

Abstract
Zhou et al. validate the expression of markers of the innate immune system in the cpk mouse model of polycystic kidney disease (PKD), in human recessive PKD, and in human autosomal dominant PKD and show that CD14 expression correlates with PKD progression, even from very early stages of disease. Moreover, they show that CD14 is expressed from the renal tubule epithelial cells, suggesting a mechanism of Toll-like receptor-4 activation in PKD prior to the infiltration of inflammatory cells.
Polycystic kidney disease (PKD) is characterized by a disruption in tubular epithelial cells that leads to inappropriate cell proliferation, increased fluid secretion, abnormal differentiation, and altered extracellular matrix that all contribute to the development of numerous renal cysts in the kidney. Countless studies have focused on the role of cell proliferation, fluid secretion, apoptosis, and extracellular matrix in the progression and pathogenesis of this devastating disease.1 These studies have led to a better understanding of the progression of the disease, and, in some cases, to potential therapeutic approaches for treating PKD.2 However, patient studies have demonstrated a remarkable heterogeneity in the onset and progression of the disease, suggesting that disease progression is influenced by other environmental and genetic factors. Recent studies have identified the innate immune system as one such biological process that influences the progression of PKD. Mrug et al.3 showed that severely affected cpk mice, compared with mildly affcted cpk mice, expressed higher levels of markers for alternatively activated macrophages, indicative of the innate immune response and effectors of fibrosis that are associated with PKD.
Autosomal dominant PKD (ADPKD) is characterized by the development of numerous renal cysts that slowly progress over the lifetime of a patient, resulting in end-stage renal disease later in life. Traditionally, the measure of glomerular filtration rate (GFR) has been used to evaluate renal function. By GFR measurements, the disease is largely asymptomatic until the third or fourth decade. Soon after the onset of symptoms, there is a rapid decline in GFR, leading to end-stage renal disease. Recently, a number of imaging studies, including the multi-institutional CRISP (Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease) study, have provided a mechanism to accurately quantify cystic disease progression.4,5 These studies make it increasingly clear that GFR is actually a poor predictor of PKD progression. While the loss of nephrons in PKD occurs in a linear progression, renal function does not decline linearly. Rather, renal function, as measured by GFR, is maintained at a fairly normal level, until it reaches a threshold at which the small number of remaining nephrons can no longer maintain a normal function. After reaching this threshold, the GFR declines precipitously, leading to end-stage renal disease. For the patient, this is viewed as a sudden acceleration of the disease, when in reality the disease has been progressing all along. Moreover, by the time the GFR declines, ADPKD has progressed to such an extent that the only treatment is transplantation. Any hope, therefore, for the treatment of PKD must come long before the disease shows symptoms. Since cysts are now known to begin during early development, perhaps even in utero, it is imperative to be able to measure cyst progression long before symptoms appear.4 The onset of symptoms means that the disease has progressed to the point that little normal renal parenchyma is left. Any treatments at this stage will likely be ineffective.
In a follow-up to Mrug et al.,3 Zhou et al.6 (this issue) validated that a well-known marker of alternatively activated macrophages, CD14, is highly upregulated in the kidneys of severely affected cpk mice. CD14 is a pattern recognition receptor that facilitates the binding of lipopolysaccharides (LPSs) to the Toll-like receptors (TLRs) to activate the innate immune system. CD14 is expressed at high levels on the cell surface of monocytes and macrophages, where it is membrane bound by glycosyl phosphatidylinositol linkage, and at lower levels in nonmyeloid cells, such as liver and kidney.7 After activation, the membrane-bound CD14 can be proteolytically shed. CD14 is also found in a soluble form that confers LPS sensitivity to cells that lack membrane-bound CD14. An increase of inflammatory cells is characteristic of late stages of PKD and is involved in the increased fibrosis observed in PKD. Zhou et al.6 found that rather than being expressed by infiltrating macrophages, the source of the elevated CD14 was the cystic and non-cystic renal tubular epithelial cells ( Figure 1 ). Moreover, cpk mice showed very high levels of the proteolytically shed variant of CD14. The elevated soluble CD14 protein was also observed by immuno-precipitation of urine from recessive PKD patients. Finally, immunoprecipitated urine from a small number of ADPKD patients showed the presence of soluble forms of CD14, which were also elevated in cyst fluid. The search for biomarkers for PKD has been ongoing. Urinary excretion of monocyte chemoattractant protein-1 (MCP-1) has been shown both in animal models of PKD and in ADPKD patients.8 However, the expression of MCP-1 is not correlative with disease progression until late stages of cystogenesis in the cpk mice. Strikingly, CD14 upregulation was significant in cpk mice as early as 5 postnatal days and increased as the disease progressed. The correlation of CD14 levels in the urine of ADPKD patients with PKD progression as measured by changes in total kidney volume over a 2-year span suggests that measurement of CD14 in the urine could be a biomarker for the early progression of ADPKD, making clinical trials feasible.
