. By comparison, the developmental stage of the lung of rats and mice is at the saccular stage. This difference will be returned to later in this review.
A number of gross structural features are species-specific in the adult lung (Table 1). Gross structural differences include lobation, airway generations, airway branching pattern, diameter of the main bronchus, and cartilage in the upper airways (bronchi). The human lung has fewer lobes, more airway generations, and the largest diameter of the main bronchus. More airway generations in the lung of humans translates to the lung's parenchyma making up a smaller calculated proportion of total lung volume in humans (∼12%) compared with mice (∼18%). The lung of humans, monkeys, and sheep also has airway branching that is symmetrical, meaning dichotomous branching of daughter airways of similar diameter. By comparison, the lung of rats and mice has airway branching that is asymmetrical (monopodial; each single daughter branch has smaller diameter than the contiguous airway). Another species difference in the lung of humans, monkeys, sheep, and rats is the wall of their upper airways has cartilage, whereas the lung of mice has cartilage that is limited to the upper one-third of the initial intrapulmonary airways. These gross structural differences may influence the distribution of airway resistance and particulate deposition along the airway tree among the five species.
Another anatomical contrast is that the lung of humans, monkeys, and sheep has many respiratory bronchioles interposed between terminal bronchioles and alveolar ducts/alveoli (Table 1). The lung of rats and mice is essentially devoid of respiratory bronchioles, or their lung may have one respiratory bronchiole that precedes alveolar ducts/alveoli. Because respiratory bronchioles subserve dual functions of conduction of air and exchange of respiratory gases (4), both physiological processes occur in different anatomic locations in the lung of humans, monkeys, and sheep compared with the lung of rats and mice.
The distribution of various cell types also is species related (Table 1). In the lung of humans, monkeys, and sheep, the principal secretory cell types along the length of the upper airways are mucus and serous epithelial cells in submucosal glands, as well as goblet cells. By comparison, the lung of rats and mice has submucosal glands that are limited to the upper third of the initial intrapulmonary airways. So, too, the number of goblet cells is limited in distribution and number in the lung of rats and mice. Instead, club cells (formerly called Clara cells; Ref. 196) are the principal secretory epithelial cell type in the upper airways of the lung of rats and mice. Club cells are located in bronchioles of all five species. These species-specific distributions of cell types may be associated with different amount and quality of airway secretions, as well as responses to exposure to xenobiotics.
Finally, the first row in Table 1 reminds us that at full-term gestation, lung development is at the transition to the alveolar stage in humans, monkeys, and sheep, whereas lung development is at the transition to the saccular stage in rats and mice. However, bronchopulmonary dysplasia occurs in human infants who are born preterm. Their lungs are at the canalicular stage or saccular stage. The structural and functional immaturity of their lungs frequently leads to respiratory distress and failure that necessitates intubation, mechanical ventilation, and surfactant replacement to keep the preterm human infants alive. Herein lays an important distinction from rats and mice because while their lungs are at the saccular stage of development at term gestation they thrive without need for respiratory support. Nonetheless, their immature lungs are susceptible to injury induced by high inspired oxygen, mechanical ventilation, and infection/inflammation. Thus the large-animal preterm models and the small-animal term models are valuable for identifying pathogenic mechanisms and testing treatment approaches.
Preterm Birth in Humans and the Adverse Outcome of Bronchopulmonary Dysplasia
The definition of preterm birth in humans is any birth, regardless of weight at birth, that occurs before 37 completed wk of gestation from the first day of menstrual cycle (11a). Pregnancies that end before 20 completed wk of gestation are referred to as miscarriages. Therefore, another definition of preterm birth is any delivery that occurs between 20 and 37 wk of gestation in humans. The rate of preterm birth in the United States has steadily declined to the most recent rate of 12% for 2012 (119). Although the rate of preterm birth has steadily declined, the number of preterm births remains ∼500,000/year (74).
When a human infant is born preterm, its lungs are structurally immature. The immature lung's developmental stage may be at the canalicular stage (17–28 wk gestation; term is ∼40 wk) or the saccular stage (24–36 wk gestation) of development (33). The canalicular stage of lung development is characterized architecturally by thick partitions of mesenchyme that separate the air canals (10). Because the mesenchyme forms thick partitions, the growing capillary bed remains physically distant from the air canals. Also, the air canals are lined by tall (columnar or cuboidal) epithelial cells. These architectural features mean that the diffusion distance from the air spaces to flowing red blood cells is large, which impedes efficient respiratory gas exchange. Furthermore, the preterm human lung at the canalicular stage of lung development is deficient in surfactant and surfactant apoproteins, creating high surface tension forces and air space instability (151). Consequently, air space collapse (atelectasis) occurs. Atelectasis contributes to poor oxygenation because of ventilation-perfusion mismatch and intrapulmonary shunt (12).
A similar scenario unfolds when a human infant is born preterm at the saccular stage of lung development. This stage's architecture is characterized by transformation of air canals into air sacs (10). The structural transformations create larger air space surface area, thinner mesenchymal partitions, shorter epithelial cells, and more capillaries that are closer to the air sacs. Therefore, these developmental transformations reduce the diffusion distance between the expanding surface areas of the gas-exchange air spaces and capillaries. However, intimate structural association between the transforming respiratory gas exchange air spaces and capillaries is not accomplished during the saccular stage.
Opening the collapsed air canals or sacs requires recruitment of collapsed air spaces, as well as air spaces yet to be recruited. Preterm human infants who do not recruit air spaces and do not adequately exchange respiratory gases during spontaneous breathing develop respiratory failure that requires endotracheal intubation and ventilation support to sustain preterm life. When high inspiratory pressures are used, however, the danger is that only the ventilated air spaces will fill with each provided breath, leading to air space distension (overinflation). Preterm human infants who have these characteristics usually develop acute lung injury (respiratory distress syndrome, RDS). Histopathologically, during the initial hours of invasive ventilation support, uneven inflation may be evident, owing to surfactant deficiency and residual, uncleared fetal lung liquid in the air spaces and mesenchyme. By 12–24 h of invasive ventilation support, necrosis of epithelial cells occurs and plasma proteins leak from the underlying blood vessels. Consequently, blankets of extravasated protein accumulate along the air space surfaces, forming hyaline membranes (10, 177). Hyaline membranes are the histopathological basis of the term hyaline membrane disease.
Survival of preterm human infants with RDS has increased since the era of antenatal glucocorticoid treatment and postnatal surfactant replacement, as well as gentler ventilation modes (83, 105, 106, 129). Survival rates at some centers are better than 90% for preterm human infants weighing 1,500 g or less at birth (36, 171). Nonetheless, more than 40% (10,000–15,000 per year) of these preterm human infants required prolonged intubation and ventilation support and therefore progress to BPD (66, 95). BPD is associated most with very low birth weight, antenatal infection, and need for sustained mechanical ventilation with oxygen-rich gas (96, 101).
The histopathology of contemporary BPD is characterized by alveolar simplification (10). Alveolar simplification is evident as distended distal air spaces that have few secondary septa, air space walls that are thick and cellular, and few capillaries that also are distant from the air spaces. A physiological consequence of reduced development of air space and capillary surface area is continued inadequate exchange of respiratory gases (hypoxemia and hypercarbia, with low blood pH).
Animal Models as Tools To Identify Pathogenic Mechanisms Leading to BPD
The first large-animal model of BPD used preterm baboons. That model, developed by Coalson and colleagues and first reported in the 1980s (46–49, 63), managed preterm baboons with invasive mechanical ventilation and oxygen-rich gas for days or weeks in the neonatal intensive care setting. Prolonged hyperoxia (100% inspired oxygen) was used in the early years of that unique model. Subsequent iterations of the preterm baboon model limited oxygen exposure to maintain adequate arterial oxygenation (60–90 mmHg).
Our group's first report using the preterm lamb model of BPD was 1997 (139). The preterm lamb model also limits oxygen exposure (<50% inspired oxygen) to maintain adequate arterial oxygenation of 60–90 mmHg. An advantage of preterm lambs is their larger size (∼3 kg at 131-day gestation) compared with preterm baboons (∼1/10th the birth weight). Larger size makes preterm lambs more amenable to frequent blood sampling across weeks of neonatal intensive care support. Another advantage related to the larger size of preterm lambs is large caliber of blood vessels and lymphatics for chronic catheters. We use the blood vessel catheters for continuous measurements of pulmonary and systemic hemodynamic parameters and the lymphatic catheter for continuous assessment of lung liquid filtration (20). A third size advantage of preterm lambs is that their organs are larger, which is valuable for postmortem sampling.
Two notable differences remain in the physiological conditions between chronically ventilated preterm lambs with evolving neonatal chronic lung disease (CLD) and chronically ventilated preterm human infants who develop BPD. One difference is that fetal lambs are delivered prematurely by Cesarean section, without prior labor and without intentional intrauterine infection. Many preterm human infants who develop BPD were born after the onset of preterm labor, often complicated by intrauterine infection (59, 159, 160). Another difference, at least during the initial years using the preterm lamb model, is that the ductus arteriosus was surgically ligated within a few days of preterm birth (6, 20, 139). The protocol was subsequently changed to let the ductus close spontaneously (5, 135, 150). Rationale for eliminating ductal ligation was to minimize surgical trauma to the lung and postoperative edema, inflammation, infection, and atelectasis. Pharmacological closure, using indomethacin (42) or ibuprofen (122), has not been used in this model. Our experience is that the ductus is physiologically closed during the first days of life.
The obvious phylogenetic advantage of baboons is their closeness to humans compared with sheep (and rats and mice). Another advantage of preterm baboons is that they are more immature at the time of operative delivery (∼134–147 days of gestation; 65–75% of gestation) than preterm lambs (125–131 days of gestation; 80–85% gestation). However, the lungs of both species are within the canalicular or saccular stage of lung development, depending on gestation age at the time of operative delivery. Both gestational ranges are at the lower limit of viability of preterm baboons and preterm lambs in the neonatal intensive care setting. The preterm baboon program ended a decade ago, leaving the preterm lamb model as the only large-animal model of BPD.
The uniqueness of both large-animal models is that they reproduce the clinical setting of preterm birth and respiratory failure requiring prolonged invasive mechanical ventilation with oxygen-rich gas for days or weeks. Both models also recapitulate the pathophysiology and pathology of evolving neonatal CLD in preterm baboons (46, 49, 50, 58, 63, 124, 199) and preterm lambs (5, 6, 20, 24, 25, 111, 139, 150). We use the term “evolving neonatal CLD” for the preterm baboon and preterm lamb models and reserve the term “BPD” for preterm human infants, to avoid confusion between large-animal models vs. the human disease.
The preterm baboon and preterm lamb models are challenging because they require intensive and lengthy life-support care 24 h per day for days or weeks. The arduous nature of these physiological studies is costly. Also, sample size is small (typically 4–6/group), in part because of cost and in part to comply with necessary guidelines for care and use of animals for biomedical research (to reduce, refine, and replace). Of these three Rs, replace is not an option because the uniqueness of both large-animal models is they recapitulate the neonatal intensive care setting because BPD occurs in preterm human infants who develop respiratory failure that requires prolonged invasive mechanical ventilation with oxygen-rich gas for days, weeks, or months. Counterbalancing the guideline to reduce the number of animals used is the expectation that study design be sufficiently powered to detect statistical differences between groups. The sample size and counterbalance to detect statistical differences also apply to models that use rats or mice.
Pathophysiology and Histopathology of Respiratory Failure
A worrisome outcome among former preterm human infants is that carbon monoxide diffusing capacity is lower than across the lung of control, healthy infants who are born at term gestation (14, 19, 132, 133). Why is diffusing capacity worse? Clues come from understanding the normal process by which the developing walls of the premature lung undergo morphogenesis to create a thin barrier across which oxygen and carbon dioxide must rapidly diffuse postnatally. Alveolar walls of the human adult lung are thin (∼1.0 μm) (4, 54, 193). Thinness facilitates rapid diffusion of oxygen and carbon dioxide, in part because water is the principal impediment to molecular diffusion, particularly oxygen diffusion (4). The structural elements of the adult alveolar wall are the alveolar epithelium, the interstitium that makes up the alveolar wall, and the alveolar endothelium.
Developing air space walls at the time of preterm birth are thick and cellular, and blood vessels are distant from the air space lining epithelial cells (7, 33, 43, 45). Thickness, cellularity, and blood vessel distance reflect the developmental stage at which preterm birth occurs. All three structural features are greater when preterm birth occurs during canalicular stage than during saccular stage. Likewise, primary or secondary septation is dependent on the developmental stage at which preterm birth occurs. Primary septation is necessary for subdivision of the air canals into air saccules. Secondary septation subdivides the air saccules into anatomic alveoli. If preterm birth occurs during the canalicular stage, and prolonged invasive mechanical ventilation is required, then primary septation is disrupted. If preterm birth happens during the saccular stage, secondary septation is disrupted by prolonged invasive mechanical ventilation. Without sequential subdivision, air space surface area is inadequate for gas exchange to support postnatal life. Presence of distal air space walls that are thick and cellular, and blood vessels that are distant from the air space-lining epithelial cells are characteristic histopathological features of alveolar simplification in preterm human infants who die with BPD (45, 90), as well as preterm baboons and preterm lambs (Fig. 1) that have evolving neonatal CLD.
Fig. 1.

Collage of radiographic and histological appearance of the head, neck, and lungs of preterm lambs supported by 2 modes of ventilation for 21 days. Left: invasive mechanical ventilation. The top radiograph shows the endotracheal tube's lumen highlighted in blue in the neck. Juxtaposed in the remainder of the left half are chest radiographic and lung histological images. The chest radiograph shows opacification of the lung fields, which obscure the heart shadow (posterior-anterior view; L, left). The terminal respiratory unit (TRU) has distal air spaces that are distended, distal air space walls that are thick and cellular (arrowhead), and secondary septa that are infrequent and stunted (arrow). Right: less invasive high-frequency nasal ventilation. The top radiograph shows the uncuffed nasal tube's lumen highlighted in blue in the nose. The chest radiograph shows better aeration and larger lung volume, which make the heart shadow obvious. The TRU has distal air spaces that are more uniformly shaped, distal air space walls that are thin and less cellular (arrowhead), and secondary septa that are numerous, long, and thin (arrow). The scale bar is 100 μm in length. Adapted and modified from Ref. 135.
Another disrupted process is capillary growth. Normally during lung development during the second half of gestation, the number and surface area of blood vessels increases in the thick, cellular mesenchymal walls that separate air canals and the subsequent air saccules (31, 163). Accompanying this growth is extension of capillaries into the mesenchymal core of secondary septa as the secondary septa sprout from the saccular walls. Preterm birth and prolonged invasive mechanical ventilation with oxygen-rich gas reduce capillary growth in the lung of preterm lambs (5, 6, 20) and preterm baboons (114). The lungs of the preterm lambs also have stunted secondary septa that lack capillaries (5). Without capillaries in the developing air space walls and stunted secondary septa, the simplified parenchyma cannot meet the need for efficient exchange of respiratory gases. This deficiency of capillary vascular surface area is compounded by fewer pre- and postcapillary arterioles and venules (6, 20). The breadth of diminished growth of pulmonary capillaries and microvessels contributes to the respiratory failure that necessitates continued invasive mechanical ventilation with oxygen-rich gas (6, 135).
