

Abstract
The ATP-binding cassette transporters are a large family (~ 48 genes divided into seven families A–G) of proteins that utilize the energy of ATP-hydrolysis to pump substrates across lipid bilayers against a concentration gradient. The ABC “A” subfamily is comprised of 13 members and transport sterols, phospholipids and bile acids. ABCA2 is the most abundant ABC transporter in human and rodent brain with highest expression in oligodendrocytes, although it is also expressed in neurons. Several groups have studied a possible connection between ABCA2 and Alzheimer’s disease as well as early atherosclerosis. ABCA2 expression levels have been associated with changes in cholesterol and sphingolipid metabolism. In this paper, we hypothesized that ABCA2 expression level may regulate esterification of plasma membrane-derived cholesterol by modulation of sphingolipid metabolism. ABCA2 overexpression in N2a neuroblastoma cells was associated with an altered bilayer distribution of the sphingolipid ceramide that inhibited acylCoA:cholesterol acyltransferase (ACAT) activity and cholesterol esterification. In contrast, depletion of endogenous ABCA2 in the rat schwannoma cell line D6P2T increased esterification of plasma membrane cholesterol following treatment with exogenous bacterial sphingomyelinase. These findings suggest that control of ABCA2 expression level may be a key locus of regulation for esterification of plasma membrane-derived cholesterol through modulation of sphingolipid metabolism.
Keywords: ABCA2, transporter, cholesterol, esterification, sphingomyelin, ceramide
1. Introduction
Cholesterol serves many diverse biological functions. It is a structural component of cellular membranes, providing a barrier function from the extracellular milieu, a major component of the myelin sheath that insulates neurons, a regulator of the activity of transporters, ion channels and intracellular signaling molecules and a precursor for the synthesis of steroid hormones, vitamins and bile acids. Cells maintain intracellular cholesterol levels within stringent tolerances since excessive cholesterol levels are toxic [1]. A number of chronic sterol-dependent disorders have been linked to aberrations in normal cholesterol homeostasis including atherosclerosis and neurodegenerative diseases Niemann-Pick Type C, Huntington’s disease, Parkinson’s disease and Smith-Lemli Optiz syndrome [2, 3]. The trafficking of cholesterol between the plasma membrane and intracellular organelles is a major focus of continuing efforts to understand the etiology of sterol-dependent disease. The identification of key modulators and their mechanisms of action that regulate intracellular cholesterol trafficking are not completely elucidated. Identification and clarification of the roles of key modulators of intracellular cholesterol trafficking will facilitate the development of therapeutics to prevent, treat, or cure sterol-dependent diseases.
The ATP-binding cassette transporters are a large family (~ 48 genes divided into seven families A–G) of proteins that utilize the energy of ATP-hydrolysis to pump substrates across lipid bilayers against a concentration gradient [4, 5]. The ABC “A” subfamily consists of 13 members and transport sterols, phospholipids and bile acids [6]. ABCA2 is the most abundant ABC transporter in human and rodent brain with highest expression in oligodendrocytes, although it is also expressed in neurons [7]. Two groups have generated ABCA2 knockout mice and phenotypic studies have described changes in myelin sheath morphology and brain lipid profile [8, 9].
Several groups have studied a possible connection between ABCA2 and Alzheimer’s disease [10–14]. A possible mechanistic role for ABCA2 in cardiovascular disease was suggested by a study describing deletion of ABCA2 in macrophages of LDL receptor knockout mice [15].
In studying the role of ABCA2 in lipid metabolism in Chinese hamster ovary (CHO) cells, the Davis lab determined that human ABCA2 overexpression decreased efflux of lipoprotein-derived cholesterol from the endolysosomal compartment and decreased esterification of lipoprotein-derived cholesterol [16]. These findings were supported in studies of ABCA2 knockout macrophages [15]. In N2a mouse neuroblastoma cells, the Davis group determined that overexpression of human ABCA2 or reduction of endogenous ABCA2 levels in N2a cells and in primary rat neurons by RNAi modulated LDL receptor level. ABCA2 expression also modulated esterification of lipoprotein and non-lipoprotein-derived cholesterol [17].
