

Abstract
Bacillus pumilus has been widely identified as a pathogen of plant and human, while the genetic information is rarely available for pathogenic B. pumilus strains. B. pumilus GR8 is a pathogen that causes ginger rhizome rot disease by invading ginger rhizome parenchymatous tissues, growing in the extracellular space, and producing plant cell wall-degrading enzymes to destroy ginger cells. In this study, the genome of GR8 was sequenced and characterized. This genome was the third completely sequenced genome of the B. pumilus species, and it exhibited high similarity to the genome of the B. pumilus strain B6033. The genome of GR8 was 3.67 Mb in length and encoded 3,713 putative ORFs. Among these predicted proteins, numerous plant cell wall-degrading enzymes and several proteins associated with invading and adapting to the environment in the extracellular space of the ginger rhizome parenchymatous tissue were found. The GR8 genome contained only one restriction-modification system and no CRISPR/Cas system. The lack of phage-resistant system suggested that phages might be potential agents for the control of GR8. The genomic analysis of GR8 provided the understanding to the pathogenesis and the phage-control strategy of pathogenic B. pumilus strains.
Bacillus pumilus is a bacterial species belonging to the Bacillus subtilis group1. Based on its abilities in producing vanillin, keratinase, xylanase, alkaline serine protease and several other bioactive substances2,3,4,5, B. pumilus is widely used in industrial processes, such as the production of several traditional fermented foods, the treatment of wastewater and the degradation of environmental pollutants6,7. More recently, B. pumilus found in healthy plant tissue was reported with properties in promoting plant growth by enhancing the uptake of nutrients, nitrogen fixation, interaction with symbiotic microorganisms and producing antimicrobial agents against pathogenic bacteria and fungi as well as by reducing metal toxicity8,9,10,11. According to previous reports, several other B. pumilus strains are also used as probiotics for animals12. However, numerous B. pumilus strains have been reported to cause plant and human diseases. Strains of B. pumilus associated with foodborne illness and from clinical environments have been isolated, and several B. pumilus strains, such as strain F4791/87 and F4552/87, have been found to produce toxicin that are harmful to humans13,14. Two B. pumilus isolates have been identified that cause severe sepsis of neonatal infants15. Six B. pumilus strains isolated from Egypt have been reported to cause mango tree disease, and one B. pumilus strain isolated from China is associated with leaf and twig dieback of Asian pear trees16,17.
In previous studies, the genomes of ten strains of B. pumilus were sequenced, with two then being fully assembled18. These ten strains mainly showed industrial or medical merits by producing bioactive substances or potential antiviral agents. However, no genome of strains associated with plant or human disease was sequenced18,19,20,21. The genomic information of pathogenic strains of B. pumilus will be important for developing an understanding on the pathogenesis and to identify potential control strategies for pathogenic B. pumilus. The B. pumilus strain GR8 isolated from Shandong Province of China is an opportunistic pathogen of ginger and can cause severe rot symptoms of ginger rhizomes22. In this study, we characterized the genome of this strain. Analysis of the GR8 genome sequence provided insight into both the pathogenesis of B. pumilus and possible novel control strategies, and made the bio-safety aspects of B. pumilus as plant growth-promoting rhizobacteria (PGPR) debatable.
Results
Genome sequence and general features of the B. pumilus strain GR8
The genome of GR8 was sequenced using the Illumina HiSeq 2500 and was de novo assembled into 10 contigs with an average sequencing depth of approximately 120-fold. The gaps between contigs were filled by primer walking. The complete circular genome of GR8 was 3,674,489 bp in length with a G+C content of 41.42% (Fig. 1), and encoded 3,713 ORFs. This genome was the third completely sequenced B. pumilus genome. A comparison of general genome features of ten sequenced strains of B. pumilus and the strain GR8 is summaried in Table 1. Compared with the other sequenced B. pumilus genomes, GR8 had a smaller genome size and possessed more tRNA and rRNA encoding genes (81 and 24, respectively). One plasmid sequence was also obtained (Fig. 1; Supplementary Fig. S1). The length of the plasmid, named pGR8, was 6,935 bp with a G+C content of 36.82%, and it encoded six ORFs. The sequence of pGR8 exhibited 53% and 61% similarity to the B. subtilis plasmid pPOD2000 and the B. pumilus plasmid pPL7065, respectively.
Figure 1.

Circular genome maps of the B. pumilus strain GR8 chromosome and the plasmid pGR8. The genome of GR8 was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) and visualized using the CGView Server.