A third histopathological characteristic of alveolar simplification is disrupted expression and accumulation of elastin. Appropriate expression of elastin and accumulation of elastic fibers are necessary for normal alveolar secondary septal formation, as demonstrated by two studies that used knockout mice constructs (108, 195). One study used mice that lacked platelet-derived growth factor-A (108). The lungs of these mice lack both myofibroblasts and elastin. The mice reach term gestation; however, they develop respiratory failure in the initial postnatal days of life. Their lungs have alveolar simplification. The other study used elastin knockout mice (195). The pups survive through the initial postnatal days of life but they are cyanotic. Their lungs have distal air spaces that are arrested at the saccular stage of lung development. Coincidently, their lungs also have fewer vascular and airway generations. The latter coincident histopathological feature suggests that elastin synthesis and deposition also regulate vascular and airway growth in the immature lung.
The lung of preterm lambs with evolving neonatal CLD has aberrant and continuously excessive expression of elastin and accumulation of elastic fibers (139). Upregulated expression of elastin mRNA occurs swiftly, by the third day of invasive mechanical ventilation. Excess and aberrant accumulation occurs in the walls lining the saccules and simplified alveoli (6, 139). Particularly conspicuous is accumulation of disorganized bundles of elastic fibers at the sites of stunted secondary septa. Expression and accumulation of elastin persists in established neonatal CLD 3–4 wk later. Persistence suggests that dysregulation of elastin gene expression and elastic fiber assembly is driven by invasive mechanical ventilation. The mechanisms driving this outcome are not known.
Less invasive ventilation leads to better lung outcomes.
Encouraging observations are emerging when less invasive modes of ventilation are used, such as nasal continuous positive airway pressure (nasal CPAP). Encouragement stems from association between less invasive modes of ventilation with lower incidence and/or severity of BPD in some preterm human infants (189). Evidence from preterm baboon and preterm lamb studies is consistent because less invasive modes of ventilation lead to less inflammation (91), more production of surfactant (131), and better alveolarization and capillary growth compared with invasive mechanical ventilation (91, 135, 150, 182).
Two recent physiological reports by our group shows that less invasive high-frequency nasal ventilation (HFNV) in spontaneously breathing preterm lambs is associated with appropriate respiratory gas exchange for 3 days or 21 days (135, 150). We use a high-frequency, flow-interruption ventilator that also provides background conventional breaths (model VDR4, Percussionaire, Sand Point, ID). This ventilator is flow regulated and time cycled and delivers high-frequency, subphysiological tidal volumes in the background of which are controlled-pressure, low-frequency breathing cycles (conventional breaths) (110). We found that significantly lower applied oxygen and lower respiratory system pressures are required to maintain physiologically targeted values of arterial partial pressure of oxygen (60–90 mmHg) and carbon dioxide (45–55 mmHg) compared with invasive mechanical ventilation.
Another physiological result in our most recent report is that intratracheal pressure is very low during less invasive HFNV (135). We measured intratracheal pressure in situ while the preterm lambs breathed spontaneously. We found that mean intratracheal pressure is ∼1/10th that during invasive mechanical ventilation. Our in situ measurements show that less invasive HFNV maintains positive mean intratracheal pressure of 1–2 cmH2O, which also means that positive end-expiratory pressure is low.
An important morphological result is that alveoli form in the lung of preterm lambs that are supported by less invasive HFNV (Fig. 1). The distal air space walls are thin and have few mesenchymal cells within them, and secondary septa are plentiful and thin and have capillaries. Each of these structural features of alveolarization is missing from the simplified architecture in the lung of preterm lambs that are supported by invasive mechanical ventilation (Fig. 1).
Why did we use a ventilator that provides pressure-limited, time-cycled conventional breaths at low frequency to deliver physiological tidal volume in the background behind high-frequency, small percussive pressures that deliver subphysiological tidal volumes (22)? Because bubble nasal CPAP did not sustain acceptable respiratory gas exchange longer than 3–4 h of postnatal life despite treating the preterm lambs antenatally with corticosteroids to promote lung development, perinatally with exogenous surfactant to increase lung compliance, and postnatally with caffeine citrate to stimulate breathing. Instead, they gradually accumulated carbon dioxide and their pH progressively decreased to unphysiological levels.
An important advantage of using less invasive HFNV for our studies is that this noninvasive mode of gentle ventilation provides positive gold standards for physiological, morphological, and molecular outcomes in the lung. Prior to using less invasive HFNV, we relied exclusively on reference lambs for normal development. The value of reference lambs is that they provide context for the stage of lung development at the time of delivering fetal lambs for preterm studies (same postconceptional age) and at the time when the preterm lamb studies end (same postconceptional age). Other reference groups provide context for postnatal age at 3 days or 21 days of life (same postnatal age), with or without invasive mechanical ventilation (25). The purpose of the latter reference groups is to differentiate the impact of invasive mechanical ventilation on the preterm lung vs. the term lung. However, none of these reference groups meet the clinical setting of preterm delivery, respiratory failure, and invasive mechanical ventilation with oxygen-rich gas. Creating the less invasive HFNV mode meets the clinical setting, using preterm lambs, with the distinction being that the outcomes are good (135, 150).
Having two ventilation modes that provide either bad outcome (invasive mechanical ventilation) vs. good outcome (less invasive HFNV) in the lung is proving to be important for identifying mechanisms that contribute to the pathogenesis of neonatal CLD. For normal alveolarization, the thick and cellular walls of the distal air spaces need to thin, in part by reducing the number of mesenchymal cells in the distal air space walls. Invasive mechanical ventilation sustains the thick and cellular mesenchymal walls of the distal air spaces (Fig. 1), in part by decreasing apoptosis and increasing proliferation of mesenchymal cells in the distal air space walls (135, 150). That is, invasive mechanical ventilation shifts the balance in favor of persistence and proliferation of mesenchymal cells, leading to persistently thick and cellular distal air space walls (135, 150). Less invasive HFNV, by comparison, leads to thin and less cellular distal air space walls (Fig. 1). These structural benefits coincide with shift toward more apoptosis, and less proliferation, of mesenchymal cells in the distal air space walls. These results may provide an explanation for why sustained inflation at birth does not protect preterm lambs from lung injury (81).
The two ventilation modes also differentially impact growth of capillaries in the lung. For normal alveolarization, capillaries grow along the thinning distal air space walls and into the sprouting secondary septa. Invasive mechanical ventilation decreases both processes (135, 150). The downregulated molecular players are vascular endothelial growth factor (VEGF), its functional receptor 2 in the lung (VEGF-R2), and midkine (150). Similar molecular downregulation occurs in the lung of preterm baboons that are supported by invasive mechanical ventilation (114). In this regard, the two physiological models are consistent. Less invasive HFNV of preterm lambs, by comparison, preserves these vascular growth factors in the thinning distal air space walls and sprouting secondary septa. Physiologically, our studies also show that less invasive HFNV is accompanied by acceptable respiratory gas exchange across the developing lung.
We conclude from these recent studies that less invasive HFNV for 3 days (150) or 21 days (135) provides acceptable respiratory gas exchange accompanied by appropriate alveolarization. Neither of these good outcomes is associated with invasive mechanical ventilation. Whether these results are translatable will require using the same type of ventilator to support preterm human infants.
Vitamin A therapy improves alveolarization in chronically ventilated preterm lambs.
A treatment approach that has potential to improve outcomes among some chronically ventilated preterm human infants is vitamin A supplementation. The rationale for supplementation is that preterm human infants are deficient in retinol (29, 85, 166, 167). However, inconsistent results have been obtained from large multicenter studies, with some studies showing that vitamin A supplementation reduces the need for oxygen at 36 wk postmenstrual age in subgroups of preterm human infants at risk of BPD, whereas other studies have not shown benefit (11, 186). Whether vitamin A supplementation affected alveolarization in the lung of the treated preterm human infants is not known because autopsy studies have not been reported.
We tested the hypothesis that vitamin A supplementation promotes alveolarization in our preterm lamb model of neonatal CLD. Supplemental vitamin A (Aquasol; 5,000 U·kg−1·day−1) given to mechanically ventilated preterm lambs for 21 days increased plasma retinol level compared with preterm lambs supported by invasive mechanical ventilation alone. Daily vitamin A supplementation improved alveolarization and alveolar capillary growth (Fig. 2) (5). Vitamin A supplementation also increased mRNA and protein levels of VEGF, its functional receptor VEGF-R2, and midkine in the same lungs. Vitamin A supplementation reduced elastin mRNA expression and elastic fiber accumulation in the developing lung parenchyma. Accompanying these beneficial structural and molecular outcomes was a trend for better respiratory gas exchange. These results are different from studies in fetal lambs that were treated with all-trans retinoic acid 3 h before intra-amniotic injection of endotoxin (100). Rationale for prior treating with all-trans retinoic acid is that chorioamnionitis reduces retinoic acid concentration. However, all-trans retinoic acid did not prevent inflammation-induced alveolar simplification. This outcome may contribute to lack of benefit of vitamin A supplementation to preterm human infants.
Fig. 2.

Quantitative histology shows that radial alveolar count (A), alveolar secondary septal volume density (B), air space capillary surface density (C), and extra-alveolar microvessel number (D) are greater in vitamin A-supplemented preterm lambs compared with vehicle controls (bracket, P < 0.05; means ± SD; n = 4/group). Term (T) lambs served as the gestation age-matched reference for the 3-wk ventilation studies. Although vitamin A supplementation improved structural development, the improvements were significantly less than the term lambs (P < 0.05) for 3 of the 4 quantitative histological parameters. IMV, invasive mechanical ventilation. Adapted from Ref. 5.
Supplemental vitamin A (Aquasol; 5,000 U·kg−1·day−1) given to mechanically ventilated preterm baboons for 2 wk did not lead to alveolarization or alveolar capillary growth, or expression of vascular growth factors (140). Nor did vitamin A treatment reduce the excessive, aberrant expression of elastin and accumulation of elastic fibers in the lung of the preterm baboons. A potential explanation for these negative outcomes is that the treatment period was short: 2 wk. Perhaps the treatment period was insufficient for downstream signaling and subsequent impact on alveolar secondary septation, which also may account for lack of improved respiratory gas exchange in the chronically ventilated preterm baboons.
Pathophysiology and Pathology of Pulmonary Hypertension
Pulmonary hypertension with associated right heart failure is a frequent complication of BPD in preterm human infants (2, 16, 35). A recent clinical study showed that ∼18% of 145 preterm human infants at 26 wk gestational age had pulmonary hypertension at any time during their hospitalization (17). Such preterm human infants have recurring episodes of cyanosis that are attributed to acute constriction of pulmonary arteries, leading to hypoxemia and bradycardia. The molecular basis of pulmonary hypertension remains incompletely understood, which may limit development of novel therapies to minimize the severity or duration of pulmonary hypertension in chronically ventilated preterm human infants.
We used our preterm lamb model to investigate the pathophysiological, histopathological, and molecular basis of pulmonary hypertension during the evolution of neonatal CLD. We ventilated the preterm lambs with either slow, deep inflations (20 breaths/min; ∼15 ml/kg tidal volume) or fast, shallow inflations (60 breaths/min; ∼5 ml/kg tidal volume) for 3–4 wk (20). The purpose of using two tidal volumes was to identify the impact of lung overdistension on outcomes. However, for the purpose of this paper, results are reported for the 60 breaths/min (∼5 ml/kg tidal volume) group because this level of targeted lung volume support is more consistent with clinical management of invasively ventilated preterm human infants. None of these preterm lambs received antenatal steroids. All of them received surfactant at birth. They subsequently had surgical ligation of their ductus arteriosus, as well as placement of vascular catheters and thermistor wire in the pulmonary artery to serially calculate pulmonary vascular resistance. Both groups of preterm lambs developed neonatal CLD based on oxygenation and ventilation parameters, serial chest radiographs, and postmortem pathology. Pathophysiological results using this study design are summarized in the next four subsections.
Evolving neonatal CLD leads to pulmonary hypertension and vascular pathology.
Normally, pulmonary vascular resistance decreases during the first postnatal month of life after birth at term gestation (2, 16). In preterm infants who develop BPD, however, a frequent occurrence is development of pulmonary hypertension (15). Insights into the pathogenesis of pulmonary hypertension are provided by our preterm lamb model of evolving neonatal CLD.
Pulmonary vascular resistance decreased postnatally in normal reference lambs that were born at term gestation and lived for 3 wk (Fig. 3). By comparison, pulmonary vascular resistance did not decrease from the first to the last (third) week of invasive mechanical ventilation (Fig. 3) (20). Accompanying development of pulmonary hypertension is persistent muscularization of pulmonary arterioles adjacent to terminal bronchioles (Fig. 3). We used terminal bronchioles as the independent structural landmark to identify the same generation of pulmonary arterioles, regardless of their diameter. Persistence was revealed by comparison with unventilated fetal and postnatal reference lambs, which provided normal developmental perspective (6, 20, 139). This comparative quantitative histological analysis revealed that greater thickness of the smooth muscle wall of pulmonary arterioles at the end of 3 wk of invasive mechanical ventilation is equivalent to that of reference fetal lambs that were matched for the gestational age when the preterm lambs were delivered. We also showed that the number of lung microvessels (Fig. 4) and surface density of capillaries (refer to Fig. 2) are significantly smaller compared with lungs of reference term lambs (20). Fewer pulmonary microvessels, including capillaries, could contribute to pulmonary hypertension through reduced total vascular surface area in the lung. These structural results are important because they demonstrate that prolonged invasive mechanical ventilation of preterm lambs decreases development of the pulmonary microcirculation, as occurs in the lung of preterm human infants who died with BPD (28, 39, 44, 82, 84, 115, 176, 183, 188).
Fig. 3.