To investigate further the mechanism of ABCA2 regulation of esterification of plasma membrane-derived cholesterol, two model systems were employed, stable human ABCA2 overexpression in a mouse N2a neuroblastoma cell line and stable depletion of endogenous ABCA2 in the rat D6P2T schwannoma cell line that expresses high levels of endogenous ABCA2. The formation of [3H]cholesteryl ester from [3H]cholesterol in metabolically radiolabeled cells was determined following enzymatic mobilization of plasma membrane cholesterol.
The key findings in this report indicate that esterification of plasma membrane-derived cholesterol correlates with ABCA2 expression level through modulation of sphingolipid metabolism. Further understanding of the cross talk between sphingolipid and intracellular cholesterol trafficking pathways in normal and disease states may provide strategies for therapeutic intervention to ameliorate disease outcomes.
2. Materials and methods
2.1. Materials
Dulbecco’s Modified Eagle medium (DMEM), Ham’s F12 and fetal bovine serum (FBS) were obtained from Hyclone. Glutamine and Penstrep were obtained from Fisher. Radiochemicals, cholesterol, [1,2-3H(N)] (40–60 Ci/mmol); [methyl14C]choline chloride (55 mCi/mmol) and [1-14C]oleoyl-CoA (58.2 mCi/mmol) were from Perkin Elmer. C6-NBD-ceramide was from Avanti Lipids and C6-NBD-ceramide complexed to BSA was from Invitrogen. D-erythro MAPP was from Enzo Life Sciences. 25-hyroxycholesterol was from MP Biomedicals.
2.2. Cell lines and culture
The N2a mouse neuroblastoma cell line was obtained from ATCC (CCL-131). Cells were grown in DMEM/Ham’s F12 (50:50) supplemented with 5% fetal bovine serum (FBS) 2 mM glutamine and 1% Penstrep at 37° C and 5% CO2. N2a cell lines stably expressing a 7.4 kilobase human ABCA2 cDNA in the pcDNA5/FRT/TO vector (Invitrogen) have been previously described [17]. D6P2T rat schwannoma cells stably expressing control lamin- or ABCA2-specific RNAi were the kind gift of Dr. Ken Tew, Medical University of South Carolina.
2.3. Western Blot
Cells were lysed in radioimmunoprecipitation (RIPA) buffer (150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 50 mM Tris-HCl, pH 7.5, 2 mM EDTA, RIPA) supplemented with HALT protease inhibitor cocktail (Pierce). Protein concentrations were determined using the DC-protein assay kit (Bio-Rad). For detection of ABCA2, 40 μg of protein were fractionated on 4–12% NuPAGE gels (Invitrogen). For detection of sphingomyelinase-2 (SMS2), 30 μg of protein were fractionated and probed. Proteins were transferred to nitrocellulose membranes and probed with a primary rabbit polyclonal antibody to the ABCA2 c-terminal 20 amino acids (1:300), a rabbit polyclonal antibody to SMS2 (Novus Biologicals 1:1000, or mouse monoclonal antibody to β-Actin (1:1000, Santa Cruz Biotechnology). Secondary antibodies were goat anti-rabbit- or anti-mouse horseradish peroxidase (HRP) (Thermo Scientific/Pierce, 1:1000). Blots were developed using the Clarity Western ECL substrate (Bio-Rad) and imaged using the ChemiDoc XRS+ imager and Image Lab software (Bio-Rad).
2.4. Mobilization of plasma membrane cholesterol to endoplasmic reticulum for esterification
On day 0, 0.4 × 106 cells were plated in 2 ml of DMEM/F12, 5% FBS, 1% Penstrep, 2 mM glutamine (medium A) in 6-well plates at 37° C 5% CO2. On day 1, the medium was supplemented with 1 μCi/ml of [3H]cholesterol and the cells were cultured for 24 hours. On day 2 the medium was replaced with complete medium supplemented with ± 0.5 units/ml bacterial sphingomyelinase (bSMase, Sigma), ± 20 μg/ml 25-hydroxycholesterol (25-HC) in ethanol and the cells were cultured for 6 hours. For cells cultured in U18666a and 25-HC, U18666a (3 μg/ml) was added and cells cultured for 4 hours before addition of 25-HC (20 μg/ml) and cells were cultured for an additional 4 hours. For cells cultured in ± D-erythro MAPP, the reagent was added (50 μg/ml) 24 hours after metabolic radiolabeling and the cells were cultured for 4 hours. At end of the incubation period, the cells were washed twice with buffer B (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mg/ml bovine serum albumin/BSA) and once with buffer C (buffer B without BSA). Lipids were extracted with hexane-isopropanol (3:2) and dried under N2. Thin-layer chromatography (TLC) on Silica G plates was performed in hexane/ethyl ether/glacial acetic acid (80:18.5:1.5). Total cholesterol and cholesteryl ester levels were measured following TLC imaging using the Bioscan 2000 thin layer chromatography (TLC) scanner. The percent esterification of [3H]cholesterol to [3H]cholesteryl ester (CE) was determined as 100 x cpm[3H]CE/(cpm[3H]CE + cpm[3H]cholesterol) and was the average of three determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05 (StatPlus, AnalystSoft).
