

SUMMARY
Cell-cell contacts inhibit cell growth and proliferation in part by activating the Hippo pathway that drives the phosphorylation and nuclear exclusion of the transcriptional coactivators YAP and TAZ. Cell density and Hippo signaling have also been reported to block transforming growth factor β (TGF-β) responses, based on the ability of phospho-YAP/TAZ to sequester TGF-β-activated SMAD complexes in the cytoplasm. Herein, we provide evidence that epithelial cell polarization interferes with TGF-β signaling well upstream and independent of cytoplasmic YAP/TAZ. Rather, polarized basolateral presentation of TGF-β receptors I and II deprives apically delivered TGF-β of access to its receptors. Basolateral ligand delivery nonetheless remains entirely effective to induce TGF-β responses. These data demonstrate that cell-type-specific inhibition of TGF-β signaling by cell density is restricted to polarized epithelial cells and reflects the polarized distribution of TGF-β receptors, which thus affects SMAD activation irrespective of Hippo pathway activation.
INTRODUCTION
Cell-cell contacts drive signals controlling the process of contact inhibition, a phenomenon whereby normal cells grown in monolayers exhibit reduced proliferation, even growth arrest, when reaching confluency. This property is often lost during neoplastic progression or in vitro transformation. Recently, clues regarding the mechanisms by which cells sense contacts with other cells have emerged. In particular, the Hippo pathway, originally identified as a mechanism controlling organ size in Drosophila via inhibition of cell proliferation and induction of apoptosis, was identified as a major player in this process (Zhao et al., 2007). Specifically, it was found that activation of Hippo signaling by cell density sensing leads to phosphorylation and nuclear exclusion of its effector molecules YAP and TAZ, thereby restraining the nuclear activity of the latter, which otherwise act as co-transcriptional activators of TEAD and other transcription factors to promote cell proliferation. In polarized cells, the apical-basal cell polarity determinant Crumbs was found to directly regulate Hippo signaling, and thus YAP/TAZ nucleo-cytoplasmic localization and function (Chen et al., 2010; Robinson et al., 2010). Remarkably, YAP and TAZ may also undergo nuclear exclusion upon mechanical stress induced by extracellular matrix rigidity and cell geometry, in a process requiring Rho GTPase signaling and the actomyosin cytoskeleton, independent from Hippo activity (Dupont et al., 2011).
Various mechanisms have been described whereby the Hippo pathway and/or its effectors YAP/TAZ interfere with the transforming growth factor beta (TGF-β)/SMAD cascade (Mauviel et al., 2012). We initially identified YAP as a SMAD7-interacting protein that cooperates with the latter to block TGF-β receptor type I (TβRI) function, thereby inhibiting TGF-β signaling (Ferrigno et al., 2002). In Drosophila, Yorkie (YAP homolog) was found to bind and potentiate Mad (SMAD homolog)-driven transcription (Alarcón et al., 2009; Oh and Irvine, 2011), while in mammalian cells, it was found that YAP and TAZ are required for efficient nuclear translocation and transcriptional activity of SMAD complexes in response to TGF-β (Varelas et al., 2008). The same authors also described a mechanism whereby phosphorylated YAP and TAZ, excluded from the cell nucleus upon cell-cell contacts, sequester active SMAD complexes in the cytoplasm, thereby suppressing cellular responses to TGF-β in cells grown at high density (Varelas et al., 2010). The latter findings suggest that confluent cells are not responsive to TGF-β, at odds with a large body of literature on TGF-β. Indeed, in the early days of TGF-β research, the use of dense, confluent cell cultures was often preferred to better understand the broad capacity of TGF-β to regulate gene expression and/or differentiation. For example, confluent skin, gingival or lung fibroblasts (Chung et al., 1996; Daniels et al., 2004; Mauviel et al., 1994; Wrana et al., 1991), keratinocytes (Kon et al., 1999; Mauviel et al., 1996), chondrocytes (Redini et al., 1991), and/or calvarial bone cells (Wrana et al., 1988), to cite a few, all respond to TGF-β with increased or decreased expression of fibrillar and non-fibrillar collagens (Chung et al., 1996; Kon et al., 1999; Mauviel et al., 1994), fibronectin (Daniels et al., 2004; Ignotz et al., 1989), SPARC (Wrana et al., 1991), laminins (Korang et al., 1995), integrins (Heino and Massagué, 1989), proteoglycans (Redini et al., 1991), metalloproteinases and protease inhibitors such as plasminogen activator inhibitor 1 (PAI-1) or tissue inhibitor of metal-loproteases (Mauviel et al., 1996; Overall et al., 1989). Likewise, early-response genes are efficiently induced by TGF-β in confluent skin fibroblasts and keratinocytes (Mauviel et al., 1993, 1996). The broad ability of TGF-β to control the turnover of extracellular matrix and cell adhesion molecules largely contributes to its capacity to orchestrate tissue homeostasis and function, to promote tissue repair, epithelial-to-mesenchymal transition (EMT), and to mediate tissue fibrosis when signaling is deregulated and excessive.
Given the conflicting results from the literature pertaining to an important aspect of TGF-β biology (i.e., interference or not of cell-cell contacts with TGF-β signaling), we undertook a broad examination of TGF-β responses as a function of cell density in a variety of cell types. Our results demonstrate that cell contact-induced nuclear exclusion of Hippo effectors YAP and TAZ does not interfere with TGF-β responses, measured as SMAD3 phosphorylation, nuclear translocation, and gene activation. Rather, loss of TGF-β responsiveness in dense cell cultures is independent of cell density per se, and appears to be restricted to polarized epithelial cells that undergo a remarkable cell density-dependent shift in TGF-β receptor (TβR) localization from pericellular at low cell density to exclusively basolateral at high cell density when tight cell-cell contacts are established. This domain-specific receptor localization, in turn, prevents SMAD phosphorylation and associated gene responses from apically delivered TGF-β ligand, while TGF-β signaling is fully efficient when initiated from the basolateral sides of polarized cells. These results shed new light on the relationship between cellular density and TGF-β responses, independent from the nucleocytoplasmic shuttling of Hippo pathway effectors.
RESULTS
In Most Cell Types, Cell Density Does Not Inhibit TGF-β Responses
We first examined how cell density affects TGF-β signaling in various cell lines from distinct lineages that have classically been used for TGF-β signaling studies, including HaCaT keratinocytes (Alarcón et al., 2009; Gomis et al., 2006; Levy and Hill, 2005; Reynisdóttir et al., 1995), 1205Lu and other human melanoma cell lines extensively characterized for functional SMAD signaling implicated in their invasive and metastatic potential (Alexaki et al., 2010; Javelaud et al., 2007; Mohammad et al., 2011; Rodeck et al., 1999), human pancreatic PANC-1 (Dennler et al., 2007; Ellenrieder et al., 2001; Nicolás and Hill, 2003) and breast MDA-MB-231 carcinoma cells (Kang et al., 2003; Yin et al., 1999), as well as mouse mammary epithelial EpH4 cells (Varelas et al., 2010). As shown in Figures 1 and S1, in all but EpH4 cells, there was no remarkable attenuation in the extent of TGF-β-induced expression of the prototypic target gene PAI-1 (Figures 1A and S1A) or activity of a SMAD3/4-specific reporter in transient cell transfection assays (Figures 1B and S1B). In fact, the extent of PAI-1 induction by TGF-β was even higher in HaCaT and 1205Lu cells grown at high density than in proliferating sparse cells.
Figure 1. Impact of Cell Density on TGF-β Signaling.
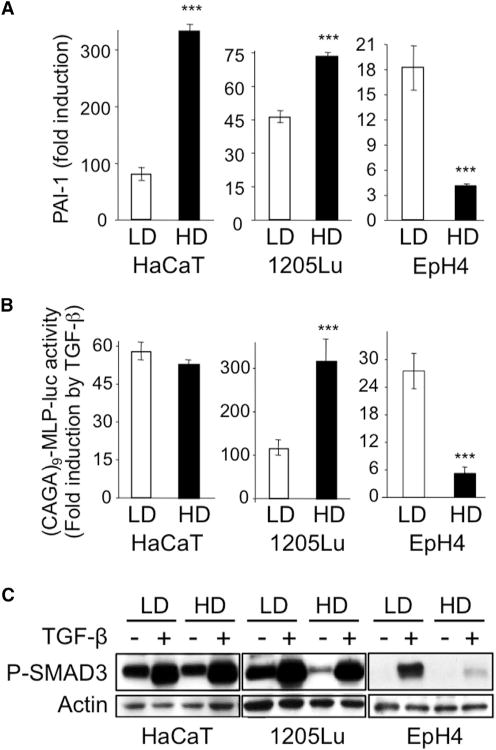
HaCaT keratinocytes, 1205Lu melanoma cells, and EpH4 mouse mammary epithelial cells were grown in either low (LD) or high (HD) density conditions prior to TGF-β (5 ng/ml) stimulation.