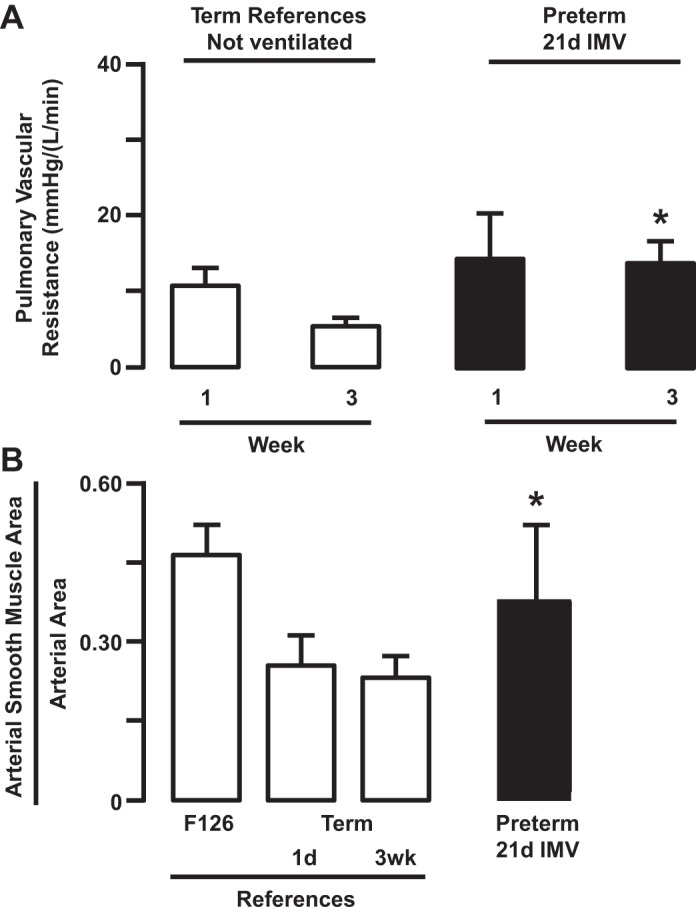
Pulmonary vascular resistance (A) decreased in term reference lambs that were not ventilated from week 1 to week 3 of postnatal life (means ± SD; n = 6–7/group). By comparison, pulmonary vascular resistance did not decrease in preterm lambs that were supported by invasive mechanical ventilation (IMV) over the same postnatal time period (*P < 0.05 compared with matched term reference). Muscularization of pulmonary arterioles (B) normally decreases postnatally in reference lambs that were not ventilated. By comparison, muscularization persisted in preterm lambs that were supported by IMV for 3 wk. 1d and 21d, 1 and 21 days, respectively. Data from Ref. 20.
Fig. 4.
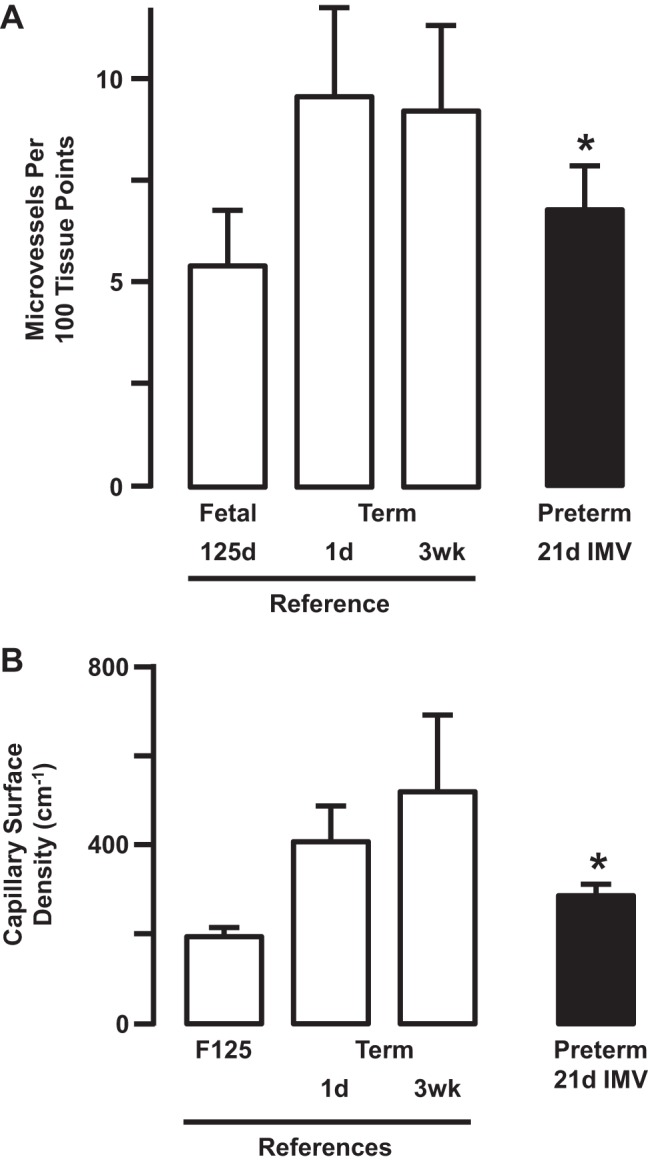
Extra-alveolar microvessel number (A) and capillary surface density (B) increased in term reference lambs that were not ventilated (means ± SD; n = 5/group). Extra-alveolar microvessel number and capillary surface density were significantly lower in preterm lambs that were supported by invasive mechanical ventilation (IMV) for 3 wk compared with both newborn references (*P < 0.05). Adapted from Ref. 20.
Evolving neonatal CLD leads to edema and pulmonary hypertension.
The pathophysiological and histopathological results in chronically ventilated preterm lambs led us to ask several pathophysiological questions. The first question is whether increased pulmonary vascular resistance led to pulmonary edema.
Postmortem extravascular lung water was significantly greater in chronically ventilated preterm lambs than in reference lambs that were born at term gestation (henceforth called reference term lambs) (Fig. 5) (20). Therefore, pulmonary edema formed in the lungs of chronically ventilated preterm lambs. Pulmonary edema was evident as cuffs of interstitial pulmonary edema liquid surrounding pulmonary blood vessels and airways, as well as within distended interlobular connective tissue septa. Lung lymphatics in both interstitial compartments were dilated. Whereas air space edema liquid was unusual in regions of hyperinflated distal air spaces, air space edema was present in regions that contained collapsed air spaces. The features were not present in the lung of reference term lambs. These results led us to ask whether the pulmonary edema is caused by increased hydrostatic pressure or increased permeability.
Fig. 5.
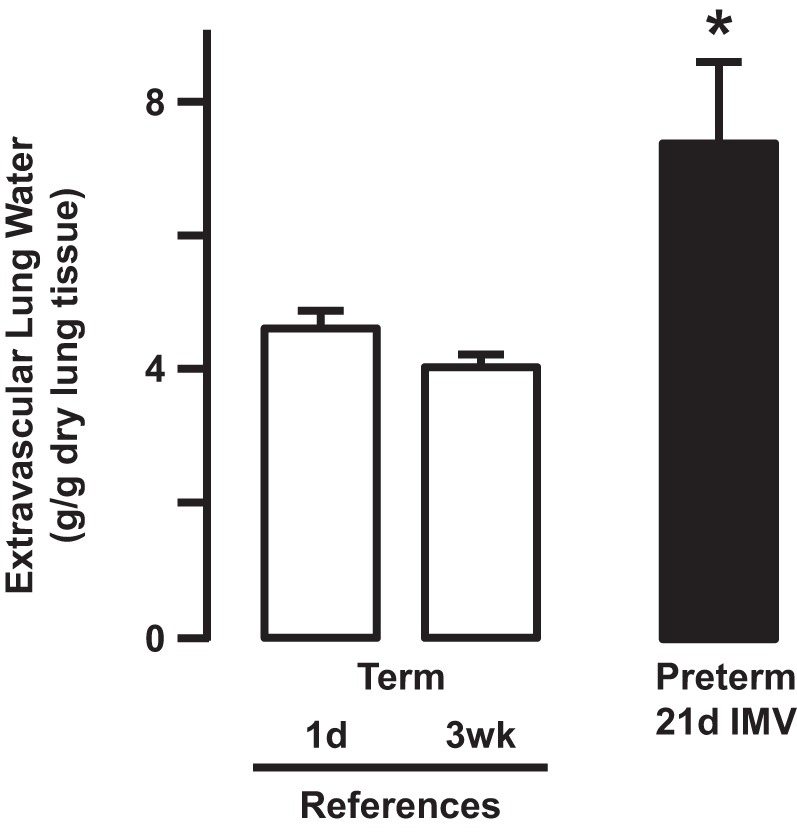
Extravascular lung water, an indicator of pulmonary edema, is significantly greater in the lung of preterm lambs supported by invasive mechanical ventilation (IMV) compared with both groups of newborn reference lambs (*P < 0.05; means ± SD; n = 6/group). Data from Ref. 20.
Evolving neonatal CLD leads to increased hydrostatic pulmonary edema.
A feature of acute lung injury, as occurs in RDS, is pulmonary edema (2, 16, 22, 82). The pulmonary edema liquid is protein rich in both the lung interstitium and air spaces (23, 37, 89). Whether neonatal CLD is associated with protein-rich edema liquid was not known at the time that we used lung lymph to investigate the pathogenesis of pulmonary edema during the evolution of neonatal CLD in chronically ventilated preterm lambs.
We addressed the uncertainty by cannulating the efferent duct of the caudal mediastinal lymph node to collect lung lymph (20). The unique lymphatic anatomy of sheep (9, 161, 174, 175) allows continuous physiological collection of lung lymph, which is used to differentiate increased hydrostatic pulmonary edema from increased permeability pulmonary edema (30, 173). Briefly, we measured lung lymph and plasma protein concentrations to calculate the lymph-to-plasma (L/P) ratio and lymph protein clearance. Decreasing or low L/P ratio is consistent with increased hydrostatic pulmonary edema. Increasing or constant L/P ratio, by comparison, is consistent with increased permeability pulmonary edema (173). We found that the L/P ratio decreased over the 3 wk of invasive mechanical ventilation of preterm lambs, although the difference from week 1 to week 3 was not statistically significant. Lymph protein clearance did not change over the same time period, owing to increased lymph flow. Thus these data are consistent with increased hydrostatic pulmonary edema in chronically ventilated preterm lambs (173).
These physiological results are unique between the two preterm large-animal models of neonatal CLD because comparable physiological measurements of cardiovascular and pulmonary lymphatic function were not made in preterm baboons. Our physiological results are informative because they demonstrate that prolonged invasive mechanical ventilation leads to pulmonary edema from increased hydrostatic pressure. Therefore, the preterm lamb model filled two important gaps in knowledge about pulmonary vascular and lymphatic function during the evolution of neonatal CLD.
Evolving neonatal CLD dysregulates the nitric oxide signaling pathway.
What molecular factors might contribute to increased pulmonary vascular resistance, persistent muscularization of pulmonary arterioles, smaller surface area of pulmonary microvessels and capillaries, and increased hydrostatic pulmonary edema in chronically ventilated preterm lambs? A pathway of interest at the time was dysregulation of the nitric oxide (NO) signaling pathway.
Several lines of evidence led us to test the NO signaling pathway. One line of evidence is that NO is important for regulating smooth muscle tone in pulmonary blood vessels and airways of newborn animals. NO diffusion into the cytoplasm of smooth muscle cells increases the concentration of guanosine 3′,5′-cyclic monophosphate (cGMP), which induces smooth muscle relaxation, and therefore leads to vasodilation (26). Diffusion of physiological doses (5–15 parts/million, ppm) of inhaled NO (iNO) into blood does not lead to systemic hypotension because NO is inactivated by tight binding to hemoglobin (73, 136, 152). The role of NO on airway smooth muscle tone will be discussed separately, after this discussion on pulmonary blood vessels.
Another line of evidence is that inhibition of NO production disrupts vascular smooth muscle tone in pulmonary blood vessels. Inhibition attenuates ∼50% of the normal postnatal decrease of pulmonary vascular resistance in newborn lambs (1, 65) and newborn piglets (147). Conversely, iNO abruptly and profoundly decreases pulmonary vascular resistance, with associated increase in arterial oxygenation, in newborn animals and human infants with pulmonary hypertension (41, 52, 57, 68, 94, 109, 131a, 153–155, 157, 203, 204).
NO also acts as a signaling molecule to inhibit growth of vascular muscle cells in vitro (69, 77, 181). In vivo studies provide supportive evidence for the latter action of NO. For example, inhibiting endogenous production of NO enhances smooth muscle growth in response to injury (162). By comparison, exogenously supplied NO, either by an NO donor or inhalation, inhibits smooth muscle growth after mechanical injury (76, 104, 164).
Clinical trials published before 2000, when our first physiological report on dysregulation of the pulmonary circulation in chronically ventilated preterm lambs was published, showed that iNO decreases pulmonary vascular resistance and improves arterial oxygenation in human newborn infants with persistent pulmonary hypertension (94, 131a, 155, 157). Other clinical trials showed that low-dose iNO given to human newborn infants with persistent pulmonary hypertension reduces the need for extracorporeal membrane oxygenation (41, 56). Likewise, experimental animal studies showed that iNO reverses pulmonary vasoconstriction following either hypoxia or prenatal closure of the ductus arteriosus (154, 203). The systemic circulation was not dilated in those studies.
Based on the aforementioned roles of NO in regulation of pulmonary vascular tone, reactivity, and smooth muscle growth, we performed three physiological studies. We 1) measured nitric oxide synthases (NOSs) and soluble guanylate cyclase (sGC) in the lung of chronically ventilated preterm lambs, 2) provided iNO for 1 h at weekly intervals to chronically ventilated preterm lambs, and 3) gave iNO continuously for 3 wk to chronically ventilated preterm lambs.
For the first assessment, we quantified protein abundance of endothelial nitric oxide synthase (eNOS), inducible NO synthase (iNOS), and sGC (24, 111). We used quantitative immunoblot analysis of third to fifth generation intrapulmonary arteries and semiquantitative immunohistochemistry of pulmonary arterioles adjacent to terminal bronchioles. Our results showed less eNOS protein in endothelial cells of intrapulmonary arteries (Fig. 6) and less immunolocalization of eNOS in pulmonary arterioles next to terminal bronchioles in the lung of the chronically ventilated preterm lambs compared with reference term lambs at 1 day of age (same postconceptional age) (24, 111). For iNOS, neither protein abundance nor localization was different compared with the reference term lambs (24, 111). We used the same analytical methods to determine abundance and localization of sGC. The latter results showed that sGC protein abundance is less in the lung of chronically ventilated preterm lambs than in reference term lambs (Fig. 6) (24, 111). Together, these results show that the normal developmental pattern of pulmonary expression of eNOS and sGC is perturbed by prolonged invasive mechanical ventilation. This perturbation also may contribute to the increased pulmonary vascular resistance that occurs in chronically ventilated preterm lambs.
Fig. 6.
