

Abstract
The use of pseudocontact shifts arising from paramagnetic metal ions in a microcrystalline protein sample is proposed as a strategy to obtain unambiguous signal assignments in solid-state NMR spectra enabling distance extraction for protein structure calculation. With this strategy, 777 unambiguous (281 sequential, 217 medium-range, and 279 long-range) distance restraints could be obtained from PDSD, DARR, CHHC, and the recently introduced PAR and PAIN-CP solid-state experiments for the cobalt(II)-substituted catalytic domain of matrix metalloproteinase 12 (159 amino acids, 17.6 kDa). The obtained structure is a high resolution one, with backbone rmsd of 1.0 ± 0.2 Å, and is in good agreement with the X-ray structure (rmsd to X-ray 1.3 Å). The proposed strategy, which may be generalized for nonmetallo-proteins with the use of paramagnetic tags, represents a significant step ahead in protein structure determination using solid-state NMR.
Introduction
The recent years have seen a rapid growth of the solid-state NMR(SSNMR) methodology applied to biomolecular systems.1–4 Indeed, SSNMR has shown to be a reliable tool for investigating the structure at atomic resolution of membrane proteins,5–9 fibril,10–16 and precipitated proteins,17 which are hardly accessible by other techniques such as X-ray diffraction or electron microscopy.
The size of the investigated proteins has progressively increased in past years, even if it still remains limited to systems smaller than 100 amino acids (AA), at least in the structures until now reported.18–22 It follows that the development of methodologies able to access larger systems is of crucial importance to SSNMR to facilitate studies of more complicated biological systems.23–25
The biomolecular SSNMR structural determination is essentially performed by collecting large number of distance restraints, in strict analogy to what is done in solution NMR with NOEs. Earlier biomolecular studies, based on REDOR or ZF TEDOR26–28 experiments and performed on short peptides, demonstrated that SSNMR is effective in determining high-resolution molecular structures. Unfortunately, these methods cannot be directly extended to proteins, which need a larger number of distance restraints for determining the folding. With the approach introduced by the Oschkinat group,2,18 a comparably large number of restraints are collected on the microcrystalline SH3 protein by inspection of PDSD29,30 and DARR31,32 spectra acquired on selectively labeled samples. The use of 1,3-13C-glycerol and 2-13C-glycerol as enriched media makes it possible to express proteins with a selective labeling scheme that allows strong spectra simplifications.2 As several types of residues are expressed with an alternate 13C labeling,18,33 PDSD and DARR spectra lack a large number of intense C–C correlations that do not provide restraints for the protein fold (corresponding to the directly covalent-bonded carbon atoms). Moreover, dipolar truncation23,24,34 and relayed transfer magnetization mechanisms19,23,24 are strongly reduced, making it possible to observe a larger number of direct long-range correlations. The main drawback of this method is the preparation of several samples with lower expression yield and a still rather expensive labeling. Consequently, alternative methods applicable also to uniformly labeled samples have been proposed. In particular, CHHC/NHHC sequences indirectly detect 1H–1H contacts through the acquisition of the more resolved 15N and 13C dimensions.35,36 These sequences provide precious restraints that make the definition of low-resolution structures of small proteins possible without selective labeling.37 A large part of the information encoded in such spectra can be only ambiguously assigned as a consequence of spectral crowding and 13C peak line-widths.20,38 This problem has been partly alleviated by extending to solid-state NMR software tools such as ARIA,20,39,40 ATNOS-CANDID,19 and PASD,21 which had already been developed in solution NMR to handle ambiguous restraints in structure calculations. The use of both ambiguous and structurally unambiguous restraints (whose ambiguity has been solved within the structure calculation procedure) increases the number of restraints by about 1 order of magnitude, achieving the determination of high-resolution structures of uniformly labeled samples.
The extension of this approach to proteins larger than 100 AA may be, however, critical because the number of ambiguous cross-peaks rapidly increases with the number of residues, while the number of unambiguous cross-peaks may decrease to only a few tens or even approach zero, limiting the determination of an accurate structure even by using dedicated software tools. These considerations point to the need of using several sources of restraints to collect a sufficiently large amount of unambiguous structural information. An increased variety of restraints does not only help in resolving assignment ambiguity, but it also improves the structure accuracy. It has been often observed that SSNMR high-resolution structures show a discrepancy between the high precision of the structure family and its comparably poorer accuracy, as judged by comparison with the X-ray crystallographic structures of the same systems.18–20,22 This problem could be originated from the lack of an accurate calibration of the cross-peak intensities due, among other causes, to relayed magnetization transfer effects that affect DARR spectra of uniformly labeled samples,19,41 but were also observed in CHHC spectra.38
An example of the need for having different sources of structural restraints is a recent study performed on the model system GB1 (56 AA), which demonstrated that, using only distance restraints, the precision of the obtained structure can reach about 1 Å, but the accuracy remains ~2 Å.22 This is observed even when a very large number of restraints are used (more than 7000 distance restraints, with more than 130 distance restraints per residue!), determined both on uniformly labeled samples (from CHHC spectra) and in selectively labeled samples (DARR spectra). To further improve the quality of the structure, other types of restraints such as dihedral angle restraints (from TALOS42) and vector angle restraints (VEAN43,44) were used, improving both precision (up to 0.3 Å) and accuracy (up to 1.4 Å), but not reducing the gap among these two quantities.
We report here the structure determination through SSNMR of the catalytic domain of matrix metalloproteinase-12 (MMP-12, 159 AA, 17.6 kDa) and demonstrate that the simultaneous use of paramagnetic pseudocontact shifts (PCS) (measured on the cobalt(II)-substituted derivative),45 along with diamagnetic restraints (measured on the native zinc(II)-containing protein), provides enough restraints to afford the structural investigation of a protein as large as 159 AA. In recent years, paramagnetic proteins in the solid state were also afforded,46–51 and paramagnetic contributions such as pseudocontact shifts52,53 or relaxation times54,55 were indicated as a further promising source of structural restraints in solid-state NMR.
The diamagnetic restraints were derived from several classical experiments (DARR,31,32 CHHC35,36) and from the recently introduced experiments PAR (proton-assisted recoupling)24,56,57 and PAIN-CP (proton-assisted insensitive nuclei cross-polarization),25 which substantially increase the number of available distance restraints.