2.5. Plasma membrane cholesterol oxidase sensitivity
On day 0, 0.4 × 106 cells were plated in 2 ml of medium A. On day 1, the medium was replaced with 1 ml of fresh medium containing 1 μCi/ml [3H]cholesterol and the cells were cultured for 24 hours. On day 2, the cells were washed 2 times with buffer B and once with buffer C. Cells were fixed with 1% glutaraldehyde in PBS for 10 min at 4° C, washed in PBS and treated for 30 min at 37° C with 0.1 units/ml bSMase followed by addition of ± 2 units/ml cholesterol oxidase (Sigma). Cells were washed twice with cold PBS. Lipids were extracted with hexane-isopropanol (3:2), dried and separated by TLC in petroleum ether/ethyl ether/acetic acid (60:40:1). TLC plates were imaged using a Bioscan AR2000 imager. The percent oxidation of [3H]cholesterol to [3H]cholestenone was determined as 100 x cpm [3H]cholestenone/(cpm[3H]cholestenone + cpm[3H]cholesterol) and was the average of three determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
2.6. Sucrose density gradient ultracentrifugation (Lipid raft)
On day 0, 4 × 106 cells were plated in 15 ml of medium a in 150 mm plates and grown to 90% confluency at 37° C 5% CO2. Cells were collected by centrifugation and the pellet was resupended in 1 ml of MBS lysis buffer (25 mM MES pH 6.5, 150 mM NaCl, 1% Triton X-100, 0.5% Lubrol WX) and HALT protease inhibitor cocktail (Pierce) and incubated on ice for 1 h with periodic vortexing. Extracts were passed through a 25-gauge needle 7 times and debris was removed by centrifugation at 10,000 × g for 3 min at 4° C. Protein concentrations were determined using the DC protein assay (Bio-Rad). Approximately 1 mg of total protein in 500 μl of MBS buffer was mixed with 500 μl of 80% sucrose in MBS and briefly mixed by vortexing. Subsequently, 2 ml of 35% sucrose in MBS and 2 ml of 5% of sucrose in MBS were added for discontinuous gradient formation and samples were centrifuged at 160,000 × g for 18 h at 4° C in an AH650 rotor. Twelve 400 μl fractions were recovered from the top of the tube and 300 μl were precipitated by the methanol chloroform method. Total precipitated proteins were fractionated on 4–12% NuPAGE gels, transferred to nitrocellulose and probed for flotillin-1 (raft, 1:2000, BD Transduction Labs), and calnexin (non-raft, 1:2000, Assay Designs). Cholesterol and sphingomyelin levels were determined on an aliquot of each fraction by fluorimetric assays (cholesterol: Amplex, Invitrogen; sphingomyelin: Amplite, AAT Bioquest) according to manufacturer’s instructions. The percent cholesterol and percent sphingomyelin in each fraction was determined as the 100 x fluorescence intensity of fraction/total fluorescence intensity of all fractions and was the average of three determinations ± SD.