(A) Quantitative RT-PCR analysis of PAI-1 expression after a 24-hr TGF-β treatment. Results are expressed as -fold induction by TGF-β in each culture condition and are the mean ± SD from three independent experiments, each measured in triplicate.
(B) Effect of TGF-β on SMAD3/4-specific transcription. Results are expressed as -fold activation of transiently transfected (CAGA)9-MLP-luc activity 18 hr after TGF-β addition to the cultures. Results are the mean ± SD of two independent experiments, each performed with triplicate samples.
(C) Western analysis of P-SMAD3 levels without or with 30 min TGF-β stimulation. Actin levels were measured as a control for the specificity of P-SMAD3 changes under each experimental condition. Results from one representative of several independent experiments are shown.
The primary signaling event downstream of activated TGF-β receptors is SMAD3 phosphorylation. Remarkably, in dense EpH4 mouse mammary cell cultures, reduction in SMAD-specific transcription and target gene activation in response to TGF-β was associated with an almost complete lack of SMAD3 phosphorylation (Figure 1C), which was not affected by cell density in any of the other five cell lines that were examined (Figures 1C and S1C).
Nuclear Translocation of SMAD2/3 in Response to TGF-β Is Independent from TAZ Nuclear Exclusion Induced by Cell Density
The previous data contrast with the report showing that TGF-β induces SMAD3 phosphorylation in confluent EpH4 cells (Varelas et al., 2010). Since Hippo pathway activation has been identified as a sensor for cell-cell contacts (Zhao et al., 2007), together with the fact that phosphorylation of SMAD3 is a prerequisite for its nuclear accumulation and subsequent gene responses, TAZ and SMAD2/3 nucleo-cytoplasmic localization were studied in parallel by indirect immunofluorescence in several cell types grown at low or high density, in the absence or presence of TGF-β. As shown in Figure 2A, HaCaT cells grown at low density exhibited both cytoplasmic and nuclear TAZ, while high-density cultures exhibited remarkable nuclear exclusion of TAZ, (red fluorescence), independent from TGF-β. Parallel examination of SMAD2/3 localization following a 30-min TGF-β stimulation of HaCaT cells grown at low or high density indicated strong nuclear accumulation of P-SMAD3 in response to TGF-β, whether at low or high density (Figure 2A, green fluorescence), without changes in TAZ localization in response to TGF-β. Similar results were obtained in 1205Lu cells (Figure 2B). Thus, in these two cell types, nuclear accumulation of P-SMAD3 occurs in response to TGF-β despite TAZ nuclear exclusion resulting from cell density sensing, indicating that the two proteins are able to independently shuttle between the cytoplasm and nucleus. Quantitation of nuclear SMAD3 and TAZ in these three cell lines at high cell density in absence or presence of TGF-β is provided in Figure S2A. Consistent with the findings in HaCaT and 1205Lu cells, efficient SMAD3 nuclear translocation was also observed in fibroblasts treated with TGF-β, whether at low or high cell density (Figure S2B).
Figure 2. Independent Regulation of SMAD2/3 Nuclear Translocation and Density-Dependent TAZ Nucleo-Cytoplasmic Localization.
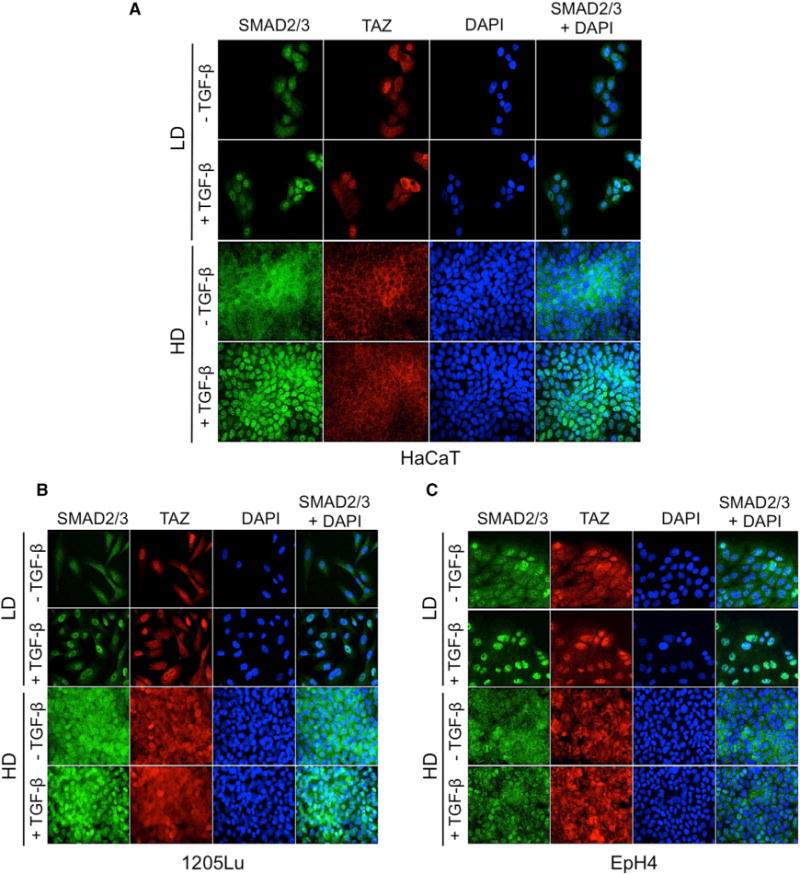
(A–C) Simultaneous detection of SMAD2/3 and TAZ in HaCaT keratinocytes (A), 1205Lu melanoma cells (B), and EpH4 mouse mammary epithelial cells (C). Cells were grown on glass coverslips in either low (LD) or high (HD) density conditions prior to TGF-β (30 min, 5 ng/ml) stimulation, then subjected to simultaneous immunofluorescent detection of SMAD2/3 (green) and TAZ (red). Experiments were repeated four to six times, depending on cell line.
In EpH4 cells, however, while TAZ was efficiently excluded from the cell nucleus at high density, SMAD3 nuclear accumulation only occurred in sparse cell cultures treated with TGF-β, not in confluent cells (Figures 2C and S2A). These data are consistent with the absence of SMAD3 phosphorylation and target gene activation observed in these cells at high density (see Figure 1) and fit the established model of TGF-β/SMAD signal transduction whereby an absence of SMAD3 phosphorylation precludes subsequent downstream signaling (reviewed in Massagué, 2012). They are, however, at odds with the model whereby active (phosphorylated) SMAD complexes are sequestered in the cytoplasm by P-TAZ in confluent cells (Varelas et al., 2010).
The Extent of TGF-β Signaling Does Not Rely upon Cellular TAZ—and YAP—Levels
It has been suggested previously that TAZ levels regulate the extent of TGF-β signaling (Varelas et al., 2008). Our data presented above do not fit this model because SMADs and TAZ localization in either the cytoplasm or nucleus of cells appear to be under distinct regulatory control mechanisms. In that these findings significantly impact current models of TGF-β signaling, the apparent lack of interference of Hippo pathway effectors with SMAD nuclear translocation observed in keratinocytes and melanoma cells (see Figures 2A and 2B) was further substantiated by simultaneous knockdown of TAZ and it paralog YAP (Nallet-Staub et al., 2014). Despite a 95% reduction in both TAZ and YAP protein levels (Figure S3A), there was no significant alteration in the extent of TGF-β target gene induction, as measured by qRT-PCR: induction of PAI-1 was essentially identical in control- and siYAP/TAZ-transfected cells (Figure S3B).