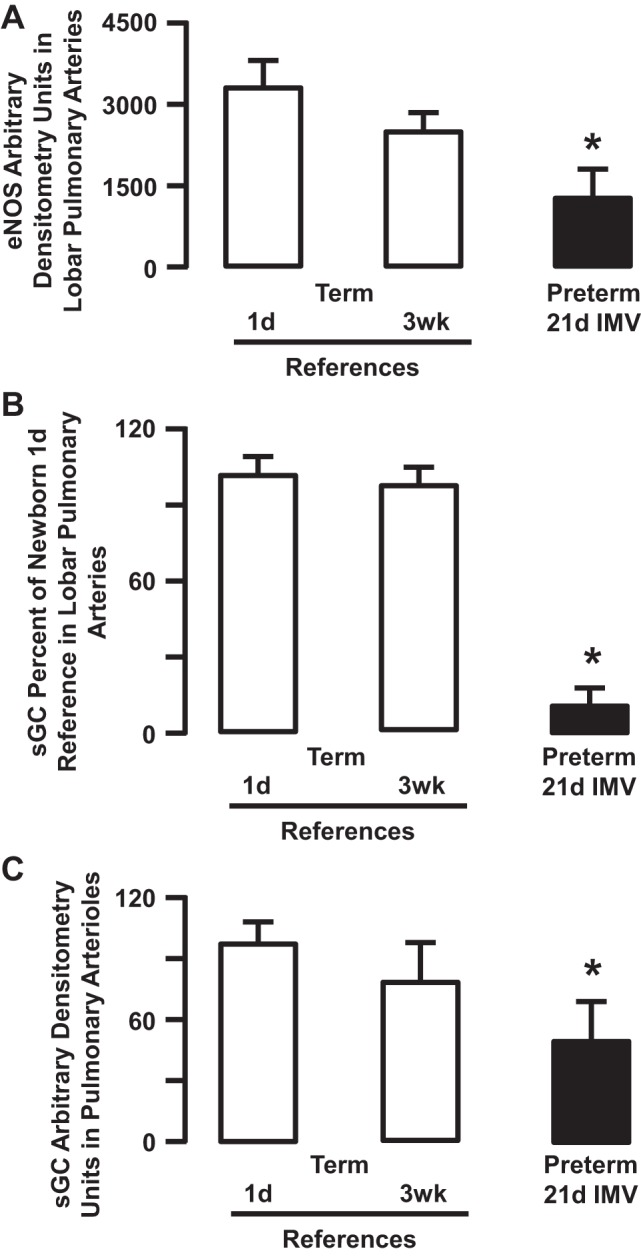
Semiquantitative densitometry for endothelial nitric oxide synthase (eNOS) (A) and soluble guanylate cyclase (sGC) (B and C). eNOS and sGC arbitrary densitometry in homogenates of 3rd to 5th generation intrapulmonary arteries (A and B), and immunohistochemical arbitrary densitometry in pulmonary arterioles landmarked next to terminal bronchioles (C) are significantly less in the lung of preterm lambs supported by invasive mechanical ventilation (IMV) for 3 wk compared with both groups of newborn reference lambs (*P < 0.05; means ± SD; n = 3–4/group). Adapted from Refs. 24 and 111.
Our second assessment used preterm lambs that had implanted catheters for measurement of pulmonary vascular pressure and blood flow to calculate pulmonary vascular resistance. During weeks 2 and 3 of invasive mechanical ventilation, we treated the preterm lambs with 5–15 parts/million (ppm) of iNO for 1 h. At 1 to 2 wk of continuous invasive mechanical ventilation, iNO for 1 h decreased pulmonary vascular resistance by ∼20% (Fig. 7). However, when we repeated the same treatment at the end of week 3 of continuous mechanical ventilation, iNO did not decrease pulmonary vascular resistance (Fig. 7). These results could be explained, however, by dysfunction of pulmonary vascular smooth muscle cells. That the pulmonary vascular smooth cells retained capacity to relax was proven by infusing 8-bromo-guanosine 3′,5′-cyclic monophosphate (8-bromo-cGMP; 150 μg·kg−1·min−1, intravenously) for 30 min in four of the same preterm lambs at the end of week 3 of mechanical ventilation (Table 2). Pulmonary vascular resistance consistently decreased by ∼30%. Besides being unique physiological results among the preterm models of neonatal CLD, these results may provide a basis for developing novel therapeutic or preventive interventions for pulmonary hypertension in chronically ventilated preterm human infants. For example, analogs of cGMP may merit investigation as potentially useful therapeutic tools in conditions where lung injury is associated with pulmonary hypertension. Further investigation would be based on our physiological results indicating that prolonged invasive mechanical ventilation of preterm lambs does not disrupt the normal apparatus for relaxation of pulmonary vascular smooth muscle, whereby cGMP activates cGMP-dependent protein kinases (38).
Fig. 7.
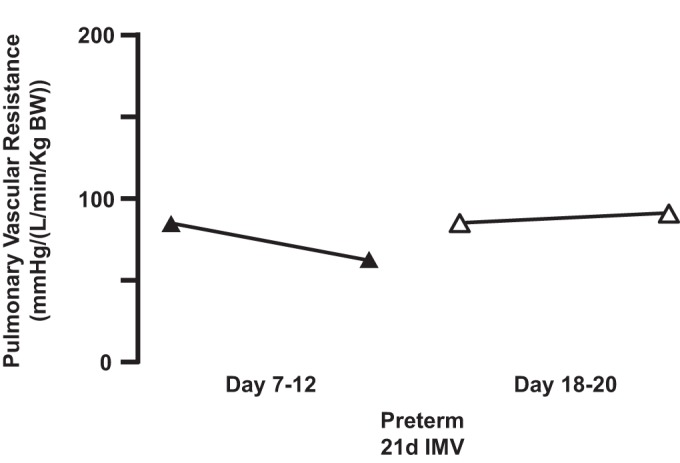
Preterm lambs were given inhaled nitric oxide (5–15 ppm) for 1 h between weeks 1 and 2 of life (days 7–12; n = 12/group) and again toward the end of week 3 of life (days 18–20; n = 8/group). Acute inhalation of nitric oxide reduced median pulmonary vascular resistance from the initial level of pulmonary vascular resistance (days 7–12). However, median pulmonary vascular resistance was slightly higher more than a week later (days 18–20). Adapted from Ref. 24.
Table 2.
Vascular smooth muscle remains responsive during prolonged invasive mechanical ventilation
| Intervention | PVR, mmHg−1·l·min−1 |
|---|---|
| Baseline | 15 ± 8 |
| 8-Bromo-cGMP (after 30-min infusion) | 10 ± 5* |
| Baseline normoxia | 7 ± 1 |
| Hypoxia | 10 ± 3* |
| 8-Bromo-cGMP (after 30-min infusion) | 6 ± 1 |
Values are means ± SD, n = 4 per group. PVR, pulmonary vascular resistance.
Significant difference compared to other group(s), P < 0.05.
Our third assessment tested whether continuous low-dose iNO from birth reduces pulmonary vascular resistance and attenuates persistent muscularization of pulmonary arterial vessels in preterm lambs with evolving neonatal CLD. Rationale for this assessment stemmed at the time from studies that used chronically hypoxic rats with pulmonary hypertension (99, 156). Continuous iNO reduced the anticipated pulmonary vascular remodeling that is associated with chronic hypoxia in rats. We hypothesized that continuous iNO starting at preterm birth would inhibit pulmonary vascular smooth muscle growth and thereby reduce pulmonary vascular resistance. To test this hypothesis, we mechanically ventilated eight preterm lambs for 3 wk, four of them with continuous iNO at 5–15 ppm, beginning at preterm birth. Contrary to our hypothesis, pulmonary vascular resistance was not significantly different between iNO-treated and untreated preterm lambs after 3 wk of continuous iNO administration during mechanical ventilation (Fig. 8). Nor did vascular smooth muscle thickness of pulmonary arterioles decrease (Fig. 8).
Fig. 8.
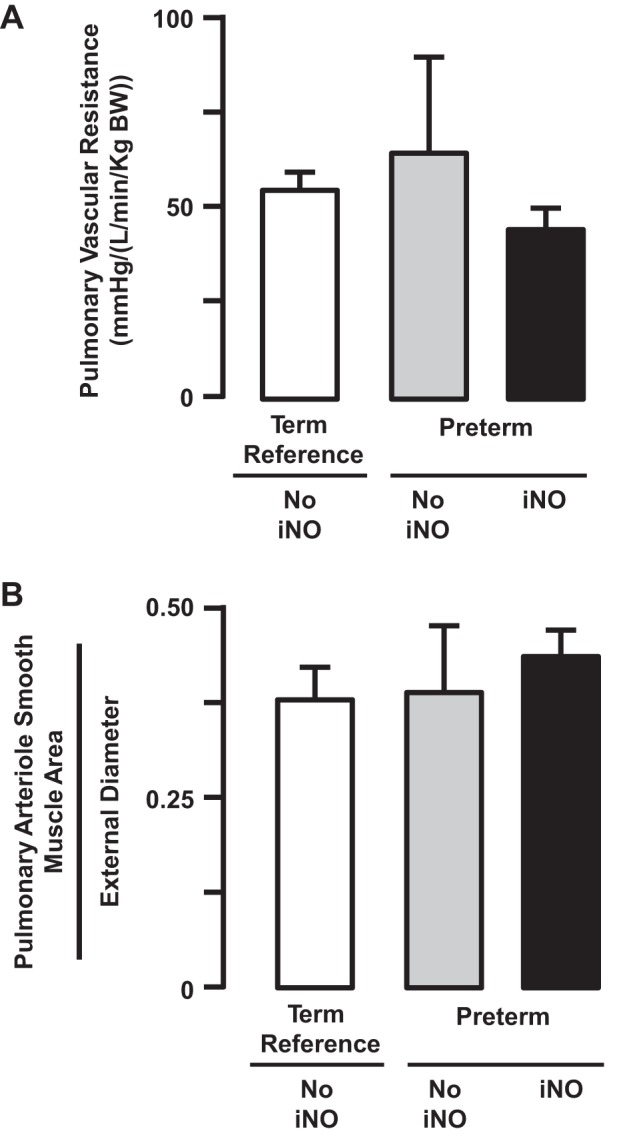
Preterm lambs were given inhaled nitric oxide (5–15 ppm) continuously for 3 wk. At the end of 3 wk of continuous inhalation of nitric oxide (iNO), pulmonary vascular resistance (A) and pulmonary arteriole smooth muscle area (B) remained the same as the respective values for preterm lambs that did not inhale nitric oxide (NO iNO) (means ± SD; n = 5/group). Adapted from Ref. 21 with permission of the American Thoracic Society.
Another surprise is the observation that eNOS protein abundance in pulmonary endothelial cells did not decrease over time. The latter result surprised us because diminished eNOS is sometimes used to explain the apparent elevation of pulmonary vascular resistance that frequently occurs when iNO therapy is discontinued. On the other hand, our surprising finding is consistent with the results of in vitro studies, which showed that NO donors increase NOS expression in cultured pulmonary artery endothelial cells derived from fetal sheep (202).
An overall conclusion from the three physiological studies is that prolonged invasive mechanical ventilation disrupts the NO signaling pathway. Among the consequences of prolonged invasive mechanical ventilation is that it decreases the normal developmental pattern of pulmonary vascular abundance of eNOS and sGC. Accompanying the decreases is persistent muscularization of pulmonary arterial vessels. We suggest that these alterations may contribute to attenuated or absent lung vascular response to continuous iNO in chronically ventilated preterm human infants.
Pathophysiology and Pathology of Excess Airway Expiratory Resistance
Preterm human infants with BPD have increased inspiratory and expiratory airway resistance, low specific compliance, and low functional residual capacity (72, 130). The mechanisms leading to reduced lung mechanics remain incompletely understood. Our initial studies using preterm lambs that we mechanically ventilated for 3–4 wk revealed that airway resistance is greater than in reference term lambs. Associated with this pathophysiological outcome is greater muscularization of airways and lower abundance of eNOS protein in airway epithelium (111). Protein abundance of sGC also is lower (21). One study at that time showed that all three isoforms of NO synthase (eNOS, iNOS, and nNOS) are expressed in airway epithelial cells of both sheep and baboons during normal fetal development (165, 168). Another contemporary study showed that eNOS protein abundance and total NOS activity are reduced in the lungs of preterm baboons after 2 wk of invasive mechanical ventilation (3). Reduced NOS activity could contribute to the increased airway resistance and abundance of airway smooth muscle, as shown in vitro (69, 77, 181, 197) and in vivo in newborn rats and piglets (120, 147).
When we began our study of the potential beneficial role of continuous iNO therapy in our preterm lamb model of evolving neonatal CLD, we anticipated that the impact of iNO would be greatest on the pulmonary vasculature. To our surprise, continuous iNO had greater impact on the structure and function of the airways. Airway resistance was significantly (∼40%) lower in the preterm lambs that received continuous iNO, even at 3 wk of mechanical ventilation (Fig. 9). Postmortem histopathology showed significantly (∼50%) less smooth muscle in the wall of distal airways in the preterm lambs that we treated with iNO compared with reference term lambs (Fig. 9). Immunoblot analysis for eNOS and sGC proteins in airways taken from iNO-treated preterm lambs showed comparable abundance to that in airways of reference term lambs. These results are informative because they reveal that continuous iNO during 3 wk of invasive mechanical ventilation prevents increased airway resistance, muscularization of airways, and loss of airway epithelial cell abundance of eNOS protein compared with the adjacent pulmonary arterial vessels.
Fig. 9.
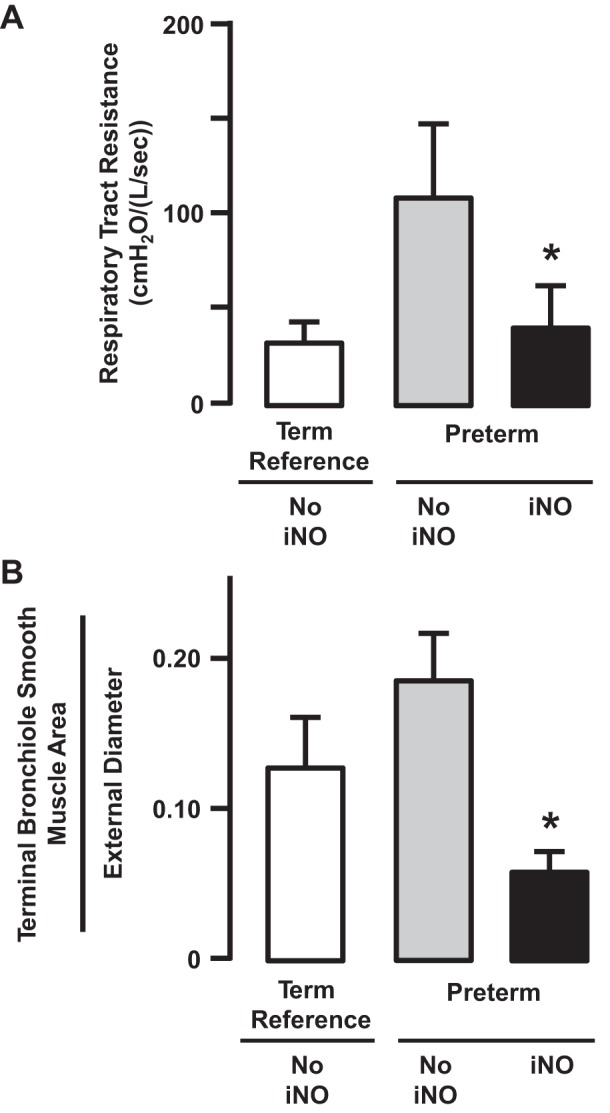
Preterm lambs were given inhaled nitric oxide (5–15 ppm) continuously for 3 wk. At the end of 3 wk of continuous inhalation of nitric oxide (iNO), respiratory tract resistance (A) and terminal bronchiole smooth muscle area (B) were significantly lower than the respective values for preterm lambs that did not inhale nitric oxide (NO iNO) (*P < 0.05; means ± SD; n = 5/group). Adapted from Ref. 21 with permission of the American Thoracic Society.