2.7. Determination of plasma membrane sphingomyelin-dependent lysenin sensitivity
On day 0, 1.5 × 106 cells were plated in 3 ml of medium A in 60 mm plates. On day 2, the medium was replaced with 2 ml of fresh growth medium supplemented with 200 ng/ml of lysenin (Sigma) and the cells were incubated for 2 hours at 37° C. After lysenin treatment, the cells were trypsinized, counted and 0.8 × 106 cells were plated in 500 μl of medium in 24-well plates. Cell viability was determined by adding 55 μl of WST-1 cell proliferation reagent (Roche) and culturing for 2 hours at 37° C. Absorbance was read at 350 nm with the reference absorbance set at 600 nm. Lysenin sensitivity was determined as percent survival ± lysenin = 100 x (Abs 440 (+) lysenin/(Abs 440 (−) lysenin of four determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
2.8. In vitro sphingomyelin synthase activity
On day 0, 4 × 106 cells were plated in 15 ml of medium A in 150 mm plates and grown to 90% confluency at 37° C 5% CO2. On day 2, cells were collected by centrifugation and the pellet was resupended in 0.75 ml of nitrogen cavitation buffer (0.25 M sucrose, 50 mM Tris-HCl pH 7.5 0.1 mM EGTA) and disrupted by nitrogen cavitation (700 psi, 20 min) followed by centrifugation at 1000 × g for 10 min. The protein concentrations were determined on an aliquot of the supernatant using the Bio-Rad DC protein assay. For the sphingomyelin assay, cell homogenates (100 μg of protein) were incubated with 2 μg/ml of C6-NBD-ceramide {N-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-D-erythro-sphingosine}, Avanti lipids) and 10 μg of L-α-phosphatidylcholine (Sigma) as the phosphocholine donor in reaction buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) in a total volume of 500 μl. The reaction proceeded for 2 hours at 37° C. Lipids were extracted by the method of Bligh and Dyer and TLC was performed in chloroform/methanol/ammonium hydroxide (70:30:5). The TLC plate was imaged on a Storm 840 phosphorimager (GE Biosciences) with PMT voltage set at 600 and digital chemifluorescent images were quantified using ImageQuant software (GE Biosciences). In vitro sphingomyelin synthase activity was determined as the ratio of fluorescence intensity of C6-NBD-sphingomyelin formation in ABCA2-overexpressing A2.1 cells or ABCA2-depleted D6P2TA compared to control N2a or D6P2TL cells respectively, and was the average duplicate determinations ± SD.
2.9. De novo sphingomyelin synthesis from [methyl 14C]choline chloride
On day 0, 0.4 × 106 cells were plated in 2 ml of medium A in 6-well plates. On day 1, the medium was replaced with fresh medium supplemented with 2 μCi of [methyl 14C]choline chloride (55–60 mCi/mmol, Perkin-Elmer) and the cells were cultured for 4 hours. Lipids were extracted with hexane-isopropanol (3:2) and TLC was performed in chloroform/methanol/ammonium hydroxide (70:30:5) and quantified using the Bioscan 2000 TLC imager. Proteins were isolated in 0.2 N NaOH and concentrations were determined using the Bio-Rad DC Protein assay. Sphingomyelin synthesis was expressed as the average nmol [14C]sphingomyelin/mg protein produced and was the average of three determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
2.10. Fluorescent ceramide kinase assay
On day 0, 1.5 × 106 cells were plated in 3 ml of medium A in 60 mm plates. On day 2, the cells were washed with ice-cold Hank’s balanced salt solution (HBBS) and the medium was replaced with ice-cold medium A lacking serum. C6-NBD-ceramide complexed to BSA (Invitrogen) was added to a final concentration of 5 μM. The cells were incubated at 4° C for 30 min in the dark. Cells were then warmed to 37° C and cultured for 2 hours in the dark. After the incubation period the cells were washed with HBBS, trypsinized and counted. Lipid extraction from equivalent numbers of cells was performed by the addition of 0.75 ml chloroform/methanol (2:1) and 0.3 ml of 50 mM Tris-HCl, pH 8.5. Following centrifugation at 15,000 × g for 1 min to separate phases. The fluorescence intensity of NBD-C1P formation was measured on a 100 μl aliquot combined with an equal volume of dimethylformamide at ex:em 460:515–580. TLC was performed in butanol/acetic acid/water (60:20:20) to confirm that the major product formed was NBD-C1P by comparing the migration of the major spot with a standard NBD-C1P (Echelon). NBD-CIP formation was expressed as the mean of four determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
2.11. Back-extraction of NBD-lipids from the plasma membrane