Last, to further confirm the independence of P-SMAD3 nuclear translocation and Hippo effectors, we took advantage of recently established human melanoma cell lines in which stable TAZ knockdown was obtained by lentiviral delivery of a specific shRNA directed against TAZ (Nallet-Staub et al., 2014). These cell lines fit the purpose of these experiments as they express much higher levels of TAZ than YAP1/2 (Nallet-Staub et al., 2014). TAZ knockdown in 1205Lu melanoma cells had no effect on TGF-β-induced SMAD3 nuclear accumulation (Figure S3C) target gene responses (Figure S3D), and induction of SMAD3/4-specific transcription measured in transient cell transfection assays with the (CAGA)9-MLP-luc reporter (Figure S3E). Likewise, in the WM852 melanoma line, knocking down TAZ did not attenuate the induction of GLI2, PAI-1, CTGF, and SMAD7 by TGF-β (Figure S3F), nor did it impact TGF-β-driven SMAD3/4-specific transcription in SKmel28 melanoma cells (Figure S3G).
Hippo pathway activation upon cell density sensing is a widespread phenomenon affecting a variety of cell types (reviewed in Zhao et al., 2010)). Yet, we found no evidence that it might interfere with SMAD nuclear accumulation in response to TGF-β as (a), altering endogenous levels of YAP/TAZ did not attenuate TGF-β responses (Figure S3) and (b), in TGF-β-responsive cells, TGF-β-driven nuclear accumulation of SMAD2/3 occurred under conditions that excluded TAZ from the nucleus (e.g., dense cell cultures; Figure 2). Specifically, out of the six cell lines tested, only Eph4 cells lost TGF-β responsiveness at high cell density. Moreover, this appeared to be due to a lack of SMAD3 phosphorylation, the primary event downstream of TGF-β receptors that initiates the signaling cascade, not to cytoplasmic retention of active SMAD complexes.
Loss of TGF-β Responsiveness in Polarized Epithelial Cells Is Restricted to Their Apical Side
Since EpH4 cells similarly exhibited density-dependent Hippo pathway activation, measured as nuclear exclusion of TAZ, yet uniquely lacked P-SMAD nuclear accumulation in response to TGF-β, we hypothesized that this cell-type specificity might be associated with their capacity to establish baso-apical polarity when grown at high density. To determine whether loss of TGF-β responsiveness due to lack of SMAD3 phosphorylation at high cell density is due to the establishment of cell polarity and/or specific alterations of TGF-β receptor localization, we performed parallel examination of TGF-β responses in EpH4, MDCK, and AKR-2B cells. Both EpH4 and MDCK cells are of epithelial origin and differ from the latter fibroblast cultures by their distinct basoapical polarized phenotype at high culture density. In a first set of experiments, monolayer cultures at low and high density were compared for SMAD3 phosphorylation in response to TGF-β. As shown in Figure 3A, a 30-min stimulation with TGF-β resulted in efficient SMAD3 phosphorylation in all three cell lines at low density while in dense cultures, significant SMAD3 phosphorylation was solely observed in the AKR-2B fibroblasts, consistent with the EpH4 results presented in Figure 1.
Figure 3. Plasma Membrane Receptor Localization Determines Cell- and Density-Specific TGF-p Responsiveness.
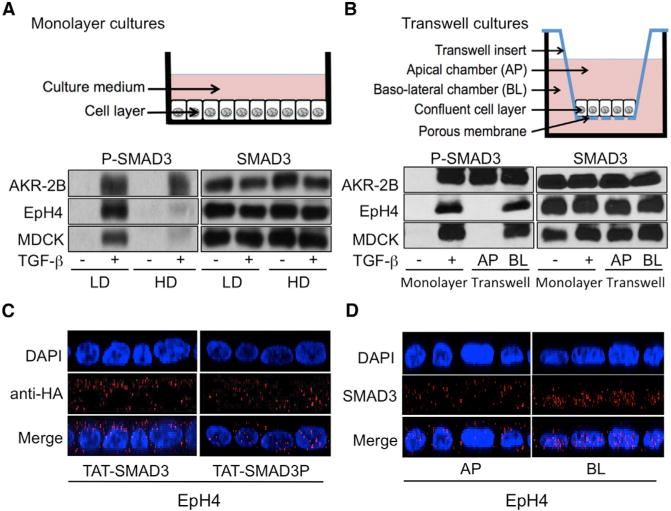
(A) Western analysis of P-SMAD3 (left) and SMAD3 (right) levels in AKR-2B fibroblasts, EpH4, and MDCK epithelial cells grown at low (LD) or high (HD) density monolayer cultures (schematic) following a 30-min TGF-β stimulation.
(B) AKR-2B, MDCK, and EpH4 cells were grown in monolayer (at LD) or Transwell cultures (schematic). P-SMAD3 (left) and SMAD3 levels (right) were determined by western blotting in monolayer cultures following 60 min stimulation in the absence (−) or presence (+) of TGF-β or subsequent to Transwell apical (AP) or basolateral (BL) TGF-β addition.
(C) Confluent monolayers of EpH4 cells were transduced with either TAT-Smad3 (left) or TAT-Smad3P (right). Forty-five minutes later, cells were fixed and subcellular localization of TAT-Smad3 and TAT-Smad3P was assessed by confocal microscopy. Representative results are shown. Nuclei (blue) were stained with DAPI.
(D) Immunofluorescent detection of SMAD3 in polarized EpH4 cells by confocal microscopy following a 30min incubation with TGF-β added either to the apical (AP, left) or basolateral (BL, right) Transwell chamber.
Because biochemical assays depend on cell density in tissue culture plates are highly sensitive to any disruption of the confluent monolayer, confluent AKR-2B, EpH4, and MDCK cells in Transwell cultures were stimulated with TGF-β from either the apical or basal reservoir, then assessed for SMAD3 phosphorylation. A schematic of the Transwell culture system is provided in Figure 3B. In AKR-2B fibroblasts, TGF-β induced SMAD3 phosphorylation from either reservoir (Figure 3B, left upper image, lanes 3 and 4). On the other hand, EpH4 and MDCK cells showed domain-specific responses, with an absence of SMAD3 phosphorylation when exposed to TGF-β on their apical side while phosphorylation was high when TGF-β was added to the basolateral (BL) compartment (Figure 3B, middle and bottom left images, compare lane 3 [apical stimulation] with lane 4 [BL stimulation]). Thus, at high density in Transwell cultures dishes, nonpolarized cells like AKR-2B fibroblasts maintain intact TGF-β signaling whereas polarized cells exhibit domain-specific SMAD3 phosphorylation. Loss of apical TGF-β responsiveness in Transwell cultures is reminiscent of what is observed in confluent monolayer epithelial cultures whereby TGF-β is essentially delivered apically to the cells (see schematic diagrams in Figures 3A and 3B).
To directly address the issue of whether active SMAD complexes may be sequestered in the cytoplasm of confluent mono-layer cultures of EpH4 cells, we took advantage of TAT-fusion cell-penetrating peptide technology and generated cell-permeable TAT-SMAD3 protein. Specifically, confluent monolayers of EpH4 cells were transduced with TAT-SMAD3, subjected or not to in vitro phosphorylation by activated TβRI (see Experimental Procedures). As shown in Figure 3C, while TAT-SMAD3 was almost exclusively localized in the cytoplasm (left image, nucleo/cytoplasmic ratio of 0.3 ± 0.05), a large portion of TAT-SMAD3P proteins directly accumulated in the nucleus of cells (right image, nucleo/cytoplasmic ratio of 1.8 ± 0.1), directly establishing that SMAD3 phosphorylation is sufficient for its nuclear accumulation. In another set of experiments, polarized EpH4 cells in Transwell cultures were treated with TGF-β added apically or basolaterally. Remarkably, efficient SMAD3 nuclear accumulation in response to TGF-β was exclusively observed when the latter was delivered basolaterally, not apically (Figure 3D, nucleo/cytoplasmic ratios of 5.5 ± 0.2 versus 0.4 ± 0.1, respectively). Thus, in these two complementary approaches, there was minimal cytoplasmic retention of P-SMAD3 in response to high cell density, because SMAD3 phosphorylation was always associated with nuclear accumulation. These data indicate that loss of apical TGF-β responsiveness in confluent EpH4 cells is due to lack of SMAD3 phosphorylation, not to cytoplasmic sequestration of P-SMAD3, as initially proposed (Varelas et al., 2010).
