

Abstract
Systems-level approaches have emerged that rely on analytical, microscopy-based technology for the discovery of novel drug targets and the mechanisms driving AR signaling, transcriptional activity, and ligand independence. Single cell behavior can be quantified by high throughput microscopy methods through analysis of endogenous protein levels and localization, or creation of biosensor cell lines that can simultaneously detect both acute and latent responses to known and unknown androgenic stimuli. The cell imaging, and analytical protocols can be automated to discover agonist/antagonist response windows for nuclear translocation, reporter gene activity, nuclear export, and subnuclear transcription events, facilitating access to a multiplex model system that is inherently unavailable through classic biochemical approaches. In this chapter, we highlight the key steps needed for developing, conducting, and analyzing high throughput screens to identify effectors of AR signaling.
Keywords: fluorescence microscopy, high content analysis, nuclear receptor, automated cytometry, androgen, endocrine disruptor, coregulator
1. Introduction
The androgen receptor (AR) is a member of the nuclear receptor (NR) superfamily and regulates gene transcription to promote male sexual development, and also cell proliferation during prostate cancer progression. Emerging functional data in several organ systems have extended the importance of AR across several endocrine targets including skeletal muscle, bone, and adipose tissue (1). As a type I NR, AR is predominantly cytoplasmic in the absence of agonists, principally testosterone (T) and 5α-dihydrotestosterone (DHT). Following ligand binding, AR sheds its interaction with heat shock proteins and translocates to the nucleus, undergoing concomitant association with coregulators and binding to promoters/enhancers of AR-regulated genes, as reviewed recently (2). Although less understood than genomic AR action, non-genomic AR effects are controlled in both ligand-dependent and -independent manners. Depending on context and stimulus, AR can contribute to gene regulation or cell adaptation through rapid, non-genomic mechanisms, whereby AR interacts with signaling elements (PI3K, Src, MAPK) or acts as scaffolds to other transcription factors (STAT family) (3).
Technical advances during the last decade have made it feasible to use microscopy as a primary tool for probing several aspects of cellular function at increasing throughput (4–10). Although still significantly slower that traditional single point readout high throughput assays, high throughput microcopy (HTM) approaches can integrate multiple coexisting variables within a cellular framework that is inherently unavailable in standard biochemical approaches (ELISA, western blotting, luciferase, ChiP). Single cell-based imaging assays allow simultaneous analysis of multiple features using fluorescent labels; which is often necessary to accurately describe underlying biological processes that are hidden in whole population-based measurements. Due to the data-rich nature of HTM applications, the approach has been broadly dubbed high content analysis (HCA, See Note 1).
AR HCA provides unique opportunities to learn important mechanistic information about AR biology. The classic steps of AR action can be quantified by image-based methods enabling researchers to correlate AR protein levels and subcellular localization with transcriptional activity. Our lab (11–16) and others (17) have used microscopy-based techniques to elucidate the actions of standard agonists and antagonists, characterize endocrine disruptors, and identify non-competitive inhibitors of AR-regulated transcription. In recent years, we have established automated microscopy and HCA as primary approaches to understand AR biology. Routinely, we are able to simultaneously quantify three central mechanistic steps that contribute to AR activation: 1) nuclear translocation via the percent localization of signal in the nucleus (e.g., the nuclear:cellular AR ratio); 2) formation of AR-rich subnuclear “speckles” that correlate with AR transcriptional activity (18); and, 3) transcription via fluorescent reporter gene activity. These assays of androgenic response have Z′ scores of 0.4 to 0.9 in several cell types, AR(−) HeLa cells that transiently or stably express GFP-AR fusion proteins and an ARR2PB-dsRED2skl reporter (13–15) and AR(+ or −) prostate cancer cell lines. Moreover, these Z′ scores have been reproduced in primary cultures generated from patients affected by various conditions of AR malfunction [i.e. genital skin fibroblasts of normal and patients affected by androgen insensitivity syndromes], where the endogenous AR is immunolabeled with antibodies (14–16).
This chapter summarizes experimental AR HCA designs used by our group to quantify nuclear receptor functions at an integrated, systems level. Importantly, these protocols are also applicable to smaller scale experiments where automated, high throughput microscopes are unavailable (e.g. using cells cells plated onto glass coverslips). The chapter closes with a discussion of typical bioinformatic approaches utilizing our central image analysis toolbox and data reduction techniques that can be used to characterize AR ligands in the context of complex biological responses.
2. Materials
2.1. Cell Culture and Stable Cell Line Creation
Phenol red-free DMEM supplemented with L-glutamine, sodium pyruvate, Penicilin/Streptomycin, and 5% fetal bovine serum (FBS). Experimental and standard culture media conditions are shown in Table 1.
Stripped-dialyzed FBS (See Note 2).
Antibiotic selection markers: hygromycin B and puromycin.
Trypsin/ethylenediamine tetraacetic acid (EDTA).
Transfection reagent of choice.
GFP-AR and ARR2PB-dsRED2-skl constructs.
Androgens and anti-androgens.
Table 1.
Growth and experimental media components for GFP-AR/ARR2PB-dsRED2skl cell culture.
| Regular culture | Experiments | |
|---|---|---|
| DMEM/phenol red free | X | X |
| Glutamine | X | X |
| Pen/Strep | X | |
| FBS (5%) | X | |
| SD-FBS (5%) | X | |
| Puromycin (1.2 ug/ml) | X | |
| Hygromycin (200 ug/ml) | X | |
| OHF (0.1 nM) | X |
2.2. Plate Handling
96- or 384-well glass bottom plates (See Note 3).
