

Summary
Eubacterium rectale is a prominent human gut symbiont yet little is known about the molecular strategies this bacterium has developed to acquire nutrients within the competitive gut ecosystem. Starch is one of the most abundant glycans in the human diet, and E. rectale increases in vivo when the host consumes a diet rich in resistant starch, although it is not a primary degrader of this glycan. Here we present the results of a quantitative proteomics study in which we identify two glycoside hydrolase 13 family enzymes, and three ABC transporter solute-binding proteins that are abundant during growth on starch and, we hypothesize, work together at the cell surface to degrade starch and capture the released maltooligosaccharides. EUR_21100 is a multidomain cell wall anchored amylase that preferentially targets starch polysaccharides, liberating maltotetraose, while the membrane associated maltogenic amylase EUR_01860 breaks down maltooligosaccharides longer than maltotriose. The three solute-binding proteins display a range of glycan-binding specificities that ensure the capture of glucose through maltoheptaose and some α1,6-branched glycans. Taken together, we describe a pathway for starch utilization by E. rectale DSM 17629 that may be conserved among other starch-degrading Clostridium cluster XIVa organisms in the human gut.
Keywords: microbiota, starch, Eubacterium rectale, solute-binding proteins, amylase, protein crystallography
Introduction
The human gut microbiota is established soon after birth and plays several important roles in human health by guiding the development of the immune (Mazmanian et al., 2005) and gastrointestinal systems (Stappenbeck et al., 2002), preventing enteric pathogen invasion (Mazmanian et al., 2008), and degrading and fermenting dietary glycans into host-absorbable short chain fatty acids (SCFAs). The composition of the gut community is largely bacterial, and despite the presence of hundreds of species, it is dominated by two bacterial phyla, the Bacteroidetes and Firmicutes (Eckburg et al., 2005, Ley et al., 2006a, Ley et al., 2006b). Key to the survival of these organisms in the mammalian intestinal tract is their ability to forage carbohydrate nutrition of dietary or host origin, making diet a controllable variable that shapes the human gut microbial community composition and metabolic output (Koropatkin et al., 2012).
Eubacterium rectale is a member of the Clostridium cluster XIVa group of the Firmicutes (Duncan & Flint, 2008), and is a common member of the human gut microbiota (Qin et al., Arumugam et al., 2011, Hold et al., 2003, Kraal et al., 2014). E. rectale as well as other members of this cluster produce the short chain fatty acid butyrate as a primary fermentation end-product (Pryde et al., 2002). Butyrate is not only the primary fuel source of colonocytes in the intestinal tract, but also a potent anti-inflammatory (Segain et al., 2000) and anti-tumorigenic molecule, thus making butyrate-producing organisms in the intestinal tract of special interest in the prevention and treatment of inflammatory bowel diseases as well as colorectal cancers (Hamer et al., 2008, Van Immerseel et al., 2010). Furthermore, the abundance of butyrate-producing organisms is observed to be markedly decreased in individuals suffering from several intestinal and systemic diseases, such as ulcerative colitis (Machiels et al., 2013, Morgan et al., 2012, Nemoto et al., 2012), Crohn’s Disease (Gevers et al., 2014, Kang et al., 2010, Willing et al., 2010, Frank et al., 2007, Frank et al., 2011), colorectal cancer (Wu et al., 2013), and diabetes (Peng et al., 2014, Larsen et al., 2010, Karlsson et al., 2013, Qin et al., 2012) suggesting the importance of these organisms in maintaining optimal health.
Compared to members of the Bacteroidetes phylum, E. rectale and other members of the Clostridium cluster XIVa, possess a smaller repertoire of glycoside hydrolases, suggesting a limited ability to degrade a variety of complex polysaccharides (El Kaoutari et al., 2013). The abundance of Clostridium cluster XIVa organisms tracks closely with the prevalence of plant polysaccharides in the diet in both human and animal studies, with the proportion of these organisms significantly decreased on low-polysaccharide diets (Duncan et al., 2007, Walker et al., 2011, Mahowald et al., 2009). More specifically, diets supplemented with resistant starch, a general term describing dietary starch that is relatively inaccessible to human (gluco)amylases due to its physical structure, cause an increase in the abundance of these organisms (Martinez et al., 2010, Conlon et al., 2012). Dietary resistant starch (RS) has been demonstrated to increase the relative butyrate production by the gut microbiota in both in vivo (Conlon et al., 2012, Abell et al., 2008, van Munster et al., 1994) and in vitro fermentation studies (Wang et al., 2004). An increase in butyrate may explain why dietary RS has been shown to be protective against DNA damage and the subsequent development of colorectal cancer in a mouse model of azoxymethane-induced colorectal cancer (Clarke et al., 2008).
One explanation for the increase in butyrate output and Clostridium cluster XIVa abundance from the fermentation of RS is that these organisms are primary degraders of the material and possess the enzymatic machinery for degradation. This hypothesis was supported by earlier work demonstrating that that these species adhere to resistant starch particles and are often associated with the insoluble dietary material found in feces (Leitch et al., 2007, Walker et al., 2008). However, more recent work has demonstrated that E. rectale in monoculture cannot utilize RS, but rather another organism such as Ruminococcus bromii is required to begin the initial degradation of the RS granules, liberating material that E. rectale can utilize (Ze et al., 2012). When R. bromii and E. rectale are grown in co-culture with resistant starch as the sole carbon source, E. rectale becomes 3-fold more abundant than R. bromii after 48 h of growth, suggesting that E. rectale is adept at scavenging the starch, maltooligosaccharides or glucose liberated by the amylotytic activity of R. bromii. However, the molecular details of nutrient uptake and scavenging by E. rectale are poorly understood. Understanding how these organisms scavenge starch and contribute to its degradation and consumption by the gut microbiome can lead to advances in the development of prebiotics to specifically increase the abundance of these dominant butyrate-producers.
In order to identify the proteins involved in starch utilization by E. rectale DSM 17629 (i.e. E. rectale A1-86) we performed a quantitative proteomics analysis on combined cell wall and membrane fractions and biochemically characterized several ATP-binding cassette (ABC) solute-binding proteins and two glycoside hydrolases abundant during cell growth on starch. Using these data we propose a model for starch utilization by E. rectale that involves the coordinated action of at least one extracellular amylase that hydrolyzes starch into maltooligosaccharides that are recognized by at least two of the ABC solute-binding proteins.
Results
Growth on starch and maltooligosaccharides
E. rectale was cultured in 96 well plates in an anaerobic chamber to determine its ability to utilize various starches and maltooligosaccharides (Figure 1). Corroborating previous reports, E. rectale grows rapidly on branched, soluble starch polysaccharides including potato amylopectin (APP), maize amylopectin (APM), and glycogen, but much less efficiently on autoclaved Hylon VII (H7), a commercial resistant corn starch comprised of 70% amylose (Duncan & Flint, 2008, Ze et al., 2012). It cannot grow at all on pullulan, a fungal cell wall component comprised of α1,6-linked maltotriose, but can grow on pullulan oligosaccharides including glucosyl-α1,6-maltotriose-α1,6-maltotriose (GMM) and glucosyl-α1,6-maltotriose (GM). While it was previously reported that E. rectale cannot grow on isomaltose, we demonstrate it can grow on the trisaccharide panose (glucose-α1,6-glucose-α1,4-glucose), although at a reduced growth rate. E. rectale also grows well on maltose, maltotriose (G3) and maltoheptaose (G7), but not on α-cyclodextrin (aCD) or β-cyclodextrin (bCD).
Figure 1. Growth of E. rectale DSM 17629 on starch and maltooligosaccharides.

Bacteria were grown in 96 well plates in a 37°C anaerobic chamber with the OD600 automatically recorded every 20 min. YCFA media was supplemented with 2 mg mL−1 of the listed carbohydrates as the sole carbon source. A. Growth on starches (APM, maize amylopectin, APP, potato amylopectin, H7, Hylon VII); water is displayed as a negative control. Note that autoclaved H7 is a turbid suspension that was not blanked in this set of experiments. B. Growth on maltooligosaccharides (G3, maltotriose; G7, maltoheptaose; aCD, α-cyclodextrin; bCD, β-cyclodextrin; GM, glucosyl-α1,6-maltotriose; GMM, glucosyl-α1,6-maltotriose-α1,6-maltotriose).
Quantitative Proteomic Analysis of Cell Wall/Membrane samples
To identify enzymes and transporters key for the growth of E. rectale on starch we performed a quantitative proteomic analysis of combined cell membrane/cell wall samples. The bacteria were grown in Hungate tubes on YCFA media containing either 2 mg mL−1 of glucose (GLU), autoclaved APM or autoclaved H7 as the sole carbon source. Amylopectin is a soluble component of corn starch by virtue of its regular α1,6-linked branch points, while H7 is an insoluble corn starch which is resistant to degradation due to its high amylose content; these starches differ in their physiochemical properties and are representative of the prominent forms of starch in the human diet. Biological triplicates of each culture grown in APM, H7 or GLU were harvested at mid-log phase (OD600 = 0.6) for combined cell membrane/cell wall extraction. Among all nine samples, a total of 828 proteins were detected (Supplemental Table S1). Seven out of nine samples had a false discovery rate (FDR) of 0%, and one H7 and one APM sample each had an FDR of 0.2%. Of the 828 proteins, 421 are predicted by PSORTb 3.0 to be localized to the cytoplasm, 6 to the cell wall, 258 to the cell membrane, 12 to extracellular environment, and 133 were annotated as having an unknown location. That just over half of the proteins identified have a predicted cytoplasmic localization was not surprising, as we intentionally used wash conditions to preserve cell membrane and cell wall associated proteins. Of the cytoplasmic proteins, 132 had predicted roles in housekeeping functions such as transcription, translation and replication. In addition, proteins of central metabolic importance were observed; such proteins have been observed to associate with the cell wall and membrane in diverse species such as Clostridium thermocellum (Yu et al., 2012), and Streptococcus oralis (Maeda et al., 2013) and thus these proteins in E. rectale may normally associate with the cell membrane.
Protein abundance was calculated using the normalized spectral abundance factor (NSAF, see Experimental Procedures), which reports the relative abundance of the identified protein within a sample, normalized for the molecular weight of the protein. Spectral counting including NSAF is a well-documented quantity for measuring the relative abundance of proteins within and between samples, and can accurately determine protein abundance changes of 2.5-fold (Colinge et al., 2005, Old et al., 2005). We therefore set a stringent cutoff of 3-fold for proteins reported as more abundant on either starch or glucose grown cells.
ABC transporters and glycoside hydrolases upregulated during growth on starch
A total of 24 proteins were identified that increased 3-fold or more in abundance during growth on either H7 or APM (or both) compared to growth on glucose (Supplemental Table S2) and eight of these have predicted functions in carbohydrate metabolism and transport (COG G; Table 1). Six of these proteins are predicted to be components of ABC transporters, and two are annotated as members of glycoside hydrolase family 13 (GH13), comprising enzymes that target glycosidic linkages in starch. ABC transporters involved in bacterial nutrient import are typically comprised of three functional proteins or domains: a solute-binding protein that binds the molecule to be imported, one or more transmembrane permeases to which the solute-binding protein docks and releases the bound molecule, and a cytosolic ATPase that hydrolyzes ATP to induce a conformational change in the permease allowing solute passage into the cytoplasm (Rees et al., 2009).
Table 1.