On day 0, 1.5 × 106 cells were plated in 3 ml of medium A in 60 mm plates. On day 2, the cells were washed with ice-cold Hank’s balanced salt solution (HBBS) and the medium was replaced with ice-cold medium A lacking serum. C6-NBD-ceramide complexed to BSA was added to a final concentration of 5 μM and cells were incubated at 4° C for 30 min in the dark. The cells were transferred to 37° C and incubated for 2 hours in the dark to promote internalization and recycling. After the incubation period, the cells were washed with HBBS and back-extracted with ± 3% BSA in serum-free DMEM/F12. The cells were trypsinized and counted and equivalent cell numbers of cells (3–4 × 106) were extracted with chloroform/methanol/0.01 N HCl (1:2:0.6). The organic phase was dried under N2 and lipids were separated by TLC in chloroform/methanol/ammonium hydroxide (70:30:5) followed by fluorescence imaging with a STORM phosphorimager (GE Healthcare) as described above. The percent of cell associated NBD lipid was calculated as 100 x ratio of cell-associated NBD lipid (NBD-Cer, NBD-GlcCer, NBD-SM) ± BSA back extraction to cell-associated lipid in N2a control cells in the absence of back extraction and was the average of four determinations ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
2.12. In vitro ACAT activity assay
On day 0, 4 × 106 cells were plated in 10 ml of medium A. On day 1, the cells were washed with HBBS and treated with 50 μg/ml D-erythro MAPP in medium A lacking serum and cultured for 5 hours. The cells were trypsinized and recovered by centrifugation. The pellet was resupended in ice-cold buffer A (20 mM Tris-HCl pH 7.4 and 1 mM EDTA) containing protease inhibitor cocktail (Pierce) and homogenized by 10 passages through a 25-gauge needle. ACAT assays were performed in a final volume of 0.2 ml containing 100 μg of protein, 50 mM potassium phosphate, pH 7.4, 2 mM dithiothreitol, and 10 mg/ml of fatty acid BSA in buffer A. Assays were incubated for 2 min at 37° C and the reaction was initiated by the addition of [1-14C]oleoyl-CoA to 100 μM and incubation for 60 min at 37° C. The reaction was terminated by adding 1 ml of chloroform and 1 ml of methanol, followed by 1 ml of H2O and 1 ml of chloroform. The lower organic phase was dried under N2 and TLC performed in hexane/ethyl ether/acetic acid (80:18.5:1.5). The chromatogram was visualized using a Bioscan 2000 TLC scanner and the ACAT activity was expressed as nmol of cholesteryl ester per mg of total protein and was the average ± SD of duplicate determinations.
2.13. Electrospray Ionization/Mass Spectrometry Lipid Analysis
Analysis of sphingolipids was performed on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple reaction monitoring positive ionization mode. Total cells, fortified with a set of internal standards, were extracted with ethyl acetate/isopropanol/water (60/30/10, v/v). The lipid extracts were dried under an N2 evaporator and reconstituted in 100 μl of methanol. The reconstituted samples were injected into the Survayor/TSQ 7000 liquid chromatography/mass spectrometry system with the BDS Hypersil C8, 150 × 3.2 mm, 3-μm particle size column, which was eluted with 1.0 mM methanolic ammonium formate/2 mM aqueous ammonium formate mobile phase system. The peaks for the target lipids and internal standards were collected and processed using an Xcalibur software system. Calibration curves were constructed by plotting peak area ratios of the target lipids to their respective internal standard against concentration, using linear regression model.
3. Results
3.1. ABCA2 expression level in N2a and D6P2T cells
N2a mouse neuroblastoma cell lines stably overexpressing expressing human ABCA2 (e.g. A2.1) have been previously described [17]. To characterize further the role of ABCA2 expression level on cholesterol trafficking, a rat immortalized cultured Schwann cell line, D6P2T, was utilized. These cells express abundant levels of endogenous ABCA2 protein and are an established model system for the study of Schwann cell biology [18]. Two cell lines, D6P2T stably expressing lamin-specific short hairpin RNAi (D6P2TL) and rat ABCA2-specific RNAi (D6P2TA) were selected for further study. In figure 1, a representative Western blot shows the relative expression of endogenous mouse ABCA2 and human ABCA2 protein in N2a and A2.1 cells and the relative expression of endogenous rat ABCA2 protein in D6P2TL and ABCA2-depleted D6P2TA cells.
Figure 1. ABCA2 protein expression in N2a and D6P2T cells.

A. A representative Western blot is shown of ABCA2 protein expression in parental N2a cells and human ABCA2 overexpressing A2.1 cells. B. A representative Western blot is shown of ABCA2 protein expression in control D6P2TL (lamin-specific RNAi) cells and ABCA2-depleted D6P2TA (ABCA2 RNAi-specific) cells. The ABCA2-specific c-terminal antibody detects human, rat and mouse ABCA2 protein.