1 mg-ml poly-D-lysine prepared in water, sterile-filter 0.22 um or 0.45 um (See Note 4).
Phosphate Buffered Saline containing Ca2+ and Mg2+ (PBS++), pH=7.4. Concentrations of constituents are as follows: 138 mM NaCl, 2.67 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4. pH must be reduced (pH=7.0) before adding CaCl2 and MgCl2 but can be re-adjusted to pH=7.4 after dissolution. Concentrations for CaCl2 and MgCl2 are 0.1 g/L.
Tris Buffered Saline (20 mM Tris-HCl, 150 mM NaCl) with 0.1% Tween 20 (TBS-T), pH=7.6.
PEM: 80 mM K-PIPES, pH=6.8; 5 mM EGTA, pH=7.0; 2 mM MgCl2.
Formaldehyde: EM-grade, 16% stock solutions (sold as paraformaldehyde by Electron Microscopy Sciences, Hatfield, PA). For fixation, prepare a 4% solution in PEM. Opened ampoules should be tightly sealed with Parafilm, and stored at 4C in the dark for only a few days. Always dispose of used formaldeyde in appropriate waste accumulation units.
Triton X-100: Prepare 10% (w/v) aliquots in PEM. Keep covered in foil or in amber-colored Eppendorf tubes. Store parent stocks at −20C. The working solution of Triton X-100 for plates is 0.5%, diluted in PEM buffer. After thawing 10% aliquots, do not refreeze. Triton X-100 dilutions are unstable and should be stored at 4C and used quickly. Use of high quality (peroxide and carbonyl free) and fresh Triton X-100 is fundamentally important if HCA experiments utilize immunofluorescence protocols.
4′,6-diamidino-2-phenylindole (DAPI) can be prepared at 1 mg/ml concentration in PEM, aliquoted in amber-colored Eppendorf tubes, and stored at 4C. Protect from light. Reasonable working solutions are 1 ug/ml in PEM.
Quench: 100 mM NH4Cl or 1 mg/ml NaBH4 prepared in PEM can be used to quench residual formaldehyde from fixation. This step reduces autofluorescence by minimizing non-specific antibody-binding to free aldehydes.
CellMask fluorescent dyes (Invitrogen, Carlsbad, CA): These different-color non-specific protein compounds are reconstituted to a final stock concentration of 10 mg/ml in DMSO. Working concentrations should be determined empirically for each experiment and cell type (See Note 5).
TiterTek Multidrop 384 (TiterTek, Huntsville, AL) or equivalent cell plating instrument.
Fluid handling robotics, e.g. BioMek NXS8 or S96 (Beckman Coulter, Mountain View, CA) coupled with plate washer module (e.g. ELx405; BioTek, Winooski, VT).
2.3. Image Acquisition, Background Subtraction, and Analysis
High throughput microscope or analogous imaging platform with automated stage and focus (See Note 6).
Three Dell workstations with the following minimum specifications: 500 GB hard drives, dual-core 2 GHz processors, and 3 GB ram.
Windows XP operating system SP2 with the following software (and their required toolboxes): Pipeline Pilot 7.5 (Accelrys, San Diego, CA) with the Imaging Toolbox, Python 2.6 with numpy, scipy and matplotlib, and R 2.10 with heatmap.plus. (See Note 7).
Workstations should be networked so computing jobs can be parallelized across machines (using an option in Pipeline Pilot)
3. Methods
Assays for HCA begin with routine standard microscopy to establish the response of both positive and negative controls of robust stable cell lines expressing GFP-AR and AR-responsive fluoro-reporter constructs. With our GFP-AR/ARR2PB-dsRED2skl cell line, in vehicle controls we look for primarily cytoplasmic localization, diffuse intranuclear organization, and low background reporter expression (dsRED2skl). Conversely, with standard AR agonists (DHT, R1881, mibolerone), positive control responses are identified by predominantly nuclear, ‘hyperspeckled’ GFP-AR and dsRED2skl-labeled peroxisomes in the cytoplasm. Once the assay is robust and highly reproducible using a coverslip-based approach, we scale the experiment up to 96- and 384-well plates. In a two-step process, we detail the creation of a double-stable cell line expressing GFP-AR and an AR-dependent reporter construct with a dsRED2skl reporter readout. Identical protocols may be used to create stable receptor/reporter cell lines in different cell backgrounds (e.g., PC3, U2OS).
3.1. Construction of GFP-AR/ARR2PB-dsRED2skl biosensor
Cotransfect GFP-AR and a linearized hygromycin marker into HeLa cells using standard chemical transfection methods. After 24 h, re-plate in media (DMEM-F12, 5% FBS) supplemented with hygromycin.
Expand surviving colonies and analyze for GFP-AR distribution and expression by wide-field fluorescence microscopy. Single cell clone positive or desired clones by limiting dilution or cell sorting (FACS).
Infect low-expressing GFP-AR HeLa cell clone(s) with a virus encoding a derivative of the widely-used androgen-responsive pARR2PB transcriptional reporter construct (19). This reporter encodes a dsRED2 protein (Clontech) fused at the C-terminus with a peroxisome targeting sequence (SKL, serine, lysine, leucine) that serves to localize and concentrate the fluorescence signal. Select for antibiotic resistant cultures with both puromycin (1.5 ug/ml) and hygromycin (200 ug/ml) for two weeks.