As compared to the other structural restraints described above, PCS are directly related to the nuclear position with respect to the metal through a simple and quantitative relationship, and a very good match between the experimental values and those predicted on the basis of a molecular structure was observed.52,53 Although PCS alone are not sufficient to define a protein structure, it can be easily determined and is helpful, together with the other diamagnetic restraints, in the structure calculation process.58,59 More importantly, it is found that when PCS are used together with even a few unambiguous restraints, a low-resolution but comparatively accurate structure can be already determined. The use of this structure allows one to solve part of the cross-peak ambiguity and to increase the number of available restraints in an iterative procedure.52
Material and Methods
Preparation of the Microcrystalline Samples
The ZnMMP-12 and the CoMMP-12 proteins complexed with the strong NNGH inhibitor (NNGH = N-isobutyl-N-[4-methoxyphenylsulphonyl]-glycyl hydroxamic acid) were prepared following the already published procedure.45,60 For each sample, the amount of protein was chosen to obtain around 10 mg of microcrystalline material for the 2.5 mm, 15 mg for the 3.2 mm, and 35–40 mg for the 4 mm rotor. All samples were crystallized following the already reported procedure.52,53,60
Solid-State NMR Spectroscopy
The NMR spectra were recorded on Bruker Avance wide-bore 700 or 850 MHz instrument or on a 900 MHz standard bore instrument operating at 16.4, 20.0, and 21.1 T, respectively (176.0, 213.8, and 226.3 MHz 13C Larmor frequency, respectively). Experiments at 16.4 T were acquired on a double/triple-channel 4.0 mm CP-MAS probehead, those at 20.0 T were acquired on a double/triple-channel 3.2 mm CP-MAS probehead, while experiments at 21.1 T were acquired on a triple-channel 2.5 mm CP-MAS probehead. The spinning frequency of the ZrO2 MAS rotors was stabilized to ±2 Hz. The 4 mm and 3.2 mm rotors were used with a PTFE spacer to reduce the effective volume, while standard 2.5 mm rotors were used for the 2.5 mm CP-MAS probe. The probe temperature was kept at a nominal temperature of 270 K for experiments performed at 11.5 kHz MAS frequency and at 260 K for experiments at 19 and 20 kHz of MAS frequency, which ensured a constant sample temperature around 280 K.
The experimental conditions for the experiments acquired on the CoMMP12 to determine PCS were already published in refs 52, 53, 61.
Standard sequences were used for cross-polarization (CP), 2D proton-driven spin diffusion (PDSD),29,30 dipolar-assisted rotational resonance (DARR),31,32 and 2D CHHC35,36 experiments. These experiments were acquired on the ZnMMP12 microcrystalline sample at 16.4 T and at MAS frequency of 11.5 kHz (4 mm CP-MAS probehead). In the CP experiments, the 1H 90° pulse was set to 2.65 μs, a 100%/50% ramp was used on the 1H channel, with 74 kHz for 100% of power level. The 13C CP power level was set to 70 kHz with a contact time of 0.75 ms. Similar parameters were also used for the standard proton-driven spin diffusion sequence (2D PDSD) and DARR experiments. The 13C 90° pulse was 4.2 μs, and the SPINAL-64 sequence62 at 92 kHz of power was used for 1H decoupling during both direct and indirect acquisition times. The PDSD and DARR spectra were acquired with variable mixing times from 50 to 800 ms; a weak 1H CW radio frequency optimized at 11.5 kHz was used during the DARR mixing time. The indirect and direct evolution times were t1max = 18.4 ms and t2max = 27.6 ms, and each experiment was acquired in about 62 h.
In the 2D CHHC experiments, the RF power level for each CP step was set to the above-reported values, the first CP was 550 μs long, the second and third CP were both 70 μs long. The 13C 90° pulse was 3.9 μs, and the SPINAL-64 1H decoupling power was 92 kHz for both direct and indirect acquisition times. A delay of 4 ms was used in the z-filter after the first CP. The acquisition times were t1max = 6 ms and t2max = 12 ms, and the whole experiment was acquired in 4 days. Mixing times of 150 and 300 μs were used in the experiments acquired for determining the distance restraints; for comparison, analogous CHHC experiments with mixing times ranging from 60 to 1200 μs were acquired.
The aliphatic 13C–13C PAR experiments24 were acquired both at 20.0 T and 19 kHz MAS frequency (3.2 mm probehead) and at 21.1 T and 20 kHz MAS frequency (2.5 mm probehead). Power level and pulse length for CP and 1H decoupling were similar to those already reported above. The aliphatic 13C–13C PAR experiments used to determine the largest number of distance restraints were acquired with 15 ms mixing time (21.1 T) and 20 ms mixing time (20.0 T), using 1H and 13C irradiation at 50 and 54 kHz, respectively. For comparison, other experiments with mixing times ranging from 5 to 20 ms were also performed.
The PAIN-CP experiments25 were performed at 21.1 T and 20 kHz MAS frequency (2.5 mm probehead). Power level and 1H pulse length for CP and 1H decoupling were similar to those already reported above. The 1H–15N Hartmann–Hahn matching was optimized with 36 kHz for the 15N B1, and a 100%/50% ramp on the 1H channel applied for 1.7 ms with 68 kHz for 100% power level. The PAIN-CP experiments used to determine the distance restraints were acquired with 15 ms mixing time, using the 15N and 13C irradiation at 50 kHz and 1H irradiation at ~48 kHz.
All PDSD, DARR, and CHHC experiments were acquired by using the States-TPPI scheme for the indirect dimension and were processed with an 8192 × 4096 matrix of points, with Gaussian and squared cosine windows functions for direct and indirect dimensions, respectively.
All of the reported 13C solid-state chemical shifts are referred to DSS as reported in the literature.63
SPARKY software64 was used in the spectra analysis, and MOLMOL65 was used for the structure analyses and figure preparation.
Intramolecular PCS for Structure Calculation
The structure was calculated using CYANA66 and its PARAMAGNETICCYANA67 module to handle paramagnetic restraints. (The patch to convert CYANA to PARAMAGNETICCYANA is available at http://www.postgenomicnmr.net) For the final calculations, the following structural restraints were used: 318 experimental intramolecular SSNMR PCS (tolerance 0.3 ppm), 186 angle restraints obtained from TALOS42 prediction on the SSNMR shifts of Cα, C′, Cβ nuclei of the fully labeled ZnMMP-12, and a total of 777 unambiguous distance restraints. Three additional restraints were introduced by linking the paramagnetic metal ion to the aromatic nitrogens of the three coordinating histidines.
The upper distance limits were determined without calibration, directly assigning values of 9.0, 7.0, and 6.5 Å for any assigned cross-peaks in the PDSD/DARR, PAR/PAIN-CP, and CHHC spectra, respectively. These numbers are analogous to the limits suggested in the literature,20,24 but increased by about 2 Å (PDSD/DARR) and about 1–1.5 Å (CHHC, PAR, PAIN-CP), to reduce the distortion due to relayed transfer effects.19 Initial paramagnetic tensor parameters Δχax and Δχrh were imposed their typical values (Δχax = 7.0 × 10−32 m3; Δχrh = −2.4 × 10−32 m3, 298 K),68 and then refined in the last steps of the structure refinement.
Up to 2000 simulated annealing minimizations were conducted using these distance restraints together with all of the 318 PCS and 186 angle restraints, collecting a family with the 20 structures with smallest target function. Table 1 summarizes all of the structural restraints used.
Table 1.