COG G Proteins upregulated +/− 3-fold or more in APM or H7 compared to GLU
| Accession number |
Protein name | Locus tag | MW (kDa) |
PSORTb 3.0 |
H7 NSAF % |
APM NSAF % |
GLU NSAF % |
Fold- change H7/GLU |
Fold- change APM/GLU |
|---|---|---|---|---|---|---|---|---|---|
| D6DYI9 | Glycosidases; GH13 | EUR_21100 | 150 | CW | 0.19 ± 0.03 | 0.12 ± 0.02 | 0.00 ± 0.00 | NA | NA |
| D6E1Y1 | Carbohydrate ABC transporter substrate- binding protein, CUT1 family | EUR_01830 | 45 | U | 2.36 ± 0.11 | 1.69 ± 0.15 | 0.36 ± 0.02 | 6.5 | 4.7 |
| D6E1Y2 | Carbohydrate ABC transporter membrane protein 1, CUT1 family | EUR_01840 | 51 | CM | 0.32 ± 0.02 | 0.24 ± 0.03 | 0.07 ± 0.01 | 4.7 | 3.6 |
| D6E1Y3 | Carbohydrate ABC transporter membrane protein 2, CUT1 family | EUR_01850 | 33 | CM | 0.16 ± 0.03 | 0.13 ± 0.05 | 0.05 ± 0.01 | 3.6 | 3.0 |
| D6E1Y4 | Glycosidases; GH13 | EUR_01860 | 63 | U | 0.17 ± 0.02 | 0.13 ± 0.02 | 0.01 ± 0.02 | 18.3 | 13.3 |
| D6DZP3 | Carbohydrate ABC transporter substrate- binding protein | EUR_31480 | 51 | U | 1.30 ± 0.02 | 0.98 ± 0.08 | 0.41 ± 0.04 | 3.1 | 2.4 |
| D6DZP5 | Carbohydrate ABC transporter membrane protein 2, CUT1 family | EUR_31500 | 34 | CM | 0.14 ± 0.03 | 0.09 ± 0.03 | 0.03 ± 0.03 | 4.5 | 2.9 |
| D6DYI1 | Carbohydrate ABC transporter substrate- binding protein, CUT1 family | EUR_21010 | 51 | U | 0.10 ± 0.02 | 0.03 ± 0.01 | 0.02 ± 0.02 | 5.7 | 2.0 |
| D6DYG6 | Glycogen phosphorylase (EC 2.4.1.1) | EUR_20850 | 94 | C | 0.00 ± 0.000 | 0.00 ± 0.01 | 0.03 ± 0.00 | NA | −7.1 |
| D6E1I9 | Phosphoenolpyruvate- protein | EUR_00180 | 61 | C | 0.00 ± 0.00% | 0.02 ± 0.02 | 0.13 ± 0.01 | NA | −5.9 |
| D6DYZ9 | 1,4-alpha-glucan branching enzyme GlgB | EUR_28650 | 76 | C | 0.00 ± 0.00% | 0.09 ± 0.03% | 0.13 ± 0.00 | NA | −1.5 |
| D6E5J9 | Glycogen synthase (EC 2.4.1.21) | EUR_16230 | 56 | U | 0.10 ± 0.05 | 0.36 ± 0.06 | 0.34 ± 0.02 | −3.4 | 1.0 |
Proteins of COG G (Carbohydrate transport and metabolism) displayed. Fold change was calculated based on the mean NSAF from three replicates. Only samples with p-value < 0.05 by student’s t-test of APM:GLU or H7:GLU reported. Only proteins with 5 or more total spectra in at least one set of samples (H7 or APM, or GLU for inverse calculation) were included. PSORTb 3.0 was used to predict cellular location: U=Unknown, CM=Cell Membrane, C=Cytoplasmic, CW=Cell Wall. NA = not applicable. Rows in bold print denote proteins that were selected for further study.
EUR_01830 is a predicted carbohydrate ABC transporter solute-binding protein, and the most abundant protein differentially expressed during growth on starch, displaying an NSAF of 2.36 ± 0.11% in H7 and 1.69 ± 0.15% in APM and fold changes of 6.5 and 4.7 on H7 and APM respectively, compared to glucose. A BLAST search of this protein against the PDB reveals ~25% sequence identity to several maltose and maltooligosaccharide binding proteins including those from Thermoactinomyces vulgaris (pdb 2ZYK, (Matsumoto et al., 2009)) and Streptococcus pneumoniae (pdb 2XD2,(Abbott et al., 2010)). Within the E. rectale DSM 17629 genome, EUR_01830 shares 50% amino acid sequence similarity with EUR_14180, which is annotated as a MalE (maltose-binding protein) homologue, although it is found at low levels in our samples, and elevated during growth on glucose (Supplemental Table S1). EUR_01830 is encoded in a putative operon with EUR_01840 and EUR_01850, both predicted ABC membrane permeases, as well as EUR_01860, a GH13 glycosidase, all of which were differentially expressed in starch-grown cells (Figure 2). A BLAST search of EUR_01860 against amino acid sequences in the PDB reveals 35% identity (47% similarity) to a characterized α-amylase from Halothermothrix orenii (pdb 1WZA, (Sivakumar et al., 2006)), as well as 33% identity (46% similarity) to the catalytic domain of the surface amylase SusG from Bacteroides thetaiotaomicron (Koropatkin & Smith, 2010). EUR_01860 is predicted to have an N-terminal transmembrane helix (TMHMM server v 2.0, http://www.cbs.dtu.dk/services/TMHMM/), suggesting it is anchored to the cell membrane (Figure 3). While the genomic organization indicates that EUR_01830 through EUR_01860 denote a single operon, EUR_01860 is upregulated several fold more than the ABC transporter proteins, with fold-changes of 18.3 and 13.3 for growth in H7 and APM respectively compared to GLU.
Figure 2. Genomic neighborhood of the genes encoding the differentially expressed proteins detected during growth on starch.

Each gene is drawn to scale as a rectangle with its orientation indicated by the closed triangle. Genes have been color coded according to annotated function (IMG: img.jgi.doe.gov), or labeled: efflux protein MATE, multi-antimicrobial efflux protein pump; DMT permease, drug metabolite transporter/permease; RNRII, ribonucleotide reductase class II activase subunit; TrxB, thioredoxin; ABC ATPase, ABC transporter ATP-binding protein.
Figure 3. Domain organization of EUR_21100 and EUR_01860.
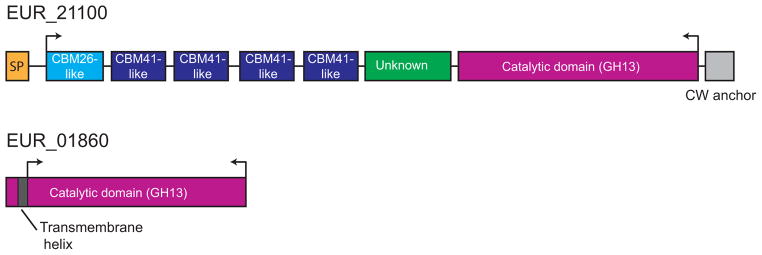
EUR_21100 is multimodular with several N-terminal domains that resemble CBMs (via BLAST search against isolated CBM sequences). Between the putative CBMs and the catalytic module (also GH13) there is a region of unknown function that does not resemble any previously characterized domains. At the C-terminal end there is a putative cell wall anchoring motif that likely attaches this protein to the peptidoglycan layer. In contrast, EUR_01860 consists of a single module, a GH13 catalytic domain, with an N-terminal transmembrane helix. Arrows indicate regions of the proteins that were cloned for biochemical characterization.
EUR_31480 is the second most abundant protein expressed during growth on starch, although the relative fold change compared to growth on glucose (3.1-fold and 2.7-fold for H7 and APM growth respectively) is lower than proteins of the EUR_01830-01860 operon. Of proteins in the PDB, EUR_31480 shares significant sequence homology (>20% identity, >35% similarity) over most of its length with the ABC xylo-oligosaccharide binding protein from Caldanaerobius polysaccharolyticus (4G68, (Han et al., 2012)) as well as the ABC acarbose-binding protein GacH from Streptomyces glaucescens (3JYR,(Vahedi-Faridi et al., 2010). The gene encoding EUR_31480 is part of a putative operon containing a predicted ABC permease (EUR_31500) and ATPase (EUR_31490), both of which were identified in the starch-grown cell samples at lower relative abundance. Just downstream of this gene cluster resides a predicted LacI-family transcriptional regulator (EUR_31510), a predicted permease (EUR_31520), a predicted multi-antimicrobial efflux (MATE) family pump (EUR_31530), and a gene of unknown function (EUR_31540). While both EUR_31520 and EUR_31540 are predicted to localize to the cell membrane, they were barely above the threshold for detection (2 unique peptides) in any of the samples.
While much less abundant than the aforementioned proteins, the solute-binding protein EUR_21010 is 5.7-fold more abundant in H7 grown cells, though only 2-fold more abundant in APM cells compared to GLU grown cells. Genomic context suggests it is also expressed within a genetic cluster encoding two predicted ABC membrane permeases (EUR_21020 and EUR_21030). Downstream of the ABC permease genes are three additional genes, which may or may not be co-transcribed with EUR_21010: EUR_21040, a predicted drug metabolite transporter (DMT); EUR_21050, a predicted ribonucleotide-triphosphate reductase class II activase subunit (RNRII); and EUR_21060, a hypothetical protein of unknown function. Upstream of EUR_21010 there is a predicted LacI family transcriptional regulator, EUR_21000, and a predicted cellobiose phosphorylase, EUR_20990. Surprisingly, despite the wealth of protein structures that have been solved for ABC solute-binding proteins, a BLAST search of the EUR_21010 amino acid sequence of to the PDB did not convey homology to any other structurally characterized solute-binding protein.
EUR_21100, a predicted GH13 family enzyme, is detected only in starch grown cells and is comprised of 1365 amino acids, with a putative cell wall sorting signal (APKSGD, residues 1329 to 1334), that may be recognized via a SrtA or SrtB pathway (Comfort & Clubb, 2004). Based upon comparative sequence analysis with known starch-binding carbohydrate-binding module (CBM) families, we predict that this amylase has several tandem CBMs most closely resembling the starch-specific families CBM26 (BLAST E-value 2e−5) and CBM41 (BLAST Evalue 1e−5–5e−7) at its N-terminus, with the catalytic domain at the C-terminus (Figure 3). While this degree of similarity is not sufficient to assign the N-terminal domains of EUR_21100 to these particular CBM families, it does suggest they perform a similar function, i.e. carbohydrate binding. The remaining genes adjacent to EUR_21100 encode a putative transcriptional regulator, a protein of unknown function, and a thioredoxin (TrxB). Of these genes, the protein product for the transcriptional regulator EUR_21110 was observed at the limit of detection (2 spectra) in two of the glucose samples, and not at all in any other samples.
While secondary to the goal of this study, we performed an inverse analysis of the proteins 3-fold or more abundant during growth on GLU as opposed to either APM or H7 (Table 1, Supplemental Table S3). We did not detect any putative glucose transporters in this analysis, but rather noted several cytoplasmic enzymes involved in the regulation of glycogen synthesis. All of these proteins are of relatively low abundance compared to the aforementioned proteins that are upregulated during cell growth on starch. EUR_00180 is a predicted phosphoenolpyruvate (PEP) phosphotransferase that cleaves phosphate from PEP, and is ultimately used to phosphorylate monosaccharides like glucose that are imported into the cytoplasm. The other proteins, EUR_20850 (glycogen phosphorylase), EUR_28650 (glycogen branching enzyme, GlgB), and EUR_16230 (glycogen synthase, GlgA), are involved in the regulation of cellular glycogen levels. Glycogen synthase EUR_16230 abundance was not significantly different between APM and glucose-grown cells, and E. rectale’s growth rate on these carbon sources is similar, while the cells grow more slowly on H7 which contains a higher percentage of RS. It is possible that during exponential growth on more readily accessible carbon sources like glucose, E. rectale, like some other bacteria, begins to synthesize the carbon storage molecule glycogen (Gibbons & Kapsimalis, 1963, Wilson et al., 2010) This may contribute to E. rectale’s prominence and survival in the gut, despite its relatively limited repertoire of carbohydrates that it can utilize compared to other gut bacteria. It should be noted that another proteomic investigation of the related bacterium Roseburia inulinovorans also revealed an upregulation of glycogen phosphorylase during growth on starch vs growth on the plant fructan inulin (Scott et al., 2011).