Following DHT treatment overnight, dual sort single-cell clones by FACS (See Note 8) by GFP-AR and ARR2PB-dsRED2-skl responsiveness (e.g. low GFP/high dsRED2). Maintain single cell clones in phenol-red free DMEM with FBS and antibiotics. Ultimately, when the double stable cell line (GFP-AR HeLa ARR2PB-dsRED2skl) is exposed to AR agonists (R1881, DHT, mibolerone), AR rapidly (~60 min) translocates to the nucleus and initiates a ‘hyperspeckled’ pattern and reporter gene transcription (13, 18). Maximal levels of dsRED2skl accumulate in cytoplasmic peroxisomes by 24–48 h (Figure 1). The quality and reproducibility of the assay should be assessed before scale-up to multi-well formats (See Note 9).
Figure 1.
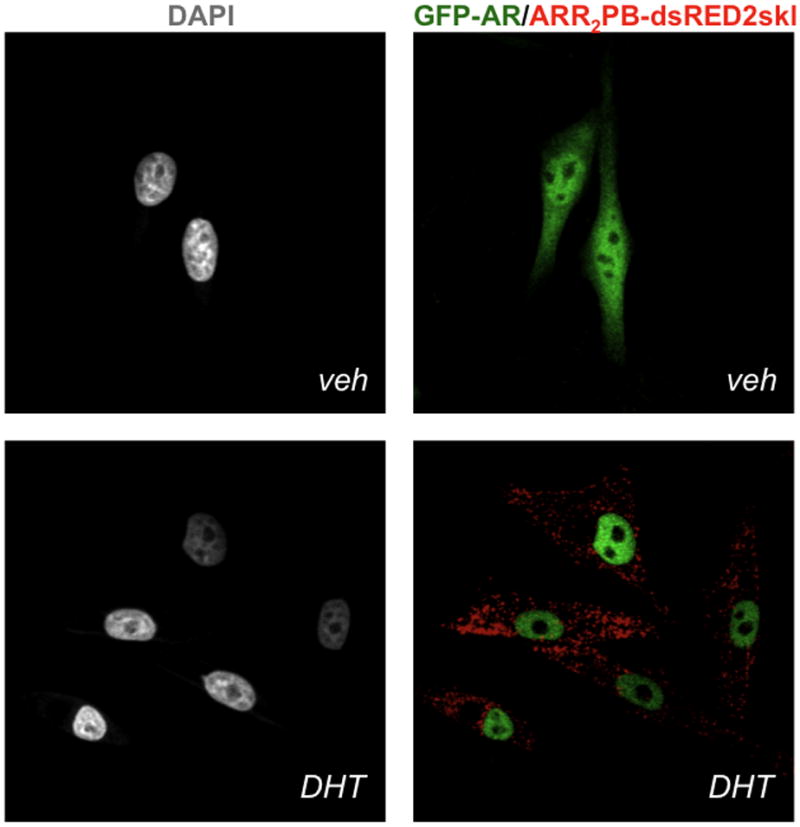
A stable HeLa AR biosensor cell line that expresses both GFP-AR and the androgen responsive fluoro-reporter construct ARR2PB-DsRED2skl. Upon 48 h 10 nM DHT exposure, AR translocates to the nucleus exhibiting visible alterations in subnuclear structure, and dsRED2 labeled peroxisomes indicative of an AR agonist response.
3.2. Surface Preparation for Plates
Coat with a 1 mg/ml, sterile-filtered solution of poly-D-lysine by adding 25 ul to each well, sealing the plate edges with Parafilm®, and incubating at room temperature four hours with gentle rocking, or overnight at 4C
Remove the PDL. To avoid toxicity, wash plates extensively (>6 times) with PBS (50 uL) to remove residual PDL. After the last wash, cells can be plated. Well plates can be sealed and stored for a few days at 4C in 20–30 uL PBS.
Alternatively, coat overnight at 37C with a 10–20% FBS solution prepared in PBS. After incubation, remove excess serum. Plates are ready to be seeded with cells. We detect no basal hormone response from residual FBS in the GFP-AR/ARR2PB-dsRED2skl cell line. For all substrate-coatings, it is critical that all solutions remain sterile.
3.3. Cell Treatment in Multi-Well Plate Formats
Below, we have summarized the steps used for detection of compound-specific effects on AR function and transcriptional activity. A schematic of this process is depicted in Figure 2. Microfluidic robotics are used for all cell processing to assure high efficiency, accuracy, and throughput.
Figure 2.
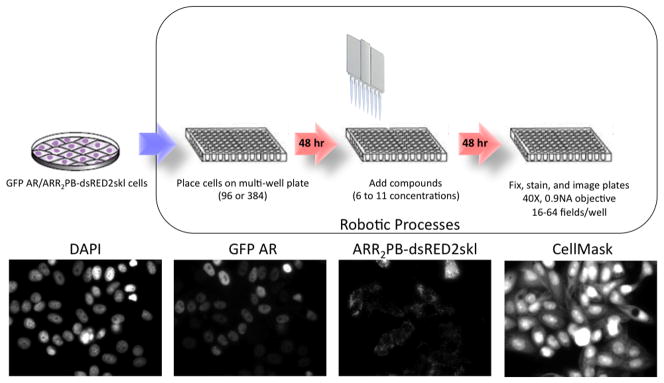
Experimental workflow to study AR activation. A. Hela cells with stable expression of GFP-AR and ARR2PB-dsRED2skl are maintained in sufficient culture size for the desired experiment. 48 h before ligand exposure, cells are trypsinized and seeded to either 96- or 384-well plates. After 48 h cell attachment and synchronization in hormone-free media, cells are treated with for 24 h-48 h with compounds of interest (6–11concentrations) and fixed and stained using robotic protocols (Biomek FXS8). Automated imaging is carried out at 40X using a dry 0.9NA objective with the Beckman IC 100 HTM. Images are collected in 4 colors, DAPI (blue) GFP-AR (green), reporter (red) and cell mask (Far red).