Summary of the Total Number and Assigned Cross-Peaks in the Investigated Spectra, and Upper Distance Limits Used for Distance Restraints from Each Spectral Typea
| type of spectra analyzed | total number of observed cross-peaks | unambiguous restraints
|
upper distance limit used | |
|---|---|---|---|---|
| from assignment | from structure | |||
| PDSD | 909 | 80 | 181 | 9.0 |
| DARR | 824 | 86 | 138 | 9.0 |
| CHHC (1) | 692 | 64 | 132 | 6.5 |
| CHHC (2) | 574 | 37 | 55 | 6.5 |
| PAR (1) | 1016 | 83 | 232 | 7.0 |
| PAR (2) | 747 | 61 | 100 | 7.0 |
| PAR (3) | 667 | 50 | 129 | 7.0 |
| PAIN-CP | 563 | 55 | 43 | 6.5 |
The different CHHC spectra were acquired with 300 μs (1) and 150 μs (2) of mixing times, and the different aliphatic 13C–13C PAR spectra were acquired with 20 ms (1) and 15 ms of mixing times at 850 MHz (2) and 900 MHz (3) of 1H Larmor frequency.
Wherever in the text reference is made to precision of the structure family, the average rmsd of the structure family to the mean structure is meant. Conversely, accuracy is defined here by the rmsd to the X-ray structure from the mean structure of the family.
ARIA Calculation
The protein calculations were done by using the ARIA 2.2.2 β-version, courtesy of M. Nilges and B. Bardiaux.69 Two sets of restraints were used: one containing 240 unambiguously assigned restraints coming from the PDSD/DARR, CHHC, PAR, and PAIN-CP spectra and using the upper-distance limits reported in Table 1, and the second containing 2098 unassigned cross-peaks obtained from the above spectra. A standard 8-iteration ARIA protocol was used with a simulated annealing in torsion angle dynamics (TAD). The simulated annealing protocol consisted of three parts: (i) 10 000 steps at high temperature (10 000 K), (ii) 100 000 steps of annealing cooling down to 1000 K, and (iii) 50 000 steps cooling down to 50 K. All ARIA calculations were performed with 186 TALOS angle restraints but without using PCS and metal link restraints.
Results
Used Restraints
The SSNMR structure of MMP-12 in the microcrystalline phase was obtained through the simultaneous use of paramagnetic PCS restraints and diamagnetic restraints. As native MMP-12 contains a Zn(II) (diamagnetic) ion in the catalytic active site, paramagnetic shifts can be observed after the replacement of the Zn(II) ion with the paramagnetic Co(II) ion.45 The pseudocontact shifts, directly measured as the difference between the shifts of the corresponding nuclei in Co(II)-MMP-12 and Zn(II)-MMP-12, are the sum of intramolecular and intermolecular PCS contributions. The latter are due to the long-distance interaction with the paramagnetic metals of the neighboring proteins in the crystal lattice.52,53 However, these contributions can be experimentally separated by diluting the paramagnetic species, and the intramolecular terms are analogous to the PCS that could be measured in liquid-state NMR. In a recent publication, up to 318 intramolecular PCS were determined and used together with 284 distance restraints (DARR and CHHC) and 152 angle restraints to obtain a low-resolution protein structure (3.1 Å backbone (bb) rmsd).52 Apparently, to achieve a high-resolution structure, a sizable increase of the number of useful diamagnetic restraints (distance and angle restraints) is needed, which is one of the reasons that we have performed PAR and PAIN-CP experiments in addition to classical experiments such as PDSD/DARR and CHHC.
The angle restraints were estimated by using the program TALOS on the basis of the backbone Cα, C′, Cβ chemical shifts; this number has been increased up to 186 angle restraints by means of an updated version of TALOS.
Increasing the number of distance restraints is a potentially difficult task for a relatively large protein due to increasing problems of assignment ambiguities. The distance restraints used at the end of the iterative assignment procedure described below (as many as 777) could be obtained from a careful analysis of the PDSD/DARR, CHHC, PAR, and PAIN-CP experiments acquired on diamagnetic U–13C,15N-ZnMMP-12 protein only by taking advantage of the paramagnetic restraints to achieve low resolution structures at an early stage of the process, and then exploiting these structures to progressively resolve assignment ambiguities. PCS restraints suffer much less from the problem of ambiguity, because they are directly obtained from the comparison of assigned cross-peaks in the spectra used for sequential assignment of the paramagnetic and diamagnetic forms.52,53 In this strategy, immediate use is thus made of all of the PCS and TALOS restraints, which are unambiguous by nature, together with the few distance restraints from cross-peaks that are also intrinsically unambiguous from the various correlation experiments. These restraints allowed us to define an initial low-resolution protein structure. This structure was then used to solve the ambiguity of part of the ambiguously assigned cross-peaks, obtaining additional “structurally unambiguous” distance restraints to be used together with the previous ones to arrive to a more resolved structure.
The restraints from all of the experiments are necessary to arrive at the final structure. However, we note that the largest number of restraints was obtained from the aliphatic 13C–13C PAR spectra. More details on the analysis of the spectra and of the number of (initial) unambiguous restraints and (final) structurally unambiguous restraints for each of the five types of experiments (PDSD, DARR, CHHC, PAIN-CP and PAR) are reported in the Supporting Information. The relevant data are summarized in Tables 1 and 2. It should be noted that the sum of all of the restraints from the various experiments largely exceeds the total of 777 diamagnetic restraints finally used in the structure calculation, because a large part of these restraints are in common to more than one experiment (see next section).
Table 2.
Summary of the Contributions of Each Different Spectra Type to the Ensemble of Distance Restraintsa
| used restraints | sequential contacts |i – j| = 1 | medium-range contacts |i – j| ≤ 4 | long-range contacts |i – j| ≥ 5 | % of total number of restraints | |
|---|---|---|---|---|---|
| PDSD/DARR | 161 | 48 | 54 | 59 | 21 |
| CHHC | 221 | 67 | 62 | 92 | 28 |
| PAR | 297 | 110 | 83 | 104 | 38 |
| PAIN-CP | 98 | 56 | 18 | 24 | 13 |
Only inter-residue contacts were converted in distance restraints and used in the structural calculations.
The present large number (777) of unambiguous restraints was achieved also because of the quality of the acquired spectra (13C line width of the order of 80–100 Hz and, for methyl, 60–70 Hz, which corresponds to 0.30–0.45 ppm at 850 MHz proton Larmor frequency, and 15N line width of the order of 30 Hz (0.35 ppm at 850 MHz)). 13C–13C J-coupling gives a significant contribution to13C line width; thus an increase in resolution should be expected by using techniques like maximum entropy or spin-state selection that remove the homo-nuclear J-coupling.70–74 Comparable quality is frequently observed on microcrystalline samples,2,4,19,20,22 but less commonly in noncrystalline ordered biological solids like fibrils.11 For samples having larger linewidths (~1.0 ppm as observed in some well-structured but noncrystalline samples),13,75,76 the number of ambiguities in the cross-peak assignment can rapidly increase, limiting the size of the investigated molecules. In these cases, PCS are expected to play an even greater role, being unambiguous from assignment. Thus, as observed here for the early steps in the structural calculation, PCS can help in providing restraints and solving ambiguities in the assignment of the diamagnetic restraints.