Loss of Apical TGF-β Responsiveness in Polarized Epithelial Cells Reflects Exclusive Basolateral TGF-β Receptor Expression
The aforementioned lack of SMAD3 phosphorylation in polarized cells exposed apically to TGF-β may reflect either an absence of receptors at the apical surface, or an inability of such receptors to couple with the signaling machinery. In initial experiments to analyze the dynamics of TβR localization as a function of cell density, AKR-2B, EpH4, and MDCK cells were grown as low-and high-density monolayer cultures and transiently transfected with tagged TβRI or TβRII expression vectors. Their respective cellular localization was analyzed by confocal microscopy. As shown in Figure 4A (upper image), no defined polarity was observed for either TβRI or TβRII in low density (LD) cultures and there was no difference in their expression pattern between fibroblasts and epithelial cells. In contrast, at high cell density (HD, Figure 4A, lower image), whereas no changes in receptor localization were observed for AKR-2B fibroblasts, both receptors showed a strict basolateral expression in EpH4 and MDCK epithelial cells. The same experiments performed in Transwell culture dishes with EpH4 cells at high density refined the data, with an exclusive localization of both TβRI and TβRII on the basolateral sides of EpH4 cells when cell-cell contacts are established (Figure 4B).
Figure 4. Loss of Apical TGF-β Responsiveness in Polarized Epithelial Cells Reflects Exclusive Basolateral TGF-β Receptor Expression.
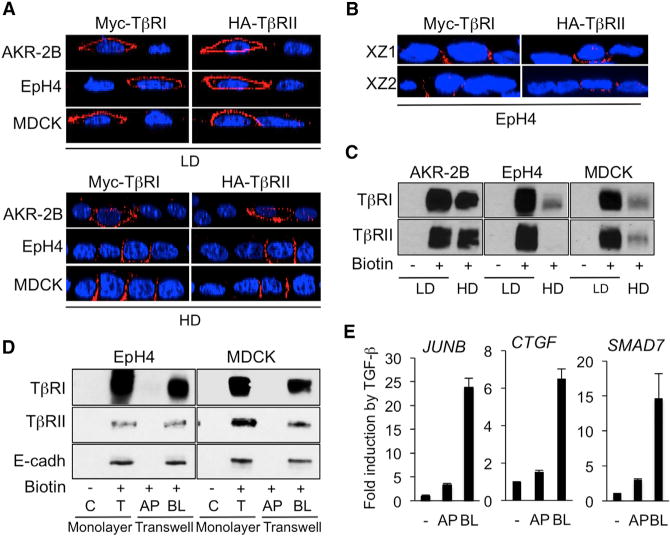
(A) IF detection of transiently transfected Myc-TβRI and HA-TβRII in low density (LD, upper) and high density (HD, lower) monolayer cultures of AKR-2B, MDCK, and EpH4 cells. Images are represented as XZ cross-sections of transfected cells.
(B) IF detection of transiently transfected Myc-TβRI and HA-TβRII in polarized Transwell cultures of EpH4 cells. Two representative XZ cross-sections of transfected cells are shown. Nuclei (blue) were stained with DAPI.
(C) Surface biotinylation assays of endogenous TβRI and TβRII in AKR-2B, EpH4, and MDCK cells grown in low- or high-density monolayer cultures.
(D) Apical (AP) and basolateral (BL) surface biotinylation assays of endogenous TβRI and TβRII in MDCK and EpH4 cells in monolayer (low-density) or Transwell cultures (high density, polarized). Cell proteins were biotinylated either from the medium (monolayer on plastic, low density) or from the apical or basolateral medium in the case of high density polarized transwell cultures. After surface biotinylation, proteins were immunoprecipitated with streptavidin-agarose and then detected by blotting with the appropriate TGF-β receptor or E cadherin antibody. Lanes C and T reflect immunoprecipitated protein in the absence of biotinylation (C) or in monolayer for total labeling (T), respectively. E-cadherin was detected to serve as a marker of basolateral cell surfaces.
(E) Quantitative reverse transcriptase–PCR analysis of JUNB, CTGF, and SMAD7 expression in polarized EpH4 cells incubated without (−) or with TGF-β (2 hr) added either in the apical (AP) or basolateral (BL) compartment of Transwells. Results are expressed as -fold induction by TGF-β and are the mean ± SD from two independent experiments, each measured in triplicate.
To validate these transient transfection findings, we next performed surface biotinylation assays and examined endogenous TβRI and TβRII in AKR-2B, EpH4, and MDCK cells grown as monolayers at low or high density. These experiments revealed a dramatic reduction in TβRI and TβRII expression levels at the surface of EpH4 and MDCK cells, not in AKR-2B fibroblasts, at high versus low density (Figure 4C), consistent with the data from Figure 3A. These results explain the dramatic drop in SMAD3 phosphorylation specifically observed in confluent EpH4 cells in response to TGF-β (Figures 1C and 3A). TβRI and TβRII levels in AKRfibroblasts remained relatively unchanged, irrespective of culture density (Figure 4C). Similar assays were next performed using EpH4 and MDCK cells grown in Transwells to promote their polarization. Polarity of the epithelial sheets formed by EpH4 and MDCK cells was verified with an inulin assay that indicated an absence of apical to basolateral flux in these cells, as opposed to what is observed in confluent fibroblasts under the same culture conditions (see Figure S4). Remarkably, unlike cells grown as low-density monolayers (T), neither EpH4 (left image) nor MDCK (right image) cells polarized in Transwell cultures expressed TβRI or TβRII on their apical (AP) surface (Figure 4D), only on their basolateral (BL) sides, in agreement with previous observations from us and others using various cell models (Murphy et al., 2004, 2007; Yakovich et al., 2010; Snodgrass et al., 2013). Consistent with SMAD3 phosphorylation and TβR localization data shown above, apically delivered TGF-β (AP) had little capacity to induce JUNB, CTGF, or SMAD7 gene expression in polarized EpH4 cells, contrasting with the solid transcriptional response that we observed when TGF-β was added to the BL compartment (Figure 4E).
As seen from the experiments above, a major discrepancy exists between the published data by Varelas et al. (2010), who observed SMAD3 phosphorylation in dense, polarized EpH4 cell monolayer cultures, without subsequent nuclear SMAD complex accumulation (Varelas et al., 2010) and ours. Specifically, our data suggest that lack of downstream signaling results from an absence of apical TGF-β receptors and subsequent SMAD3 phosphorylation in polarized EpH4 cells, not YAP/TAZ driven SMAD3 cytoplasmic sequestration. Although all these experiments were performed under conditions that closely followed those described in (Varelas et al., 2010; TGF-β stimulation 48 hr/60 hr after plating at a density of 250,000cells/well in 24-well plates), it was possible that these divergent conclusions might result from discrete differences in experimental conditions whereby their data may have been obtained when cells were at an intermediate/early stage of cell polarization whereas ours are fully polarized. To address that issue, kinetic experiments were performed that combined parallel examination of TβRI, TβRII, and E-cadherin surface expression, together with the capacity of TGF-β to induce both SMAD3 phosphorylation and target gene induction at various time points following cell seeding. EpH4 cells were seeded at a density equivalent to that of 250,000cells/24-well plate in monolayer cultures and TβRI, TβRII, and E-cadherin levels were determined at 8, 24, 48, and 72 hr post-plating by surface biotinylation assays. This approach thus allows a determination of the amount of receptors available for apically delivered TGF-β over the course of cell polarization. As shown in Figure 5A, surface expression of both TβRI and TβRII was maximal at the 8-hr time-point, slightly decreased 24 hr after plating, and was absent from the cell surface at both 48 and 72 hr after cell seeding (Figures 4C and 4D). Remarkably, E-cadherin in the same samples was detectable up to 48 hr, indicating that in EpH4 cells, TGF-β receptors relocalize basolaterally faster than E-cadherin does. In other words, TβRs disappear from the apical cell surface before cells achieve full polarity. Phosphorylation of SMAD3 in response to TGF-β and induction of target gene expression (JUNB, CTGF, and SMAD7) followed the same pattern observed for TβR surface expression: both were maximal at both 8 and 24 hr after cell seeding, and largely attenuated at the later time points (Figures 5B and 5C, respectively). This is consistent with the data presented in Figures 3 and 4 obtained 48 hr after cell plating. Thus, these kinetic experiments did not identify a time-point following TGF-β receptor binding whereby SMAD3 phosphorylation would occur while downstream events would be blunted in response to cell density driven Hippo activation, as reported by Varelas et al. (2010). Rather, they support our experiments described above that TGF-β-induced SMAD3 phosphorylation is followed by nuclear accumulation of activated SMAD complexes and subsequent target gene activation. The absence of apical TGF-β signaling in polarized cells, as is observed at 48 hr after cell plating, reflects the basolateral relocalization of TGF-β receptors that takes place during the establishment of polarity.
Figure 5. TβR Basolateral Relocalization during EpH4 Cell Polarization: Implication for TGF-β Signaling.