Because some enzymes produce glucose directly during starch digestion, we looked for putative glucose-specific transporters in our data sets that fell below the stringent 3-fold cut off (Table 2). Several proteins encoding phosphotransferase system (PTS) components (EUR_10290, and EUR_28570) are found in samples with an NSAF of 0.1% or greater and slightly higher in abundance on glucose compared to starch grown cells. These proteins are annotated as components of glucose and fructose-targeting PTS systems respectively. The predicted ABC solute-binding proteins EUR_31580 and EUR_14180 are nearly 2-fold more abundant in GLU compared to APM grown cells. As mentioned earlier, EUR_14180 is annotated as MalE (maltose-binding protein), but the target ligand for EUR_31580 cannot be predicted. EUR_08690 and EUR_08700 comprise parts of a PTS that are slightly more abundant in starch-grown cells over glucose-grown cells. EUR_08700 is annotated as a putative PTS IIC permease that may recognize the sugars maltose or glucose for transport across the cell membrane. Of the ABC transport solute-binding proteins, EUR_01240, a putative glucose-galactose binding protein, displays a relatively high abundance in all three growth conditions, though somewhat elevated in starch-grown cells, and so we included this protein in our list of potential maltose/malto-oligosaccharide targeting transporters. A BLAST search of the EUR_01240 amino acid sequence to the PDB revealed it is 30% identical (44% similar) to the ABC glucose-binding protein from E. coli (pdb 2HPH) and 31% identical (44% similar) to the ABC glucose/galactose-binding protein from S. typhimurium (pdb 1GCA) (Mowbray et al., 1990). Curiously EUR_01240 has an extra ~100 amino acids at its C-terminus compared to these well-characterized binding proteins. Like the aforementioned solute-binding proteins, Eur_01240 is part of a putative operon encoding both an ABC ATPase and membrane permease (Figure 2).
Table 2.
Additional proteins identified in all data sets with a predicted role in carbohydrate uptake
| Accession number |
Protein name | Locus tag | MW (kDa) |
PSORTb 3.0 |
H7 NSAF % |
APM NSAF % |
GLU NSAF % |
Fold- change H7/GLU |
Fold- change APM/GLU |
H7:GLU t-test p-value |
APM:GLU t-test p-value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D6E1S8 | Glucose/Galactose ABC transporter substrate-binding protein | EUR_01240 | 46 | U | 1.156 ± 0.04 | 0.763 ± 0.042 | 0.522 ± 0.048 | 2.21 | 1.46 | 0.000 | 0.003 |
| D6E1S9 | Monosaccharide ABC transporter ATP-binding protein | EUR_01250 | 56 | CM | 0.691 ± 0.061 | 0.355 ± 0.042 | 0.471 ± 0.065 | 1.47 | 0.75 | 0.013 | 0.061 |
| D6E1T0 | Monosaccharide ABC transporter membrane protein | EUR_01260 | 60 | CM | 0.754 ± 0.092 | 0.490 ± 0.015 | 0.285 ± 0.083 | 2.65 | 1.72 | 0.003 | 0.013 |
| D6E3P2 | PTS system IIA component, Glc family | EUR_08690 | 17 | CM | 0.134 ± 0.012 | 0.104 ± 0.023 | 0.120 ± 0.027 | 1.12 | 0.87 | 0.466 | 0.488 |
| D6E3P3 | PTS system N- acetylglucosamine- specific IIB component, Glc/ maltose family | EUR_08700 | 52 | CM | 0.762 ± 0.055 | 0.592 ± 0.051 | 0.306 ± 0.056 | 2.49 | 1.94 | 0.001 | 0.003 |
| D6E440 | PTS system IIA component, Glc family | EUR_10290 | 80 | CM | 0.219 ± 0.01 | 0.149 ± 0.011 | 0.304 ± 0.031 | 0.72 | 0.49 | 0.011 | 0.001 |
| D6E521 | Carbohydrate ABC transporter substrate-binding protein | EUR_14180 | 46 | U | 0.081 ± 0.01 | 0.067 ± 0.009 | 0.113 ± 0.042 | 0.72 | 0.59 | 0.271 | 0.138 |
| D6E581 | ABC transporter substrate-binding protein (MalE) | EUR_14810 | 57 | U | 0.154 ± 0.024 | 0.077 ± 0.017 | 0.067 ± 0.020 | 2.32 | 1.16 | 0.008 | 0.519 |
| D6DYZ3 | PTS system D- fructose-specific IIA component | EUR_28570 | 67 | CM | 0.125 ± 00.026 | 0.105 ± 0.003 | 0.133 ± 0.022 | 0.94 | 0.80 | 0.720 | 0.107 |
| D6DZQ3 | Carbohydrate ABC transporter substrate-binding protein | EUR_31580 | 64 | U | 0.117 ± 0.022 | 0.103 ± 0.025 | 0.179 ± 0.023 | 0.65 | 0.58 | 0.027 | 0.017 |
| D6DZZ0 | PTS system, lactose/cellobiose family IIC component | EUR_32520 | 47 | CM | 0.010 ± 0.017 | 0.000 ± 0.00 | 0.000 ± 0.00 | H7 | H7 | 0.374 | N/A |
Only proteins of COG G (Carbohydrate transport and metabolism) displayed. Fold change was calculated based on the mean NSAF from three replicates. Only proteins that had 5 or more total spectra in at least one set of samples (H7 or APM) were included. PSORTb 3.0 was used to predict cellular location: U=Unknown, CM=Cell Membrane, C=Cytoplasmic, CW=Cell Wall. NA = not applicable. Rows in bold print denote proteins that were selected for further study.
GH13 enzymes EUR_01860 and EUR_21100 target different starch molecules
In order to clarify the roles of the two enzymes that are most upregulated in the presence of starch they were expressed, purified and biochemically characterized (Table 3). EUR_01860 displays its maximal activity against β-cyclodextrin (100%) and maltotriose (67%), with only weak activity against the soluble starch polymer amylopectin (23%) and trace activity on amylose (5%) and insoluble corn starch (1%). This is perhaps unsurprising as this enzyme lacks CBMs that facilitate activity against polysaccharides, though it is currently unknown if this enzyme possesses a surface binding site, which are often found to either replace or work in conjunction with CBMs in GH13 enzymes (Nielsen et al., 2009, Koropatkin & Smith, 2010, Cockburn & Svensson, 2013). While EUR_01860 has detectable activity on maltotriose as determined by a glucose oxidase assay (Table 3), it seems to prefer oligosaccharides with a degree of polymerization (dp) of four or longer. Thin layer chromatography (TLC) on the digestion products of EUR_01860 with maltotetraose through maltoheptaose (Figure 4A) demonstrates that the enzyme has approximately equal activity on these substrates, while products from a similar assay with maltotriose can barely be detected (data not shown). EUR_01860, has weak, but detectable activity against α1,6 linkages, demonstrated by the low level production of maltotriose from GMM (Figure 4B), and is able to hydrolyze cyclodextrins (Figure 4B). The result of prolonged incubation of EUR_01860 with maltotetraose, maltohexaose or glycogen is maltose is along with a small amount of maltotriose (Supplementary Figure S1). This set of characteristics most closely resembles the maltogenic α-amylases/cyclodextrinases, although EUR_01860 lacks the N-terminal domain that is the main identifying feature of these enzymes (Lee et al., 2002). Furthermore, BLAST searches against the GH13 enzymes that have been assigned to subfamilies revealed that EUR_01860 is most closely related to the subfamily 31 enzymes (α1,6 glucosidases) rather than the maltogenic α-amylases/cyclodextrinases found in subfamilies 2 and 20 (Stam et al., 2006).
Table 3.
Specific activities for EUR_01860 and EUR_21100
| Substrate | EUR_01860 | EUR_21100 | ||
|---|---|---|---|---|
| SA ± SD (μmol min−1 mg−1) | % Maximal Activity | SA ± SD (μmol min−1 mg−1) | % Maximal Activity | |
| Amylopectin (0.3%) | 1.43 ± 0.15 | 23 | 62.57 ± 2.46 | 100 |
| Glycogen (0.3%) | 2.60 ± 0.21 | 42 | 56.37 ± 2.54 | 90 |
| Amylose (0.3%) | 0.29 ± 0.03 | 5 | 34.76 ± 3.82 | 56 |
| Pullulan (0.3%) | 0.58 ± 0.49 | 9 | 1.09 ± 0.26 | 2 |
| Dextran (0.3%) | 0.12 ± 0.06 | 2 | 0.00 ± 0.14 | 0 |
| β-cyclodextrin (0.3%) | 6.14 ± 0.51 | 100 | 7.19 ± 0.76 | 11 |
| α-cyclodextrin (0.3%) | 0.33 | 5 | 1.86 ± 0.41 | 3 |
| Corn Starch (0.3%) | 0.09 ± 0.07 | 1 | 4.24 ± 0.23 | 7 |
| Hylon VII (H7, 0.3%) | ND | ND | 5.08 ± 1.60 | 8 |
| Maltose (5 mM) | 0.12 ± 0.33 | 2 | 0.09 | 0 |
| Maltotriose (5 mM) | 4.11 ± 0.46 | 67 | 0.31 | 0 |
Average of three assays per substrate reported.
Figure 4. Thin layer chromatography of enzyme hydrolysis products.

Each reaction was analyzed at four time points: 0, 10, 20, 30 min. Lanes denoted C are TLC controls consisting of 2 nmol each of glucose, maltose and maltotriose. A) EUR_01860 hydrolysis of maltooligosaccharides. B) EUR_01860 hydrolysis of GMM and BCD. Lane Pan is a 5 nmol panose standard, and lane GM is a 5 nmol glucose-α1,6-maltotriose (GM) standard. C) EUR_21100 hydrolysis of amylopectin. Lane G4 is a 5 nmol maltotetraose standard. D) EUR_21100 hydrolysis of maltooligosaccharides. Abbreviations are as follows: β-cyclodextrin (BCD), glucose (Glc), maltose (G2), maltotriose (G3), maltotetraose (G4), maltopentaose (G5), maltohexaose (G6), maltoheptaose (G7), glucose-α(1,6)-maltotriose (GM), glucose-α(1,6)-maltotriose-α(1,6)-maltotriose (GMM), panose (Pan).
In contrast to EUR_01860, EUR_21100 is most active against polymeric substrates, displaying maximal activity against the soluble starch component amylopectin with high activity on glycogen (90%) and amylose (56%) as determined by reducing sugar assays. However, this enzyme can also degrade intact starch granules as demonstrated by its activity on granular corn starch (7%) and H7 (8%). TLC analysis reveals that the main product of starch degradation by EUR_21100 is maltotetraose, with significant amounts of maltopentaose (Figure 4C). Not surprisingly, EUR_21100 has no detectable activity against maltose, maltotriose (Table 3), or maltotetraose, but displays weak activity on maltopentaose, and can hydrolyze longer maltooligosaccharides such as maltohexaose and maltoheptaose (Figure 4D). This enzyme is thus classified as a maltotetraose producing α-amylase (Yoshioka et al., 1997) even though BLAST searches suggest it is more closely related to Type I pullulanases (α1,6 hydrolyzing enzymes) in subfamilies 12 and 14 of GH13 (Stam et al., 2006).