Before experiments, grow cells in the appropriate growth media and serum to 90–95% confluency. Change media 1 day prior to subculturing the cells for experimentation.
Trypsinize the cells with trypsin/EDTA and wash cultures three times with phenol red-free DMEM containing 5% SD-FBS.
Determine cell density. We use 20,000 cells/well (100 uL volume) for 96-wells and 4,000 cell/well (30 uL) in 384-well plates. It is critical to determine the appropriate cell number that results in a subconfluent cell density; confluent cultures are incompatible with HCA for multiple reasons, including heighted probability of washing away during pipetting, altered immunofluorescence results, and greatly reduced ability to perform accurate automated cell segmentation for image analysis. If available, cells can be seeded using the TiterTek Multidrop (TiterTek) or comparable equipment. Otherwise, multi-channel pipettors can be used, albeit with less throughput and accuracy.
Allow cells to attach and incubate in experimental media for 48 h.
In multi-well format, expose cells to 10–11 compounds with 8–11 doses. An example plate layout for 384 wells is shown in Figure 3. Columns 21, 22, 23 are used for agonist (10 nM R1881), antagonist (1 uM OHF), and vehicle controls. The other 20 rows across the top are used for 10 different compounds. In this example, we exposed GFP-AR/ARR2PB-dsRED2skl cells to 10 common AR agonists, antagonists, and endocrine disruptors. A maximum concentration is prepared for row 1 and dilutions are performed using the BioMek S8 (Beckman Coulter, Brea, CA), all at 2X concentrations; we also use the BioMek S8 to transfer compounds (30 uL) to each well.
Following compound transfer, incubate cells for 24–48 h to allow for simultaneous detection of AR translocation and reporter expression (See Note 10).
Figure 3.
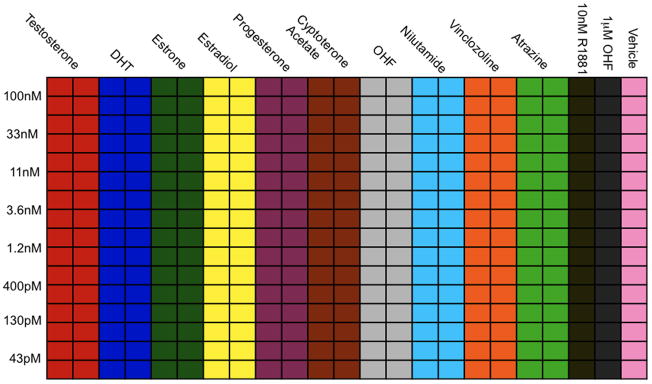
Example plate layout for HCA of AR agonists, antagonists, and endocrine disruptors. Concentrations decrease from rows A-H, while the ligands vary across columns 1–20. Each experimental treatment is performed in quadruplicate.
3.4. Plate Processing and Image Acquisition by HTM
Subsequent to compound treatment, plates are transferred to a microfluidic robot for plate processing. The buffers and solutions needed for the robotic protocols are described above. All solutions are filter sterilized and stored at 4C until needed. PBS and TBS-T buffers can be pipetted from basins; also, upside down tip box lids can suffice. Other reagents such as formaldehyde, quench, Triton X-100, and DAPI are set up in modular basins that fit the microfluidic robot. For plate processing protocols, we regularly make 2 L PEM, connected directly to the plate washer. After buffers are prepared and organized in appropriate basins, the following steps are used to maximize signal integrity and minimize background noise:
Microfluidic robots and plate washer can be programmed to achieve final aspirate heights that leave an approximate residual volume of 5–7 ul. This is the lowest the aspiration can go without disrupting the cell monolayer. Aspiration of each well is performed in the back right corner to minimize disruption of the cell monolayers by fluid transfers.
Transfer plates to robot pods, where all aspects of the protocol will be controlled by software. All volumes added to wells are 25 uL.
Wash two times with cold PBS++, add 4% PFA in PEM (initially cold to minimize cell autolysis, then allow to warm to RT). Incubate 20 min at RT.
Wash three times with PEM, add 100 mM NH4Cl (quench). Incubate 15 min at RT. A shorter incubation time can be used with an alternate quenching agent, NaBH4 (5 min); however, NaBH4 is a naturally bubbling agent that can complicate pipetting. Both solutions should be prepared fresh.
Wash three times with PEM, add 0.5% Triton-X 100, and incubate 5 min RT.
Wash three times with PEM, and once with TBS-T.
Stain cells with DAPI and CellMask FarRed 1:200000 or CellMask Blue in PEM. Incubate 45 min at RT (See Note 5).