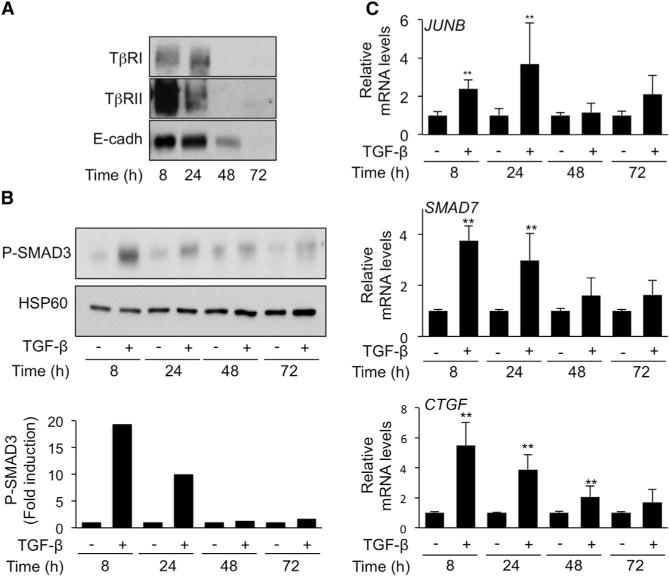
EpH4 mouse mammary epithelial cells were grown in high-density conditions (see Experimental Procedures).
(A) Surface biotinylation assays of endogenous TβRI, TβRII, and E-cadherin performed at various time points following cell seeding. One of two experiments with sub-identical results is shown.
(B) Western analysis of P-SMAD3 levels following a 2-hr TGF-β stimulation at various time points following cell seeding. Upper panel: representative blot from one of five experiments that gave similar results. Lower: Densitometric analysis of P-SMAD3 levels. Results are expressed as -fold induction by TGF-β at each time-point.
(C) Quantitative reverse transcriptase–PCR analysis of JUNB, CTGF, and SMAD7 expression in EpH4 cells incubated without (−) or 2 hr with TGF-β at various time points following cell seeding. Results, expressed as -fold induction by TGF-β at each time point are the mean ± SD from five independent experiments, each measured in triplicate.
Disruption of Cell-Cell Contacts in Polarized EpH4 Cells by Ca2+ Depletion Restores SMAD3 Phosphorylation and Downstream Target Gene Activation in Response to Apically Delivered TGF-β
Polarized epithelial cells establish cell-cell contacts that include Ca2+-dependent tight and adherens junctions. These junctions prevent intercellular diffusion of large molecules and thus, are likely to prevent apically delivered TGF-β from binding its basolateral receptors. To test this hypothesis and further confirm the dissociation of density per se, and SMAD3 signaling, we assessed whether disruption of these junctions may be sufficient to restore SMAD3 phosphorylation upon apical TGF-β delivery. As shown in Figure 6A, Ca2+ depletion had no effect on TGF-β-induced SMAD3 phosphorylation in sparse EpH4 cultures (LD). On the other hand, disruption of cell-cell contacts from confluent EpH4 cells (HD) by Ca2+ depletion restored SMAD3 phosphorylation in response to “apically delivered” TGF-β. These data establish that cell-cell contacts in polarized epithelial cells control the access of apically available TGF-β ligands to basolateral TβRs. We next tested whether Ca2+ depletion could also restore efficient TGF-β-induced target gene expression in confluent EpH4 cells. As shown in Figure 6B, consistent with the data from Figure 1, confluent EpH4 cells in Ca2+-containing culture conditions exhibited very weak activation of target genes (e.g., 2- to 3-fold). A 30-min Ca2+ depletion, however, was sufficient to allow solid transcriptional responses, as evidenced by induction of JUNB, CTGF, and SMAD7 10- to 25-fold above control values. Given the basolateral localization of TβRs in confluent EpH4 cells (see Figures 4A–4D), these data suggest that disruption of Ca2+-dependent cell-cell contacts allows paracellular diffusion of apically delivered TGF-β to activate signaling.
Figure 6. Effect of Ca2+ Depletion on TGF-β Response in High-Density Monolayer Cultures of EpH4 Cells.
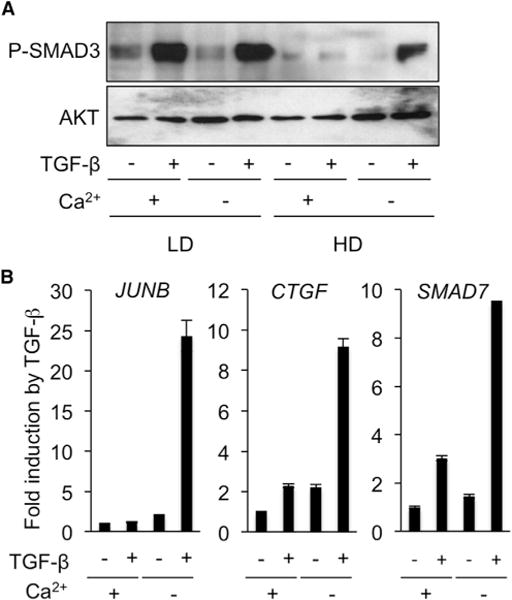
(A) Western analysis of P-SMAD3 levels in EpH4 cells treated without (−) or with (+) TGF-β for 30 minat low (LD) or high (HD) density in monolayer cultures in the presence (+) or absence (−) of Ca2+.
(B) Quantitative reverse transcriptase-PCR analysis of JUNB, CTGF, and SMAD7 expression in confluent monolayers of EpH4 cells incubated without (−) or with TGF-β (2 hr) in medium with (+) or depleted of (−) Ca2+. Results are expressed as -fold induction by TGF-β and are the mean ± SD from two independent experiments, each measured in triplicate.
DISCUSSION
In summary, we have confirmed previous observations that Hippo pathway activation occurs as a result of cell density sensing (Zhao et al., 2007), and demonstrated that it occurs in a wide variety of cell types, irrespective of their lineage. Second, we have demonstrated that most cell types exhibit functional TGF-β signaling, whether they are at low or high cell density, consistent with a large body of literature. Our data also document that SMAD nuclear accumulation in response to TGF-β is independent from YAP/TAZ levels. Most importantly, we demonstrate that density-driven loss of TGF-β responsiveness is exclusively observed in polarized epithelial cells and reflects the establishment of Ca2+-dependent cell-cell contacts that prevent ligand binding to, and activation of, basolaterally expressed TGF-β receptors. This, in turn, prevents SMAD phosphorylation, subsequent nuclear accumulation, and gene responses. A model depicting these findings is shown in Figure 7.
Figure 7. Schematic Summary of TAZ and SMAD3 Nucleo-Cytoplasmic Shuttling in Response to Cell Density or TGF-β Stimulation in Non-Polarized or Polarized Cells at Low or High Density.
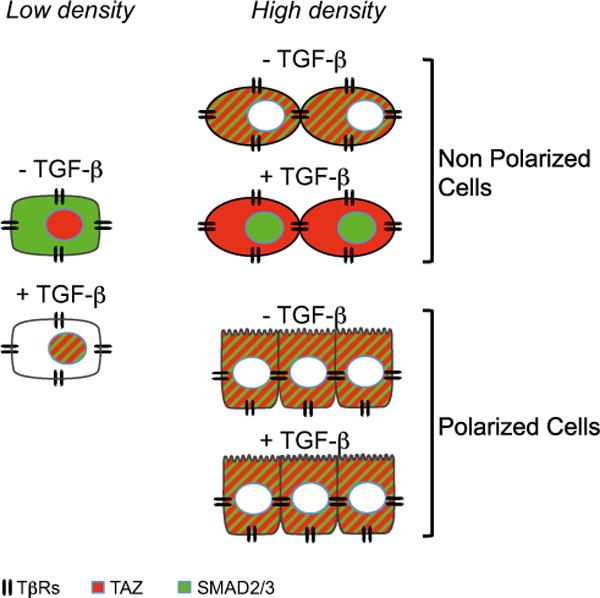
At low density (LD), TAZ (red) is nuclear and SMAD3 (green) is cytoplasmic. TGF-β receptors are distributed throughout the cell surface. In response to TGF-β, SMAD3 accumulates in the nucleus where TAZ is located. At high density (HD), TAZ is excluded from the nucleus, as a result of cell density sensing. In non-polarized cells, TGF-β induces SMAD3 nuclear accumulation, while TAZ remains cytoplasmic. In polarized cells (schematically represented with apical villi), apical delivery of TGF-β is without effect on SMAD localization because TβRs are exclusively basolateral and TGF-β access to its receptors is blocked by intercellular junctions.