Table 1. Comparison of general genome features of sequenced strains of B. pumilus.
| Feature |
Bacillus pumilus
strains |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GR8 | B603318 | SAFR-03247 | S-12 | 7p20 | CCMA-56019 | Fairview55 | 3-19a,56 | ATCC706147 | INR756 | ||
| Origin of strain | China | Inda | USA | China | Russia | Brazil | Australia | Russia | USA | Korea | China |
| Chromosome size (bp) | 3,674,489 | 3,763,493 | 3,704,465 | 3,692,073 | 3,573,949 | 3,848,811 | 3,838,013 | 3,573,949 | 3,833,998 | 3,681,709 | 3,747,698 |
| G+C content (%) | 41.4 | 41.4 | 41.3 | 41.3 | 41.8 | 41.5 | 41.9 | 41.7 | 41.3 | 41.3 | |
| Number of genes | 3,713 | 3,807 | 3,822 | 3,708 | 3,596 | 3,594 | 3,784 | 3,563 | 4,162 | 3,725 | 6,756 |
| No. of CDS | 3,592 | 3,659 | 3,679 | 3,643 | 3,460 | 3,460 | 3,717 | 3,363 | 3,900 | 3,625 | 3,676 |
| No. of pseudogenes | 15 | 42 | 50 | 11 | 57 | 41 | 123 | 190 | 30 | 12 | |
| No. of rRNA | 24 | 24 | 21 | 4 | 4 | 4 | 3 | 4 | 7 | 4 | 21 |
| No. of tRNA | 81 | 81 | 69 | 49 | 72 | 72 | 22 | 72 | 58 | 65 | 46 |
| No. of other RNA | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 7 | 1 | 1 | |
| GenBank accession | CP009108 | CP007436 | CP000813 | AGBY00000000 | JHUD00000000 | AUYP01000000 | JFBY01000000 | JOJX00000000 | ABRX00000000 | AYTK00000000 | AMDH00000000 |
a3-19 is derived from B. pumilus strain 7P.
Comparative genomic analysis
The eleven strains of B. pumilus from which genomes have been sequenced had a wide biogeographical distribution, while they exhibited highly similar genome features (Table 1). Comparative genomic analysis showed that the genome of GR8 exhibited high similarity to the genome of strain B6033 isolated from India (Fig. 2; Supplementary Fig. S2). Sequence comparisons among the three completely sequenced genomes of the B. pumilus (strains GR8, B6033 and SAFR-032), showed widespread co-linearity, except for a 24-kbp genome rearrangement from 976,679 nt to 1,000,676 nt in GR8 genome. The rearrangement region in GR8 genome was predicted to encode proteins with functions of genome replication and nucleotide metabolism. The encoding genes of two recombinases, RecF and RecR, were found at the 5’-terminus and the 3’-terminus of the rearrangement region, respectively, and might be essential for the recombination of this region.
Figure 2.
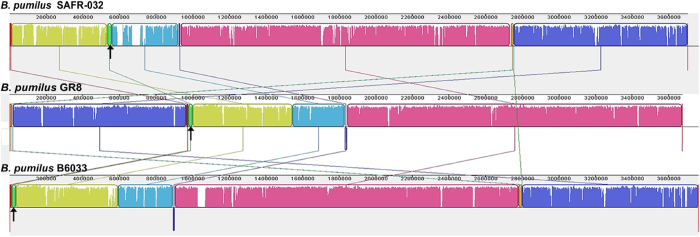
Comparative genome analysis of three completely sequenced strains of B. pumilus. The genome sequences of SAFR-032, GR8 and B6033 were compared using progressive Mauve. The rearrangement regions in the genomes are indicated with black arrows.
Plant cell wall-degrading enzymes
Plant pathogens infect plants by employing plant cell wall-degrading enzymes (PCWDEs) to destroy plant cells23. Numerous putative proteins associated with the degradation of the plant cell wall were predicted to be encoded by the genome of the ginger rhizome rot disease pathogen GR8. The PCWDEs were mainly CAZymes, wiht 452 CAZyme genes identified in the GR8 genome. The predicted CAZymes included five categories as follows: carbohydrate esterase (CE), glycosyltransferase (GT), glucoside hydrolases (GH), polysaccharide lyase (PL) and carbohydrate-binding molecules (CBMs) (Supplementary Fig. S3). In the CE class, the abundance of the acetyl xylan esterases, CE1 and CE4 (representing 22% and 15%, respectively), was high, but the abundance of CE1 and CE4 was less than that of CE11 with an abundance of 29%. Cellulose (GT2) was the most abundant (representing 54%) in the GT class, and chitinase (GH18) represented the most abundant glucoside hydrolase (GH) followed by galactanase (GH53), mannosidase (GH92 and GH1), and glucosidase (GH1), which all degrade plant cell wall components23. Polysaccharide lyase (PL) represented the CAZymes that degraded pectin, the major component of plant cell walls24. Three types of the predicted PLs (PL1, PL9 and PL10) were all pectate lyases. Carbohydrate-binding molecules (CBMs) are responsible for the attaching of enzymes to cellulose, the major component of plant cell walls. CBM2 and CBM3 were the second and third most abundant CBMs identified. Overall, there were fewer CAZyme genes in the GR8 genome compared to the genomes of strains B6033 and SAFR-032 (Supplementary Fig. S4).