Aspirate wells, add 60 ul PBS++/0.01% sodium azide. Seal plates with adhesive or with parafilm to reduce evaporation. Plates are now ready to image. To hold plates more than a day prior to imaging, include 0.4% formaldehyde as a preservative.
Acquire images using HTM or microscope with automated stage with either laser- or image-based auto-focus. Images for the GFP-AR/ARR2PB-dsRED2skl HeLa cell line are collected with a 40x/0.9NA S-Fluor objective (or 40x/0.95NA) (Nikon, Melville, NY). 8-bit images, written as bitmap files, are generated with 2×2 binning (672×512 pixels, 0.344 × 0.344 um2/pixel). For 384 well-plates, 16 fields can be acquired (4×4), while 96-well formats permit the collection of 8×8 fields.
3.6. Image Analysis
Export data to image analysis server/database (See Note 7).
Remove background fluorescence from the images by subtraction of the minimum pixel intensity (MIP) plus noise (the square root of the MIP) from each image. This also corrects for variable background across different wells, and helps to ensure measurements across images are comparable (See Note 11).
Segment nuclei to permit both whole cell segmentation and nucleus measurement extraction. Apply a white top-hat transform to the DAPI images to extract peaks in the image corresponding to the nuclei. Threshold the image using a global, K-means threshold. Apply binary morphology operations to smooth out the resulting objects, giving tight nucleus regions.
Segment cells to extract single-cell level features can from the whole cell. Invert CellMask image and use seeded watershed manipulations, with seeds defined by the nucleus regions. This splits the image into single cell regions. The original CellMask image is thresholded to foreground from background, and this binary image is masked by the watershed result to produce tightly segmented cells. Binary morphology operations are performed to smooth the cell regions; the cytoplasm is segmented by subtracting the nucleus from cell regions.
After segmentation, cells touching the edge of the image field are removed. A sample segmentation schematic is shown in Figure 4.
Figure 4.
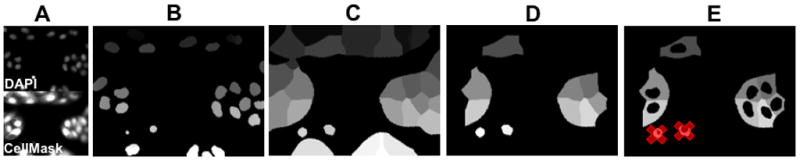
Image segmentation using Pipeline Pilot. A) Raw images are grouped into an image reader. B) Nuclear and C) whole cell masks are created to extract cell-by-cell measurements D) with removal of edge touching regions. E) Cytoplasmic masks are generated by subtraction the nuclear mask from the whole cell masks. Filtering (gray strikethroughs) is used to discard dead cells or mis-segmented objects.
3.7. Feature Extraction in Pipeline Pilot
Extract androgen receptor (AR) object and intensity features using the AR images in the image region shape statistics component (IRSC) and image region intensity statistics component (IRISC).
Extract AR in nucleus-object intensity and object colocalization features using AR and nucleus-regions images in the IRISC and the object colocaliztion component (OCC).
Extract AR in cytoplasm-object intensity and object colocalization features using the AR and cytoplasm-regions images in the IRISC and OCC.
Extract the IRISC and OCC features for the reporter in the whole cell using the reporter and cell-regions images.
For filtering purposes, extract IRSC features from the nucleus- and cell-regions images.
Remove cells that have misshapen nuclei (too large, too small, not ellipsoidal enough) or that are expressing AR outside of the physiological range (See Note 12).
From the features extracted, derive translocation, hyperspeckling, and reporter activity features. The nuclear/cytoplasmic ratio, representative of AR translocation, is determined by measuring the percent of total AR signal (sum of pixel intensities) within the nucleus mask. Nuclear ‘hyperspeckling’ is the statistical variance in AR pixel intensity within the nucleus mask. Accumulation of the AR-sensitive ARR2PB-dsRED2skl fluorescent reporter protein is measured as the sum of pixel intensities under the cytoplasmic mask. These measurements are highlighted in Figure 5A for all 10 compounds using a heat map representation (generated in Python).
Figure 5.

Example data generated from exposure of the AR biosensor cell line to a group of AR effectors. Dose response data is depicted for the 3 central measurements: A) nuclear AR/cellular AR, B) nuclear hyperspeckling, and C) ARR2PB-dsREDskl activity. Note that agonist, antagonist, and vehicle treatments are denoted on the color bars by black, gray, and white arrows, respectively. D) Hierarchical cluster analysis and associated heat map responses of image-derived features over the dose regime of the 10 compounds in the experimental design. Four PCA features (PC0, PC1, PC2, PC3) capture 80% of the dataset variance.
3.7. Data Reduction
In order to define treatment “fingerprints,” we perform hierarchical clustering using responses at the treatment level (single cell-based measurements of features are averaged at the well level across treatments). Sorting through hundreds of features can be challenging; furthermore, not all features are informative in defining a unique treatment type. In order to produce a compact fingerprint, principal component analysis (PCA; (20) can be used to reduce measurements to a smaller set of features useful for grouping compound treatments. Features derived from cytological measurements and object/shape colocalization functions are highly effective in distinguishing between subcellular patterns as has been established for location proteomics studies (21). Increasingly these features are being integrated into assays for mechanism- and/or screening-based applications. Considerable effort has been put in to developing and using phenotype-based assays for siRNA or other screens (22). Open source-based feature processing and selection was performed in Python 2.6.4, while clustering was done in R 2.10, using the heatmap.plus package. Our group has wrapped these methods in Pipeline Pilot to produce graphics-based workflows that can be utilized and modified by end users. Other recent examples (5–7, 10, 23) of this type of analytical approach have been used in a similar manner to maintain data-rich features, but provide a simpler interpretation of a handful of condensed variables as opposed to 100s of esoteric measurements. A basic approach for data reduction of HCA datasets is as follows:
Calculate the average (median) feature values across each field (using the cell-level features). Then, calculate the average feature values across each well (using the field-level features). Finally calculate the average feature values across each treatment (using the well-level data). This step can reduce the dataset to a more manageable number of samples (dealing with dozens-to-thousands of treatments instead of hundred-of-thousands-to-tens of millions of cells).