These data are at odds with some of those from a previous report that concluded that activated SMAD complexes are sequestered in the cytoplasm by P-TAZ (Varelas et al., 2010). In the latter report, the authors gave priority to their finding of a physical interaction between TAZ and SMAD3. Several conclusions were then drawn. For example, Ca2+ depletion was found to both drive TAZ nuclear accumulation and restore TGF-β signaling. It is well established that Ca2+ is required for the integrity of epithelial cell contacts, which themselves directly control TAZ nucleo-cytoplasmic localization (Chen et al., 2010; Robinson et al., 2010; Zhao et al., 2007). Thus, while we agree with both observations, we believe that the occurrence of the two phenomena is temporally parallel but mechanistically independent. Given the relatively similar levels of expression of YAP/TAZ (and SMADs) in all cell types, if P-YAP/TAZ were to retain P-SMADs in the cytoplasm, this phenomenon would be rather widespread across various cell types, irrespective of polarization, because cell-cell contact-driven YAP/TAZ nuclear exclusion seems to be a general phenomenon not restricted to polarized cells. This is not the case, as attested by the plethora of data published using confluent cells to study TGF-β responses (see examples and corresponding references in the Introduction) and by our demonstration that (a) cell-permeable TAT-PSMAD3 accumulates in the nucleus of confluent transduced EpH4 cells while TAT-SMAD3 does not (Figure 3C), and (b) basolateral stimulation of polarized cells with TGF-β not only leads to phosphorylation, but also to rapid nuclear accumulation of SMAD3 (Figures 3B and 3D), indicating that phosphorylation of SMAD3 is sufficient to initiate the process of its nuclear translocation.
Physical interactions between YAP/TAZ and SMADs have been studied by various groups. While we originally identified a direct YAP/SMAD7 interaction (Ferrigno et al., 2002), a few research groups have focused their attention on the interactions between YAP/TAZ and receptor-associated SMADs. Massagué’s group showed in immunoprecipitation experiments of over-expressed proteins that YAP interacts strongly with SMAD1, which allows for an enhancement of BMP-dependent transcription, while weakly interacting with SMAD3 (Alarcón et al., 2009). This was confirmed by another study with tagged proteins that showed weak affinity of YAP for SMAD3 as compared to SMAD1, yet identified a large transcriptional complex comprising YAP, SMAD3, TEAD, and p300 involved in the upregulation of a subset of TGF-β target genes including CTGF (Fujii et al., 2012). Likewise, in human embryonic stem cells, TGF-β was recently found to recruit SMAD2/3 to TEAD/YAP/TAZ complexes in association with OCT4, contributing to the regulation of pluripotency genes (Beyer et al., 2013). The latter have been shown independently to be targets for the p300/CBP-interacting protein (p/CIP), which is required for transcriptional activity of various CBP/p300-dependent transcription factors (Chitilian et al., 2014). These data are consistent with the demonstration that external signaling and the core transcriptional machinery form tightly integrated regulatory networks in embryonic stem cells (Chen et al., 2008). Altogether, it would appear that cooperation between YAP/TAZ and SMAD3 to regulate a subset of TGF-β target genes primarily reflects a nuclear action linked to their shared capacity to bind CBP/p300. Similarly, a recent paper by Varelas’ group reported that nuclear YAP/TAZ and SMAD2/3/4 drive invasion and metastasis via shared p300-dependent mechanisms (Hiemer et al., 2014). On the other hand, they concluded that when the Hippo pathway is active (i.e., YAP/TAZ are cytoplasmic), TGF-β-driven tumor suppression and cytostasis occurs via SMAD2/3/4-driven transcription independent of YAP/TAZ, consistent with our data presented herein.
A critical difference between the works from Wrana’s group (Varelas et al., 2008, 2010) and ours resides in the determination of early events regulating TGF-β signaling in relation to cell density. We identify a defect in SMAD3 phosphorylation due to TGF-β receptor localization to basolateral membranes, exclusively in dense polarizable cell lines such as EpH4 or MDCK cells, which then lack downstream TGF-β signaling and gene responses. Surface biotinylation experiments aimed at defining the kinetics of receptor basolateral relocalization during establishment of polarity showed that TβRs disappear from the apical surface of EpH4 cells more rapidly than E-cadherin, suggesting that they establish basolateral domain localization before cells have fully established adherens junctions, (i.e., are not fully polarized). In these experiments, target gene transcription in response to apical TGF-β occurred at all time points when SMAD3 was phosphorylated (in other words, when TβRs were still available on the apical surface). We were unable to define a time in the polarization process whereby high density, per se, could have resulted in the cytoplasmic sequestration of P-SMAD complexes. While our findings are fully consistent with previously published observations (Murphy et al., 2004, 2007) of basolateral positioning of TβRs in polarized epithelial cells, we also provide evidence for the restriction of this process to polarized cells because fibroblasts do not undergo any domain-specific reorganization of cell surface receptors in relation to cell density. This may be particularly important as in some models polarized epithelial cells secrete TGF-β ligands from their apical side (Murphy et al., 2004) and luminal TGF-β is commonly observed. As such, it is highly plausible that expression of TGF-β receptors restricted to the basolateral compartment may allow Ca2+-dependent cellular junctions, tight and/or adherens, to provide polarized epithelia with a resistance mechanism against TGF-β-induced EMT by preventing TGF-β binding to its receptors. This, in turn, would maintain epithelial integrity and organ function. Loss of polarity, as seen during early carcinogenesis may accelerate TGF-β-induced EMT, leading to cell dissociation and metastatic dissemination. Further experiments will be needed to verify these hypotheses.
To conclude, we have uncovered cell-type specific regulation of TGF-β responses by cell density, whereby epithelial cells with basoapical polarity exhibit strict and exquisite density-dependent redistribution of TβRs to their basolateral sides, an event that precludes TGF-β responsiveness from their apical compartment. This phenomenon appears to be specific for polarized epithelial cells, as fibroblasts grown to confluency did not undergo any detectable repositioning of TβRs on their cell surface. We also provide evidence that TAZ nuclear exclusion induced by cell-cell contacts is ubiquitous and that mechanisms driving nucleo-cytoplasmic localization of TAZ and P-SMAD are independent, eventually leading to situations whereby both proteins are fortuitously localized in the same cellular compartment, without obvious functional consequences for TGF-β responses.
EXPERIMENTAL PROCEDURES
Cell Culture
Human lung Wi26 and mouse embryonic AKR-2B fibroblasts, human immortalized HaCaT keratinocytes, human MDA-MB-231 and mouse EpH4 breast carcinoma cell lines, canine MDCK kidney epithelial cells, as well as human PANC-1 pancreatic carcinoma cell lines were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). Human 1205Lu, WM852, and SKmel28 melanoma cell lines (Alexaki et al., 2008, 2010; Javelaud et al., 2007, 2011a; Rodeck et al., 1999) were grown in RPMI 1640 (Invitrogen) supplemented with 10% FBS. For experiments on monolayer cultures, cells were plated at low or high density, 25,000 cells or 250,000 cells in 24-well dishes or proportionally in accordance to dish surface. Twenty-four hours later, medium was replaced to reduce FBS concentration to 1%. TGF-β treatment (30 min to 2 hr, depending on endpoint) was performed either the next day or4hr later in the case of kinetic experiments. For Transwell cultures (0.4-μm Costar polycarbonate membranes), cells were plated at a density of 5 × 104 cells/ml in 0.5 ml of culture medium and grown for 3 days prior to experiments. Fully polarized monolayers of MDCK and EpH4 cells were obtained after 3 days, as verified by serial measurements of trans-epithelial resistance with a Millipore EMR2 device, consistent with our previous work (Murphy et al., 2004, 2007; Yin et al., 2013).
Cell-Permeable TAT-Smad3 and Tat-Smad3P Protein Transduction and Detection
TAT-Smad3 or TAT-Smad3P proteins (see Supplemental Experimental Procedures) containing an amino terminal HA epitope tag were added to high density monolayer cultures of EpH4 cells at 37°C for 45 min. Cells were then fixed in 4% paraformaldehyde, permeabilized with 0.1%Triton X-100, and blocked by goat serum. TAT-Smad3 and TAT-Smad3P were detected with an anti-HA primary antibody (Cell Signaling) and Cy3-conjugated secondary antibody (Jackson ImmunoResearch Laboratories).