Structure Calculations Protocol
An overall initial number of 240 unambiguously assigned inter-residue cross-peaks was determined from the analysis of the PDSD, DARR, CHHC, PAR, and PAIN-CP spectra, and converted into distance restraints (Figure 1, right-hand side). Unfortunately, any attempts to define an accurate calibration of the cross-peak intensity in term of internuclear distances produced unsatisfactory results (we defer to the following section for a more detailed discussion of the calibration criteria used to define distance restraints). Thus, distance restraints were obtained by defining a unified upper distance limit for all of the restraints coming from the same spectra (DARR, CHHC, PAR, and PAIN-CP). The used values are reported in Table 1. Figure 1 shows how the initial number of 240 restraints is arrived at: essentially, restraints that were in common between two different spectra were taken from the more restrictive one, that is, CHHC over PAR, and PAR over DARR/PDSD (C–N restraints from PAIN-CP being of course not in common with the other spectra).
Figure 1.
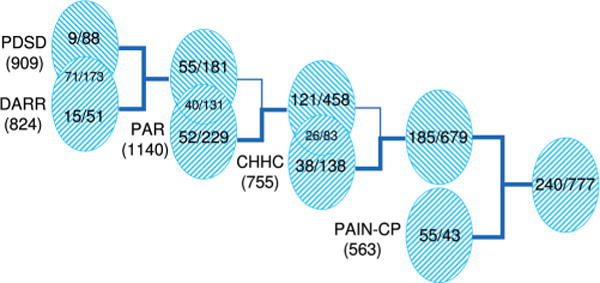
Summary of SSNMR experiments used to obtain internuclear distance restraints, and of the number of the resulting unambiguous distance restraints obtained in the initial and final stages of the structure calculations. The numbers in parentheses under the acronym of each experiment represent the total number of cross-peaks obtained by peak picking from that class of experiments. The numbers enclosed in the ellipses and separated by slashes represent the unambiguous cross-peaks assigned in the initial (left) and final (right) stages of the calculations for that experiment. The numbers in the intersections of the ellipses represent the cross-peaks in common between the two corresponding experiments, while the numbers outside the intersections are the cross-peaks uniquely present in that experiment. The tighter restraints were always selected when in common between two experiments.
Because the upper distance limits are relatively large, ranging from 6.5 to 9.0 Å, a large number of intraresidue cross-peaks would be translated in essentially ineffective restraints, and thus they were not included in the structural calculation. This “crude” method loses part of the structural information that could be implicitly included in the cross-peak intensity (or volume). Consequently, a larger number of restraints need to be collected to define the protein folding. On the other hand, by using these conservative upper distance limits, we reduce to a minimum the number of violations/structural distortions that could be introduced through possible miscalibration of internuclear contacts.19
The above introduced unambiguous distance restraints were used in a simulated annealing structure calculation in CYANA 2.1 software, together with the TALOS angle restraints. The family of 20 structures with smaller target functions showed an rmsd within the family of 9.3 ± 2.0 Å, and the distance of the family mean structure from the X-ray structure was 6.5 Å. The resolution of this preliminary structure is too poor to allow one to manually resolve a significant number of cross-peak ambiguities and further extend the structural refinement.
At this point, the 318 PCS were introduced as further structural restraints. The use of PCS in the refinement of the protein structure calculation is well-known in solution NMR of proteins. PCS can be converted into distance information once the axial and rhombic components of the anisotropy of the magnetic susceptibility tensor are known. These are usually determined through an iterative procedure together with the structural refinement once the structure has reached a resolution of the order of at least 2 Å. In the absence of such a structure (i.e., until the SSNMR structure has reached a sufficiently high resolution, see later), typical values reported for Co(II)-replaced metalloproteins68 (Δχax = 7.0 × 10−32 m3; Δχrh = −2.4 × 10−32 m3, 298 K) have been used.
By taking these initial tensor parameters together with the 318 intramolecular PCS restraints, the quality of the structure improves significantly. The resulting family of 20 structures has an rmsd of 3.0 ± 0.7 Å and an accuracy of 5.0 Å to X-ray. At this point, the structure was resolved enough to allow us to proceed with the further assignment of additional structurally unambiguous restraints.
With the newly assigned restraints, another structural calculation was performed including PCS, and this procedure was iterated until we reached a structure with a backbone resolution around 2 Å. This resolution is high enough to introduce the refinement of the tensor parameters in the iterative procedure: therefore, the structure itself has been used for back-calculating the tensor parameters from the experimental PCS values, and the new values were used again in another structural calculation. This procedure converged both to a further slightly more refined structure and to tensor values closer to the published, and highly refined, values for this system.52,53 Within this procedure of structure refinement, the position of the paramagnetic Co(II) ion becomes more and more accurate, because all PCS contributes to define the metal position. When the family rmsd is of the order of 2 Å, the structure is resolved enough to unambiguously identify a set of three, plausible, coordinating histidines. This makes it possible to determine three additional metal links in the structural calculation.
From this point on, the search for new structurally unambiguous distance restraints and the refinement of the tensor parameters were done in parallel. At the end of the procedure, up to 537 new restraints were obtained (Figure 1), for a total of 777 unambiguous distance restraints. The final lower-energy family of structures calculated including also PCS and TALOS restraints shows a backbone rmsd to the mean (precision) of 1.0 ± 0.2 Å, which reduces to 0.9 ± 0.2 Å when only the secondary-structure elements were considered. (The same calculation performed without PCS showed 1.3 Å of backbone rmsd (1.6 Å of accuracy), which reduced to 1.1 Å on the secondary-structural elements.) The rmsd between the mean of this structure family and the crystallographic X-ray structure is reasonably close to the precision, being 1.3 Å for the whole backbone and 1.0 Å for secondary-structure elements. Figure 2 reports the final structure family and its comparison with the crystallographic X-ray structure (pdb code: 1RMZ60). The refined tensor values are Δχax = (9.9 ± 0.3) × 10−32 m3 and Δχrh = (−2.2 ± 0.3) × 10−32 m3, which are in very good agreement with the values determined from the X-ray crystallographic structure.52,53
Figure 2.
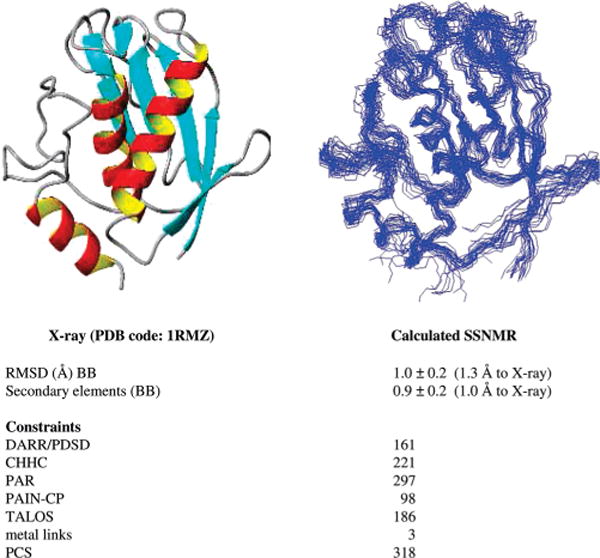
SSNMR structure family (right), comparison with the X-ray structure (left), and summary of the restraints used.