Standardize features by subtracting from them their mean and then dividing them by their standard deviation.
Normalize treatment samples by dividing them by their magnitude.
Perform PCA to reduce measurements to a smaller set of features that are useful for grouping compound treatments. Set the PCA to return features that capture 75% of the variance in the dataset. Increasing the variance parameter in PCA causes it to return more features.
Using the PCA features, perform hierarchical clustering on processed features. Cluster using the Euclidean distance metric (Figure 5B).
Acknowledgments
This work was funded by NIH 5R01DK055622, the Hankamer Foundation, DOD Prostate Cancer Research Program (DAMD W81XWH-10-1-0390) and pilot grant and equipment support from the John S. Dunn Gulf Coast Consortium for Chemical Genomics (MA Mancini). Additional funding was provided by NIH 1F32DK85979 (SM Hartig), 5T32HD007165 (BW O’Malley) and 5K12DK083014 (DJ Lamb). Imaging resources were supported by SCCPR U54 HD-007495 (BW O’Malley), P30 DK-56338 (MK Estes), P30 CA-125123 (CK Osborne), and the Dan L. Duncan Cancer Center of Baylor College of Medicine. The authors thank the members of the Mancini Lab for thoughtful discussion and J.H. Price (Vala Sciences) and Tim Moran (Accelrys) for longstanding support in automated cytometry.
Footnotes
HTM and HCA are increasingly being adopted as standardized approaches to understand the complex biology of cell signaling, not just nuclear receptors and gene regulation. Further ease-of-use and speed-associated developments in auto-focus, image analysis, illumination, and data reduction/visualization will continue to progress HCA. As other, more advanced technologies, like FRET, FLIM, and RNA FISH, become integrated into HCA approaches, the untapped biology of the genome will be more readily made accessible. Despite the progress of HCA, the use of complex data reduction approaches to understand AR cytology, and other proteins for that matter, remains in its infancy. A merger of structure activity relationships with multiplex measurement capability, available only by HCA, should provide detailed mechanistic insights to guide pharmacological leads that modulate nuclear receptor activity and other difficult drug discovery bottlenecks.
For all AR response experiments, cells are plated and treated in phenol red-free DMEM with 5% charcoal stripped, dialyzed FBS (SD-FBS). SD-FBS can be prepared from dialyzed FBS with dextran/charcoal stripping of hormones.
When using HCA as a means to quantify cellular response, the choice of well-plate is critical. We have found that Greiner SensoPlate Plus 96- and 384- low skirt glass plates provide both excellent image quality (low background in FITC/GFP and dsRED2/Texas Red/mCherry) and appear free of intrinsic estrogenic activity from plate plastic, sealant or adhesive derivatives. The issue of multiwell plate-derived estrogenic activities from (presumably) plasticizers (24) like bisphenol A is a much unappreciated problem in general cell culture, and we have utilized our sensitive estrogen receptor HCA model (11, 25) to empirically eliminate “estrogenic” plates from all of our steroid receptor studies. For strong antibody or XFP signals, Aurora 384 optical bottom plastic plates are a markedly less-expensive alternative, do not possess estrogenic activity, and do not require pre-experiment coating; low antibody or XFP signals, however, can be obscured by a intrinsic high background signal specifically in the dsRED2/Texas Red/mCherry wavelengths.
Many cell types, including HeLa or HeLa stables, do not adhere well to glass surfaces. Hence, we have used both poly-D-lysine (PDL) and FBS coatings to facilitate optimal cell adhesion. For experiments involving several 96- or 384-well plates, coatings can be added using the Titer-Tek Multidrop or equivalent cell plating systems. In brief, coating protocols involve overnight treatment with PDL or FBS prior to cell seeding; additional details on acid etching and coating of coverslips can be found in a previous methods chapter (12).
We predominantly use CellMask Blue or CellMask FarRed to allow segmentation of cell bodies. It is essential to determine empirical working concentrations for each experiment and cell type. We find that CellMask Blue generally works best at 0.1 ug/ml (in PEM) while CellMask FarRed at 5 ng/ml (in PEM). It is important to indicate that for the FarRed CellMask variant, excessive incubation (time or concentration) can result in bleeding of the dyes into dsRED2/Texas Red/mCherry fluorescence filter sets. CellMask Blue has the advantage of opening up the Cy5 channel for antibodies while using the UV channel specifically for detecting the cell body features. When staining is optimal, lower exposures can be used to detect only the nucleus outline from DAPI while longer exposures slightly saturate the DAPI signal while allowing the cell edge detection with CellMask Blue. It is important to note that combinations of CellMask Blue and DAPI may be detrimental for detecting both cell border/size properties and cell cycle position as CellMask dyes also label the nucleus.