RNA Extraction and Gene Expression Analysis by qRT-PCR
RNA extraction and quantitative RT-PCR using the SYBR Green technology have been described previously (Javelaud et al., 2011b; Mohammad et al., 2011). For experiments comparing TGF-β in low- (LD) versus high-density (HD) cultures, cells were seeded at a ratio of 1 to 10 between LD and HD, with cell numbers in HD conditions determined in prior experiments to be sufficient for cell confluency immediately after cell attachment. TGF-β treatment was done 24 hr after cell seeding and luciferase activity was measured 16 hr later. Experiments were performed in triplicate to validate gene expression data in each cell line. Primers used are listed in the Supplemental Experimental Procedures.
Cell Transfections
For reporter assays, cells in monolayer cultures were transfected with the polycationic compound FuGENE (Roche Diagnostics) at 70% to 80% confluency in fresh medium with 200 ng of the SMAD3/4-specific firefly luciferase reporter construct (CAGA)9-MLP-luc (Dennler et al., 1998) and 50 ng of Renilla luciferase expression vector to estimate transfection efficiencies. For experiments comparing TGF-β in LD versus HD cultures, transfections were performed in 10 cm diameter dishes. Cells were split the next day and seeded at a ratio of 1 to 10 between LD and HD. TGF-β treatment was done 24 hr after cell seeding and luciferase was measured 16 hr later. Transient transfection of fully polarized EpH4 cell monolayers in 12-mm Transwells was performed on day 3 after plating, using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). Details of the procedure and description of the Myc-tagged TbRI expression vector used for confocal microscopy experiments may be found in (Murphy et al., 2004). HA-tagged TβRII expression vector was a gift from Dr. Jeffrey Wrana (University of Toronto, Ontario). Luciferase activities were determined with a Dual-Glo luciferase assay kit (Promega) using a Fluoroskan Ascent FL (Thermo Labsystems). All experiments were performed at least three times.
For transient gene silencing, HaCaT cells were transfected with two distinct siRNAs specifically targeting YAP or TAZ (Sigma-Aldrich human YAP or TAZ Mission siRNAs SASI_Hs01_00182403 and SASI_Hs01_00124479, respectively). The siRNA sequences were different from those from the shRNA lentiviral vectors. Sequence details may be found in (Nallet-Staub et al., 2014). A non-targeting siRNA (Sigma-Aldrich Mission Universal Negative control siRNA #2) was used as control. For experiments requiring RNA or protein extraction, cells were seeded at 2 × 105 cells/well in six-well plates and transfected 24 hr later in fresh medium containing 1% fetal calf serum (FCS) with siRNA (150 ng/well) using 12 ml of HiPerfect reagent. Sample processing occurred 48 hr later. Stable TAZ knockdown in melanoma cell lines has been described previously (Nallet-Staub et al., 2014).
Biotinylation of Cell Surface Receptors
Briefly, freshly made sulfo-NHS-SS biotin/HBSS (1 mg/ml; Thermo Scientific) was added to either a monolayer culture in a six-well plate (1 ml) to assess total (T) labeling, or domain-specific receptor expression was determined by sulfo-NHS-SS biotin/HBSS addition to either the apical (0.5 ml) or basolateral (1 ml) reservoirs of Transwell dishes. Biotin-bound proteins were pulled down with Streptavidin-agarose and resolved on 10% SDS-PAGE. TβRI and TβRII levels were examined with western blotting. Details may be found in (Yin et al., 2013).
Western Blotting
Whole cell extracts and procedures for western analyses have been described previously (Alexaki et al., 2010; Dennler et al., 2009; Yin et al., 2013). For experiments comparing SMAD3 phosphorylation in response to TGF-β in LD versus HD cultures, cells were seeded at a ratio of 1 to 10 between LD and HD, with cell numbers in HD conditions determined in prior experiments to be sufficient for cell confluency immediately after cell attachment. TGF-β treatment was done 24 hr after cell seeding and proteins extracted 30min later. Antibody descriptions, references, and dilutions are provided in the Supplemental Experimental Procedures.
Immunofluorescence
Cells grown on poly-lysine-coated coverslips were fixed with 4% paraformaldehyde for 10 min and permeabilized with 1 × PBS, 0.1% Triton X-100, 5% FCS for 1 hr at room temperature then blocked with goat serum. Cells were then incubated overnight at 4°C with primary antibodies raised against SMAD2/3 or TAZ (see Supplemental Experimental Procedures for details). Nuclei were stained with Hoechst 37342 (1:1,000, Sigma-Aldrich). After washing with PBS, appropriate Alexa Fluor (Life Technologies) anti-goat (488), anti-rabbit (555), or anti-mouse (467) fluorescent secondary antibodies (1:1,000) were used for immunofluorescent visualization using either an inverted fluorescent microscope (Leica DM-IRB, Leica Microsystems) equipped with a digital video camera (Princeton Instruments), or with a 100× objective on a Zeiss LSM 510 confocal system. Image capture was performed using MetaMorph software version 7.7 (Molecular Devices). Fluorescence signals were analyzed and images merged using ImageJ software tools.
Statistics
All quantitative data are expressed as mean ± SD. The p values for experiments were obtained with the Mann-Whitney U-test. Differences between groups were considered significant for p ≤ 0.05. **p < 0.01; ***p < 0.001.
Supplementary Material
Highlights.
Hippo pathway activation upon cell density sensing does not inhibit TGF-b signaling
Cell density interferes with TGF-b signaling solely in polarized epithelial cells
Apical loss of TGF-β responsiveness reflects basolateral TGF-β receptor expression
Cytoplasmic YAP/TAZ does not interfere with TGF-β signaling
Acknowledgments
This paper is dedicated to the memory of Anita B. Roberts, great mentor and friend. Supported by the Donation Henriette et Emile Goutière, Institut National du Cancer (INCa, PLBIO12-020, and MELA13-002), INSERM, CNRS, Ligue Nationale Contre le Cancer (Comitéde la Manche, Equipe Labellisée LIGUE EL-2011AM), Université Paris XI (to A.M.), France-Berkeley Fund (to D.J.), and Public Health Service Grants GM-54200 and GM-55816 from the National Institute of General Medical Sciences and the Mayo Foundation (to E.B.L.). F. N.-S. was the recipient of a Ligue Nationale Contre le Cancer doctoral studentship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2015.01.011.
AUTHOR CONTRIBUTIONS
A.M. and E.B.L. designed and planned the project, supervised data acquisition, and wrote the manuscript. F.N.S., X.Y., C.G., V.M., S.B.M., and D.J. carried out the experimental work. All authors discussed the results and commented on and approved the manuscript.