While from Table 3 it appears that EUR_21100 is much more active against the soluble components of starch than against starch granules, it should be kept in mind that these values were all acquired at 0.3% w/v of substrate and that for a given weight starch granules will have a far lower accessible area than soluble amylopectin. Using 10% granular starch the specific activity rises to 81.66 μmol min−1 mg−1, significantly higher than the 62.57 μmol min−1 mg−1 seen for amylopectin, which does not increase when the substrate concentration is raised to 1%. Interestingly, EUR_21100 had nearly identical activity towards H7 as towards normal corn starch. However, these are only initial activities and likely reflect that H7 is composed of approximately 50% resistant starch (Jiang & Liu, 2002, Vonk et al., 2000), so it is unsurprising that it resembles regular corn starch in the initial stages of degradation.
EUR_01830 targets maltooligosaccharides of three glucose units and longer
To determine how the abundant carbohydrate-binding protein EUR_01830 assists in nutrient capture during growth on starch, we expressed and purified this protein (residues 24–422, removing the signal peptide) for biochemical characterization. Via isothermal titration calorimetry (ITC), EUR_01830 binds maltooligosaccharides of three to seven glucose units with relatively equal affinity (Kd range 288 – 550 nM, Table 4). EUR_01830 displays no detectable affinity for maltose, suggesting that at least three subsites within the binding pocket must be occupied for efficient binding. A 100-fold reduction in affinity was observed for binding to α-cyclodextrin (Kd = 39 900 ± 2 430 nM), suggesting that this is not a biologically relevant substrate; likewise, E. rectale cannot grow on α- or β-cyclodextrin. While E. rectale cannot grow on pullulan, it can grow on oligosaccharides generated from the breakdown of pullulan. EUR_01830 can bind the short pullulan oligosaccharide GM (Kd = 9 080 ± 618 nM), most likely because the maltotriose moiety engages the predicted three essential subsites required for binding, although there is a >10-fold reduction in affinity compared to α1,4-linked maltooligosaccharides. We obtained an isotherm with GMM, but these data could not be fit well enough to obtain an accurate Kd; thus this substrate is unlikely to be biologically relevant. Like some acarbose-recycling solute-binding proteins, EUR_01830 can bind acarbose (Kd 302 ± 21.8 nM) with similar affinity as other maltooligosacharides (Vahedi-Faridi et al., 2010). To examine the molecular basis of maltooligosaccharide discrimination, we determined the crystal structures of EUR_01830 complexed with either maltotriose (1.54Å, Rwork=13.7%, Rfree = 16.1%) or acarbose (1.6Å, Rwork=14.4%, Rfree=17.7%; Table 5, Figure 5A,B). The structure of EUR_01830 displays a Type II two-domain solute-binding fold with a ligand-binding site located at the interface between the N- and C-terminal domains, connected by three β-strands, residues 152–157, 300–309, and 371–374 (Figure 5B). The closest structural homolog of EUR_01830 via a DALI search (Holm & Sander, 1995) is the maltose-maltodextrin binding protein from Alicyclobacillus acidocaldarius (1URG, Z=39.2, rmsd=2.0Å for 350 residues, (Schafer et al., 2004)), though numerous maltose- and maltodextrin-binding proteins display striking similarity (Z ≥33, RMSD ≤2.5Å), including MalX from Streptococcus pneumoniae (Abbott et al., 2010) and MalE from E. coli (Oldham et al., 2013), both of which can accommodate sugars as long as maltoheptaose.
Table 4.
Affinity of ABC solute-binding proteins for maltooligosaccharides and monosaccharides
| EUR_01830 | EUR_31480 | EUR_01240 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ka (μM−1) | n | Kd (nM) | Ka (μM−1) | n | Kd (nM) | Ka (μM−1) | n | Kd (nM) | |
| Glucose | -- | -- | -- | NB | NB | NB | 0.64 ± 0.13 | 0.56 ± 0.035 | 1900 ± 460 |
| Galactose | -- | -- | -- | -- | -- | -- | 2.41 ± 0.12 | 0.56 ± 0.045 | 415 ± 20.2 |
| Maltose | NB | 1.79 ± 0.021 | 0.88 ± 0.003 | 563 ± 66.4 | NB | ||||
| Isomaltose | NB | NB | -- | ||||||
| Panose | NB | NB | -- | ||||||
| Maltotriose | 1.82 ± 0.05 | 0.88 ± 0.06 | 550 ± 14 | 2.82 ± 0.0035 | 0.87 ± 0.024 | 355 ± 0.4 | -- | ||
| Maltotetraose | 9.10 ± 1.15 | 1.12 ± 0.08 | 112 ± 19.8 | 0.60 ±0.072 | 0.96 ± 0.063 | 1670 ± 199 | -- | ||
| Maltopentaose | 5.26 ± 0.85 | 0.77 ± 0.10 | 194 ± 34.8 | NB | -- | ||||
| Maltohexaose | 6.27 ± 0.03 | 0.80 ± 0.10 | 160 ± 6 | NB | -- | ||||
| Maltoheptaose | 3.47 ± 0.07 | 0.82 ± 0.01 | 288 ± 5.9 | NB | -- | ||||
| Acarbose | 3.33 ± 0.24 | 0.81 ± 0.04 | 302 ± 21.8 | NB | -- | ||||
| GM | 0.11 ± 0.008 | 0.87 ± 0.08 | 9080 ± 618 | NB | -- | ||||
Average results of 2–3 titrations reported.
NB = No observable binding
-- = Experiment was not performed.
Table 5.
Data collection and refinement statistics of Eur_01830
| 01830 - maltotriose | 01830 -acarbose | |
|---|---|---|
| PDB ID | 4UA8 | 4UAC |
| Resolution range (Å)1 | 37.01 – 1.542 (1.597 – 1.542) | 31.85 – 1.6 (1.657 – 1.6) |
| Space group | P 21 21 2 | P 21 21 2 |
| Unit cell | 99.691 110.495 38.515 90 90 90 | 99.805 110.408 38.473 90 90 90 |
| Total reflections1 | 472084 (30736) | 296981 (14671) |
| Unique reflections1 | 63371 (6152) | 55134 (4348) |
| Multiplicity1 | 7.4 (5.0) | 5.4 (3.4) |
| Completeness (%)1 | 99.54 (98.12) | 96.52 (77.70) |
| Mean I/sigma(I)1 | 30.19 (7.16) | 23.56 (4.23) |
| Wilson B-factor | 10.89 | 13.8 |
| R-merge1 | 0.08121 (0.1752) | 0.07917 (0.2257) |
| R-meas | 0.08573 | 0.08776 |
| CC1/21 | 0.995 (0.974) | 0.995 (0.926) |
| CC*1 | 0.999 (0.993) | 0.999 (0.981) |
| R-work1 | 0.1370 (0.1476) | 0.1443 (0.1571) |
| R-free1 | 0.1613 (0.1906) | 0.1769 (0.1862) |
| Number of atoms | 3490 | 3441 |
| macromolecules | 2916 | 2899 |
| ligands | 45 | 63 |
| water | 529 | 479 |
| Protein residues | 383 | 383 |
| RMS(Å) | 0.012 | 0.01 |
| RMS(°) | 1.4 | 1.39 |
| Ramachandran favored (%) | 99 | 98 |
| Ramachandran outliers (%) | 0 | 0 |
| Clashscore | 0.69 | 1.56 |
| Average B-factor | 15.3 | 17.4 |
| macromolecules | 13.4 | 15.8 |
| ligands | 13.7 | 18.7 |
| solvent | 25.7 | 26.8 |
Values in parenthesis are for the highest resolution shell.
Figure 5. Structure of the maltodextrin-binding protein EUR_01830.

A. Omit maps (σ = 3.0) displaying representative electron density for maltotriose and acarbose. B. Overlay of the structures of EUR_01830 complexed with acarbose (grey ribbon) and with maltotriose (blue ribbon). Acarbose is displayed as black sticks and maltotriose as blue sticks. The aromatic residues that cradle the maltodextrins are shown as sticks. C. Close-up of the binding pocket of EUR_01830. Direct hydrogen-bonding interactions between the protein and maltrotriose/acarbose are displayed as dashed lines. K91 and D384 have been omitted for clarity. Waters involved in water-mediated hydrogen-bonds are shown as spheres. D. Close-up end view of the hydrogen-bonding network that confers recognition of the reducing end sugar. Acarbose and the relevant amino acids are shown as sticks, while waters involved in water-mediated hydrogen bonds are shown as red spheres. Dashed lines indicate interactions within hydrogen bonding distance, and distances are displayed in Å. Here E246, though unlikely to contribute to hydrogen-bonding, is shown for reference. E. Close-up side view of EUR_01830 binding pocket demonstrating the manner in which E246, S87 and D88 may occlude the front of the binding pocket. The loop of residues 242–248 is shown in black ribbon, and E246, S87 and D88 are shown in black sticks. Note that S87 was observed in two conformations in the crystal structure. F. Superposition of the coordinates of the acarbose-bound EUR_01830 (grey) and MalX of S. pneumoniae (blue, pdb 2XD3). The C-terminal strand-loop-helix comprising residues 236–259 of EUR_01830 is displayed in black, and the corresponding helix-loop-helix comprising residues 240–266 of MalX is displayed in dark blue. Acarbose is displayed as black sticks and maltoheptaose is displayed as blue sticks.
The structure of the EUR_01830 maltotriose-bound and acarbose-bound structures are nearly identical (0.1 Å rmsd for 383 amino acids; Figure 5B). Like other maltodextrin-binding proteins, the binding pocket consists of three aromatic residues, here W267, W193 and W383, that stack with Glc1– 3 (Figure 5C). An additional hydrogen-bonding network between the residues that line the innermost hinge region of the binding cleft and the O2 and O3 hydroxyls of these Glc residues further facilitate binding. The reducing end sugar (Glc1) is recognized by the protein via both water mediated and direct hydrogen bonding (Figure 5D). The O2 and O3 hydroxyls of Glc1 are proximal to the side chains E86 and K305, and the O2 hydroxyl is involved in a water-mediated hydrogen bond to R339. The reducing end O1 is coordinated with both the hydroxyl side chain of S85, as well as with E51 via an ordered water molecule. The binding cleft at the non-reducing end is more open, which likely aids in accommodating the pseudotetrasaccharide acarbose. The O3 of the non-reducing end of the acarviosine moiety is proximal to the side chains of D91and K90, while the O6 makes water-mediated hydrogen-bonds to the side chain of D384 and the peptidic O of W383. However, these interactions apparently do not contribute to the affinity for the sugar, as ITC experiments demonstrate a nearly identical Kd for both maltotriose and acarbose. The structures of many ABC maltooligosaccharide-binding proteins have been determined, revealing that ligand binding induces a closure of the two domains over the sugar, mediated by a flexible hinge region between the two domains. That EUR_01830 cannot bind maltose suggests that all three tryptophan-containing subsites must be occupied in order to induce and stabilize closure of the two domains around the sugar.
From these structures, it is clear that binding the oligosaccharide GM, which contains an α1,6-linked glucose at the nonreducing end of the sugar would necessarily push the binding cleft open, as well as disrupt the hydrogen-bonding between side chain of N191 and the O6 of the nonreducing end of maltotriose (Figure 5C). However, it is difficult to envision how a much longer oligosaccharide like maltoheptaose is accommodated with high affinity. A loop created by residues 242–248 from the C-terminus closes the opening to the binding cleft (Figure 5E & F), with E246 extending upwards towards the middle of the oligosaccharide curve, and both S87 and D88 from the N-terminal domain projects downward to the middle of the oligosaccharide and in front of the binding cleft. Presumably EUR_01830 would have to open to accommodate a longer helical maltooligosaccharide, similar to the manner in which MalX is somewhat propped open to accommodate the natural helix created by the α1,4-linkages in maltoheptaose (Abbott et al., 2010). In the MalX structure, a helix-loop-helix created by residues 240–266 is positioned away from the binding cleft allowing maltoheptaose access to the binding site. In the EUR_01830 structure an equivalently positioned β-strand-loop-helix created by residues 236–259 would have to move similarly to accommodate the longer oligosaccharide (Figure 5F).