Plant pathogenicity gene candidates
The putative functional classifications of the proteins encoded by GR8 genome were analyzed using KAAS25. In total, 1,553 putative proteins (representing 43.2% of ORFs) were associated with the processes of metabolism, environmental information processing and genetic information processing (Fig. 3).
Figure 3.

Functional classification of proteins encoded by the GR8 genome. The functions of the proteins were assigned using KAAS. The number of each class is indicated above the corresponding column.
Four putative proteins from the GR8 genome, including three flagellin FliC proteins and one elongation factor Tu (EF-Tu), were classified as environmental adaptation proteins. These proteins exhibit functions in the interaction between bacterial pathogens and plants. They can also induce the immunity of the plant host to bacterial pathogens, and the flagellin also facilitates the adherence of plant pathogens to plants by absorbing plant lignin26,27.
Two putative proteases, with similarity to the human pathogen protease responsible for degradation of protein in the mucus layer during infection28, were found to be encoded by the GR8 genome. The GroEL and LepA encoding genes were also found in the GR8 genome. GroEL is a surface-located chaperonin which plays a role in pathogen entry or the entry of the bacteria-containing vacuole to the host cell29,30. The LepA protein aids the exit of bacteria from the host cell to allow infection of neighboring cells29. Similar proteins encoded by the GR8 genome might facilitate the invasion of the strain into ginger rhizome parenchyma cells.
Numerous proteins, which may be associated with the adaptation of the strain to the ginger parenchyma tissue extracellular space environment, were predicted from the GR8 genome as follows: NsrR, Hmp, RocF, DltA, DltB, DltC, DltD, MprF, BceA, BceB, PstS, DnaK, LuxS and SdhA. NsrR, Hmp and RocF might inhibit the production of NO or transform NO into nitro. The accumulation of NO in plant cells acts to induce the plant defense system against plant pathogens, and NO is also a toxicant to pathogenic bacteria31,32. The production of antimicrobial peptides is a crucial way for plant hosts to prevent infection and colonization by bacterial plant pathogens. A three-component system that provides Gram-positive strains with resistance to plant-produced antimicrobial peptides has been well characterized33. The system is composed of a dlt system (DltA/DltB/DltC/DltD), an MprF protein and an ABC transporter (BceA/BceB). The related genes of the three-component system were found in the genome of GR8 (Table 2). The GR8 genome also encoded PstS and DnaK proteins, which may help GR8 to adapt to the low phosphorus environment of the extracellular space of ginger parenchymatous tissue34. The formation of biofilm is a strategy bacterium utilize to enhance infective capability, and LuxS has been reported to mediate biofilm formation35. Biofilm formed by GR8 was observed during daily culture of the strain. According to previous reports, GR8 could invade parenchymatous cells and live within the host by using the nutrition of the cells22. Two proteins, PstS and SdhA, which inhibit host cell apoptosis30, were encoded by the GR8 genome and these might benefit the intracellular life cycle of GR8.
Table 2. Plant pathogenic gene candidates in the B. pumilus strain GR8 genome and corresponding genes in the genomes of strains B6033 and SAFR-032.