3.2. ABCA2 expression modulates cholesterol esterfication in N2a cells
ABCA2 expression has been associated with the modulation of cholesteryl ester formation by acyl-CoA:cholesterol acyltransferase activity (ACAT) in the endoplasmic reticulum. In order to clarify the effects ABCA2 of expression on mobilization of plasma membrane-derived cholesterol for esterification by ACAT, cells were treated with bacterial sphingomyelinase, which hydrolyzes plasma membrane sphingomyelin to ceramide. Cholesterol associates with sphingomyelin in the plasma membrane and depletion of sphingomyelin releases cholesterol that traffics to the endoplasmic reticulum for esterification by ACAT [19]. Cells were metabolically radiolabeled to equilibrium for 24 hours with [3H]cholesterol, and treated with agents to mobilize cholesterol from the plasma membrane to the endoplasmic reticulum for esterification. In untreated cells, the basal level of [3H] cholesteryl ester formation was reduced from 18% of total [3H]cholesterol ± SD 0.42 in N2a cells to 8.5% ± SD 0.68 in A2.1 cells; p < 0.001 (*) Fig. 2A. Addition of bacterial sphingomyelinase and culture for 6 h increased cholesteryl ester formation to 27% ± SD 1.87 in N2a cells but only to 16% ± SD 0.56 in A2.1 cells; p < 0.009 (**). The oxysterol, 25-hydroxycholesterol (25-HC) is reported to trigger cholesterol movement to the endoplasmic reticulum and increase cholesterol esterification by ACAT [20, 21]. Treatment of cells with 25-hydroxycholesterol for 6 h increased cholesteryl ester formation to 31% ± SD 0.97 in N2a cells and to 37% ± SD 2.35 in A2.1 cells; p < 0.025 (***). A slight additive effect on cholesterol esterification was observed by addition of both bSMase and 25-HC. Cholesteryl ester formation increased to 38% ± SD 3.02 in N2a cells and to 43% ± SD 2.51 in A2.1 cells; p < 0.11. These results indicate that cholesterol esterification is reduced by ABCA2 overexpression and that displacement of cholesterol from membrane lipids with 25-HC can overcome the effects of ABCA2 expression to increase cholesterol esterification.
Figure 2. ABCA2 expression modulates cholesterol esterification in N2a cells.

A. Cells were metabolically radiolabeled to equilibrium with [3H]cholesterol for 24 hours. Bacterial sphingomyelinase (0.5 units), 25-hydroxycholesterol (20 μg/ml), or both were added and the cells were cultured for 6 hours before lipid extraction and TLC.
B. Cells were radiolabeled as described above and treated with U18666a (3 mg/ml) for 4 hours before addition of ± 25-hydroxycholesterol (20 μg/ml) and the cells were cultured for an additional 4 hours before lipid extraction in hexane/isopropanol (3:2) and thin-layer chromatography (TLC) in hexane/ethyl ether/acetic acid (80:18.5:1.5). [3H]cholesteryl ester levels were determined using a Bioscan 2000 thin layer chromatography scanner.
To evaluate the role of cholesterol trafficking in the modulating the effects of 25-HC, cells were treated with the cholesterol trafficking inhibitor U18666a. U18666a decreases cholesterol movement from late-endosomal/lysosomal compartments to the plasma membrane and movement of cholesterol from the plasma membrane to the endoplasmic reticulum [22, 23]. Addition of 3 μg/ml U18666a and culture for 5 hours decreased cholesteryl ester formation from 14% ± SD 0.19 to 6.4 % ± SD 0.59 in N2a cells; p < 0.002 and from 6.38% to 3.75% ± SD 0.42 % in A2.1 cells; p < 0.001 (*) (Fig. 2B). Addition of both U18666a and 25-HC decreased cholesteryl ester formation from 21.38% ± SD 1.53 to 13.7% ± SD 0.74; p < 0.004 in N2a cells and from 24.66 % ± SD 0.96 to 14.11% ± SD 0.50 in A2.1 cells; p < 0.001 (**). These results demonstrate that the 25-HC mobilization of cholesterol for esterification is sensitive to inhibition of cholesterol trafficking by U18666a and U18666a-mediated inhibition is independent of ABCA2 overexpression.