EUR_31480 and EUR_01240 provide binding for smaller maltooligosaccharides and glucose
To understand how the additional ABC solute-binding proteins observed in our starch-grown cells may contribute to starch utilization, we expressed and purified EUR_01240, EUR_31480, and EUR_21010 for determination of their maltooligosaccharide binding specificity (Table 4). EUR_31480 can bind maltose (Kd = 563 ± 66 nM) and maltotriose (Kd = 355 ± 0.4 nM) with similar affinity, but binds maltotetraose with slightly less affinity (Kd = 1670 ± 199 nM) and cannot bind longer maltoligosaccharides, or any oligosaccharides containing an α1,6 linkage including GM and GMM. While the xylotriose-binding protein from C. polysaccharolyticus displays the best sequence similarity of structures in the PDB, we did not observe binding of EUR_31480 to xylotriose.
EUR_01240 is a glucose and galactose-binding protein, and cannot bind maltose or lactose. The affinity of this protein for galactose (Kd = 415 ± 20.2 nM) is nearly 5-fold better than for glucose (Kd = 1 900 ± 460 nM), and overall about 10-fold worse than the affinities reported for the E. coli and S. typhimurium homologs (Zukin et al., 1977). Despite trying different methods of measuring the protein concentration for ITC experiments, our data consistently fit a binding stoichiometry of n ~0.5, which might suggest that two molecules of protein bind one molecule of monosaccharide. While this stoichiometry has not been observed in any other solute-binding protein to the best of our knowledge, some solute-binding proteins such as the lactate-binding protein of the tripartite ATP-independent periplasmic (TRAP) transporter in Thermus thermophilus HB8 can dimerize (Akiyama et al., 2009). Due to the stoichiometry observed and the fact that the EUR_01240 protein displays an extra ~100 amino acids at its C-terminus, we decided to investigate the multimeric state of this protein. After inconsistent results were obtained with size exclusion chromatography, we employed analytical ultracentrifugation to determine whether EUR_01240, and for comparison both EUR_31480 and EUR_01830, could dimerize in the presence or absence of substrate. Diffusion corrected van Holde – Weischet sedimentation coefficient distributions indicated a clear shift to slightly larger values when maltooligosaccharides were added at saturating concentrations (1 mM) to each of the proteins, however these shifts are consistent with maltooligosaccharide binding, and not protein-protein dimerization (Supplemental Figure S2; Supplemental Table S4.) The solution characteristics of EUR_01240 at both high and low concentrations and in the presence or absence of glucose suggest that it is a monomer like other characterized ABC glucose/galactose-binding proteins. Thus we conclude that the unexpected stoichiometry with EUR_01240 during ITC is simply due to about 50% of the protein failing to bind in these experiments, despite the fact that the protein remained soluble and otherwise behaved normally.
We also purified and tested EUR_21010 for binding to glucose and maltooligosaccharides, but did not observe binding. A predicted cellobiose phosphorylase encoding gene upstream of EUR_21010 prompted us to test for binding to cellooligosaccharides and revealed that EUR_21010 has affinity for both cellobiose (Kd = 542 nM, n = 0.846 ) and cellotetraose (Kd = 3 009 nM, n = 1.009). As mentioned earlier, this protein was observed at much lower relative abundance compared to the other tested ABC solute-binding proteins, and therefore we did not think it was likely to contribute significantly to starch utilization. This finding may suggest that E. rectale has the ability to scavenge cellooligosaccharides generated by the cellulose-degrading organisms that have recently been identified in the human gut microbiota (Chassard et al., 2012, Robert & Bernalier-Donadille, 2003).
Transcriptional analysis of select genes
In order to further explore the response of E. rectale to growth on starch, we examined the transcriptional profile of the genes encoding our five selected proteins during exponential growth on glucose, APM and H7 (Table 6). The transcripts for EUR_01830, EUR_01860 and EUR_21100 are upregulated during growth on starch compared to glucose, although the observed fold increase is higher than that in the proteomic data. Discrepancies in the fold changes for transcriptomic and proteomic data from cells cultured in the same conditions are well - reported for a variety of organisms, and are due to a variety of technical and biological factors (Nie et al., 2007). Currently little is known about the post-transcriptional and post-translational regulatory processes of E. rectale, preventing a direct comparison between fold changes observed on a transcriptional vs proteomic level. Furthermore, limitations and differences in how transcriptional vs proteomic data is measured, as well as the turnover and stability of the transcripts and proteins will affect how well these two types of information can be correlated (Haider & Pal, 2013).
Table 6.
Fold-change in transcripts relative to growth in glucose
| Gene | APM | H7 | p-value* |
|---|---|---|---|
| EUR_21100 | 30.9 ± 3.8 | 37.1 ± 3.1 | 0.110 |
| EUR_01860 | 11.04 ± 0.2 | 33.2 ± 4.9 | 0.011 |
| EUR_01830 | 10.9 ± 2.6 | 113.3 ± 49.2 | 0.049 |
| EUR_31480 | −1.43 ± 0.5 | 3.6 ± 1.1 | 0.015 |
| EUR_01240 | −25.3 ± 2.8 | −9.0 ± 4.1 | 0.022 |
Probability associated with a Student’s t-Test comparing APM and H7.
More difficult to explain are the changes in the transcript levels of EUR_31480 and EUR_01240 compared to the proteomic data. The transcript for EUR_31480 appears to be slightly upregulated during growth on H7 and slightly repressed during growth on APM compared to glucose, although the overall fold changes are quite modest. Significantly less transcript for EUR_01240 was observed during growth on starch compared to glucose, although more of these proteins are detected in the starch-grown cells. Note that cells were grown overnight in YCFA plus glucose, then back-diluted 15-fold the following morning into YCFA containing glucose, APM or H7 for the transcriptional and proteomic experiments. It is possible that while new transcripts for EUR_01240 are not being made during growth in starch, slow turnover of the synthesized proteins and existing transcripts allow for relatively stable levels of the proteins in both glucose and starch grown cells. Excluding EUR_21100, more transcript is detected in the H7 versus APM samples, as suggested by the higher fold changes compared to glucose. H7 is a more insoluble and resistant form of starch than APM, by virtue of its higher amylose content, and the cell may be responding to these subtle differences in starch structure.
Discussion
Previous reports have indicated that despite the fact that E. rectale cannot degrade resistant starch, it is typically associated with starch granules in culture, and its abundance can increase in the mammalian gut when the host consumes RS. In this study we identified two GH13 enzymes that participate in starch degradation as well as three ABC glycan-binding proteins involved in scavenging the liberated maltooligosaccharides and glucose, allowing us to propose a model for how E. rectale targets starch in the intestinal tract (Figure 6). This model is similar to that for glycogen uptake in Streptococcus pneumoniae (Abbott et al., 2010); in S. pneumoniae, the cell wall anchored pullulanase SpuA degrades glycogen into a ladder of malto-oligosaccharides, most of which are specifically recognized by MalX for import into the cell. In E. rectale, the maltotetraose-producing, cell-wall tethered α-amylase EUR_21100 likely participates in the initial anchoring of the bacterium to the starch polymer, a process facilitated by the five predicted starch-binding CBMs found at the N-terminus of the protein. The ability to attach to starch particles in the intestinal tract is advantageous to E. rectale, and may serve to place it in close proximity to resistant starch degrading bacteria such as R. bromii. Prolonged hydrolysis of resistant starch by R. bromii liberates many short maltooligosaccharides and glucose (Ze et al., 2012) which could be taken up by E. rectale via various solute-binding proteins or a PTS. However, the degradation of starch granules is generally initiated by attack of the surface, eventually exposing internal amorphous regions that are more vulnerable to hydrolysis (Foresti et al., 2014) and it is likely that R. bromii performs this function as well, making the starch accessible to E. rectale. This would allow E. rectale to grow efficiently on resistant starch in vivo, despite its relatively small repertoire of proteins that are upregulated in the presence of starch.
Figure 6. Model of starch digestion and maltooligosaccharide uptake by Eubacterium rectale.

The multidomain EUR_21100 is tethered to the peptidoglycan layer, and may anchor the bacterium to starch via its five N-terminal CBMs (predicted CBM family number as indicated, U= unknown domain). Released oligosaccharides may be targeted by EUR_01830 and EUR_31480 for import into the cell. The location of EUR_01860 is unknown. This maltogenic amylase produces some glucose, as suggested by its breakdown of maltotriose, and its presence on the surface of the cell may generate glucose and maltose that can be captured by EUR_01240 and EUR_31480 respective
Starch degradation by EUR_21100 results primarily in maltotetraose and maltotriose, both of which are recognized by the abundant cell surface proteins EUR_01830 and EUR_31480; previous studies have demonstrated the ability of E. rectale to grow on maltotetraose (Ze et al., 2012). EUR_01830 and EUR_31480 display both overlapping and distinct maltooligosaccharide binding profiles. EUR_01830 is the most abundant upregulated protein during growth on starch, and targets longer maltooligosaccharides from maltotriose through maltoheptaose with nearly equal affinity. Although it has weaker affinity for GM, EUR_01830 may contribute to E. rectale’s ability to grow on this branched oligosaccharide.
A key question that we aim to address in future studies is the localization of EUR_01860. This glucosidase has a predicted N-terminal transmembrane helix, though whether it is exported to the outside of the cell and contributes to initial oligosaccharide processing or it is located inside of the cell and responsible for breakdown of the imported maltooligosaccharides is unknown. The substrate profile of EUR_01860 suggests that this enzyme targets maltooligosaccharides longer than maltotriose, which could be imported via EUR_01830. However, EUR_01860 produces maltose and maltotriose, which can be harvested by EUR_31480 and EUR_01830 respectively. Having EUR_01860 on the surface of the cell may be most advantageous to E. rectale by allowing it to process maltooligosaccharides generated by other bacteria, including those with α1,6-linkages.
In addition to EUR_21100 and EUR_01860, E. rectale has eleven other GH13 enzymes and one GH77 enzyme as annotated in the CAZy database, all of which are predicted to have a cytoplasmic location. Of these, several are predicted to have glucosidase activity on α1,4 and/or α1,6 linked maltooligosaccharides and disaccharides to generate glucose. These glycoside hydrolases likely contribute to the final processing of imported oligosaccharides as well as to the breakdown of internal glycogen stores.
The relative abundance of the glucose-binding protein EUR_01240 in all samples, as well as its relative fold increase during growth on starch was somewhat surprising as neither EUR_21100 or EUR_01860 generates glucose as a major product, although many other starch-utilizing gut bacteria including R. bromii and B. thetaiotaomicron have surface enzymes that generate glucose as a product of starch digestion (Ze et al., 2012, Koropatkin & Smith, 2010). Our qPCR data suggests that transcript levels for EUR_01240 are higher during growth on glucose, although the protein is present and modestly elevated during growth on starch. As little is known about transcriptional and translational regulation in E. rectale, we can only speculate that this protein may experience less turnover during growth on starch. It is possible that glucose is the most preferred carbon source of E. rectale, and thus the omnipresence of EUR_01240 in the membrane ensures glucose capture. This idea is supported by the lack of a dramatic increase in abundance of predicted glucose transporters during cell growth on glucose.