Figure 1. A potential mechanism of TLR4 activation in PKD.
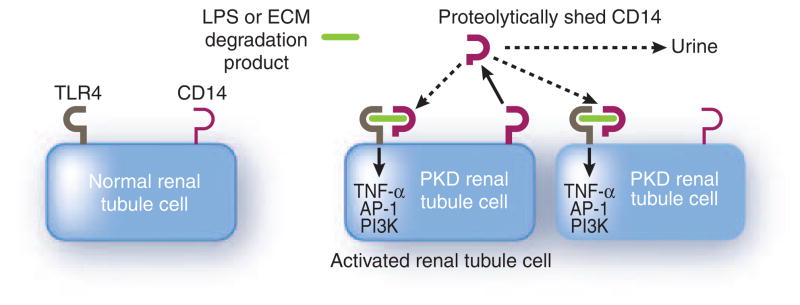
Toll-like receptor-4 (TLR4) is present on renal epithelial cells, including cells of the proximal tubule and collecting ducts. CD14 is expressed at high levels in macrophages and monocytes, and low levels in nonmyeloid cells, including cells of the kidney and liver, where it is membrane bound through glycosyl phosphatidylinositol linkage. Normally, CD14 facilitates the binding of lipopolysaccharides (LPSs) to TLR to activate the innate immune response. However, TLR4 signaling can also be activated by interaction with extracellular matrix (ECM) degradation products. In PKD, there is an upregulation of CD14 expression and increased levels of the proteolytically shed CD14 variant, indicative of immunological activation or cell injury. Thus, the renal tubule-derived CD14 could activate TLR4 signaling either locally or in more distal segments of the nephron, long before the infiltration of inflammatory cells. In addition, proteolytically shed CD14 is found in the urine of both recessive PKD and autosomal dominant PKD (ADPKD) patients, where, in the case of ADPKD, the levels correlate with PKD progression. Thus, CD14 could be a biomarker for the early progression of PKD. Abbreviations: AP-1, activator protein 1; PI3K, phosphatidylinositol-3′-kinase; TNF-α, tumor necrosis factor-α.
CD14 functions in the innate immune system to bind LPSs and, together with LPS-binding protein (LBP), to bind and activate TLR signaling. One of the TLR proteins, TLR4, is expressed throughout the nephron, including the proximal tubule and collecting duct. 7 Thus, increased CD14 protein in PKD, shed from renal tubular epithelial cells, could interact with TLR4 in the proximal tubule or collecting duct to activate signaling. Interestingly, TLR4 is a candidate modifier gene of cystic kidney disease. As Zhou et al.6 very nicely describe, TLR4 fulfills all of the major modifier-gene criteria established by the Complex Trait Community. In addition, TLR4 signaling can be activated not only by LPSs, but also by heparan sulfate, fibrinogen, and other extracellular matrix degradation products.9,10 In the case of fibrinogen, this activation can be blocked with anti-CD14 antibodies.10 Thus, the elevated CD14, in addition to being a biomarker for PKD, could be a therapeutic target for the treatment of PKD.
Footnotes
DISCLOSURE
The author declared no competing interests.
References
- 1.Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 2.Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4:1140–1150. doi: 10.2215/CJN.00790209. [DOI] [PubMed] [Google Scholar]
- 3.Mrug M, Zhou J, Woo Y, et al. Overexpression of innate immune response genes in a model of recessive polycystic kidney disease. Kidney Int. 2008;73:63–76. doi: 10.1038/sj.ki.5002627. [DOI] [PubMed] [Google Scholar]
- 4.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 5.Kistler AD, Poster D, Krauer F, et al. Increases in kidney volume in autosomal dominant polycystic kidney disease can be detected within 6 months. Kidney Int. 2009;75:235–241. doi: 10.1038/ki.2008.558. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Ouyang X, Cui X, et al. Renal CD14 expression correlates with the progression of cystic kidney disease. Kidney Int. 2010;78:550–560. doi: 10.1038/ki.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders HJ, Schlöndorff D. Toll-like receptors: emerging concepts in kidney disease. Curr Opin Nephrol Hypertens. 2007;16:177–183. doi: 10.1097/MNH.0b013e32803fb767. [DOI] [PubMed] [Google Scholar]
- 8.Zheng D, Wolfe M, Cowley BD, Jr, et al. Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2003;14:2588–2595. doi: 10.1097/01.asn.0000088720.61783.19. [DOI] [PubMed] [Google Scholar]
- 9.Johnson GB, Brunn GJ, Kodaira Y, et al. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 10.Kuhns DB, Priel DA, Gallin JI. Induction of human monocyte interleukin (IL)-8 by fibrinogen through the toll-like receptor pathway. Inflammation. 2007;30:178–188. doi: 10.1007/s10753-007-9035-1. [DOI] [PubMed] [Google Scholar]