Our results for airways are supported by results of other studies. One study showed that iNO decreased airway resistance in anesthetized newborn piglets (147). Another study showed that iNO decreased airway resistance in anesthetized adult guinea pigs during induced bronchoconstriction (61). A cellular consequence of NO is that it reduces proliferation of airway smooth muscle cells in vitro (77, 181). In aggregate, these results indicate that NO positively influences airway smooth muscle tone and abundance during postnatal development, as well as in adults.
An overall conclusion is that prolonged invasive mechanical ventilation also disrupts the NO signaling pathway in airways. However, unlike the neighboring pulmonary arteries in the same lungs, continuous iNO improves airway development. Perhaps the lack of effect on the neighboring pulmonary arteries is due to diffusion distance from the lumen of airways, where iNO concentration is high, to pulmonary arterial endothelial and smooth muscle cells, where the iNO concentration would be lower or nil. Nonetheless, the positive influence of continuous iNO on airway development is encouraging because of the potential to improve peripheral distribution of ventilation and reduce airway hyperreactivity in chronically ventilated preterm human infants.
Continuous iNO therapy improves alveolarization in chronically ventilated preterm lambs.
Returning to our study that provided continuous iNO for 3 wk, another surprising outcome is improved alveolarization (Fig. 10) (21). We found that radial alveolar count and secondary septal volume density are greater, whereas distal air space walls are thinner, than in the lung of untreated, mechanically ventilated preterm lambs (Fig. 10). Our structural results are consistent with results of studies using rats and mice that had various insults (13, 62, 78, 102, 107, 179, 200).
Fig. 10.
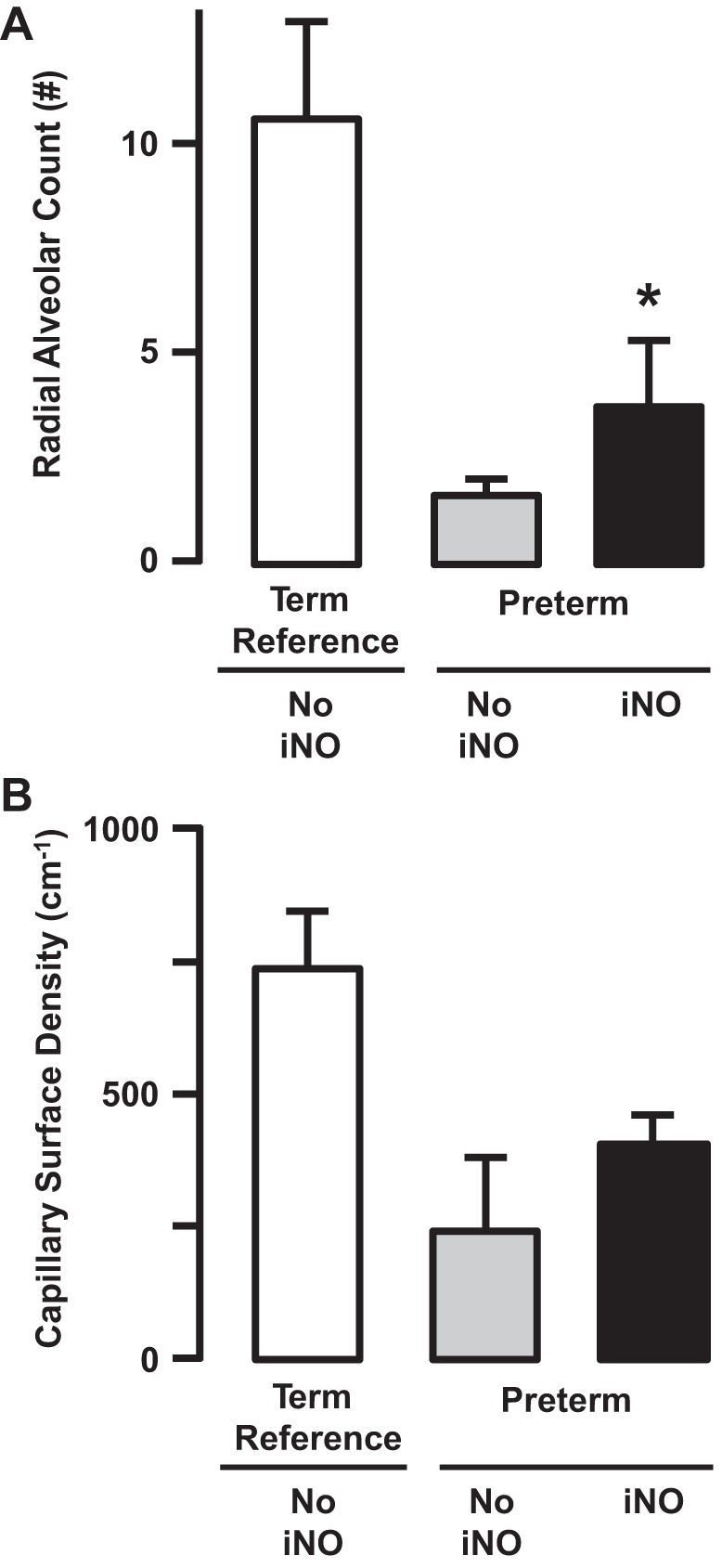
Preterm lambs were given inhaled nitric oxide (5–15 ppm) continuously for 3 wk. At the end of 3 wk of continuous inhalation of nitric oxide (iNO), radial alveolar count (A) and capillary surface density (B) were significantly greater than the respective values for preterm lambs that did not inhale nitric oxide (NO iNO) (*P < 0.05; means ± SD; n = 5/group). Radial alveolar count also was significantly lower than the term reference group. Capillary surface density also was significantly greater than the term reference group. Adapted from Ref. 21.
A potential mechanism for improved alveolarization may be via the role of NO on expression of vascular growth factor signaling molecules. For example, NO induces synthesis of VEGF in vascular smooth muscle cells (60). Relevance of the signaling cascade from NO to VEGF in neonatal CLD is that expression of VEGF and its functional receptor VEGF-R2 are diminished in the lung of preterm human infants who died of BPD (18) and in the preterm baboon model of this disease (114). Both VEGF and VEGF-R2 are important regulators of alveolarization through their impact on capillary growth (87, 103). Therefore, a possibility is that delivery of iNO leads to downstream signaling of VEGF and VEGF-R2 in the preterm lung during prolonged invasive mechanical ventilation. This possibility is consistent with our observation that continuous iNO therapy preserves pulmonary vascular eNOS protein abundance in chronically ventilated preterm lambs (21).
Different outcome followed treating chronically ventilated preterm baboons with continuous iNO (123). The lung of the treated preterm baboons did not have structural evidence of alveolarization. Why might the experiment using preterm baboons have different outcome compared with preterm lambs? One reason may be that the preterm baboons received lower concentration of continuous iNO (5 ppm). Our preterm lambs received 5–15 ppm iNO continuously. Also, the preterm baboons were ventilated and treated for less time (2 wk). Our preterm lambs were ventilated and treated for 3–4 wk. We suggest that the lower concentration of iNO and its shorter duration of treatment may account for the lack of efficacy in chronically ventilated preterm baboons compared with chronically ventilated preterm lambs.
Mechanisms Leading to Long-Term Outcomes Among Survivors of Neonatal CLD
Persistent pulmonary morbidities such as pulmonary hypertension (2, 16, 17, 35), excess airway expiratory resistance and airway hyperreactivity (64, 72, 130), and lower carbon monoxide diffusing capacity (14, 19, 132, 133) later in life are important outcomes for survivors of preterm birth and BPD. Some mechanistic insights into long-term outcomes are provided by the preterm baboon model. The lungs of former preterm baboons that were mechanically ventilated for 14 days and recovered for 33 wk had persistently enlarged air spaces and less parenchymal tissue than control baboons (48). Thirty-three weeks of life for baboons is equivalent to ∼2 yr of postnatal age for humans. Much remains to be learned, mechanistically, about persistent morbidities of the pulmonary vasculature and airways in survivors of BPD and ways to minimize the morbidities.
Summary
This review focused on unique insights provided by the preterm lamb physiological model of neonatal CLD. The preterm lamb model and the preterm baboon model recapitulate the clinical setting of preterm birth and respiratory failure that require prolonged ventilation support with oxygen-rich gas. The impact of both models is that they provide mechanistic insights into the pathogenesis of neonatal CLD. A particular advantage of the preterm lamb model is the large size of preterm lambs, which facilitates pathophysiological studies during the evolution of neonatal CLD. To this pathophysiological advantage is linked an array of morphological, biochemical, and molecular analyses that together provide a bigger picture of the mechanisms leading to neonatal CLD. Our results indicate that the mode of ventilation is related to lung outcomes. Specifically, poor outcomes are the result from invasive mechanical ventilation, whereas better outcomes are the result of less invasive high-frequency nasal ventilation. Our integrative studies also show that daily supplementation with vitamin A during invasive mechanical ventilation partially improves lung outcomes. The new insights that are being gained may have important translational implications about the pathogenesis and treatment of BPD in preterm human infants.
GRANTS
Portions of this work were supported by National Heart, Lung, and Blood Institute Grants HL110002 and HL062875.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.H.A. conception and design of research; performed experiments; analyzed data; interpreted results of experiments; prepared figures; drafted manuscript; edited and revised manuscript; approved final version of manuscript.
ACKNOWLEDGMENTS
The author expresses appreciation to the late Dr. Norman C. Staub (1929–2014) for memories and impact on this work. Appreciation is expressed to Mar Janna Dahl for more than a decade of dedication to our chronically ventilated preterm lamb program. Thanks are extended to the numerous technicians and hundreds of undergraduate and medical students who contributed to our studies as tenders of the preterm lambs and in research projects over the past two-plus decades. Appreciation also is expressed to Dr. Richard D. Bland, Dr. David P. Carlton, Dr. Bradley A. Yoder, and Dr. Donald M. Null for leadership and contributions. Thank you is extended to Diana Lim for creating the digital figure. Dr. Rachel Reader also is thanked for assisting with compilation of Table 1.