Our lab has predominantly used the Cell Lab IC 100 Image Cytometer (IC 100) platform (Beckman Coulter) for high-resolution HTM. The imaging platform consists of: 1) Nikon Eclipse TE2000-U inverted microscope (Nikon, Melville, NY) with a Chroma 82000 triple band filter set (Chroma, Brattleboro, VT); 2) Hamamatsu ORCA-ER digital CCD camera (Hamamatsu, Bridgewater, NJ); and, 3) Photonics COHU progressive scan camera (Photonics, Oxford, MA). Due to auto-focus feedback loops, the IC 100 performs accurate and fast image-based focus, minimizing image acquisition times and providing more accurate focus compared to laser reflection methods.
We use the Pipeline Pilot data management, workflow and analysis software platform (Accelrys, San Diego, CA; www.accelrys.com). Other platforms are available that perform similar functions include CyteSeer (26, 27); Vala Sciences, San Diego, CA; www.valasciences.com) and CellProfiler (28, 29); www.cellprofiler.org; Broad Institute, MIT), in addition to software that comes with equipment manufacturers. We choose to use Pipeline Pilot because it provides us with a single software package that is highly customizable for all image and data analysis methods that further allows us to report data internally or to 3rd party software. As many industrial, biotech and research environments have adopted Pipeline Pilot, protocols can be easily transferred and exchanged. However, research groups engaging in HCA generally find it necessary for HTM users to devote sufficient time learning the functionalities to apply PLP or similar software adequately and efficiently.
Upon single cell sorting the GFP-AR/ARR2-dsRED2skl biosensor cell line, 81 single cells clones were isolated, with two clones exhibiting marked androgen responsiveness; the clone that responded maximally to DHT was used for all additional studies, and maintained in phenol red-free DMEM/5% FBS growth media with 1.5 ug/ml puromycin and 200 ug/ml hygromycin. We additionally added 0.1 uM o-hydroxyflutamide (OHF) to cultures during routine maintenance to minimize basal dsRED2skl expression, and we removed OHF 24 h prior to experimental setup.
| EQUATION 1 |
To detect unknown antagonists, titrate compounds against 0.5–10 nM DHT or R1881 as described previously (15). As anti-androgen concentration is increased against constant agonist, an inhibition of transcriptional activity is detected. Depending on the antagonist, reduced transcriptional competence may accompany effects on AR subnuclear organization and localization. This approach is particularly useful both in terms of defining a transcriptional antagonist, but also, simultaneously, report on possible mechanisms in play. Here, the time and cost efficiency of an HCA approach is readily apparent.
We have additionally created alternative background subtraction methods that remove background signal from all images using plate- and channel-specific correction images. An average of >500 random randomly selected images for each channel are amplified with a correction factor based on the median pixel intensity of experimental images. The combination of correction factor and average image (from randomly selected images) allows for a pixel-by-pixel subtraction that compensates any consistent, uneven background artifacts in the image set. Object detection algorithms are highly dependent on the specific fluorescent marker morphologies and signal to noise properties of the HCA assay Reliable identification methods and background subtraction methods are key for accurate quantification of image data sets.
Data streams, containing cell feature data, can be filtered to remove spurious sources of error by removing cell aggregates, mitotic cells, cellular debris, or events with expression levels that are outside the physiological range. The applied filters are based upon cell morphological measurements derived from cell and nucleus segmentation: nucleus area, nucleus circularity, nucleus/cell size ratio, and DNA content (derived from DAPI). Additional filters are used to remove cells with non-physiological expression of GFP-AR expression, using levels found in LnCaP as a reference (14, 16). Generally, these filters remove 10–15% of the cells in our GFP-AR HeLa cell line with individual limits determined by analyzing the Gaussian distribution of pixel intensities (e.g. 5% outliers); in transient GFP-AR transfection assays, up to ~70% of the cells are generally discarded (14–16).