| Function during GR8 infection | Protein | Gene for GR8 | Corresponding gene for B6033 | Corresponding gene for SAFR-032 | Description |
|---|---|---|---|---|---|
| Induce host immunity | FliC | ID12_11065 | BW16_06360 | BPUM_0150 | Induce host immunity and aid adherence27 |
| FliC | ID12_11180 | BW16_06550 | BPUM_1149 | ||
| FliC | ID12_11185 | BW16_06555 | BPUM_1152 | ||
| EF-Tu | ID12_05735 | BW16_00715 | BPUM_0099 | ||
| Invasion | Protease | ID12_17480 | BW16_12860 | BPUM_2370 | Degrade protein28 |
| Protease | ID12_17485 | BW16_12865 | BPUM_2371 | ||
| GroEL | ID12_08120 | BW16_03100 | BPUM_0535 | ||
| LepA | ID12_17000 | BW16_12350 | BPUM_2284 | ||
| Resistance to NO | NsrR | ID12_09830 | BW16_04850 | BPUM_0893 | NO detoxification32 |
| Hmp | ID12_09835 | BW16_04855 | BPUM_0894 | ||
| RocF | ID12_06065 | BW16_01045 | BPUM_0159 | ||
| Resistance to antimicrobial peptides | BceA | ID12_18605 | BW16_13990 | BPUM_2594 | Antimicrobial peptide transport33 |
| BceB | ID12_18610 | BW16_13995 | BPUM_2595 | ||
| DltA | ID12_18030 | BW16_13390 | BPUM_2479 | ||
| DltB | ID12_18025 | BW16_13395 | BPUM_2478 | ||
| DltC | ID12_18020 | BW16_13400 | BPUM_2477 | ||
| DltD | ID12_18015 | BW16_13405 | BPUM_2476 | ||
| MprF | ID12_09195 | BW16_04235 | BPUM_0787 | ||
| Phosphorus starvation tolerance | PstS | ID12_16725 | BW16_12070 | BPUM_2227 | Phosphate binding34 |
| DnaK | ID12_16980 | BW16_12330 | BPUM_2280 | ||
| Biofilm forming | LuxS | ID12_19195 | BW16_14590 | BPUM_2705 | Mediate biofilm forming35 |
| Inhibition of host cell apoptosis | PstS | ID12_16725 | BW16_12070 | BPUM_2227 | Inhibit host cell apoptosis30 |
| SdhA | ID12_18145 | BW16_13530 | BPUM_2502 |
Phage-resistance system and prophage regions of GR8
Because phage and phage endolysin can lyse bacteria, they are thought of as potential antimicrobial agents against plant bacterial pathogen36,37. To avoid phage infection, bacteria have also evolved defense systems, such as resistance to phage absorption, penetration blocks, restriction modification (R-M) system and clustered regularly interspaced short palindromic repeats (CRISPR/Cas) system38. The CRISPR/Cas system is a general adaptive immunity system that protects the bacterium from foreign phage DNA and widely exists in the prokaryotic organism39,40. The GR8 genome was analyzed by the CRISPR finder online service, and no CRISPR/Cas system was found. The R-M system in the GR8 genome was also analyzed by comparing the genome sequence with the REBASE database, and the result showed that only one type I R-M system was found. The R-M system recognized the specific sites on the foreign DNA and cleaved the DNA without modification41. According to previous reports, strain GR8 could be lyzed by kinds of phages42. The reason might be the lack of phage defense system in the GR8 genome. This result suggested that phage might be a promising antimicrobial agent for the control of ginger rhizome rot disease caused by strain GR8.
The prophage region in the GR8 genome was evaluated using PHAST and three putative prophages, including an intact prophage (region 1), an incomplete prophage (region 2) and a questionable prophage (region 3), were found (Fig. 4A). The intact prophage region was located between sites 12,281 to 39,856 nt with a G+C content of 41.47%, which was similar to the G+C content of the GR8 genome, and it exhibited 87% genomic similarity to the genome of the defective phage, PBP180, from B. pumilus strain CCTCC AB9418043. The GR8 strain was inducible by mitomycin C, but the induced phage could not infect the GR8 strain (Supplementary Fig. S5). The induced supernatants were observed by transmission electron microscopy (TEM). Phage particles had an isometric head with a diameter of 27 nm, a neck (19 nm in length), a contractile tail (189 nm in length and 16 mm in width), and a tail needle (60 nm in length) (Fig. 4B). The phage particles were purified, and the DNA was extracted as described44. Restriction enzyme digestions of the DNA extracted from the phage particles showed that the DNA could not be digested into separated bands (Supplementary Fig. S6). According to previous reports, the prophage PBSX induced from Bacillus subtilis could randomly pack chromosomal DNA45. We speculated that the induced phage from GR8 exhibited the same feature as prophage PBSX and the capsids packed the chromosomal DNA randomly.
Figure 4.

Analysis of prophages in the GR8 genome. The prophage genome in the GR8 chromosome was predicted using PHAST. (A) Schematic diagrams of the three predicted prophage regions (region 1, region 2, and region 3) in the GR8 genome. The location of each prophage region in the GR8 genome and the functional classification of the predicted prophage proteins are indicated. (B) Morphology of phage particles induced from GR8. The phage particle was negatively stained with 2% potassium phosphotungstate and observed by transmission electron microscopy.