REFERENCES
- 1.Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol Heart Circ Physiol 259: H1921–H1927, 1990. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Wolfe RR, Accurso FJ, Koops BL, Bowman CM, Wiggins JW Jr. Pulmonary vascular response to oxygen in infants with severe bronchopulmonary dysplasia. Pediatrics 75: 80–84, 1985. [PubMed] [Google Scholar]
- 3.Afshar S, Gibson LL, Yuhanna IS, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC, Shaul PW. Pulmonary NO synthase expression is attenuated in a fetal baboon model of chronic lung disease. Am J Physiol Lung Cell Mol Physiol 284: L749–L758, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Albertine KH. Anatomy of the lungs. In: Murray & Nadel's Textbook of Respiratory Medicine (5th ed), edited by Saunders NR. Philadelphia: Elsevier, 2010, p. 3–25. [Google Scholar]
- 5.Albertine KH, Dahl MJ, Gonzales LW, Wang ZM, Metcalfe D, Hyde DM, Plopper CG, Starcher BC, Carlton DP, Bland RD. Chronic lung disease in preterm lambs: effect of daily vitamin A treatment on alveolarization. Am J Physiol Lung Cell Mol Physiol 299: L59–L72, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, Carlton DP, Bland RD. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med 159: 945–958, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Albertine KH, Pysher TJ. Impaired lung growth after injury in premature lung. In: Fetal and Neonatal Physiology (4th ed), edited by Polin RA, Fox WW, and Abman S. New York: Elsevier Sciences, 2011, p. 1039–1047. [Google Scholar]
- 8.Albertine KH, Wiener-Kronish JP, Roos PJ, Staub NC. Structure, blood supply, and lymphatic vessels of the sheep's visceral pleura. Am J Anat 165: 277–294, 1982. [DOI] [PubMed] [Google Scholar]
- 9.Albertine KH, Wiener-Kronish JP, Staub NC. Blood supply of the caudal mediastinal lymph node in sheep. Anat Rec 212: 129–131, 154–155, 1985. [DOI] [PubMed] [Google Scholar]
- 10.Albertine KH, Yoder B. Pulmonary consequences of preterm birth. In: The Lung: Development, Aging and the Environment, edited by Hardin R and Pinkerton K. New York: Academic, 2015, p. 311–327. [Google Scholar]
- 11.Ambalavanan N, Tyson JE, Kennedy KA, Hansen NI, Vohr BR, Wright LL, Carlo WA; National Institute of Child Health and Human Development Neonatal Research Network. Vitamin A supplementation for extremely low birth weight infants: outcome at 18 to 22 months. Pediatrics 115: e249–e254, 2005. [DOI] [PubMed] [Google Scholar]
- 11a.American Academy of Pediatrics and American College of Obstetricians and Gynecologists. Guidelines for Perinatal Care (7th ed). Elk Grove Village, IL: American Academy of Pediatrics; Washington, DC: American College of Obstetricians and Gynecologists, 2012. [Google Scholar]
- 12.Avery ME, Mead J. Surface properties in relation to atelectasis and hyaline membrane disease. Am J Dis Child 97: 517–523, 1959. [DOI] [PubMed] [Google Scholar]
- 13.Balasubramaniam V, Tang JR, Maxey A, Plopper CG, Abman SH. Mild hypoxia impairs alveolarization in the endothelial nitric oxide synthase-deficient mouse. Am J Physiol Lung Cell Mol Physiol 284: L964–L971, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Balinotti JE, Chakr VC, Tiller C, Kimmel R, Coates C, Kisling J, Yu Z, Nguyen J, Tepper RS. Growth of lung parenchyma in infants and toddlers with chronic lung disease of infancy. Am J Respir Crit Care Med 181: 1093–1097, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkelhamer SK, Mestan KK, Steinhorn RH. Pulmonary hypertension in bronchopulmonary dysplasia. Semin Perinatol 37: 124–131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman W Jr, Yabek SM, Dillon T, Burstein R, Corlew S. Evaluation of infants with bronchopulmonary dysplasia using cardiac catheterization. Pediatrics 70: 708–712, 1982. [PubMed] [Google Scholar]
- 17.Bhat R, Salas AA, Foster C, Carlo WA, Ambalavanan N. Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics 129: e682–e689, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 164: 1971–1980, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Bhutani VK, Abbasi S. Long-term pulmonary consequences in survivors with bronchopulmonary dysplasia. Clin Perinatol 19: 649–671, 1992. [PubMed] [Google Scholar]
- 20.Bland RD, Albertine KH, Carlton DP, Kullama L, Davis P, Cho SC, Kim BI, Dahl M, Tabatabaei N. Chronic lung injury in preterm lambs: abnormalities of the pulmonary circulation and lung fluid balance. Pediatr Res 48: 64–74, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Bland RD, Albertine KH, Carlton DP, MacRitchie AJ. Inhaled nitric oxide effects on lung structure and function in chronically ventilated preterm lambs. Am J Respir Crit Care Med 172: 899–906, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bland RD, Carlton D. Pulmonary edema in chronic lung disease of early infancy. In: Chronic Lung Disease in Early Infancy, edited by Bland RD and Coalson J. New York: Dekker, 1999, p. 711–749. [Google Scholar]
- 23.Bland RD, Carlton DP, Scheerer RG, Cummings JJ, Chapman DL. Lung fluid balance in lambs before and after premature birth. J Clin Invest 84: 568–576, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bland RD, Ling CY, Albertine KH, Carlton DP, MacRitchie AJ, Day RW, Dahl MJ. Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 285: L76–L85, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Bland RD, Xu L, Ertsey R, Rabinovitch M, Albertine KH, Wynn KA, Kumar VH, Ryan RM, Swartz DD, Csiszar K, Fong KS. Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 292: L1370–L1384, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Bloch KD, Filippov G, Sanchez LS, Nakane M, de la Monte SM. Pulmonary soluble guanylate cyclase, a nitric oxide receptor, is increased during the perinatal period. Am J Physiol Lung Cell Mol Physiol 272: L400–L406, 1997. [DOI] [PubMed] [Google Scholar]
- 27.Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of Clara cells in normal human airway epithelium. Am J Respir Crit Care Med 159: 1585–1591, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Bonikos DS, Bensch KG, Northway WH Jr, Edwards DK. Bronchopulmonary dysplasia: the pulmonary pathologic sequel of necrotizing bronchiolitis and pulmonary fibrosis. Hum Pathol 7: 643–666, 1976. [DOI] [PubMed] [Google Scholar]
- 29.Brandt RB, Mueller DG, Schroeder JR, Guyer KE, Kirkpatrick BV, Hutcher NE, Ehrlich FE. Serum vitamin A in premature and term neonates. J Pediatr 92: 101–104, 1978. [DOI] [PubMed] [Google Scholar]
- 30.Brigham KL, Woolverton WC, Blake LH, Staub NC. Increased sheep lung vascular permeability caused by pseudomonas bacteremia. J Clin Invest 54: 792–804, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burri PH. Fetal and postnatal development of the lung. Annu Rev Physiol 46: 617–628, 1984. [DOI] [PubMed] [Google Scholar]
- 32.Burri PH. The postnatal growth of the rat lung. 3. Morphology. Anat Rec 180: 77–98, 1974. [DOI] [PubMed] [Google Scholar]
- 33.Burri PH. Structural aspects of prenatal and postnatal development and growth of the lung. In: Lung Growth and Development, edited by McDonald JA. New York: Dekker, 1997, p. 1–35. [Google Scholar]
- 34.Burri PH, Dbaly J, Weibel ER. The postnatal growth of the rat lung. I. Morphometry. Anat Rec 178: 711–730, 1974. [DOI] [PubMed] [Google Scholar]
- 35.Bush A, Busst CM, Knight WB, Hislop AA, Haworth SG, Shinebourne EA. Changes in pulmonary circulation in severe bronchopulmonary dysplasia. Arch Dis Child 65: 739–745, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrne BJ, Mellen BG, Lindstrom DP, Cotton RB. Is the BPD epidemic diminishing? Semin Perinatol 26: 461–466, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Carlton DP, Albertine KH, Cho SC, Lont M, Bland RD. Role of neutrophils in lung vascular injury and edema after premature birth in lambs. J Appl Physiol 83: 1307–1317, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol 184: 409–420, 2000. [DOI] [PubMed] [Google Scholar]
- 39.Chambers HM, van Velzen D. Ventilator-related pathology in the extremely immature lung. Pathology 21: 79–83, 1989. [DOI] [PubMed] [Google Scholar]
- 40.Chang LY, Mercer RR, Stockstill BL, Miller FJ, Graham JA, Ospital JJ, Crapo JD. Effects of low levels of NO2 on terminal bronchiolar cells and its relative toxicity compared to O3. Toxicol Appl Pharmacol 96: 451–464, 1988. [DOI] [PubMed] [Google Scholar]
- 41.Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, Roy BJ, Keszler M, Kinsella JP. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med 342: 469–474, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Clyman RI, Campbell D. Indomethacin therapy for patent ductus arteriosus: when is prophylaxis not prophylactic? J Pediatr 111: 718–722, 1987. [DOI] [PubMed] [Google Scholar]
- 43.Coalson JJ. Pathology of bronchopulmonary dysplasia. Semin Perinatol 30: 179–184, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Coalson JJ. Pathology of chronic lung disease of early infancy. In: Chronic Lung Disease in Early Infancy, edited by Bland RD and Coalson JJ. New York: Dekker, 2000, p. 85–124. [Google Scholar]
- 45.Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol 8: 73–81, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Coalson JJ, Kuehl TJ, Escobedo MB, Hilliard JL, Smith F, Meredith K, Null DM Jr, Walsh W, Johnson D, Robotham JL. A baboon model of bronchopulmonary dysplasia. II. Pathologic features. Exp Mol Pathol 37: 335–350, 1982. [DOI] [PubMed] [Google Scholar]
- 47.Coalson JJ, Kuehl TJ, Prihoda TJ, deLemos RA. Diffuse alveolar damage in the evolution of bronchopulmonary dysplasia in the baboon. Pediatr Res 24: 357–366, 1988. [DOI] [PubMed] [Google Scholar]
- 48.Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med 152: 640–646, 1995. [DOI] [PubMed] [Google Scholar]
- 49.Coalson JJ, Winter VT, Gerstmann DR, Idell S, King RJ, Delemos RA. Pathophysiologic, morphometric, and biochemical studies of the premature baboon with bronchopulmonary dysplasia. Am Rev Respir Dis 145: 872–881, 1992. [DOI] [PubMed] [Google Scholar]
- 50.Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med 160: 1333–1346, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Conradi C, Burri PH, Kapanci Y, Robinson FR, Weibel ER. Lung changes after beryllium inhalation: ultrastructural and morphometric study. Arch Environ Health 23: 348–358, 1971. [DOI] [PubMed] [Google Scholar]
- 52.Cornfield DN, Maynard RC, deRegnier RA, Guiang SF 3rd, Barbato JE, Milla CE. Randomized, controlled trial of low-dose inhaled nitric oxide in the treatment of term and near-term infants with respiratory failure and pulmonary hypertension. Pediatrics 104: 1089–1094, 1999. [DOI] [PubMed] [Google Scholar]
- 53.Costeloe K, Hennessy E, Gibson AT, Marlow N, Wilkinson AR. The EPICure study: outcomes to discharge from hospital for infants born at the threshold of viability. Pediatrics 106: 659–671, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respir Dis 128: S42–S46, 1983. [DOI] [PubMed] [Google Scholar]
- 55.Cutz E, Conen PE. Ultrastructure and cytochemistry of Clara cells. Am J Pathol 62: 127–141, 1971. [PMC free article] [PubMed] [Google Scholar]
- 56.Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, Rhines J, Chang CT. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study Group. Pediatrics 101: 325–334, 1998. [DOI] [PubMed] [Google Scholar]
- 57.Day RW, Lynch JM, White KS, Ward RM. Acute response to inhaled nitric oxide in newborns with respiratory failure and pulmonary hypertension. Pediatrics 98: 698–705, 1996. [PubMed] [Google Scholar]
- 58.DeLemos RA, Coalson JJ. The contribution of experimental models to our understanding of the pathogenesis and treatment of bronchopulmonary dysplasia. Clin Perinatol 19: 521–539, 1992. [PubMed] [Google Scholar]
- 59.DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, Kim CJ, Erez O, Edwin S, Relman DA. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS One 3: e3056, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wojtowicz A, Szuba A, Cooke JP. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 20: 659–666, 2000. [DOI] [PubMed] [Google Scholar]
- 61.Dupuy PM, Shore SA, Drazen JM, Frostell C, Hill WA, Zapol WM. Bronchodilator action of inhaled nitric oxide in guinea pigs. J Clin Invest 90: 421–428, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edwards YS, Sutherland LM, Murray AW. NO protects alveolar type II cells from stretch-induced apoptosis. A novel role for macrophages in the lung. Am J Physiol Lung Cell Mol Physiol 279: L1236–L1242, 2000. [DOI] [PubMed] [Google Scholar]
- 63.Escobedo MB, Hilliard JL, Smith F, Meredith K, Walsh W, Johnson D, Coalson JJ, Kuehl TJ, Null DM Jr, Robotham JL. A baboon model of bronchopulmonary dysplasia. I. Clinical features. Exp Mol Pathol 37: 323–334, 1982. [DOI] [PubMed] [Google Scholar]
- 64.Fawke J, Lum S, Kirkby J, Hennessy E, Marlow N, Rowell V, Thomas S, Stocks J. Lung function and respiratory symptoms at 11 years in children born extremely preterm: the EPICure study. Am J Respir Crit Care Med 182: 237–245, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fineman JR, Wong J, Morin FC 3rd, Wild LM, Soifer SJ. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Invest 93: 2675–2683, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finer NN, Carlo WA, Walsh MC, Rich W, Gantz MG, Laptook AR, Yoder BA, Faix RG, Das A, Poole WK, Donovan EF, Newman NS, Ambalavanan N, Frantz ID 3rd, Buchter S, Sanchez PJ, Kennedy KA, Laroia N, Poindexter BB, Cotten CM, Van Meurs KP, Duara S, Narendran V, Sood BG, O'Shea TM, Bell EF, Bhandari V, Watterberg KL, Higgins RD. Early CPAP versus surfactant in extremely preterm infants. N Engl J Med 362: 1970–1979, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fox JG, Barthold S, Davisson M, Newcomer C, Quimby F, Smith A. The Mouse in Biomedical Research: Diseases. New York: Academic, 2006. [Google Scholar]
- 68.Frostell C, Fratacci MD, Wain JC, Jones R, Zapol WM. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 83: 2038–2047, 1991. [DOI] [PubMed] [Google Scholar]
- 69.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83: 1774–1777, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gehr P, Bachofen M, Weibel ER. The normal human lung: ultrastructure and morphometric estimation of diffusion capacity. Respir Physiol 32: 121–140, 1978. [DOI] [PubMed] [Google Scholar]