References
- 1.Zitzmann M. Testosterone deficiency, insulin resistance and the metabolic syndrome. Nat Rev Endocrinol. 2009;5:673–681. doi: 10.1038/nrendo.2009.212. [DOI] [PubMed] [Google Scholar]
- 2.Echeverria PC, Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta-Mol Cell Res. 2010;1803:641–649. doi: 10.1016/j.bbamcr.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Rahman F, Christian HC. Non-classical actions of testosterone: an update. Trends Endocrinol Metab. 2007;18:371–378. doi: 10.1016/j.tem.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Bakal C, Aach J, Church G, Perrimon N. Quantitative morphological signatures define local signaling networks regulating cell morphology. Science. 2007;316:1753–1756. doi: 10.1126/science.1140324. [DOI] [PubMed] [Google Scholar]
- 5.Loo LH, Lin HJ, Steininger RJ, Wang YQ, Wu LF, Altschuler SJ. An approach for extensibly profiling the molecular states of cellular subpopulations. Nat Methods. 2009;6:759–U718. doi: 10.1038/nmeth.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nat Methods. 2007;4:445–453. doi: 10.1038/nmeth1032. [DOI] [PubMed] [Google Scholar]
- 7.Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 8.Ramadan N, Flockhart I, Booker M, Perrimon N, Mathey-Prevot B. Design and implementation of high-throughput RNAi screens in cultured Drosophila cells. Nat Protoc. 2007;2:2245–2264. doi: 10.1038/nprot.2007.250. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka M, Bateman R, Rauh D, Vaisberg E, Ramachandani S, Zhang C, Hansen KC, Burlingame AL, Trautman JK, Shokat KM, Adams CL. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005;3:764–776. doi: 10.1371/journal.pbio.0030128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young DW, Bender A, Hoyt J, McWhinnie E, Chirn GW, Tao CY, Tallarico JA, Labow M, Jenkins JL, Mitchison TJ, Feng Y. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat Chem Biol. 2008;4:59–68. doi: 10.1038/nchembio.2007.53. [DOI] [PubMed] [Google Scholar]
- 11.Berno V, Amazit L, Hinojos CA, Zhong J, Mancini MG, Sharp ZD, Mancini MA. Activation of estrogen receptor-alpha by E2 or EGF induces temporally distinct patterns of large-scale chromatin modification and mRNA transcription. PLOS One. 2008;3:e2286. doi: 10.1371/journal.pone.0002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berno V, Hinojos CA, Amazit L, Szafran AT, Mancini MA. High-resolution, high-throughput microscopy analyses of nuclear receptor and coregulator function, in. Measuring Biological Responses With Automated Microscopy. 2006:188–210. doi: 10.1016/S0076-6879(06)14011-2. [DOI] [PubMed] [Google Scholar]
- 13.Marcelli M, Stenoien DL, Szafran AT, Simeoni S, Agoulnik IU, Weigel NL, Moran T, Mikic I, Price JH, Mancini MA. Quantifying effects of ligands on androgen receptor nuclear translocation, intranuclear dynamics, and solubility. J Cell Biochem. 2006;98:770–788. doi: 10.1002/jcb.20593. [DOI] [PubMed] [Google Scholar]
- 14.Szafran AT, Hartig S, Sun H, Szwarc M, Chen Y, Mediwala S, Bell J, McPhaul MJ, Mancini MA, Marcelli M. Androgen receptor mutations associated with androgen insensitivity syndrome: A high content analysis approach leading to personalized medicine. PLOS One. 2009;4:e8179. doi: 10.1371/journal.pone.0008179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szafran AT, Szwarc M, Marcelli M, Mancini MA. Androgen receptor functional analyses by high throughput imaging: Determination of ligand, cell cycle, and mutation-specific effects. PLOS One. 2008;3:e6205. doi: 10.1371/journal.pone.0003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szafran AT, Szwarc M, Marcelli M, Mancini MA. High throughput multiplex image analyses for androgen receptor function. Proc Intl Symp Biomed Imaging. 2008;5:320–324. [Google Scholar]
- 17.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, Hann B, Diamond MI. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad U S A. 2009;106:7233–7238. doi: 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, Dubbink HJ, Verschure PJ, Trapman J, Houtsmuller AB. Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JF, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141:4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 20.Wall ME, Rechtsteiner A, Rocha LM. Singular value decomposition and principal component analysis. In: Berrar DP, Dubitzky W, Granzow M, Norwell MA, editors. A practical approach to microarray data analysis. Kluwer; 2003. pp. 91–109. [Google Scholar]
- 21.Glory E, Murphy RF. Automated subcellular location determination and high-throughput microscopy. Dev Cell. 2007;12:7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, Cetin C, Sieckmann F, Pau G, Kabbe R, Wunsche A, Satagopam V, Schmitz MHA, Chapuis C, Gerlich DW, Schneider R, Eils R, Huber W, Peters JM, Hyman AA, Durbin R, Pepperkok R, Ellenberg J. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newberg J, Murphy RF. A framework for the automated analysis of subcellular patterns in human protein atlas images. J Proteome Res. 2008;7:2300–2308. doi: 10.1021/pr7007626. [DOI] [PubMed] [Google Scholar]
- 24.McDonald GR, Hudson AL, Dunn SMJ, You HT, Baker GB, Whittal RM, Martin JW, Jha A, Edmondson DE, Holt A. Bioactive contaminants leach from disposable laboratory plasticware. Science. 2008;322:917–917. doi: 10.1126/science.1162395. [DOI] [PubMed] [Google Scholar]
- 25.Sharp ZD, Mancini MG, Hinojos CA, Dai F, Berno V, Szafran AT, Smith KP, Lele TT, Ingber DE, Mancini MA. Estrogen-receptor-alpha exchange and chromatin dynamics are ligand- and domain-dependent. J Cell Sci. 2006;119:4101–4116. doi: 10.1242/jcs.03161. [DOI] [PubMed] [Google Scholar]
- 26.McDonough PM, Agustin RM, Ingermanson RS, Loy PA, Buehrer BM, Nicoll JB, Prigozhina NL, Mikic I, Price JH. Quantification of Lipid Droplets and Associated Proteins in Cellular Models of Obesity via High-Content/High-Throughput Microscopy and Automated Image Analysis. Assay Drug Dev Technol. 2009;7:440–460. doi: 10.1089/adt.2009.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prigozhina NL, Zhong L, Hunter EA, Mikic I, Callaway S, Roop DR, Mancini MA, Zacharias DA, Price JH, McDonough PM. Plasma membrane assays and three-compartment image cytometry for high content screening. Assay Drug Dev Technol. 2007;5:29–48. doi: 10.1089/adt.2006.024. [DOI] [PubMed] [Google Scholar]
- 28.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:30. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler(TM): free, versatile software for automated biological image analysis. Biotechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 30.Zhang JH, Chung TD, Oldenburg KH. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]