Discussion
As a widely used bacterium for producing bioactive substances with industrial merits or used as a plant growth-promoting rhizobacterium, the biosafety of B. pumilus has been scarcely evaluated11. Although several strains of B. pumilus have been identified as pathogens of plants and humans, the pathogenesis and the genetic foundation of pathogenic B. pumilus was unknown11,15,16. However, the genetic analysis of pathogenic strains is helpful for understanding pathogenesis and to identify novel control strategies.
According to previous report, the GR8 was identified as an opportunistic pathogen of ginger, gaining access via wounds and subsequently invading the cells, and causing the rotting of the rhizome22. The genomic analysis of GR8 provided an understanding to the pathogenesis of the strain. Several genes were found to be involved in the invasion of GR8 to the ginger rhizome (Fig. 5; Supplementary Fig. S7). After invasion, the GR8 strain was located in the extracellular space of the parenchymatous tissue, and numerous genes were responsible for the adaptation of GR8 in the extracellular space. During the growth of GR8 in the extracellular space, the expression and the secretion of enzymes belonging to the CAZyme families caused the degradation of ginger rhizome cells and finally the breakdown of the ginger rhizome tissue around the wounded site. The gene candidates associated with the pathogenesis of GR8 also existed in the genomes of the other completely sequenced B. pumilus strains, B6033 and SAFR-032 (Table 2) and numerous genes among them were associated with human infective diseases caused by bacteria28,30,35. Comparative genomic analysis revealed that the genomes among GR8, B6033 and SAFR-032 were highly similar. The B. pumilus strain B6033 was selected for bio-catalyzing the stereospecific oxidation of β-lactams18. The SAFR-032 B. pumilus strain was isolated from the Spacecraft Assembly Facility at the NASA Jet Propulsion Laboratory, and spores produced by this strain exhibited unusual resistance to UV radiation46. The high genome similarity and the wide existence of the putative pathogenic associated genes in the other strains of B. pumilus used for industrial purposes make the use of B. pumilus debatable, and highlights the need to determine the biosafety of the strains before further industrial use, especially before the use of such strains as plant growth-promoting rhizobacteria.
Figure 5.

Pathogenicity of B. pumilus strain GR8 to ginger rhizome. The putative proteins involved in the infection of ginger rhizome by GR8 are indicated at each stage, and the functions are also shown.
A three-component system, which might provide resistance to antimicrobial peptides, was found based on the genomic analysis of strain GR8, so the microbe-produced antimicrobial peptides might be useless for controlling ginger rhizome rot disease caused by GR8. Thus, development of effective strategies for controlling the disease caused by strain GR8 is necessary. In the past few years, phage was thought to be a promising novel antimicrobial agent against plant pathogenic bacteria, but the defense systems of the bacteria prevent the infection of phage and cause failure of phage therapy47,48. The CRISPR/Cas system and R-M system are two general defense systems of the prokaryotic organism against phage infection. Only a type I R-M system was detected in the GR8 genome, and no CRISPR/Cas system was identified. According to a previous report, phage therapy can efficiently control the ginger rhizome rot caused by GR842. The phage defense systems were also absent in the other genome-sequenced strains of B. pumilus, thus suggesting that phage therapy might also be also useful in the control of human and plant diseases caused by B. pumilus.
Methods
Bacterial strain and culture conditions
The ginger rhizome rot disease pathogen B. pumilus strain GR8 was isolated from soil collected around rotten ginger rhizome located in Weifang City of Shandong Province in China and stored by our laboratory. The strain was grown in Luria-Bertani (LB) broth at 30 °C for 12 h with moderate shaking. The cells were then harvested for genomic DNA extraction. The pathogenesis of B. pumilus strain GR8 to the ginger rhizome was analyzed as previously described22.
Genome sequencing and annotation
The genome of GR8 was extracted as previously described49, and randomly sheared and purified to construct a paired-end library with an insert size of 300 bp. The constructed genome library was then sequenced using Illumina HiSeq2500 (Illumina, California, USA). The genome was assembled into contigs using Velvet v1.2.0750 software, and the gaps between contigs were filled by primer walking. The complete genome of GR8 was annotated using the Prokaryotic Genomes Annotation Pipeline (PGAP; http://www.ncbi.nlm.nih.gov/genome/annotation_prok/) at NCBI.