It should be pointed out again that the relatively high final number of distance restraints could be achieved by exploiting the initial low-resolution structures obtained thanks to the presence of the PCS to resolve assignment ambiguities. To further explore this aspect, sample calculations were performed using the ARIA20,39,40 software to check whether the initial 240 unambiguous restraints could be used to initiate an iterative search for additional restraints without the help of PCS. The final structure provided a poor rmsd within the family (7.3 ± 1.0 Å), and a corresponding poor rmsd from the X-ray structure (8.6 Å). Therefore, the present number of unambiguous restraints (240) is insufficient for ARIA to converge to a high-resolution structure (details in the Supporting Information). It follows that PCS has been absolutely necessary to overcome the initial restraint ambiguity. It is conceivable that incorporation of PCS into ARIA could further improve the accuracy of the final structure.
Discussion
Accuracy of the Structure and the Calibration Problem
Determining the accuracy of a given protein structure requires the availability of an independent experimental reference. Atomic resolution X-ray structures, when available as in the present case, are good candidates for an accurate reference. However, it should be considered that even when a microcrystalline SSNMR sample is prepared under the same conditions of the X-ray one, the NMR structure is determined under conditions that are quite different from those used to determine the X-ray structure. In this respect, MMP-12 may be a critical case because it has been demonstrated that the crystallographic structures of either the full-length protein77 or the catalytic domain60 differ from the solution structures because of localized conformational dynamics. As the temperature used in the SSNMR spectra is not far from room temperature and much higher than that used in X-ray crystallography, it cannot be excluded that conformational dynamics within the crystal lattice may be still operative under the SSNMR conditions. Thus, some limited structural differences between the X-ray structure and the investigated system might be possible.
PCS offer the possibility to assess whether or not the X-ray structure is still an accurate reference for the protein structure in microcrystalline samples under the SSNMR conditions. If the available X-ray structure is used to fit the experimental data, calculated and experimental PCS agree with one another with an rmsd of 0.19 ppm, which is comparable with the a priori estimated experimental error of PCS. This does not only demonstrate the quality of the measured PCS, but also that the X-ray structure is a reliable reference in the SSNMR conditions.
The quality of NMR structures does not only depend on the number of contacts (mainly long-range) that can be measured, but also on the accuracy of the distance information extracted from the experimental data (intensity, volume, chemical shift…). Indeed, the less accurate is the distance information, the larger are the number of contacts needed to determine the protein folding. This is even more relevant in SSNMR, where several multidimensional correlation experiments (DARR, CHHC…) make the determination of long-range contacts possible, but the relationship between the experimental information and the effective distance is often complex and affected by the errors introduced by dipolar truncation or relayed transfer effects.19,41 This problem is especially relevant in uniformly labeled samples: for example, in DARR spectra, the relayed magnetization transfer effects are strong, and often a single class of distance restraints should be used in view of the poor correlation between the observed cross-peak intensities and the effective distances.20 This problem is significantly reduced in the PAR and PAIN-CP spectra in view of the particular third-spin assisted recoupling (TSAR) method.24,25
If only relatively few restraints per-residue are available, even a limited number of miscalibrated restraints could produce a distortion in the protein structure rather than an increase in the number of violations. For such a reason, we investigated the accuracy of the assigned restraints to derive the proper upper-distance limits to be used in converting the observed cross-peaks in distance restraints.
Any attempt to plot observed SSNMR cross-peaks intensity (or volume) against the crystallographic internuclear distance produced (for each assigned spectra) uncorrelated plots. The DARR spectrum is the spectrum with the largest dispersion: almost all of the cross-peaks correspond to nuclei within 9 Å, with only a few of them exceeding this threshold up to 10 Å. Limiting the maximum upper-limit distance restraints to 7 Å, as it is done in small proteins, appears improper here because many cross-peaks with significant intensity correspond to distances that are still farther than 7 Å.
By performing the same analysis on the CHHC, PAR, and PAIN-CP spectra again, uncorrelated intensity/distance plots are observed, but with much less dispersion than in the DARR spectra, because only a very low number of cross-peaks is found above 7 Å. This limit can be reduced for CHHC, where almost no contact farther than 6.5 Å is observed in the 1H–1H internuclear distances. This analysis should be largely independent of the nature of the protein, so that the resulting upper distance limits of 9, 7, and 6.5 Å for the various classes of spectra can be assumed to be fairly general under similar experimental conditions. To evaluate the impact of these assigned upper-distance limits on the calculated structure, we ran the same structure calculation performed above but limiting the DARR restraints to 7 Å, the PAR/PAIN-CP restraints to 6 Å, and the CHHC restraints to 5 Å, as is used in small protein SSNMR. A structure with rmsd within the family of 0.87 ± 0.2 Å was obtained, but the rmsd of the mean SSNMR structure to the X-ray structure was 1.7 Å. These results indicate that, despite the good precision of the structure family, there is an increased gap between precision and accuracy, with an evident structure over-refinement. The too tight restraints force the protein chain to assume a less correct fold.
Analogously, the categorization of the distance restraints into different classes on the basis of the “strong”, “intermediate”, and “weak” intensity of the cross-peaks appears incorrect, with several evident miscalibrations. On the other hand, the reconstruction of the magnetization buildup through the acquisition of spectra with different mixing times does not seem a practical solution. In fact, it requires a large amount of machine time (acquiring at least 4–5 spectra with different mixing times for each PDSD or DARR, CHHC, PAR, PAIN-CP, …). Moreover, the correct build-up curve can be accurately reconstructed only for sufficiently intense and resolved peaks, and, where possible, different behaviors are observed depending on the type of carbon (number of attached protons) or the presence of dynamics.
Structure Analysis
Figure 2 reports the 3D SSNMR structure of the catalytic domain of MMP-12. This domain is able to bind five metal ions: two Zn(II) ions and three Ca(II) ions. The protein folding is analogous to that of the other MMP’s and is composed by three helices, a five-stranded β-sheet and eight intervening loops (L1–L8). The comparably high size (159 AA, 17.6 kDa) with respect to the other proteins studied by SSNMR so far, and the small amount of β-sheet (only 17%), represent a significant problem in the structure investigation of this protein. The helices represent only 28% of the whole protein folding, which is composed by more than 55% of loops, one of these (L8) being extremely long (around 27 AA).
As reported above, the overall backbone rmsd is calculated to be 1.0 Å. In Figure 3, the backbone rmsd per residue is reported. It can be appreciated that both precision and accuracy vary significantly along the sequence, being generally larger in loops than in the helices and β-sheet. The trend of the per-residue rmsd within the family is directly corresponding to the trend of the accuracy. Wherever the rmsd in the family is large, the discrepancy with respect to the X-ray structure is also large. Figure 3 correlates the trends of precision and accuracy per-residue with the number of restraints per residue. It is evident that the protein regions whose structure is determined with smaller accuracy are generally those with the smaller number of restraints. For the same reason, in the protein regions where a higher number of restraints is concentrated, the precision of the structure is of the order of that typically obtained in solution NMR.
Figure 3.