Between the three sequenced genomes of E. rectale (strains DSM 17629, ATCC 33656, and M104/1), syntenic loci for the putative operons encoding EUR_01830 – EUR_01860, EUR_31480-31540, and EUR_01240–EUR_01260 are conserved, with ≥96% amino acid identity between the individual protein homologs for EUR_01830, EUR_01860, EUR_31480 and EUR_01240. The predicted locus within the ATCC strain that is homologous to the EUR_21100 – 21130 of our isolate, displays an insertion after the EUR_21100 homolog (EUBREC_1081), and includes a predicted DNA invertase and transposase that is not seen in the other two strains. While the homologous protein to EUR_21100 in M104/1 (ERE_20420) is 99% identical, the amino acid sequence for EUBREC_1081 is more divergent, at 48% identical to EUR_21100, with most of these differences taking place in the predicted CBM region at the N-terminus of the protein. Overall it seems that the proteins involved in starch uptake in E. rectale DSM 17629 are conserved among other isolates.
Transcriptional and proteomic data collected on the related Clostridium cluster XIVa bacterium Roseburia inulinovorans during growth on inulin and starch has been previously reported, demonstrating that polysaccharide exposure induces enzyme and ABC transporter expression (Scott et al., 2011). During growth on starch a solute-binding protein and amylase from a similar operon structure as EUR_01830-01860 were observed, as well as another larger amylase of 1315 amino acids, ROSEINA2194_03334, that is likely anchored to the cell wall. Interestingly, this larger amylase, designated AmyB, has a predicted domain structure with the catalytic domain comprising residues ~316 – 747, rather than at the C-terminus of the protein as in EUR_21100. These types of large, multidomain cell-wall anchored amylases can be found in many of the Clostridium cluster XIVa organisms and have a variety of domain organizations, while seemingly performing similar functions; the purified catalytic domains of R. inulinovorans and Butyrvibrio fibrisolvens both display amylase activity (Ramsay et al., 2006), similar to EUR_21100. Thus the starch degrading and oligosaccharide import pathway described here for E. rectale may describe a conserved pathway by which many human gut Clostridium cluster XIVa organisms take advantage of the abundant dietary carbohydrate starch.
Experimental Procedures
Reagents
Primers used for cloning were synthesized by IDT DNA Technologies and are listed in Supplemental Table S5. Hylon VII (H7) was kindly provided by Ingredion Inc, Bridgewater, NJ. All other chemicals were purchased from Sigma Aldrich, with the exception of GM, GMM and pullulan that were purchased from Megazyme.
Media and Growth Conditions
Eubacterium rectale DSM 17629 was routinely grown in modified YCFA media (Ze et al., 2012) supplemented with 0.2% – 0.5% glucose, H7 or APM as the sole carbon source. Media was prepared aerobically, filter sterilized, and equilibrated in a Coy anaerobic chamber (85% N2, 10% H2, 5 % CO2) for 1 h prior to dispensing into sterile Hungate tubes for growth experiments.
Proteomic sample preparation
For proteomics, 1mL of an overnight culture of E. rectale grown in YCFA plus 2 mg mL−1 glucose was added to 14 mL of YCFA media containing 2 mg mL−1 of glucose, APM, or H7 as the sole carbon source in 15 mL screw-cap Hungate tubes. Cultures were dispensed and sealed in the anaerobic chamber then removed and grown at 37 °C on a tube roller to provide constant agitation. When the OD600 reached ~0.6, the cultures were harvested by centrifugation and quickly frozen in liquid N2. Each culture was grown in triplicate. For combined cell wall/cell membrane (CMW) samples, cells were first thawed on ice in 8 mL of cold PBS. Cells were lysed by three passages through a French press at 2 000 psi, then centrifuged at 10 000 x g for 15 min to remove unbroken cells. The supernatant was then centrifuged at 10 000 x g for another 10 min and the remaining supernatant containing the cell wall and membrane was subjected to ultracentrifugation at 200 000 x g for 2 h. The pellet was then resuspended in 1 mL of high-salt buffer (25 mM NaH2PO4, 5 mM EDTA, 1M NaCl, pH 8.0) and incubated on ice for 5 min, then centrifuged again at 200 000 x g for 30 min. This high-salt buffer wash and spin was repeated a second time, and the final pellet was resuspended in 0.15 mL of PBS then frozen in liquid N2 until all samples were ready for proteomic analysis.
Quantitative Mass Spectroscopy
The CMW samples were submitted to MS Bioworks LLC (www.msbioworks.com, Ann Arbor, MI) for Quant-works Label-free Unfractionated proteomic analysis. Briefly, samples were diluted 1:1 with 0.1% SDS, heated at 65 °C for 10 min and quantified using Qubit fluorimetry (Invitrogen). Samples were then mixed with LDS buffer and 10 μg of each sample was loaded into separate lanes of a 10% SDS-PAGE gel and separated ~1 cm. The gel was stained with Coomassie and the entire mobility region of the gel was excised. Trypsin digestion was performed using the ProGest Automated Protein Digestion robot (Digilab). The excised gel was washed with 25 mM ammonium bicarbonate then acetonitrile, followed by treatment with 10 mM DTT at 60 °C. The samples were then alkylated with 50 mM iodoacetamide at room temperature, then digested with trypsin (Promega) for 4 h at 37 °C. The reaction was quenched with formic acid and the supernatants were analyzed by nano LC/MS/MS with a Waters NanoAcquity HPLC system interfaced to a Thermofisher Q Exactive. Peptides were loaded on a trapping column and eluted over a 75 mm analytical column at 350 nL min−1 for a 1 h gradient. Both columns were packed with Jupiter Proteo resin (Phenomenex). Mass spectrophotometry was performed in data-dependent mode with MS and MS/MS performed in the Orbitrap at 70 000 FWHM and 17 500 FWHM resolution respectively. The fifteen most abundant ions were selected for MS/MS.
The resulting proteomic mass spectrum data were searched using a local copy of Mascot against the Uniprot Eubacterium rectale DSM 17629 proteome (combined forward and reverse plus common contaminants such as keratin). A peptide mass tolerance of 10 ppm, with a fragment mass tolerance of 0.015 Da and a maximum of two missed cleavages was included. Mascot DAT files were parsed into the Scaffold (version 3.5.1) software to create a non-redundant list of proteins per sample. Data were filtered using a minimum protein value of 90%, a minimum peptide value of 50% (Prophet scores), and requiring at least two unique peptides per protein. Proteins were quantified by calculating the normalized spectral abundance factor (NSAF) of each protein as NSAF = (SpC/MW)/Σ(SpC/MW)N, where SpC = Spectral Counts, MW = Protein MW in kDa, and N = Total Number of Proteins. NSAF was used to determine the abundance of proteins within and across samples. ANOVA on the three groups (GLU, H7 and APM) was also performed and p-values are included in Supplemental Table S1. The cellular localization of identified proteins was predicted using PSORTb 3.0 (Yu et al., 2010).
Cloning and protein expression
The genes encoding EUR_01830 (residues 25–423), EUR_01240 (residues 23–427), EUR_31480 (residues 27–462), and EUR_21010 (residues 26–464), were PCR amplified from genomic DNA for ligation-independent cloning into the pETite N-His vector (Lucigen Madison, WI) according to the manufacturer’s instructions. The N-terminal primer contained a TEV cleavage site immediately downstream of the complementary 18 bp overlap (encoding the His tag) to create a TEV-cleavable His-tagged protein. EUR_21100 (residues 45–1329) and EUR_01860 (residues 43–564) were PCR amplified from genomic DNA for ligation-independent cloning into the pETite C-His vector (Lucigen Madison, WI) according to the manufacturer’s instructions. The resulting pETite plasmids were transformed into Rosetta(DE3) pLysS cells, plated on LB agar containing 50 μg mL−1 kanamycin (Kan) and incubated overnight at 37 °C. For native protein expression, these plates were used to inoculate 2 × 1 L of terrific broth media supplemented with 50 μg mL−1 Kan and 30 ug mL−1 of chloramphenicol (Cm) for protein expression. Cells were grown at 37 °C to an OD600 of ~0.6, IPTG was added to a final concentration of 0.5 mM, and the cells were grown at room temperature (20–23 °C) for an additional 16 h. Cells were then harvested by centrifugation and flash-frozen in liquid N2.
Selenomethionine-substituted EUR_01830 was expressed via the methionine inhibition method (Van Duyne et al., 1993). Freshly transformed Rosetta(DE3) pLysS cells grown overnight on LB/Kan agar plates were used to inoculate 100 mL starter cultures of M9 minimal media containing 50 μg mL−1 Kan and 30 μg mL−1 Cm for growth overnight at 37 °C. After 16 h, 50 mL of the starter culture was used to inoculate 2 × 1 L of M9 minimal media plus 50 μg mL−1 Kan, 30 μg mL−1 Cm and 5 μg ml−1 thiamine. Cultures were grown at 37 °C to an OD600 of ~0.45, before adjusting the temperature to 23°C for the remainder of the growth period. At this time, each 1 L of media was supplemented with 100 mg each of L-lysine, L-threonine, L-phenylalanine, and 50 mg each of L-leucine, L-isoleucine, L-valine, and L-selenomethionine. After 20 additional min of growth, the cells were induced with 0.5 mM IPTG, and allowed to grow for 16 h. Cells were then harvested by centrifugation and flash-frozen in liquid N2.
Purification of native and selenomethionine-substituted solute-binding proteins
His-tagged proteins were purified using a 5 mL Hi-Trap metal affinity cartridge (GE Healthcare) according to the manufacturer’s instructions. The cell lysate was applied to the column in His Buffer (25 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole pH 7.4) and proteins were eluted with an imidazole (20 – 300 mM) gradient. The His-tag was removed by incubation with TEV protease (1:100 molar ratio relative to protein) at room temperature for 2 h, then overnight at 4 °C while dialyzing against His Buffer. The cleaved protein was then re-purified on the 5 mL Ni column to remove undigested target protein, the cleaved His-tag, and His-tagged TEV protease. Purified proteins were dialyzed against 20 mM HEPES / 100 mM NaCl (pH 7.0) prior to crystallization, and concentrated using Vivaspin 15 (10 000 MWCO) centrifugal concentrators (Vivaproducts, Inc.).
Protein Crystallization and Data Collection
EUR_01830 was initially screened using a variety of commercially available sparse matrix crystallization screens using a 96-well hanging drop format at both room temperature and 4°C. Diffraction quality crystals of native EUR_01830 with maltotriose complex were obtained by mixing the protein solution (OD280 = 31 with 1mM maltotriose) 1:1 with the well solution containing 35–40% pentaerythritol propoxylate 5/4 PO/OH, 0.2 M NaCl, and 0.1 M 2-(N-morpholino)ethanesulfonic acid pH 5.0 in hanging drop experiment at 4 °C. For data collection, crystals were frozen by quickly transferring into cryoprotectant made by mixing 80 μL of mother liquor with 20 μL of ethylene glycol then plunging into liquid nitrogen. Native crystals of the EUR_01830/acarbose complex were obtained by mixing the protein solution (OD280 = 31 with 2 mM acarbose) 1:1 with a well solution of 40% pentaerythritol propoxylate 5/4 PO/OH, 0.2 M NaCl, and 0.1 M 2-(N-morpholino)ethanesulfonic acid pH 6.0 in a hanging drop experiment at 4 °C. These crystals were also frozen by quickly swiping through cryoprotectant containing 80% crystallization solution plus 20% ethylene glycol before flash-freezing in liquid nitrogen. Selenmethionine-substituted protein crystals with acarbose were grown and frozen in an identical manner as the native protein plus acarbose. SAD x-ray data for all seleno-methionine substituted crystals with acarbose were collected at the Life Sciences Collaborative Access Team (LS-CAT) ID-D beamline, while the native X-ray data sets were collected at LS-CAT ID-F beamline at the Advanced Photon Source at Argonne National Labs, Argonne, IL. All X-ray data were processed with HKL3000 and scaled with SCALEPACK (Otwinowski & Minor, 1997). The structure of EUR_01830 was determined from the SAD data using the AutoSol subroutine within the Phenix software package (Adams et al., 2002, Zwart et al., 2008), including automatic model tracing. This initial model of EUR_01830 was then utilized for molecular replacement in Phaser (McCoy et al., 2007) against the native X-ray data sets. Successive rounds of manual model building in Coot (Emsley & Cowtan, 2004) and refinement in Phenix were employed. Data collection and refinement statistics are presented in Table 5.