Bioinformatic analysis
The functions of the proteins were classified by searching the amino acid sequences of the putative proteins against the KEGG Automatic Annotation Server (KAAS, http://www.genome.jp/tools/kaas/). Comparative genomic analysis of the genomes was performed using progressive Mauve51. The CRISPR/Cas system in the genome was identified using the CRISPR Finder server52, and the R-M system in the genome was analyzed based on the data of REBASE41. Carbohydrate-active enzymes (CAZymes) were predicted with the CAZymes Analysis Toolkit (CAT v1.8)53. The genome was visualized using CGView54.
Prophage induction
Mitomycin C (ranging from 0.1 to 1.0 μg/ml with intervals of 0.1 μg/ml) was added to the culture of GR8 at the exponential growth stage in LB broth with a turbidity of 0.8 at 600 nm. After growing for 8 hours, the turbidity at 600 nm of the cultures was detected, and the culture supernatants were collected. The infective ability of the induced phages in the supernatants was determined using the double agar overlay assay method as described before44. The induced phage particles were negative stained with 2% potassium phosphotungstate and were observed by transmission electron microscopy (TEM) (H-7000FA, HITACHI, Tokyo, Japan).
Accession numbers
The chromosome sequence of B. pumilus GR8 and the plasmid sequence of the B. pumilus GR8 plasmid (pGR8) were deposited in the NCBI database under the accession numbers CP009108 and CP009109, respectively.
Author Contributions
M.G. conceived and designed the project; Y.H. performed the experiments and analyzed the data; Y.H. drafted the manuscript; M.G. reviewed, revised, and edited the manuscript. All authors have read and approved the final manuscript.
Additional Information
How to cite this article: Yuan, Y. and Gao, M. Genomic analysis of a ginger pathogen Bacillus pumilus providing the understanding to the pathogenesis and the novel control strategy. Sci. Rep. 5, 10259; doi: 10.1038/srep10259 (2015).
Supplementary Material
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No.31170123) and the projects of Chinese Academy of Sciences (KSZD-EW-Z-021-2-2).
References
- Fritze D. Taxonomy of the genus Bacillus and related genera: The aerobic endospore-forming bacteria. Phytopathology 94, 1245–1248 (2004). [DOI] [PubMed] [Google Scholar]
- Hua D. L. et al. Biotransformation of isoeugenol to vanillin by a newly isolated Bacillus pumilus strain: Identification of major metabolites. J. Biotechnol. 130, 463–470 (2007). [DOI] [PubMed] [Google Scholar]
- Huang Q., Peng Y., Li X., Wang H. F. & Zhang Y. Z. Purification and characterization of an extracellular alkaline serine protease with dehairing function from Bacillus pumilus. Curr. Microbiol. 46, 169–173 (2003). [DOI] [PubMed] [Google Scholar]
- Rajput R. & Gupta R. Expression of Bacillus pumilus keratinase rK(27) inBacillus subtilis: enzyme application for developing renewable flocculants from bone meal. Ann. Microbiol. 64, 1257–1266 (2014). [Google Scholar]
- Thomas L., Ushasree M. V. & Pandey A. An alkali-thermostable xylanase from Bacillus pumilus functionally expressed in Kluyveromyces lactis and evaluation of its deinking efficiency. Bioresource. Technol. 165, 309–313 (2014). [DOI] [PubMed] [Google Scholar]
- Nwokoro O. & Dibua M. E. U. Degradation of soil cyanide by single and mixed cultures of Pseudomonas stutzeri and Bacillus subtilis. Arh. Hig. Rada Toksiko 65, 113–119 (2014). [DOI] [PubMed] [Google Scholar]
- Saranya P., Kumari H. S., Rao B. P. & Sekaran G. Lipase production from a novel thermo-tolerant and extreme acidophile Bacillus pumilus using palm oil as the substrate and treatment of palm oil-containing wastewater. Environ. Sci. Pollut. R 21, 3907–3919 (2014). [DOI] [PubMed] [Google Scholar]
- Ramzan N., Noreen N. & Shahzad S. Inhibition of in Vitro Growth of Soil-Borne Pathogens by Compost-Inhabiting Indigenous Bacteria and Fungi. Pak. J. Bot. 46, 1093–1099 (2014). [Google Scholar]
- Myresiotis C. K., Vryzas Z. & Papadopoulou-Mourkidou E. Enhanced root uptake of acibenzolar-S-methyl (ASM) by tomato plants inoculated with selected Bacillus plant growth-promoting rhizobacteria (PGPR). Appl. Soil Ecol. 77, 26–33 (2014). [Google Scholar]