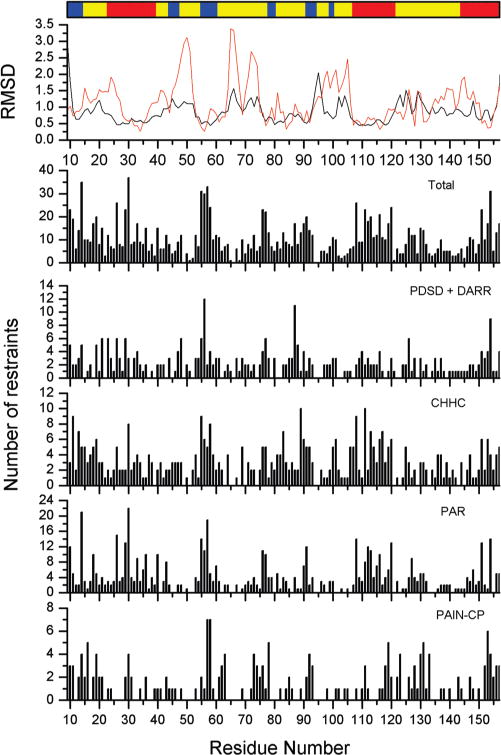
Correlation between the rmsd per-residue within the family and number of restraints per-residue used (residues 9–157 of the catalytic domain of MMP-12). The bar at the top of the figure indicates the secondary structure elements along the sequence as helices (red), β-sheets (blue), and loops (yellow). The first panel at the top reports the trend of the per-residue rmsd to the mean (precision, black line) and the rmsd between the mean structure and the X-ray structure (accuracy, red line).
The structure of the catalytic domain of the MMP-12 protein was also solved by solution NMR,60 with 0.7 ± 0.1 Å bb rmsd within the family. In this case, 2641 distance restraints were used (with 2215 inter-residue restraints) together with angle restraints (167) and RDC, as far as permitted by the solution dynamics observed for this protein. With respect to these numbers, it is not surprising to achieve a 1.0 ± 0.2 Å backbone rmsd in the SSNMR structure obtained by using, besides 186 angle restraints and 318 PCS, 3 metal links, and only 777 distance restraints, the latter also being looser than those used in solution NMR.
Besides being crucial to arrive at the determination of 777 unambiguous distance restraints, PCS intrinsically improve both precision and accuracy (by more than 20%) with respect to the structure obtained by using only angle and distance restraints. Their effect is much more pronounced when a small number of distance restraints are used to obtain low-resolution structure. Indeed, as long as the resolution is low, PCS act by guiding the corresponding nuclei towards their correct position in the structure, improving precision and accuracy, and also helping the other restraints to roughly define the protein fold.
Individual Contributions of Diamagnetic Distance Restraints
To investigate the role, and the effective contribution to the structural calculation, of the distance restraints from each spectrum type, several structural calculations were performed by excluding the distance restraints derived from one or two spectral types. Paramagnetic restraints were retained in all cases. Table 3 shows, for each combination of sets of restraints, the precision and the accuracy obtained in the structural calculation. It can be observed that all of the restraint types are important to obtain an accurate high-resolution structure. The DARR internuclear correlations were categorized into an upper distance limit class of 9 Å, and when these restraints are used alone they are too loose to define an accurate protein folding. Conversely, CHHC and PAR restraints are much stronger restraints, and when they are used alone, a low-resolution protein structure can be already defined. Despite this, if all of the DARR/PDSD restraints are removed from the structure calculation (i.e., using only CHHC and PAR/PAIN-CP restraints), the rmsd within the family increases to 1.4 ± 0.2 and to 2.4 Å from the mean to the X-ray structure, so the DARR restraints remain important to improve the accuracy. Indeed, while around 48% of the cross-peaks assigned in the DARR/PDSD spectra (151 cross-peaks) was also observed in the CHHC and PAR spectra, there are still 161 internuclear correlations not found in the other spectra. Among them, 48 (30%) are sequential, 54 (33%) are intermediate, and 59 (37%) are long-range restraints (Table 2). We can appreciate that the DARR spectra, despite yielding the most loose distance restraints, are still providing several long-range restraints that were not determined otherwise. Figure 3 shows how PDSD/DARR provides restraints for loops (like that one around residue 65) that are not obtained in the other spectra.
Table 3.
Precision and Accuracy of the Structure Obtained through Several Combinations of Restraintsa
| PDSD/DARR | CHHC | PAR | PAIN-CP | precision (Å) | accuracy (Å) |
|---|---|---|---|---|---|
| X | X | X | X | 1.0 ± 0.2 | 1.3 |
| X | X | X | 1.4 ± 0.2 | 2.35 | |
| X | X | X | 1.2 ± 0.2 | 1.3 | |
| X | X | X | 1.5 ± 0.2 | 1.75 | |
| X | X | X | 1.05± 0.1 | 1.4 |
All of the calculations were performed with 318 PCS and 186 dihedral angle restraints.
As mentioned above, the MMP12 catalytic domain is binding five metals, and more than 50% of the protein is composed by loops. Noticeably, it is possible to obtain a high-resolution protein structure without imposing metal links (apart for the paramagnetic metal, where they can be determined within the structural calculation), and with an accurate definition of the loop regions. On the other side, the intrinsic difficulty of this structure requires the use of several experimental restraints.
By comparing the CHHC and PAR spectra, we found only a limited number of common cross-peaks (63). The CHHC spectra provide 138 restraints that are not observed in any of the other spectra, while PAR provides 201 exclusively assigned restraints. By superimposing CHHC and PAR spectra (Figure S5), it can be appreciated that more than 48% of the PAR cross-peaks fall in the 0–22 ppm region, and 169 of the PAR restraints are assigned to intermolecular correlations involving methyl 13C nuclei. Overall, from PAR spectra, 110 sequential, 83 intermediate, and 104 long-range restraints are obtained. On the contrary, CHHC has more than 75% of the restraints in the aliphatic region from 22 to 70 ppm, and overall 67 are sequential restraints, and there are 62 and 92 intermediate and long-range restraints, respectively.
The PAIN-CP spectrum provides N–C restraints; most of the spectrum shows intraresidue correlations, but up to 98 inter-residue correlations can be observed. A large part of these are sequential restraints (57%), but a small number of important intermediate and long-range restraints can be assigned (18% and 25%, respectively).
Finally, we have also addressed the problem of the relative significance of the distance restraints involving the same two residues, as this might be an issue especially when the upper distance limits are large. However, in the course of the structural refinement described above, we have noted that the addition of new unambiguous restraints, even when involving residues already connected by other restraints, was generally providing both a decrease in the rmsd and an improvement in the target function.
Conclusions
The high-resolution SSNMR structure of the catalytic domain of MMP-12 has been determined through a combined use of the paramagnetic (PCS) and diamagnetic restraints. PCS are accurate and easily determined, but they are not sufficient to determine a protein structure alone. Diamagnetic distance restraints can be now determined in a sufficiently large number by using the structural information derived from PAR and PAIN-CP spectra in addition to the known PDSD/DARR and CHHC spectra. The early availability of unambiguous PCS restraints is a key feature that allows the progressive removal of ambiguities in the various 13C–13C and 13C–15N correlation spectra and the achievement of a relatively high number of unambiguous distance restraints through an iterative procedure. The combined use of all of these different types of restraints made it possible to achieve a structure with a bb rmsd of 1.0 ± 0.2 Å, and with a comparable accuracy to X-ray (1.3 Å). The accuracy of this structure is comparable with the other published systems even if the size of the MMP-12 protein is much higher (159 AA, 17.6 kDa). The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2krj).