- 71.Gehr P, Mwangi DK, Ammann A, Maloiy GM, Taylor CR, Weibel ER. Design of the mammalian respiratory system. V. Scaling morphometric pulmonary diffusing capacity to body mass: wild and domestic mammals. Respir Physiol Neurobiol 44: 61–86, 1981. [DOI] [PubMed] [Google Scholar]
- 72.Gerhardt T, Hehre D, Feller R, Reifenberg L, Bancalari E. Serial determination of pulmonary function in infants with chronic lung disease. J Pediatr 110: 448–456, 1987. [DOI] [PubMed] [Google Scholar]
- 73.Gibson QH, Roughton FJ. The kinetics of dissociation of the first ligand molecule from fully saturated carboxyhaemoglobin and nitric oxide haemoglobin in sheep blood solutions. Proc R Soc Lond B Biol Sci 147: 44–56, 1957. [DOI] [PubMed] [Google Scholar]
- 74.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet 371: 75–84, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo JP, Panday MM, Consigny PM, Lefer AM. Mechanisms of vascular preservation by a novel NO donor following rat carotid artery intimal injury. Am J Physiol Heart Circ Physiol 269: H1122–H1231, 1995. [DOI] [PubMed] [Google Scholar]
- 77.Hamad AM, Johnson SR, Knox AJ. Antiproliferative effects of NO and ANP in cultured human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 277: L910–L918, 1999. [DOI] [PubMed] [Google Scholar]
- 78.Han RN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res 94: 1115–1123, 2004. [DOI] [PubMed] [Google Scholar]
- 79.Harkema JR, Mariassy AT, St George JA. Epithelial cells of the conducting airways: a species comparison. In: The Airway Epithelium: Physiology, Pathophysiology and Pharmacology, edited by Farmer CG and Hay OW. New York: Dekker, 1991, p. 3–39. [Google Scholar]
- 80.Hayatdavoudi G, O'Neil JJ, Barry BE, Freeman BA, Crapo JD. Pulmonary injury in rats following continuous exposure to 60% O2 for 7 days. J Appl Physiol 51: 1220–1231, 1981. [DOI] [PubMed] [Google Scholar]
- 81.Hillman NH, Kemp MW, Noble PB, Kallapur SG, Jobe AH. Sustained inflation at birth did not protect preterm fetal sheep from lung injury. Am J Physiol Lung Cell Mol Physiol 305: L446–L453, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hislop AA, Haworth SG. Pulmonary vascular damage and the development of cor pulmonale following hyaline membrane disease. Pediatr Pulmonol 9: 152–161, 1990. [DOI] [PubMed] [Google Scholar]
- 83.Hodson WA. Ventilation strategies and bronchopulmonary dysplasia. In: Chronic Lung Disease in Early Infancy, edited by Bland RD and Coalson JJ. New York: Dekker, 2000, p. 173–208. [Google Scholar]
- 84.Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol 29: 710–717, 1998. [DOI] [PubMed] [Google Scholar]
- 85.Hustead VA, Gutcher GR, Anderson SA, Zachman RD. Relationship of vitamin A (retinol) status to lung disease in the preterm infant. J Pediatr 105: 610–615, 1984. [DOI] [PubMed] [Google Scholar]
- 86.Hyde DM, Hubbard WC, Wong V, Wu R, Pinkerton K, Plopper CG. Ozone-induced acute tracheobronchial epithelial injury: relationship to granulocyte emigration in the lung. Am J Respir Cell Mol Biol 6: 481–497, 1992. [DOI] [PubMed] [Google Scholar]
- 87.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279: L600–L607, 2000. [DOI] [PubMed] [Google Scholar]
- 88.Jarkovska D. Ultrastructure of the epithelium of the respiratory bronchioles in man. Folia Morphol (Warsz) 18: 352–358, 1970. [PubMed] [Google Scholar]
- 89.Jobe A, Ikegami M, Jacobs H, Jones S, Conaway D. Permeability of premature lamb lungs to protein and the effect of surfactant on that permeability. J Appl Physiol 55: 169–176, 1983. [DOI] [PubMed] [Google Scholar]
- 90.Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr 23: 167–172, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jobe AH, Kramer BW, Moss TJ, Newnham JP, Ikegami M. Decreased indicators of lung injury with continuous positive expiratory pressure in preterm lambs. Pediatr Res 52: 387–392, 2002. [DOI] [PubMed] [Google Scholar]
- 92.Johanson WG Jr, Pierce AK. Lung structure and function with age in normal rats and rats with papain emphysema. J Clin Invest 52: 2921–2927, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaplan HP, Robinson FR, Kapanci Y, Weibel ER. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. I. Clinical and light microscopic studies. Lab Invest 20: 94–100, 1969. [PubMed] [Google Scholar]
- 94.Kinsella JP, Neish SR, Shaffer E, Abman SH. Low-dose inhalation nitric oxide in persistent pulmonary hypertension of the newborn. Lancet 340: 819–820, 1992. [DOI] [PubMed] [Google Scholar]
- 95.Kirpalani H, Millar D, Lemyre B, Yoder B, Chiu A, Roberts RS for the NIPPV Study Group. A Trial Comparing Noninvasive Ventilation Strategies in Preterm Infants. N Engl J Med 369: 611–620, 2013. [DOI] [PubMed] [Google Scholar]
- 96.Klinger G, Sokolover N, Boyko V, Sirota L, Lerner-Geva L, Reichman B; Israel Neonatal Network. Perinatal risk factors for bronchopulmonary dysplasia in a national cohort of very-low-birthweight infants. Am J Obstet Gynecol 208: 115.e1–e9, 2013. [DOI] [PubMed] [Google Scholar]
- 97.Koblinger L, Hofmann W. Analysis of human lung morphometric data for stochastic aerosol deposition calculations. Phys Med Biol 30: 541–556, 1985. [DOI] [PubMed] [Google Scholar]
- 98.Koblinger L, Hofmann W. Stochastic morphological model of the rat lung. Anat Rec 221: 533–539, 1988. [DOI] [PubMed] [Google Scholar]
- 99.Kouyoumdjian C, Adnot S, Levame M, Eddahibi S, Bousbaa H, Raffestin B. Continuous inhalation of nitric oxide protects against development of pulmonary hypertension in chronically hypoxic rats. J Clin Invest 94: 578–584, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kramer BW, Albertine KH, Moss TJ, Nitsos I, Ladenburger A, Speer CP, Newnham JP, Jobe AH. All-trans retinoic acid and intra-amniotic endotoxin-mediated effects on fetal sheep lung. Anat Rec 291: 1271–1277, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laughon MM, Langer JC, Bose CL, Smith PB, Ambalavanan N, Kennedy KA, Stoll BJ, Buchter S, Laptook AR, Ehrenkranz RA, Cotten CM, Wilson-Costello DE, Shankaran S, Van Meurs KP, Davis AS, Gantz MG, Finer NN, Yoder BA, Faix RG, Carlo WA, Schibler KR, Newman NS, Rich W, Das A, Higgins RD, Walsh MC. Prediction of bronchopulmonary dysplasia by postnatal age in extremely premature infants. Am J Respir Crit Care Med 183: 1715–1722, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Le Cras TD, Kim DH, Markham NE, Abman AS. Early abnormalities of pulmonary vascular development in the Fawn-Hooded rat raised at Denver's altitude. Am J Physiol Lung Cell Mol Physiol 279: L283–L291, 2000. [DOI] [PubMed] [Google Scholar]
- 103.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 283: L555–L562, 2002. [DOI] [PubMed] [Google Scholar]
- 104.Lee JS, Adrie C, Jacob HJ, Roberts JD Jr, Zapol WM, Bloch KD. Chronic inhalation of nitric oxide inhibits neointimal formation after balloon-induced arterial injury. Circ Res 78: 337–342, 1996. [DOI] [PubMed] [Google Scholar]
- 105.LeFlore JL, Salhab WA, Broyles RS, Engle WD. Association of antenatal and postnatal dexamethasone exposure with outcomes in extremely low birth weight neonates. Pediatrics 110: 275–279, 2002. [DOI] [PubMed] [Google Scholar]
- 106.Lemons JA, Bauer CR, Oh W, Korones SB, Papile LA, Stoll BJ, Verter J, Temprosa M, Wright LL, Ehrenkranz RA, Fanaroff AA, Stark A, Carlo W, Tyson JE, Donovan EF, Shankaran S, Stevenson DK. Very low birth weight outcomes of the National Institute of Child Health and Human Development Neonatal Research Network, January 1995 through December 1996. NICHD Neonatal Research Network. Pediatrics 107: E1, 2001. [DOI] [PubMed] [Google Scholar]
- 107.Leuwerke SM, Kaza AK, Tribble CG, Kron IL, Laubach VE. Inhibition of compensatory lung growth in endothelial nitric oxide synthase-deficient mice. Am J Physiol Lung Cell Mol Physiol 282: L1272–L1278, 2002. [DOI] [PubMed] [Google Scholar]
- 108.Lindahl P, Karlsson L, Hellstrom M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124: 3943–3953, 1997. [DOI] [PubMed] [Google Scholar]
- 109.Lonnqvist PA, Winberg P, Lundell B, Sellden H, Olsson GL. Inhaled nitric oxide in neonates and children with pulmonary hypertension. Acta Paediatr 83: 1132–1136, 1994. [DOI] [PubMed] [Google Scholar]
- 110.Lucangelo U, Fontanesi L, Antonaglia V, Pellis T, Berlot G, Liguori G, Bird FM, Gullo A. High frequency percussive ventilation (HFPV). Principles and technique. Minerva Anestesiol 69: 841–848, 2003. [PubMed] [Google Scholar]
- 111.MacRitchie AN, Albertine KH, Sun J, Lei PS, Jensen SC, Freestone AA, Clair PM, Dahl MJ, Godfrey EA, Carlton DP, Bland RD. Reduced endothelial nitric oxide synthase in lungs of chronically ventilated preterm lambs. Am J Physiol Lung Cell Mol Physiol 281: L1011–L1020, 2001. [DOI] [PubMed] [Google Scholar]
- 112.Madl P, Hofmann W, Oldham MJ, Asgharian B. Stochastic morphometric model of the BALB/c mouse lung. Anat Rec 293: 1766–1775, 2010. [DOI] [PubMed] [Google Scholar]
- 113.Madurga A, Mizikova I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 305: L893–L905, 2013. [DOI] [PubMed] [Google Scholar]
- 114.Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Respir Cell Mol Biol 282: L811–L823, 2002. [DOI] [PubMed] [Google Scholar]
- 115.Margraf LR, Tomashefski JF Jr, Bruce MC, Dahms BB. Morphometric analysis of the lung in bronchopulmonary dysplasia. Am Rev Respir Dis 143: 391–400, 1991. [DOI] [PubMed] [Google Scholar]
- 116.Mariassy AT. Epithelial cells of the trachea and bronchi. In: Treatise on Pulmonary Toxicology: Comparative Biology of the Normal Lung, edited by Parent RA. Boca Raton: CRC, 1991, p. 63–76. [Google Scholar]
- 117.Mariassy AT, Plopper CG. Tracheobronchial epithelium of the sheep: I. Quantitative light-microscopic study of epithelial cell abundance, and distribution. Anat Rec 205: 263–275, 1983. [DOI] [PubMed] [Google Scholar]
- 118.Martin JA, Hamilton BE, Osterman MJ, Curtin SC, Ma M, Mathews TJ. Births: final data for 2012. Nat Vital Stat Rep 62(9): 1–68, 2013. [PubMed] [Google Scholar]
- 119.Martin JA, Osterman MJ; Centers for Disease Control and Prevention. Preterm births — United States, 2006 and 2010. MMWR Surveill Summ 62, Suppl 3: 136–138, 2013. [PubMed] [Google Scholar]
- 120.Martin RJ, Mhanna MJ, Haxhiu MA. The role of endogenous and exogenous nitric oxide on airway function. Semin Perinatol 26: 432–438, 2002. [DOI] [PubMed] [Google Scholar]
- 121.McBride JT. Architecture of the tracheobronchial tree, edited by Parent RA. Boca Raton: CRC, 1991, p. 49–61. [Google Scholar]
- 122.McCurnin D, Seidner S, Chang LY, Waleh N, Ikegami M, Petershack J, Yoder B, Giavedoni L, Albertine KH, Dahl MJ, Wang ZM, Clyman RI. Ibuprofen-induced patent ductus arteriosus closure: physiologic, histologic, and biochemical effects on the premature lung. Pediatrics 121: 945–956, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McCurnin DC, Pierce RA, Chang LY, Gibson LL, Osborne-Lawrence S, Yoder BA, Kerecman JD, Albertine KH, Winter VT, Coalson JJ, Crapo JD, Grubb PH, Shaul PW. Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 288: L450–L459, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McGreal EP, Chakraborty M, Winter VT, Jones SA, Coalson JJ, Kotecha S. Dynamic expression of IL-6 trans-signalling molecules in the lungs of preterm baboons undergoing mechanical ventilation. Neonatology 100: 130–138, 2011. [DOI] [PubMed] [Google Scholar]
- 125.Mercer RR, Crapo JD. Three-dimensional reconstruction of the rat acinus. J Appl Physiol 63: 785–794, 1987. [DOI] [PubMed] [Google Scholar]
- 126.Mercer RR, Russell ML, Crapo JD. Alveolar septal structure in different species. J Appl Physiol 77: 1060–1066, 1994. [DOI] [PubMed] [Google Scholar]
- 127.Mercer RR, Russell ML, Crapo JD. Radon dosimetry based on the depth distribution of nuclei in human and rat lungs. Health Phys 61: 117–130, 1991. [DOI] [PubMed] [Google Scholar]
- 128.Mercer RR, Russell ML, Roggli VL, Crapo JD. Cell number and distribution in human and rat airways. Am J Respir Cell Mol Biol 10: 613–662, 1994. [DOI] [PubMed] [Google Scholar]
- 129.Merritt TA, Hallman M, Berry C, Pohjavuori M, Edwards DK 3rd, Jaaskelainen J, Grafe MR, Vaucher Y, Wozniak P, Heldt G, Rapold J. Randomized, placebo-controlled trial of human surfactant given at birth versus rescue administration in very low birth weight infants with lung immaturity. J Pediatr 118: 581–594, 1991. [DOI] [PubMed] [Google Scholar]
- 130.Morray JP, Fox WW, Kettrick RG, Downes JJ. Improvement in lung mechanics as a function of age in the infant with severe bronchopulmonary dysplasia. Pediatr Res 16: 290–294, 1982. [DOI] [PubMed] [Google Scholar]
- 131.Mulrooney N, Champion Z, Moss TJ, Nitsos I, Ikegami M, Jobe AH. Surfactant and physiologic responses of preterm lambs to continuous positive airway pressure. Am J Respir Crit Care Med 171: 488–493, 2005. [DOI] [PubMed] [Google Scholar]
- 131a.Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N Engl J Med 336: 597–604, 1997. [DOI] [PubMed] [Google Scholar]
- 132.Northway WH., Jr Bronchopulmonary dysplasia: twenty-five years later. Pediatrics 89: 969–973, 1992. [PubMed] [Google Scholar]
- 133.Northway WH Jr, Moss RB, Carlisle KB, Parker BR, Popp RL, Pitlick PT, Eichler I, Lamm RL, Brown BW Jr. Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med 323: 1793–1799, 1990. [DOI] [PubMed] [Google Scholar]
- 134.Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med 276: 357–368, 1967. [DOI] [PubMed] [Google Scholar]
- 135.Null DM, Alvord J, Leavitt W, Wint A, Janna Dahl M, Presson AP, Lane RH, Digeronimo RJ, Yoder BA, Albertine KH. High-frequency nasal ventilation for 21 d maintains gas exchange with lower respiratory pressures and promotes alveolarization in preterm lambs. Pediatr Res 75: 507–516, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oda H, Kusumoto S, Nakajima T. Nitrosyl-hemoglobin formation in the blood of animals exposed to nitric oxide. Arch Environ Health 30: 453–456, 1975. [DOI] [PubMed] [Google Scholar]
- 137.Pack RJ, Al-Ugaily LH, Morris G. The cells of the tracheobronchial epithelium of the mouse: a quantitative light and electron microscope study. J Anat 132: 71–84, 1981. [PMC free article] [PubMed] [Google Scholar]
- 138.Pack RJ, Al-Ugaily LH, Morris G, Widdicombe JG. The distribution and structure of cells in the tracheal epithelium of the mouse. Cell Tissue Res 208: 65–84, 1980. [DOI] [PubMed] [Google Scholar]