- Tiwari S., Singh S. N. & Garg S. K. Bacterial Reduction of Cr(VI) and Fe(III) in In Vitro Conditions. Bioremediat. J. 18, 158–168 (2014). [Google Scholar]
- Logan N. A. Bacillus and relatives in foodborne illness. J. Appl. Microbiol. 112, 417–429 (2012). [DOI] [PubMed] [Google Scholar]
- Larsen N. et al. Characterization of Bacillus spp. strains for use as probiotic additives in pig feed. Appl. Microbiol. Biot. 98, 1105–1118 (2014). [DOI] [PubMed] [Google Scholar]
- Nieminen T. et al. Toxinogenic Bacillus pumilus and Bacillus licheniformis from mastitic milk. Vet. Microbiol. 124, 329–339 (2007). [DOI] [PubMed] [Google Scholar]
- Suominen I. et al. Toxic Bacillus pumilus from indoor air, recycled paper pulp, Norway spruce, food poisoning outbreaks and clinical samples. Syst. Appl. Microbiol. 24, 267–276 (2001). [DOI] [PubMed] [Google Scholar]
- Kimouli M. et al. Two cases of severe sepsis caused by Bacillus pumilus in neonatal infants. J. Med. Microbiol. 61, 596–599 (2012). [DOI] [PubMed] [Google Scholar]
- Galal A. A., A.A.E.-B.a.J.J. Bacillus pumilus, a new pathogen on mango plants. Egypt. J. Phytopathol. 34, 17–29 (2006). [Google Scholar]
- Li B. et al. Association of a Bacillus Species with Leaf and Twig Dieback of Asian Pear (Pyrus Pyrifolia) in China. J. Plant. Pathol. 91, 705–708 (2009). [Google Scholar]
- Villanueva J., Switala J., Ivancich A. & Loewen P. C. Genome Sequence of Bacillus pumilus MTCC B6033. Genome Announc. 2, e00327–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos D. F. et al. Draft Genome Sequence of Bacillus pumilus CCMA-560, Isolated from an Oil-Contaminated Mangrove Swamp. Genome Announc. 1, e00707–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagimardanova E. I. et al. Draft Genome Sequence of Bacillus pumilus 7P, Isolated from the Soil of the Tatarstan Republic, Russia. Genome Announc. 2, e00599–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C. W., Wang H. Y., Zhang Y. Z. & Feng H. Draft genome sequence of Bacillus pumilus BA06, a producer of alkaline serine protease with leather-dehairing function. J. Bacteriol. 194, 6668–6669 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q., Yuan Y. & Gao M. Bacillus pumilus, a Novel Ginger Rhizome Rot Pathogen in China. Plant Dis. 97, 1308–1315 (2013). [DOI] [PubMed] [Google Scholar]
- Brunner P. C., Torriani S. F., Croll D., Stukenbrock E. H. & McDonald B. A. Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol. Biol. Evol. 30, 1337–1347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott D. W. & Boraston A. B. Structural biology of pectin degradation by Enterobacteriaceae. Microbiol. Mol. Biol. R. 72, 301–316 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A. C. & Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–185 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T., Inagaki H., Takai R., Hirai H. & Che F. S. Two distinct EF-Tu epitopes induce immune responses in rice and Arabidopsis. Mol. Plant Microbe In. 27, 113–124 (2014). [DOI] [PubMed] [Google Scholar]
- Rossez Y. et al. Flagella interact with ionic plant lipids to mediate adherence of pathogenic Escherichia coli to fresh produce plants. Environ. Microbiol. 16, 2181–2195 (2014). [DOI] [PubMed] [Google Scholar]
- Martinez-Climent J. A. The origin and targeting of mucosa-associated lymphoid tissue lymphomas. Curr. Opin. Hematol. 21, 309–319 (2014). [DOI] [PubMed] [Google Scholar]
- Shin S. & Roy C. R. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell. Microbiol. 10, 1209–1220 (2008). [DOI] [PubMed] [Google Scholar]
- Newton H. J., Ang D. K. Y., van Driel I. R. & Hartland E. L. Molecular Pathogenesis of Infections Caused by Legionella pneumophila. Clin. Microbiol. Rev. 23, 274–298 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mur L. A. J., Carver T. L. W. & Prats E. NO way to live; the various roles of nitric oxide in plant-pathogen interactions. J. Exp. Bot. 57, 489–505 (2006). [DOI] [PubMed] [Google Scholar]
- Tucker N. P., Le Brun N. E., Dixon R. & Hutchings M. I. There’s NO stopping NsrR, a global regulator of the bacterial NO stress response. Trends Microbiol. 18, 149–156 (2010). [DOI] [PubMed] [Google Scholar]