Isothermal Titration Calorimetry
ITC measurements for EUR_01830 with maltoheptaose, maltotriose, α-cyclodextrin and acarbose were performed on a MicroCal VP-ITC. The protein was dialyzed against 20 mM HEPES, 100 mM NaCl pH 7.0 and oligosaccharide solutions were prepared using the dialysis buffer. Protein (215 μM) was placed in the sample cell and the reference cell filled with deionized water. After the temperature was equilibrated to 25 °C, a first injection of 2 μL was performed followed by 29 subsequent injections of 10 μL each of substrate (3 mM) α-cyclodextrin, maltoheptaose, maltotriose or acarbose. Calorimetry was also attempted twice with maltose but no binding was observed. The solution was stirred at 305 rpm and the resulting heat of reaction was measured. Data were analyzed using the Origin software package (MicroCal).
The remaining ITC measurements for EUR_01830 as well as all of those for EUR_31480, EUR_01240 and EUR_21010 were performed on a low-volume (250 μL sample cell) TA instruments NanoITC. A single trial repeat of the previously obtained values for EUR_01830 on the VP-ITC verified that the measurements did not vary instrument to instrument. Proteins were dialyzed against 20 mM HEPES, 100 mM NaCl pH 7.0 and oligosaccharide solutions were prepared using the dialysis buffer. Protein (30 – 150 μM) was placed in the sample cell and the reference cell filled with deionized water. After the temperature was equilibrated to 25 °C, a first injection of 0.75 μL was performed followed by 27 subsequent injections of 1.75 μL of 0.5 – 5 mM of the substrates listed in Table 5, plus cellobiose and cellotetraose (EUR_21010 only), and the pullulan oligosaccharide GMM (EUR_01830 only). The solution was stirred at 350 rpm and the resulting heat of reaction was measured. Data were analyzed using the NanoAnalyze software package (TA instruments).
Enzymatic assays
Activity towards maltose and maltotriose was measured using the glucose oxidase (GO) kit from Sigma-Aldrich, according to the manufacturer’s instructions. Activity was measured at 37 °C with the enzyme concentration at 1 μg mL−1and substrate concentration at 0.3%. The reactions were performed in 25 mM HEPES pH 6.5, 150 mM NaCl, 0.02% Tween 80. Measurements were performed for 30 min taking samples every 10 min. Reactions were stopped at each time point through heating at 80 °C for 10 min.
Activity towards polysaccharides was measured by determination of generated reducing ends through the BCA method (Waffenschmidt & Jaenicke, 1987), using glucose as a standard. EUR_01860 was used at 1 μg mL−1 in all assays, while EUR_21100 was used at 50 ng mL−1 for amylopectin, glycogen, amylose and starch and 1 μg mL−1 for all other substrates. Amylopectin (potato), amylose (potato) and pullulan were all initially made as concentrated solutions in dimethyl sulfoxide (DMSO) before dilution with water, resulting in a final DMSO concentration of 7%. All other substrates were dissolved in water. All assays were performed at 37 °C in 25 mM HEPES pH 6.5, 150 mM NaCl, 0.02% Tween 80. Measurements were performed for 30 min taking samples every 10 min. For soluble polysaccharides reactions were stopped through the addition of the BCA reagent. For starch samples, reactions were stopped through the addition of NaOH to 50 mM. Samples were then subjected to centrifugation at 20 000 x g for 5 min followed by removal of the supernatant for reducing end measurement.
Thin layer chromatography
Assays for TLC analysis were performed at 37 °C in 25 mM HEPES pH 6.5, 150 mM NaCl, 0.02% Tween 80. The reactions were allowed to proceed for 30 min and samples were taken every 10 min and stopped by the addition of NaOH to 50 mM. These reactions were spotted onto TLC Silica gel 60 F254 20×20 cm glass plates (Millipore) and separated with the solvent acetonitrile:ethyl acetate:isopropanol:water (85:20:50:50) until the solvent front was within 1 cm of the top of the plate. The sugars were then stained with 0.3% (w/v) N-(1-napthyl)ethylenediamine, 5% (v/v) sulfuric acid in methanol and heated until spots developed.
Analytical Ultracentrifugation (Sedimentation velocity measurements)
The maltooligosaccharide binding proteins EUR_01240, EUR_01830, and EUR_31480 were measured in the presence and absence of saturating (1 mM) glucose, maltotriose and maltose respectively. Experiments were performed in a Beckman Optima XL-I analytical ultracentrifuge at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio. All samples were measured at 35 000 rpm and scanned at 280 nm. Data were acquired in intensity mode. All samples were measured in a buffer containing 20 mM HEPES pH 7, and 100 mM NaCl. Experimental data were collected at 20 °C using 1.2 cm epon 2-channel centerpieces (Beckman-Coulter), using an An60Ti rotor. All conditions were measured at two loading concentrations, OD280 = 0.3 and OD280 = 0.9 (see Supplemental Table S4). Hydrodynamic corrections for buffer density and viscosity were estimated by UltraScan to be 1.00293 g mL−1 and 1. 01295 cP. The partial specific volume was estimated by UltraScan from protein sequence analogous to methods outlined previously (Laue et al., 1992) and found to be 0.7225 mL g−1 for EUR_01830, 0.716 mL g−1 for EUR_01240 and 0.7191 mL g−1 for EUR_31480. All data were analyzed with UltraScan-III ver. 3.1, release 1796 (Demeler et al., 2014), according to method described in (Demeler, 2010). Optimization was performed by 2-dimensional spectrum analysis (2DSA) (Brookes et al., 2010) with simultaneous removal of time- and radially-invariant noise contributions (Schuck & Demeler, 1999). Diffusion-corrected integral sedimentation coefficient distributions were obtained from the enhanced van Holde – Weischet analysis (Demeler & van Holde, 2004). All calculations were performed on the Lonestar cluster at the Texas Advanced Computing Center at the University of Texas at Austin.
Quantitative PCR (qPCR)
For qPCR of the genes encoding EUR_21100, EUR_01860, EUR_01830, EUR_31480 and EUR_01240, cells were grown as described above for the proteomic study, except cells (OD600 = 0.6) were arrested by adding 3 mL of culture into 6 mL of RNA protect, incubating for 5 min, centrifuged at 5 500 x g for 5 min and the cell pellet was stored at −80 °C until RNA extraction. Total RNA was isolated using the RNeasy Minikit (Qiagen) according to the manufacturer’s instructions with the exception that the lysis buffer was supplemented with 1% lysozyme and resuspended cells were lysed by bead-beating with acid-washed glass beads (Sigma Aldrich) using a vortex at intervals of 20 s on and 20 s off for 10 min. DNA digestion was performed with TURBO™ DNase I (Ambion) via two digests of 20 min each. Following DNA digestion, control PCR experiments were performed to ensure that there was no DNA contamination in the RNA preparations. RNA Reverse transcription was performed with 2 μg of RNA, 1 pmol random primers (Invitrogen), and SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Synthesized cDNA was purified using a Gel-Extraction Kit (Qiagen) according to the manufacturer’s instructions. cDNA quantification was performed with a Mastercycler® ep realplex (Eppendorf), using KAPA SYBR® FAST qPCR Master Mix and 500 nM primers for 40 cycles of 95 °C for 3 s, 55 °C for 8 s, 72 °C for 20 s, followed by a melting step to determine amplicon purity. Primers used for qPCR are listed in Supplemental Table S5. The cDNA quantification was normalized across samples via amplification of gapA encoding glyceraldehyde-3-phosphate dehydrogenase, a glycolytic enzyme. The GapA protein was observed at the same abundance in all proteomic samples.
Supplementary Material
Acknowledgments
We wish to thank Christine Pelker at Ingredion (formerly National Starch) for the generous gift of Hylon VII. We also thank Eric Martens from the University of Michigan Medical School for the use of his anerobic chamber and 96 well plate reader for routine culturing of E. rectale. Many thanks as well to Richard Jones of MSBioworks (Ann Arbor) for helpful discussions and processing of the proteomic data. We acknowledge the support of the San
Antonio Cancer Institute grant P30 CA054174 for the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center at San Antonio. This work was supported by funds from a pilot/feasibility grant from the University of Michigan Gastrointestinal Peptides Research Center (DK 034933) awarded to N.M.K., as well as the Host Microbiome Initiative at the University of Michigan Medical School (N.M.K.). B.D. acknowledges support from the National Science Foundation grant NSF-ACI-1339649. Supercomputer time allocations were provided through National Science Foundation grant TG-MCB070039 (to B.D.).