The method developed here can be extended to proteins that do not bind paramagnetic metals, once they are endowed with specially designed metal-binding tags. Several systems have been proposed in solution NMR, including synthetic tags78,79 or metal-binding peptides to be expressed in fusion with the investigated protein.80 Recently, EDTA-like metal-binding tags have been used in SSNMR to functionalize the GB1 protein.55 In this application, the tag was used to exploit the paramagnetic enhancement of nuclear relaxation rates, but tags can be used also to generate PCS contributions by using metals endowed with sizable magnetic susceptibility anisotropy.
Supplementary Material
Acknowledgments
We thank M. Nilges and B. Bardiaux for providing us a preliminary version of ARIA 2.2.2. This work was supported by EC contracts 201640(SFMET), LSHB-CT-2005-019102 (Nano4Drugs), 026145 (EU-NMR JRA2 and JRA3), Bio-DNP 011721, SPINE2-COMPLEXES 031220, by MIUR-RBAU013NSB, MIUR-RBLA032M7, and MIUR-RBIP06LSS2, and by Ente CR Firenze.
Footnotes
Supporting Information Available: Details about the analysis of the spectra, peak-picking, and the ARIA calculation, as well as significant examples of 2D DARR, CHHC, PAR, and PAIN-CP spectra (Figures S1–S6). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Griffin RG. Nat Struct Biol. 1998;5:508–512. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 2.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 3.McDermott AE. Curr Opin Struct Biol. 2004;14:554–561. doi: 10.1016/j.sbi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Böckmann A. Angew Chem Int Ed. 2008;47:6110–6113. doi: 10.1002/anie.200801352. [DOI] [PubMed] [Google Scholar]
- 5.Ketchem RR, Hu W, Cross TA. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 6.Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 7.Etzkorn M, Kneuper H, Dünnwald P, Vijayan V, Krämer J, Griesinger C, Becker S, Unden G, Baldus M. Nat Struct Mol Biol. 2008;10:1031–1039. doi: 10.1038/nsmb.1493. [DOI] [PubMed] [Google Scholar]
- 8.Cady SD, Hong M. Proc Natl Acad Sci USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi M, Cross TA, Zhou HX. Proc Natl Acad Sci USA. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tycko R. Protein Pept Lett. 2006;13:229–234. doi: 10.2174/092986606775338470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wasmer C, Lange A, van Melckebeke H, Siemer AB, Riek R, Meier BH. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 12.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paravastu AK, Leapman RD, Yau W-M, Tycko R. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizzi B, Ishii Y. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 15.Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Proc Natl Acad Sci USA. 2008;105:6284–6289. doi: 10.1073/pnas.0711716105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Proc Natl Acad Sci USA. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jehle S, van Rossum B, Stout JR, Noguchi SM, Falber K, Rehbein K, Oschkinat H, Klevit RE, Rajagopal P. J Mol Biol. 2009;5:1481–1497. doi: 10.1016/j.jmb.2008.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellani F, van Rossum BJ, Diehl A, Rehbein K, Oschkinat H. Biochemistry. 2003;42:11476–11483. doi: 10.1021/bi034903r. [DOI] [PubMed] [Google Scholar]
- 19.Manolikas T, Herrmann T, Meier BH. J Am Chem Soc. 2008;130:3959–3966. doi: 10.1021/ja078039s. [DOI] [PubMed] [Google Scholar]
- 20.Loquet A, Bardiaux B, Gardiennet C, Blanchet C, Baldus M, Nilges M, Malliavin T, Boeckmann A. J Am Chem Soc. 2008;130:3579–3589. doi: 10.1021/ja078014t. [DOI] [PubMed] [Google Scholar]
- 21.Korukottu J, Schneider R, Vijayan V, Lange A, Pongs O, Becker S, Baldus M, Zweckstetter M. PLoS ONE. 2008;3:e2359. doi: 10.1371/journal.pone.0002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franks WT, Wylie BJ, Schmidt HL, Nieuwkoop AJ, Mayrhofer RM, Shah GJ, Graesser DT, Rienstra CM. Proc Natl Acad Sci USA. 2008;12:4621–4626. doi: 10.1073/pnas.0712393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Paëpe G, Lewandowski JR, Griffin RG. J Chem Phys. 2008;128:124503. doi: 10.1063/1.2834732. [DOI] [PubMed] [Google Scholar]
- 24.De Paëpe G, Lewandowski JR, Loquet A, Bo¨ckmann A, Griffin RG. J Chem Phys. 2008;24:245101. doi: 10.1063/1.3036928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewandowski JR, De Paëpe G, Griffin RG. J Am Chem Soc. 2007;129:728–729. doi: 10.1021/ja0650394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa PR, Sun BQ, Griffin RG. J Am Chem Soc. 1997;119:10821–10830. [Google Scholar]
- 27.Rienstra CM, Tucker-Kellogg L, Jaroniec CP, Hohwy M, Reif B, McMahon MT, Tidor B, Lozano-Pérez T, Griffin RG. Proc Natl Acad Sci USA. 2002;16:10260–10265. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. Proc Natl Acad Sci USA. 2004;3:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloembergen N. Physica. 1949;15:386–426. [Google Scholar]
- 30.Szeverenyi NM, Sullivan MJ, Maciel GE. J Magn Reson. 1982;47:462–475. [Google Scholar]
- 31.Takegoshi K, Nakamura S, Terao T. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 32.Takegoshi K, Nakamura H, Terao T. Chem Phys Lett. 1999;307:295–302. [Google Scholar]
- 33.LeMaster DM, Kushlan DM. J Am Chem Soc. 1996;118:9255–9264. [Google Scholar]
- 34.Hodgkinson P, Emsley L. J Magn Reson. 1999;139:46–59. doi: 10.1006/jmre.1999.1759. [DOI] [PubMed] [Google Scholar]
- 35.Lange A, Luca S, Baldus M. J Am Chem Soc. 2002;124:9704–9705. doi: 10.1021/ja026691b. [DOI] [PubMed] [Google Scholar]
- 36.Lange A, Seidel K, Verdier L, Luca S, Baldus M. J Am Chem Soc. 2003;125:12640–12648. doi: 10.1021/ja034555g. [DOI] [PubMed] [Google Scholar]
- 37.Lange A, Becker S, Seidel K, Giller K, Pongs O, Baldus M. Angew Chem Int Ed. 2005;44:2089–2092. doi: 10.1002/anie.200462516. [DOI] [PubMed] [Google Scholar]