References
- Alarcón C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexaki VI, Javelaud D, Mauviel A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment Cell Melanoma Res. 2008;21:429–438. doi: 10.1111/j.1755-148X.2008.00466.x. [DOI] [PubMed] [Google Scholar]
- Alexaki VI, Javelaud D, Van Kempen LC, Mohammad KS, Dennler S, Luciani F, Hoek KS, Juàrez P, Goydos JS, Fournier PJ, et al. GLI2-mediated melanoma invasion and metastasis. J Natl Cancer Inst. 2010;102:1148–1159. doi: 10.1093/jnci/djq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, Wrana JL. Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013;5:1611–1624. doi: 10.1016/j.celrep.2013.11.021. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, Halder G. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci USA. 2010;107:15810–15815. doi: 10.1073/pnas.1004060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitilian JM, Thillainadesan G, Manias JL, Chang WY, Walker E, Isovic M, Stanford WL, Torchia J. Critical components of the pluripotency network are targets for the p300/CBP interacting protein (p/CIP) in embryonic stem cells. Stem Cells. 2014;32:204–215. doi: 10.1002/stem.1564. [DOI] [PubMed] [Google Scholar]
- Chung KY, Agarwal A, Uitto J, Mauviel A. An AP–1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem. 1996;271:3272–3278. doi: 10.1074/jbc.271.6.3272. [DOI] [PubMed] [Google Scholar]
- Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, André J, Alexaki I, Li A, Magnaldo T, ten Dijke P, Wang XJ, Verrecchia F, Mauviel A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6986. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- Dennler S, André J, Verrecchia F, Mauviel A. Cloning of the human GLI2 Promoter: transcriptional activation by transforming growth factorbeta via SMAD3/beta-catenin cooperation. J Biol Chem. 2009;284:31523–31531. doi: 10.1074/jbc.M109.059964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Ellenrieder V, Hendler SF, Ruhland C, Boeck W, Adler G, Gress TM. TGF-beta-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int J Cancer. 2001;93:204–211. doi: 10.1002/ijc.1330. [DOI] [PubMed] [Google Scholar]
- Ferrigno O, Lallemand F, Verrecchia F, L’Hoste S, Camonis J, Atfi A, Mauviel A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-beta/Smad signaling. Oncogene. 2002;21:4879–4884. doi: 10.1038/sj.onc.1205623. [DOI] [PubMed] [Google Scholar]
- Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, Ito H, Murakami H, Kondo Y, Kondo E, et al. TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J Exp Med. 2012;209:479–494. doi: 10.1084/jem.20111653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis RR, Alarcón C, He W, Wang Q, Seoane J, Lash A, Massagué J. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA. 2006;103:12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heino J, Massagué J. Transforming growth factor-beta switches the pattern of integrins expressed in MG-63 human osteosarcoma cells and causes a selective loss of cell adhesion to laminin. J Biol Chem. 1989;264:21806–21811. [PubMed] [Google Scholar]
- Hiemer SE, Szymaniak AD, Varelas X. The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem. 2014;289:13461–13474. doi: 10.1074/jbc.M113.529115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignotz RA, Heino J, Massagué J. Regulation of cell adhesion receptors by transforming growth factor-beta. Regulation of vitronectin receptor and LFA-1. J Biol Chem. 1989;264:389–392. [PubMed] [Google Scholar]
- Javelaud D, Mohammad KS, McKenna CR, Fournier P, Luciani F, Niewolna M, André J, Delmas V, Larue L, Guise TA, Mauviel A. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67:2317–2324. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Alexaki VI, Pierrat MJ, Hoek KS, Dennler S, Van Kempen L, Bertolotto C, Ballotti R, Saule S, Delmas V, Mauviel A. GLI2 and M-MITF transcription factors control exclusive gene expression programs and inversely regulate invasion in human melanoma cells. Pigment Cell Melanoma Res. 2011a;24:932–943. doi: 10.1111/j.1755-148X.2011.00893.x. [DOI] [PubMed] [Google Scholar]
- Javelaud D, van Kempen L, Alexaki VI, LeScolan E, Luo K, Mauviel A. Efficient TGF-β/SMAD signaling in human melanoma cells associated with high c-SKI/SnoN expression. Mol Cancer. 2011b;10:2. doi: 10.1186/1476-4598-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kon A, Vindevoghel L, Kouba DJ, Fujimura Y, Uitto J, Mauviel A. Cooperation between SMAD and NF-kappaB in growth factor regulated type VII collagen gene expression. Oncogene. 1999;18:1837–1844. doi: 10.1038/sj.onc.1202495. [DOI] [PubMed] [Google Scholar]
- Korang K, Christiano AM, Uitto J, Mauviel A. Differential cytokine modulation of the genes LAMA3, LAMB3, and LAMC2, encoding the constitutive polypeptides, alpha 3, beta 3, and gamma 2, of human laminin 5 in epidermal keratinocytes. FEBS Lett. 1995;368:556–558. doi: 10.1016/0014-5793(95)00740-z. [DOI] [PubMed] [Google Scholar]
- Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor beta (TGF-beta) target genes and distinguishes TGF-beta-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauviel A, Qiu Chen Y, Dong W, Evans CH, Uitto J. Transcriptional interactions of transforming growth-factor-beta with pro-inflammatory cytokines. Curr Biol. 1993;3:822–831. doi: 10.1016/0960-9822(93)90216-b. [DOI] [PubMed] [Google Scholar]
- Mauviel A, Lapière JC, Halcin C, Evans CH, Uitto J. Differential cytokine regulation of type I and type VII collagen gene expression in cultured human dermal fibroblasts. J Biol Chem. 1994;269:25–28. [PubMed] [Google Scholar]
- Mauviel A, Chung KY, Agarwal A, Tamai K, Uitto J. Cell-specific induction of distinct oncogenes of the Jun family is responsible for differential regulation of collagenase gene expression by transforming growth factor-beta in fibroblasts and keratinocytes. J Biol Chem. 1996;271:10917–10923. doi: 10.1074/jbc.271.18.10917. [DOI] [PubMed] [Google Scholar]
- Mauviel A, Nallet-Staub F, Varelas X. Integrating developmental signals: a Hippo in the (path)way. Oncogene. 2012;31:1743–1756. doi: 10.1038/onc.2011.363. [DOI] [PubMed] [Google Scholar]
- Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, Duong V, Dunn LK, Mauviel A, Guise TA. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011;71:175–184. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SJ, Doré JJ, Edens M, Coffey RJ, Barnard JA, Mitchell H, Wilkes M, Leof EB. Differential trafficking of transforming growth factor-beta receptors and ligand in polarized epithelial cells. Mol Biol Cell. 2004;15:2853–2862. doi: 10.1091/mbc.E04-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SJ, Shapira KE, Henis YI, Leof EB. A unique element in the cytoplasmic tail of the type II transforming growth factor-beta receptor controls basolateral delivery. Mol Biol Cell. 2007;18:3788–3799. doi: 10.1091/mbc.E06-10-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallet-Staub F, Marsaud V, Li L, Gilbert C, Dodier S, Bataille V, Sudol M, Herlyn M, Mauviel A. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol. 2014;134:123–132. doi: 10.1038/jid.2013.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolás FJ, Hill CS. Attenuation of the TGF-beta-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-beta-induced growth arrest. Oncogene. 2003;22:3698–3711. doi: 10.1038/sj.onc.1206420. [DOI] [PubMed] [Google Scholar]
- Oh H, Irvine KD. Cooperative regulation of growth by Yorkie and Mad through bantam. Dev Cell. 2011;20:109–122. doi: 10.1016/j.devcel.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, Wrana JL, Sodek J. Independent regulation of collagenase, 72-kDa progelatinase, and metalloendoproteinase inhibitor expression in human fibroblasts by transforming growth factor-beta. J Biol Chem. 1989;264:1860–1869. [PubMed] [Google Scholar]
- Redini F, Daireaux M, Mauviel A, Galera P, Loyau G, Pujol JP. Characterization of proteoglycans synthesized by rabbit articular chondrocytes in response to transforming growth factor-beta (TGF-beta) Biochim Biophys Acta. 1991;1093:196–206. doi: 10.1016/0167-4889(91)90123-f. [DOI] [PubMed] [Google Scholar]
- Reynisdóttir I, Polyak K, Iavarone A, Massagué J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- Robinson BS, Huang J, Hong Y, Moberg KH. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol. 2010;20:582–590. doi: 10.1016/j.cub.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeck U, Nishiyama T, Mauviel A. Independent regulation of growth and SMAD-mediated transcription by transforming growth factor beta in human melanoma cells. Cancer Res. 1999;59:547–550. [PubMed] [Google Scholar]
- Snodgrass SM, Cihil KM, Cornuet PK, Myerburg MM, Swiatecka-Urban A. Tgf-beta1 inhibits cftr biogenesis and prevents functional rescue of ∆f508-cftr in primary differentiated human bronchial epithelial cells. PLoS One. 2013;8:e63167. doi: 10.1371/journal.pone.0063167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, Wrana JL. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev Cell. 2010;19:831–844. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Maeno M, Hawrylyshyn B, Yao KL, Domenicucci C, Sodek J. Differential effects of transforming growth factor-beta on the synthesis of extracellular matrix proteins by normal fetal rat calvarial bone cell populations. J Cell Biol. 1988;106:915–924. doi: 10.1083/jcb.106.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Overall CM, Sodek J. Regulation of the expression of a secreted acidic protein rich in cysteine (SPARC) in human fibroblasts by transforming growth factor beta. Comparison of transcriptional and post-transcriptional control with fibronectin and type I collagen Eur J Biochem. 1991;197:519–528. doi: 10.1111/j.1432-1033.1991.tb15940.x. [DOI] [PubMed] [Google Scholar]
- Yakovich AJ, Huang Q, Du J, Jiang B, Barnard JA. Vectorial TGFbeta signaling in polarized intestinal epithelial cells. J Cell Physiol. 2010;224:398–404. doi: 10.1002/jcp.22135. [DOI] [PubMed] [Google Scholar]
- Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretionbybreast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Murphy SJ, Wilkes MC, Ji Y, Leof EB. Retromer maintains basolateral distribution of the type II TGF-β receptor via the recycling endosome. Mol Biol Cell. 2013;24:2285–2298. doi: 10.1091/mbc.E13-02-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.