References
- Abbott DW, Higgins MA, Hyrnuik S, Pluvinage B, Lammerts van Bueren A, Boraston AB. The molecular basis of glycogen breakdown and transport in Streptococcus pneumoniae. Mol Microbiol. 2010;77:183–199. doi: 10.1111/j.1365-2958.2010.07199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abell GC, Cooke CM, Bennett CN, Conlon MA, McOrist AL. Phylotypes related to Ruminococcus bromii are abundant in the large bowel of humans and increase in response to a diet high in resistant starch. FEMS Microbiol Ecol. 2008;66:505–515. doi: 10.1111/j.1574-6941.2008.00527.x. [DOI] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta crystallographica. Section D, Biological crystallography. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Akiyama N, Takeda K, Miki K. Crystal structure of a periplasmic substrate-binding protein in complex with calcium lactate. J Mol Biol. 2009;392:559–565. doi: 10.1016/j.jmb.2009.07.043. [DOI] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Junwang, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Consortium M, Weissenbach J, Ehrlich SD, Bork P. Enterotypes of the human gut microbiome. Nature. 2011 doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Cao W, Demeler B. A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. European Biophysical Journal. 2010;39:405–414. doi: 10.1007/s00249-009-0413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassard C, Delmas E, Robert C, Lawson PA, Bernalier-Donadille A. Ruminococcus champanellensis sp. nov., a cellulose-degrading bacterium from human gut microbiota. Int J Syst Evol Microbiol. 2012;62:138–143. doi: 10.1099/ijs.0.027375-0. [DOI] [PubMed] [Google Scholar]
- Clarke JM, Topping DL, Bird AR, Young GP, Cobiac L. Effects of high-amylose maize starch and butyrylated high-amylose maize starch on azoxymethane-induced intestinal cancer in rats. Carcinogenesis. 2008;29:2190–2194. doi: 10.1093/carcin/bgn192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn DW, Svensson B. Surface binding sites in carbohydrate active enzymes: and emerging picture of structural and functional diversity. In: Lindhorst TK, Rauter AP, editors. Carbohydrate Chemistry: Chemical and Biological Approaches. Cambridge: Royal Society of Chemistry; 2013. pp. 204–221. [Google Scholar]
- Colinge J, Chiappe D, Lagache S, Moniatte M, Bougueleret L. Differential proteomics via probabilistic peptide identification scores. Anal Chem. 2005;77:596–606. doi: 10.1021/ac0488513. [DOI] [PubMed] [Google Scholar]
- Comfort D, Clubb RT. A comparative genome analysis identifies distinct sorting pathways in gram-positive bacteria. Infect Immun. 2004;72:2710–2722. doi: 10.1128/IAI.72.5.2710-2722.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon MA, Kerr CA, McSweeney CS, Dunne RA, Shaw JM, Kang S, Bird AR, Morell MK, Lockett TJ, Molloy PL, Regina A, Toden S, Clarke JM, Topping DL. Resistant starches protect against colonic DNA damage and alter microbiota and gene expression in rats fed a Western diet. J Nutr. 2012;142:832–840. doi: 10.3945/jn.111.147660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeler B. Methods for the design and analysis of sedimentation velocity and sedimentation equilibrium experiments with proteins. Current Protocols in Protein Science. 2010:7.13.11–17.13.24. doi: 10.1002/0471140864.ps0713s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeler B, Gorbet G, Zollars D, Dubbs B, Brookes E, Cao WD. UltraScan-III version 3.1: A comprehensive data analysis software package for analytical ultracentrifugation experiments. 2014. [Google Scholar]
- Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Anal Biochem. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, Lobley GE. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol. 2007;73:1073–1078. doi: 10.1128/AEM.02340-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Flint HJ. Proposal of a neotype strain (A1-86) for Eubacterium rectale. Request for an opinion. Int J Syst Evol Microbiol. 2008;58:1735–1736. doi: 10.1099/ijs.0.2008/004580-0. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Foresti ML, del Williams MP, Martinez-Garcia R, Vazquez A. Analysis of a preferential action of alpha-amylase from B. licheniformis towards amorphous regions of waxy maize starch. Carbohydr Polym. 2014;102:80–87. doi: 10.1016/j.carbpol.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, Pace NR, Li E. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, Gonzalez A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, Kapsimalis B. Synthesis of intracellular iodophilic polysaccharide by Streptococcus mitis. Arch Oral Biol. 1963;8:319–329. doi: 10.1016/0003-9969(63)90024-4. [DOI] [PubMed] [Google Scholar]
- Haider S, Pal R. Integrated analysis of transcriptomic and proteomic data. Curr Genomics. 2013;14:91–110. doi: 10.2174/1389202911314020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- Han Y, Agarwal V, Dodd D, Kim J, Bae B, Mackie RI, Nair SK, Cann IK. Biochemical and structural insights into xylan utilization by the thermophilic bacterium Caldanaerobius polysaccharolyticus. J Biol Chem. 2012;287:34946–34960. doi: 10.1074/jbc.M112.391532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hold GL, Schwiertz A, Aminov RI, Blaut M, Flint HJ. Oligonucleotide probes that detect quantitatively significant groups of butyrate-producing bacteria in human feces. Appl Environ Microbiol. 2003;69:4320–4324. doi: 10.1128/AEM.69.7.4320-4324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Jiang G, Liu Q. Characterization of residues from partially hydrolyzed potato and high amylose corn starches by pancreatic alpha-amylase. Starch. 2002;54:527–533. [Google Scholar]
- Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin NM, Smith TJ. SusG: A unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure. 2010;18:200–215. doi: 10.1016/j.str.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Kraal L, Abubucker S, Kota K, Fischbach MA, Mitreva M. The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS One. 2014;9:e97279. doi: 10.1371/journal.pone.0097279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Peltier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Cambridge: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- Lee HS, Kim MS, Cho HS, Kim JI, Kim TJ, Choi JH, Park C, Lee HS, Oh BH, Park KH. Cyclomaltodextrinase, neopullulanase, and maltogenic amylase are nearly indistinguishable from each other. J Biol Chem. 2002;277:21891–21897. doi: 10.1074/jbc.M201623200. [DOI] [PubMed] [Google Scholar]
- Leitch EC, Walker AW, Duncan SH, Holtrop G, Flint HJ. Selective colonization of insoluble substrates by human faecal bacteria. Environ Microbiol. 2007;9:667–679. doi: 10.1111/j.1462-2920.2006.01186.x. [DOI] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006a;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006b;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2013 doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- Maeda K, Nagata H, Kuboniwa M, Ojima M, Osaki T, Minamino N, Amano A. Identification and characterization of Porphyromonas gingivalis client proteins that bind to Streptococcus oralis glyceraldehyde-3-phosphate dehydrogenase. Infect Immun. 2013;81:753–763. doi: 10.1128/IAI.00875-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, Cantarel BL, Coutinho PM, Henrissat B, Crock LW, Russell A, Verberkmoes NC, Hettich RL, Gordon JI. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106:5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5:e15046. doi: 10.1371/journal.pone.0015046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Yamada M, Kurakata Y, Yoshida H, Kamitori S, Nishikawa A, Tonozuka T. Crystal structures of open and closed forms of cyclo/maltodextrin-binding protein. FEBS J. 2009;276:3008–3019. doi: 10.1111/j.1742-4658.2009.07020.x. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowbray SL, Smith RD, Cole LB. Structure of the periplasmic glucose/galactose receptor of Salmonella typhimurium. Receptor. 1990;1:41–53. [PubMed] [Google Scholar]
- Nemoto H, Kataoka K, Ishikawa H, Ikata K, Arimochi H, Iwasaki T, Ohnishi Y, Kuwahara T, Yasutomo K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig Dis Sci. 2012;57:2955–2964. doi: 10.1007/s10620-012-2236-y. [DOI] [PubMed] [Google Scholar]
- Nie L, Wu G, Culley DE, Scholten JC, Zhang W. Integrative analysis of transcriptomic and proteomic data: challenges, solutions and applications. Crit Rev Biotechnol. 2007;27:63–75. doi: 10.1080/07388550701334212. [DOI] [PubMed] [Google Scholar]
- Nielsen MM, Bozonnet S, Seo ES, Motyan JA, Andersen JM, Dilokpimol A, Abou Hachem M, Gyemant G, Naested H, Kandra L, Sigurskjold BW, Svensson B. Two secondary carbohydrate binding sites on the surface of barley alpha-amylase 1 have distinct functions and display synergy in hydrolysis of starch granules. Biochemistry. 2009;48:7686–7697. doi: 10.1021/bi900795a. [DOI] [PubMed] [Google Scholar]
- Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Oldham ML, Chen S, Chen J. Structural basis for substrate specificity in the Escherichia coli maltose transport system. Proc Natl Acad Sci U S A. 2013;110:18132–18137. doi: 10.1073/pnas.1311407110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L. Long term effect of gut microbiota transfer on diabetes development. J Autoimmun. 2014 doi: 10.1016/j.jaut.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217:133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, LePaslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Ramsay AG, Scott KP, Martin JC, Rincon MT, Flint HJ. Cell-associated alpha-amylases of butyrate-producing Firmicute bacteria from the human colon. Microbiology. 2006;152:3281–3290. doi: 10.1099/mic.0.29233-0. [DOI] [PubMed] [Google Scholar]
- Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Bernalier-Donadille A. The cellulolytic microflora of the human colon: evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol Ecol. 2003;46:81–89. doi: 10.1016/S0168-6496(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Schafer K, Magnusson U, Scheffel F, Schiefner A, Sandgren MO, Diederichs K, Welte W, Hulsmann A, Schneider E, Mowbray SL. X-ray structures of the maltose-maltodextrin-binding protein of the thermoacidophilic bacterium Alicyclobacillus acidocaldarius provide insight into acid stability of proteins. J Mol Biol. 2004;335:261–274. doi: 10.1016/j.jmb.2003.10.042. [DOI] [PubMed] [Google Scholar]
- Schuck P, Demeler B. Direct Sedimentation Boundary Analysis of Interference Optical Data in Analytical Ultracentrifugation. Biophysical Journal. 1999;76:2288–2296. doi: 10.1016/S0006-3495(99)77384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KP, Martin JC, Chassard C, Clerget M, Potrykus J, Campbell G, Mayer CD, Young P, Rucklidge G, Ramsay AG, Flint HJ. Substrate-driven gene expression in Roseburia inulinivorans: importance of inducible enzymes in the utilization of inulin and starch. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4672–4679. doi: 10.1073/pnas.1000091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segain JP, Raingeard de la Bletiere D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blottiere HM, Galmiche JP. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47:397–403. doi: 10.1136/gut.47.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar N, Li N, Tang JW, Patel BK, Swaminathan K. Crystal structure of AmyA lacks acidic surface and provide insights into protein stability at poly-extreme condition. FEBS Lett. 2006;580:2646–2652. doi: 10.1016/j.febslet.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Stam MR, Danchin EG, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of alpha-amylase-related proteins. Protein Eng Des Sel. 2006;19:555–562. doi: 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi-Faridi A, Licht A, Bulut H, Scheffel F, Keller S, Wehmeier UF, Saenger W, Schneider E. Crystal structures of the solute receptor GacH of Streptomyces glaucescens in complex with acarbose and an acarbose homolog: comparison with the acarbose-loaded maltose-binding protein of Salmonella typhimurium. J Mol Biol. 2010;397:709–723. doi: 10.1016/j.jmb.2010.01.054. [DOI] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Van Immerseel F, Ducatelle R, De Vos M, Boon N, Van De Wiele T, Verbeke K, Rutgeerts P, Sas B, Louis P, Flint HJ. Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. J Med Microbiol. 2010;59:141–143. doi: 10.1099/jmm.0.017541-0. [DOI] [PubMed] [Google Scholar]
- van Munster IP, Tangerman A, Nagengast FM. Effect of resistant starch on colonic fermentation, bile acid metabolism, and mucosal proliferation. Dig Dis Sci. 1994;39:834–842. doi: 10.1007/BF02087431. [DOI] [PubMed] [Google Scholar]
- Vonk RJ, Hagedoorn RE, de Graaff R, Elzinga H, Tabak S, Yang YX, Stellaard F. Digestion of so-called resistant starch sources in the human small intestine. Am J Clin Nutr. 2000;72:432–438. doi: 10.1093/ajcn/72.2.432. [DOI] [PubMed] [Google Scholar]
- Waffenschmidt S, Jaenicke L. Assay of reducing sugars in the nanomole range with 2,2′-bicinchoninate. Anal Biochem. 1987;165:337–340. doi: 10.1016/0003-2697(87)90278-8. [DOI] [PubMed] [Google Scholar]
- Walker AW, Duncan SH, Harmsen HJ, Holtrop G, Welling GW, Flint HJ. The species composition of the human intestinal microbiota differs between particle-associated and liquid phase communities. Environ Microbiol. 2008;10:3275–3283. doi: 10.1111/j.1462-2920.2008.01717.x. [DOI] [PubMed] [Google Scholar]
- Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. Isme J. 2011;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Zhu YH, Li DF, Wang Z, Jensen BB. In vitro fermentation of various fiber and starch sources by pig fecal inocula. J Anim Sci. 2004;82:2615–2622. doi: 10.2527/2004.8292615x. [DOI] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854 e1841. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- Wilson WA, Roach PJ, Montero M, Baroja-Fernandez E, Munoz FJ, Eydallin G, Viale AM, Pozueta-Romero J. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol Rev. 2010;34:952–985. doi: 10.1111/j.1574-6976.2010.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, Chen Y, Yang F, Lu N, Wang Z, Luan C, Liu Y, Wang B, Xiang C, Wang Y, Zhao F, Gao GF, Wang S, Li L, Zhang H, Zhu B. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66:462–470. doi: 10.1007/s00248-013-0245-9. [DOI] [PubMed] [Google Scholar]
- Yoshioka Y, Hasegawa K, Matsuura Y, Katsube Y, Kubota M. Crystal structures of a mutant maltotetraose-forming exo-amylase cocrystallized with maltopentaose. J Mol Biol. 1997;271:619–628. doi: 10.1006/jmbi.1997.1222. [DOI] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Xu X, Peng Y, Luo Y, Yang K. Cell wall proteome of Clostridium thermocellum and detection of glycoproteins. Microbiol Res. 2012;167:364–371. doi: 10.1016/j.micres.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6:1535–1543. doi: 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukin RS, Strange PG, Heavey R, Koshland DE. Properties of the galactose binding protein of Salmonella typhimurium and Escherichia coli. Biochemistry. 1977;16:381–386. doi: 10.1021/bi00622a007. [DOI] [PubMed] [Google Scholar]
- Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods in molecular biology. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.