- 38.Gardiennet C, Loquet A, Etzkorn M, Heise H, Baldus M, Bo¨ckmann A. J Biomol NMR. 2008;40:239–250. doi: 10.1007/s10858-008-9229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilges M, O’Donoghue SI. Prog NMR Spectrosc. 1998;32:107–139. [Google Scholar]
- 40.Linge JP, O’Donoghue SI, Nilges M. Methods Enzymol. 2001;339:71–90. doi: 10.1016/s0076-6879(01)39310-2. [DOI] [PubMed] [Google Scholar]
- 41.Grommek A, Meier BH, Ernst M. Chem Phys Lett. 2006;427:404–409. [Google Scholar]
- 42.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 43.Hohwy M, Jaroniec CP, Reif B, Rienstra CM, Griffin RG. J Am Chem Soc. 2000;122:3218–3219. [Google Scholar]
- 44.Rienstra CM, Hohwy M, Mueller LJ, Jaroniec CP, Reif B, Griffin RG. J Am Chem Soc. 2002;124:11908–11922. doi: 10.1021/ja020802p. [DOI] [PubMed] [Google Scholar]
- 45.Bertini I, Fragai M, Lee YM, Luchinat C, Terni B. Angew Chem Int Ed. 2004;43:2254–2256. doi: 10.1002/anie.200353453. [DOI] [PubMed] [Google Scholar]
- 46.Lee H, Ortiz de Montellano PR, McDermott AE. Biochemistry. 1999;38:10808–10813. doi: 10.1021/bi990463l. [DOI] [PubMed] [Google Scholar]
- 47.Jovanovic T, McDermott AE. J Am Chem Soc. 2005;127:13816–13821. doi: 10.1021/ja0438314. [DOI] [PubMed] [Google Scholar]
- 48.Pintacuda G, Giraud N, Pierattelli R, Böckmann A, Bertini I, Emsley L. Angew Chem Int Ed. 2007;46:1079–1082. doi: 10.1002/anie.200603093. [DOI] [PubMed] [Google Scholar]
- 49.Laage S, Sachleben JR, Steuernagel S, Pierattelli R, Pintacuda G, Emsley L. J Magn Reson. 2009;196:133–141. doi: 10.1016/j.jmr.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 50.Wickramasinghe NP, Shaibat MA, Jones CR, Casabianca LB, de Dios AC, Harwood JS, Ishii Y. J Chem Phys. 2008;128:052210. doi: 10.1063/1.2833574. [DOI] [PubMed] [Google Scholar]
- 51.Wickramasinghe NP, Parthasarathy S, Jones CR, Bhardwaj C, Long F, Kotecha M, Mehboob S, Fung LW, Past J, Samoson A, Ishii Y. Nat Methods. 2009;6:215–218. doi: 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balayssac S, Bertini I, Lelli M, Luchinat C, Maletta M. J Am Chem Soc. 2007;129:2218–2219. doi: 10.1021/ja068105a. [DOI] [PubMed] [Google Scholar]
- 53.Balayssac S, Bertini I, Bhaumik A, Lelli M, Luchinat C. Proc Natl Acad Sci USA. 2008;105:17284. doi: 10.1073/pnas.0708460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nadaud PS, Helmus JJ, Hofer N, Jaroniec CP. J Am Chem Soc. 2007;129:7502–7503. doi: 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- 55.Nadaud PS, Helmus JJ, Kall SL, Jaroniec CP. J Am Chem Soc. 2009;131:8108–8120. doi: 10.1021/ja900224z. [DOI] [PubMed] [Google Scholar]
- 56.Lewandowski JR, De Paëpe G, Eddy MT, Griffin RG. J Am Chem Soc. 2009;131:5769–5776. doi: 10.1021/ja806578y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewandowski JR, De Paëpe G, Eddy MT, Struppe J, Maas W, Griffin RG. J Phys Chem B. 2009;113:9062–9069. doi: 10.1021/jp810280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertini I, Donaire A, Jimenez B, Luchinat C, Parigi G, Piccioli M, Poggi L. J Biomol NMR. 2001;21:85–98. doi: 10.1023/a:1012422402545. [DOI] [PubMed] [Google Scholar]
- 59.Barbieri R, Luchinat C, Parigi G. ChemPhysChem. 2004;5:797–806. doi: 10.1002/cphc.200301058. [DOI] [PubMed] [Google Scholar]
- 60.Bertini I, Calderone V, Cosenza M, Fragai M, Lee YM, Luchinat C, Mangani S, Terni B, Turano P. Proc Natl Acad Sci USA. 2005;102:5334–5339. doi: 10.1073/pnas.0407106102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balayssac S, Bertini I, Falber K, Fragai M, Jehle S, Lelli M, Luchinat C, Oschkinat H, Yeo KJ. ChemBioChem. 2007;8:486–489. doi: 10.1002/cbic.200600408. [DOI] [PubMed] [Google Scholar]
- 62.Fung BM, Khitrin AK, Ermolaev K. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 63.Morcombe CR, Zilm KW. J Magn Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 64.Goddard DT, Kneller GD. SPARKY. 2007:3. [Google Scholar]
- 65.Koradi R, Billeter M, Wu¨thrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 66.Guntert P, Mumenthaler C, Wuthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 67.Balayssac S, Bertini I, Luchinat C, Parigi G, Piccioli M. J Am Chem Soc. 2006;128:15042–15043. doi: 10.1021/ja0645436. [DOI] [PubMed] [Google Scholar]
- 68.Bertini I, Luchinat C, Parigi G, Pierattelli R. ChemBioChem. 2005;6:1536–1549. doi: 10.1002/cbic.200500124. [DOI] [PubMed] [Google Scholar]
- 69.Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 70.Straus SK, Bremi T, Ernst RR. Chem Phys Lett. 1996;262:709–715. [Google Scholar]
- 71.Scholz I, Jehle S, Schmieder P, Hiller M, Eisenmenger F, Oschkinat H, van Rossum BJ. J Am Chem Soc. 2007;129:6682–6683. doi: 10.1021/ja070849g. [DOI] [PubMed] [Google Scholar]
- 72.Duma L, Hediger S, Lesage A, Emsley L. J Magn Reson. 2003;164:187–195. doi: 10.1016/s1090-7807(03)00187-3. [DOI] [PubMed] [Google Scholar]
- 73.Duma L, Hediger S, Brutscher B, Böckmann A, Emsley L. J Am Chem Soc. 2003;125:11816–11817. doi: 10.1021/ja036893n. [DOI] [PubMed] [Google Scholar]
- 74.Laage S, Lesage A, Emsley L, Bertini I, Felli IC, Pierattelli R, Pintacuda G. J Am Chem Soc. 2009;131:10816–10817. doi: 10.1021/ja903542h. [DOI] [PubMed] [Google Scholar]
- 75.Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 76.Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Proc Natl Acad Sci USA. 2009;18:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bertini I, Calderone V, Fragai M, Jaiswal R, Luchinat C, Melikian M, Mylonas E, Svergun DI. J Am Chem Soc. 2008;130:7011–7021. doi: 10.1021/ja710491y. [DOI] [PubMed] [Google Scholar]
- 78.Ikegami T, Verdier L, Sakhaii P, Grimme S, Pescatore B, Saxena K, Fiebig KM, Griesinger C. J Biomol NMR. 2004;29:339–349. doi: 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- 79.John M, Pintacuda G, Park AY, Dixon NE, Otting G. J Am Chem Soc. 2006;128:12910–12916. doi: 10.1021/ja063584z. [DOI] [PubMed] [Google Scholar]
- 80.Wohnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. J Am Chem Soc. 2003;125:13338–13339. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.