- 139.Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: disordered pulmonary elastin deposition. Am J Physiol Lung Cell Mol Physiol 272: L452–L460, 1997. [DOI] [PubMed] [Google Scholar]
- 140.Pierce RA, Joyce B, Officer S, Heintz C, Moore C, McCurnin D, Johnston C, Maniscalco W. Retinoids increase lung elastin expression but fail to alter morphology or angiogenesis genes in premature ventilated baboons. Pediatr Res 61: 703–709, 2007. [DOI] [PubMed] [Google Scholar]
- 141.Plopper CG, Chu FP, Haselton CJ, Peake J, Wu J, Pinkerton KE. Dose-dependent tolerance to ozone. I. Tracheobronchial epithelial reorganization in rats after 20 months' exposure. Am J Pathol 144: 404–420, 1994. [PMC free article] [PubMed] [Google Scholar]
- 142.Plopper CG, Hatch GE, Wong V, Duan X, Weir AJ, Tarkington BK, Devlin RB, Becker S, Buckpitt AR. Relationship of inhaled ozone concentration to acute tracheobronchial epithelial injury, site-specific ozone dose, and glutathione depletion in rhesus monkeys. Am J Respir Cell Mol Biol 19: 387–399, 1998. [DOI] [PubMed] [Google Scholar]
- 143.Plopper CG, Heidsiek JG, Weir AJ, George JA, Hyde DM. Tracheobronchial epithelium in the adult rhesus monkey: a quantitative histochemical and ultrastructural study. Am J Anat 184: 31–40, 1989. [DOI] [PubMed] [Google Scholar]
- 144.Plopper CG, Hill LH, Mariassy AT. Ultrastructure of the nonciliated bronchiolar epithelial (Clara) cell of mammalian lung. III. A study of man with comparison of 15 mammalian species. Exp Lung Res 1: 171–180, 1980. [DOI] [PubMed] [Google Scholar]
- 145.Plopper CG, Hyde DM. The non-human primate as a model for studying COPD and asthma. Pulm Pharmacol Ther 21: 755–766, 2008. [DOI] [PubMed] [Google Scholar]
- 146.Plopper CG, Mariassy AT, Hill LH. Ultrastructure of the nonciliated bronchiolar epithelial (Clara) cell of mammalian lung: I. A comparison of rabbit, guinea pig, rat, hamster, and mouse. Exp Lung Res 1: 139–154, 1980. [DOI] [PubMed] [Google Scholar]
- 147.Potter CF, Dreshaj IA, Haxhiu MA, Stork EK, Chatburn RL, Martin RJ. Effect of exogenous and endogenous nitric oxide on the airway and tissue components of lung resistance in the newborn piglet. Pediatr Res 41: 886–891, 1997. [DOI] [PubMed] [Google Scholar]
- 149.Raabe OG, Yeh HC, Schum GM, Phalen RF. Tracheobronchial Geometry: Human, Dog, Rat, Hamster. Albuquerque, NM: Lovelace Foundation for Medical Education and Research, 1976. Lovelace Foundation Report LF-53. [Google Scholar]
- 150.Reyburn B, Li M, Metcalfe DB, Kroll NJ, Alvord J, Wint A, Dahl MJ, Sun J, Dong L, Wang ZM, Callaway C, McKnight RA, Moyer-Mileur L, Yoder BA, Null DM, Lane RH, Albertine KH. Nasal ventilation alters mesenchymal cell turnover and improves alveolarization in preterm lambs. Am J Respir Crit Care Med 178: 407–418, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Reynolds EO, Roberton NR, Wigglesworth JS. Hyaline membrane disease, respiratory distress, and surfactant deficiency. Pediatrics 42: 758–768, 1968. [PubMed] [Google Scholar]
- 152.Rimar S, Gillis CN. Selective pulmonary vasodilation by inhaled nitric oxide is due to hemoglobin inactivation. Circulation 88: 2884–2887, 1993. [DOI] [PubMed] [Google Scholar]
- 153.Roberts JD., Jr Inhaled nitric oxide for treatment of pulmonary artery hypertension in the newborn and infant. Crit Care Med 21: S374–S376, 1993. [DOI] [PubMed] [Google Scholar]
- 154.Roberts JD Jr, Chen TY, Kawai N, Wain J, Dupuy P, Shimouchi A, Bloch K, Polaner D, Zapol WM. Inhaled nitric oxide reverses pulmonary vasoconstriction in the hypoxic and acidotic newborn lamb. Circ Res 72: 246–254, 1993. [DOI] [PubMed] [Google Scholar]
- 155.Roberts JD Jr, Fineman JR, Morin FC 3rd, Shaul PW, Rimar S, Schreiber MD, Polin RA, Zwass MS, Zayek MM, Gross I, Heymann MA, Zapol WM. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med 336: 605–610, 1997. [DOI] [PubMed] [Google Scholar]
- 156.Roberts JD Jr, Roberts CT, Jones RC, Zapol WM, Bloch KD. Continuous nitric oxide inhalation reduces pulmonary arterial structural changes, right ventricular hypertrophy, and growth retardation in the hypoxic newborn rat. Circ Res 76: 215–222, 1995. [DOI] [PubMed] [Google Scholar]
- 157.Roberts JD, Polaner DM, Lang P, Zapol WM. Inhaled nitric oxide in persistent pulmonary hypertension of the newborn. Lancet 340: 818–819, 1992. [DOI] [PubMed] [Google Scholar]
- 158.Rodriguez M, Bur S, Favre A, Weibel ER. Pulmonary acinus: geometry and morphometry of the peripheral airway system in rat and rabbit. Am J Anat 180: 143–155, 1987. [DOI] [PubMed] [Google Scholar]
- 159.Rojas MA, Gonzalez A, Bancalari E, Claure N, Poole C, Silva-Neto G. Changing trends in the epidemiology and pathogenesis of neonatal chronic lung disease. J Pediatr 126: 605–610, 1995. [DOI] [PubMed] [Google Scholar]
- 160.Romero R, Gomez R, Chaiworapongsa T, Conoscenti G, Kim JC, Kim YM. The role of infection in preterm labour and delivery. Paediatr Perinat Epidemiol 15, Suppl 2: 41–56, 2001. [DOI] [PubMed] [Google Scholar]
- 161.Roos PJ, Wierner-Kronish JP, Albertine KH, Staub NC. Removal of abdominal sources of caudal mediastinal node lymph in anesthetized sheep. J Appl Physiol 55: 996–1001, 1983. [DOI] [PubMed] [Google Scholar]
- 162.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 101: 731–736, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schachtner SK, Wang Y, Baldwin HS. Qualitative and quantitative analysis of embryonic pulmonary vessel formation. Am J Respir Cell Mol Biol 22: 157–165, 2000. [DOI] [PubMed] [Google Scholar]
- 164.Seki J, Nishio M, Kato Y, Motoyama Y, Yoshida K. FK409, a new nitric-oxide donor, suppresses smooth muscle proliferation in the rat model of balloon angioplasty. Atherosclerosis 117: 97–106, 1995. [DOI] [PubMed] [Google Scholar]
- 165.Shaul PW, Afshar S, Gibson LL, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC. Developmental changes in nitric oxide synthase isoform expression and nitric oxide production in fetal baboon lung. Am J Physiol Lung Cell Mol Physiol 283: L1192–L1199, 2002. [DOI] [PubMed] [Google Scholar]
- 166.Shenai JP, Chytil F. Vitamin A storage in lungs during perinatal development in the rat. Biol Neonate 57: 126–132, 1990. [DOI] [PubMed] [Google Scholar]
- 167.Shenai JP, Chytil F, Stahlman MT. Vitamin A status of neonates with bronchopulmonary dysplasia. Pediatr Res 19: 185–188, 1985. [DOI] [PubMed] [Google Scholar]
- 168.Sherman TS, Chen Z, Yuhanna IS, Lau KS, Margraf LR, Shaul PW. Nitric oxide synthase isoform expression in the developing lung epithelium. Am J Physiol Lung Cell Mol Physiol 276: L383–L390, 1999. [DOI] [PubMed] [Google Scholar]
- 169.Shijubo N, Itoh Y, Yamaguchi T, Imada A, Hirasawa M, Yamada T, Kawai T, Abe S. Clara cell protein-positive epithelial cells are reduced in small airways of asthmatics. Am J Respir Crit Care Med 160: 930–933, 1999. [DOI] [PubMed] [Google Scholar]
- 170.Shijubo N, Itoh Y, Yamaguchi T, Shibuya Y, Morita Y, Hirasawa M, Okutani R, Kawai T, Abe S. Serum and BAL Clara cell 10 kDa protein (CC10) levels and CC10-positive bronchiolar cells are decreased in smokers. Eur Respir J 10: 1108–1114, 1997. [DOI] [PubMed] [Google Scholar]
- 171.Smith VC, Zupancic JA, McCormick MC, Croen LA, Greene J, Escobar GJ, Richardson DK. Trends in severe bronchopulmonary dysplasia rates between 1994 and 2002. J Pediatr 146: 469–473, 2005. [DOI] [PubMed] [Google Scholar]
- 172.St George JA, Harkema JR, Hyde DM, Plopper C. Cell populations and structure/function relationships of cells in the airways. In: Toxicology of the Lung, edited by Gardner D, Crapo JD, and Massaro EJ. New York: Raven, 1988, p. 71–102. [Google Scholar]
- 173.Staub NC. Pulmonary edema. Physiol Rev 54: 678–811, 1974. [DOI] [PubMed] [Google Scholar]
- 174.Staub NC, Albertine KH. Biology of lung lymphatics. In: Experimental Biology of the Lymphatic Circulation, edited by Johnston M. Amsterdam: Elsevier Science, 1985, p. 305–325. [Google Scholar]
- 175.Staub NC, Bland RD, Brigham KL, Demling R, Erdmann AJ 3rd, Woolverton WC. Preparation of chronic lung lymph fistulas in sheep. J Surg Res 19: 315–320, 1975. [DOI] [PubMed] [Google Scholar]
- 176.Stocker JT. Pathologic features of long-standing “healed” bronchopulmonary dysplasia: a study of 28 3- to 40-month-old infants. Hum Pathol 17: 943–961, 1986. [DOI] [PubMed] [Google Scholar]
- 177.Stocker JT. The respiratory tract. In: Pediatric Pathology (2nd ed.), edited by Dehner LP and Stocker JT. Philadelphia, PA: Lippincott Williams, 2001, p. 445–517. [Google Scholar]
- 178.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sanchez PJ, O'Shea TM, Goldberg RN, Van Meurs KP, Faix RG, Phelps DL, Frantz ID 3rd, Watterberg KL, Saha S, Das A, Higgins RD. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126: 443–456, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tang JR, Markham NE, Lin YJ, McMurtry IF, Maxey A, Kinsella JP, Abman SH. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am J Physiol Lung Cell Mol Physiol 287: L344–L351, 2004. [DOI] [PubMed] [Google Scholar]
- 180.Tenney SM, Remmers JE. Comparative quantitative morphology of the mammalian lung: diffusing area. Nature 197: 54–56, 1963. [DOI] [PubMed] [Google Scholar]
- 181.Thomae KR, Nakayama DK, Billiar TR, Simmons RL, Pitt BR, Davies P. The effect of nitric oxide on fetal pulmonary artery smooth muscle growth. J Surg Res 59: 337–343, 1995. [DOI] [PubMed] [Google Scholar]
- 182.Thomson MA, Yoder BA, Winter VT, Martin H, Catland D, Siler-Khodr TM, Coalson JJ. Treatment of immature baboons for 28 days with early nasal continuous positive airway pressure. Am J Respir Crit Care Med 169: 1054–1062, 2004. [DOI] [PubMed] [Google Scholar]
- 183.Tomashefski JF Jr, Oppermann HC, Vawter GF, Reid LM. Bronchopulmonary dysplasia: a morphometric study with emphasis on the pulmonary vasculature. Pediatr Pathol 2: 469–487, 1984. [DOI] [PubMed] [Google Scholar]
- 184.Tyler NK, Plopper CG. Morphology of the distal conducting airways in rhesus monkey lungs. Anat Rec 211: 295–303, 1985. [DOI] [PubMed] [Google Scholar]
- 185.Tyler WS, Julian MD. Gross and subgross anatomy of the lungs, pleura, connective tissue septa, distal airways, and structural units, edited by Parent RA. Boca Raton: CRC, 1991, p. 37–48. [Google Scholar]
- 186.Tyson JE, Wright LL, Oh W, Kennedy KA, Mele L, Ehrenkranz RA, Stoll BJ, Lemons JA, Stevenson DK, Bauer CR, Korones SB, Fanaroff AA. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med 340: 1962–1968, 1999. [DOI] [PubMed] [Google Scholar]
- 187.Valerius KP. Size-dependent morphology of the conductive bronchial tree in four species of myomorph rodents. J Morphol 230: 291–297, 1996. [DOI] [PubMed] [Google Scholar]
- 188.Van Lierde S, Cornelis A, Devlieger H, Moerman P, Lauweryns J, Eggermont E. Different patterns of pulmonary sequelae after hyaline membrane disease: heterogeneity of bronchopulmonary dysplasia? A clinicopathologic study. Biol Neonate 60: 152–162, 1991. [DOI] [PubMed] [Google Scholar]
- 189.Van Marter LJ, Allred EN, Pagano M, Sanocka U, Parad R, Moore M, Susser M, Paneth N, Leviton A. Do clinical markers of barotrauma and oxygen toxicity explain interhospital variation in rates of chronic lung disease? The Neonatology Committee for the Developmental Network. Pediatrics 105: 1194–1201, 2000. [DOI] [PubMed] [Google Scholar]
- 190.Van Winkle LS, Fanucchi MV, Miller LA, Baker GL, Gershwin LJ, Schelegle ES, Hyde DM, Evans MJ, Plopper CG. Epithelial cell distribution and abundance in rhesus monkey airways during postnatal lung growth and development. J Appl Physiol 97: 2355–2363; discussion 2354, 2004. [DOI] [PubMed] [Google Scholar]
- 191.Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, De Langhe S, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E. Lung organogenesis. Curr Top Dev Biol 90: 73–158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Weibel ER. Morphological basis of alveolar-capillary gas exchange. Physiol Rev 53: 419–495, 1973. [DOI] [PubMed] [Google Scholar]
- 193.Weibel ER. Morphometry of the Lung. New York: Academic, 1963. [Google Scholar]
- 194.Weibel ER, Federspiel WJ, Fryder-Doffey F, Hsia CC, Konig M, Stalder-Navarro V, Vock R. Morphometric model for pulmonary diffusing capacity. I. Membrane diffusing capacity. Respir Physiol 93: 125–149, 1993. [DOI] [PubMed] [Google Scholar]
- 195.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 23: 320–326, 2000. [DOI] [PubMed] [Google Scholar]
- 196.Winkelmann A, Noack T. The Clara cell: a “Third Reich eponym”? Eur Respir J 36: 722–727, 2010. [DOI] [PubMed] [Google Scholar]
- 197.Yang W, Ando J, Korenaga R, Toyo-oka T, Kamiya A. Exogenous nitric oxide inhibits proliferation of cultured vascular endothelial cells. Biochem Biophys Res Commun 203: 1160–1167, 1994. [DOI] [PubMed] [Google Scholar]
- 198.Yeh HC, Schum GM, Duggan MT. Anatomic models of the tracheobronchial and pulmonary regions of the rat. Anat Rec 195: 483–492, 1979. [DOI] [PubMed] [Google Scholar]
- 199.Yoder BA, Siler-Khodr T, Winter VT, Coalson JJ. High-frequency oscillatory ventilation: effects on lung function, mechanics, and airway cytokines in the immature baboon model for neonatal chronic lung disease. Am J Respir Crit Care Med 162: 1867–1876, 2000. [DOI] [PubMed] [Google Scholar]
- 200.Young SL, Evans K, Eu JP. Nitric oxide modulates branching morphogenesis in fetal rat lung explants. Am J Physiol Lung Cell Mol Physiol 282: L379–L385, 2002. [DOI] [PubMed] [Google Scholar]
- 201.Young SL, Fram EK, Randell SH. Quantitative three-dimensional reconstruction and carbohydrate cytochemistry of rat nonciliated bronchiolar (Clara) cells. Am Rev Respir Dis 133: 899–907, 1986. [PubMed] [Google Scholar]
- 202.Yuhanna IS, MacRitchie AN, Lantin-Hermoso RL, Wells LB, Shaul PW. Nitric oxide (NO) upregulates NO synthase expression in fetal intrapulmonary artery endothelial cells. Am J Respir Cell Mol Biol 21: 629–636, 1999. [DOI] [PubMed] [Google Scholar]
- 203.Zayek M, Cleveland D, Morin FC 3rd. Treatment of persistent pulmonary hypertension in the newborn lamb by inhaled nitric oxide. J Pediatr 122: 743–750, 1993. [DOI] [PubMed] [Google Scholar]
- 204.Zayek M, Wild L, Roberts JD, Morin FC 3rd. Effect of nitric oxide on the survival rate and incidence of lung injury in newborn lambs with persistent pulmonary hypertension. J Pediatr 123: 947–952, 1993. [DOI] [PubMed] [Google Scholar]