- Li M. et al. Gram-positive three-component antimicrobial peptide-sensing system. Proc. Natl. Acad. Sci. USA 104, 9469–9474 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre P. et al. Three different putative phosphate transport receptors are encoded by the Mycobacterium tuberculosis genome and are present at the surface of Mycobacterium bovis BCG. J. Bacteriol. 179, 2900–2906 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trappetti C., Potter A. J., Paton A. W., Oggioni M. R. & Paton J. C. LuxS Mediates Iron-Dependent Biofilm Formation, Competence, and Fratricide in Streptococcus pneumoniae. Infect, Immun, 79, 4550–4558 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh B., Jones J. B., Iriarte F. B. & Momol M. T. Phage Therapy for Plant Disease Control. Curr. Pharm. Biotechno. 11, 48–57 (2010). [DOI] [PubMed] [Google Scholar]
- Jones J. B. et al. Bacteriophages for plant disease control. Annu. Rev. Phytopathol. 45, 245–262 (2007). [DOI] [PubMed] [Google Scholar]
- Gasiunas G., Sinkunas T. & Siksnys V. Molecular mechanisms of CRISPR-mediated microbial immunity. Cell. Mol. Life. Sci. 71, 449–465 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F. & Xu P. Genomic analysis of thermophilic Bacillus coagulans strains: efficient producers for platform bio-chemicals. Sci. Rep. 4, 3926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q. L., Tun H. M., Leung F. C. C. & Shah N. P. Genomic insights into high exopolysaccharide-producing dairy starter bacterium Streptococcus thermophilus ASCC 1275. Sci. Rep. 4, 4974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. J., Vincze T., Posfai J. & Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 38, D234–D236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y. et al. Effects of Actin-Like Proteins Encoded by Two Bacillus pumilus Phages on Unstable Lysogeny, Revealed by Genomic Analysis. Appl. Environ.l Microbiol. 81, 339–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T. T. et al. Biological and genomic analysis of a PBSX-like defective phage induced from Bacillus pumilus AB94180. Arch. Virol. 159, 739–752 (2014). [DOI] [PubMed] [Google Scholar]
- Yuan Y. H., Gao M. Y., Wu D. D., Liu P. M. & Wu Y. Genome Characteristics of a Novel Phage from Bacillus thuringiensis Showing High Similarity with Phage from Bacillus cereus. Plos One 7, e37557 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh S., OReilly M., Nolan N. & Devine K. M. The phage-like element PBSX and part of the skin element, which are resident at different locations on the Bacillus subtilis chromosome, are highly homologous. Microbiol-Uk 142, 2031–2040 (1996). [DOI] [PubMed] [Google Scholar]
- Tirumalai M. R. et al. Candidate Genes That May Be Responsible for the Unusual Resistances Exhibited by Bacillus pumilus SAFR-032 Spores. Plos One 8, e66012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y. H., Peng Q. & Gao M. Y. Characteristics of a broad lytic spectrum endolysin from phage BtCS33 of Bacillus thuringiensis. BMC Microbiol. 12, 297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermoso J. A., Garcia J. L. & Garcia P. Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr. Opin. Microbiol. 10, 461–472 (2007). [DOI] [PubMed] [Google Scholar]
- Gao M. Y., Li R. S., Dai S. Y., Wu Y. & Yi D. Diversity of Bacillus thuringiensis strains from soil in China and their pesticidal activities. Biol. Control. 44, 380–388 (2008). [Google Scholar]
- Zerbino D. R. & Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. C. E., Mau B., Blattner F. R. & Perna N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I., Vergnaud G. & Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B. H., Karpinets T. V., Syed M. H., Leuze M. R. & Uberbacher E. C. CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20, 1574–1584 (2010). [DOI] [PubMed] [Google Scholar]
- Stothard P. & Wishart D. S. Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539 (2005). [DOI] [PubMed] [Google Scholar]
- Vockler C. J., Greenfield P., Tran-Dinh N. & Midgley D. J. Draft Genome Sequence of Bacillus pumilus Fairview, an Isolate Recovered from a Microbial Methanogenic Enrichment of Coal Seam Gas Formation Water from Queensland, Australia. Genome Announc. 2, e00279–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T., Ciufo S., Fedorov B., O’Neill K. & Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42, D553–559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.