3.3. Cholesterol oxidase sensitivity of plasma membrane cholesterol in N2a cells
Susceptibility of plasma membrane cholesterol to oxidation to cholestenone by the enzyme cholesterol oxidase in fixed cells is an established method for measuring relative plasma membrane cholesterol levels [24]. To determine whether ABCA2 expression modulated cholesterol distribution in cholesterol oxidase sensitive compartments, cells were radiolabeled with [3H]cholesterol, fixed with glutaraldehyde, treated with bacterial sphingomyelinase and cholesterol oxidase. The percent [3H]cholestenone formation in N2a cells was 71% ± SD 0.37 and in ABCA2-overexpressing A2.1 cells, 61.6 % ± SD 4.04; p < 0.057. The modest decrease in cholesterol oxidase susceptibility in A2.1 cells suggest that it may not be the major mechanism for the decrease in cholesterol esterification observed in N2a cells.
3.4. Reduction of endogenous ABCA2 increases cholesterol esterification in D6P2T cells
To evaluate whether depletion of endogenous ABCA2 would result in increased cholesteryl ester formation, the rat schwannoma cell line D6PT2 cell line that expresses high levels of endogenous ABCA2 was utilized. Control D6P2T cells stably expressed lamin-specific short-hairpin RNAi (D6P2TL) and ABCA2-depleted D6P2T cells stably expressed rat-specific ABCA2 short-hairpin RNAi (D6P2TA). In D6P2TL cells, the basal level of cholesteryl ester formation was 4.27% ± SD 0.37 and in D6P2TA cells 7.2% ± SD 0.32; p < 0.001 (*) (Fig. 3). Treatment with bSMase for 5 hours increased cholesteryl ester formation to 13.64% in D6P2TL cells ± SD 1.62 and 23.42% ± SD 2.93 in D6P2TA cells; p < 0.0149 (**). These results indicate that depletion of endogenous ABCA2 increases esterification of plasma membrane derived cholesterol.
Figure 3. Depletion of endogenous ABCA2 increases cholesterol esterification in D6P2T cells.
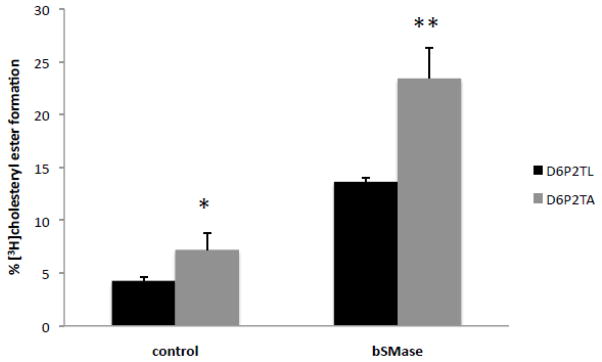
Cells were metabolically radiolabeled to equilibrium with [3H]cholesterol for 24 hours. Bacterial sphingomyelinase (0.5 units) was added and the cells were cultured for 5 hours before lipid extraction and TLC performed as described in Methods.
3.5. Distribution of cholesterol and sphingomyelin in lipid raft compartments in D6P2T cells
Lipid rafts are described as ordered regions of the lipid bilayer that are enriched in cholesterol and sphingolipids [25]. A possible mechanism for ABCA2 regulation of cholesterol esterification may involve the distribution of cholesterol in lipid raft compartments. A previous examination of cholesterol distribution in N2a cells demonstrated that ABCA2 expression did not significantly modulate the cholesterol content in lipid raft compartments [17]. To determine whether depletion of endogenous ABCA2 in D6P2TA cells alters cholesterol levels in lipid rafts, cell homogenates were prepared in Triton X-100 and Lubrol WX and subjected to density gradient ultracentrifugation. Fractions were collected and analyzed for cholesterol content using the Amplex Red cholesterol assay kit as described in methods. ABCA2 depletion in D6P2T cells did not significantly alter the cholesterol content in lipid raft compartments (fractions 4, 5, 6) of 46.17 % in D6P2TL and 47.73 % in D6P2TA respectively (Fig. 4). These results indicate that the distribution of cholesterol in lipid raft compartments may not be the major mechanism for the increase in cholesterol esterification observed in ABCA2-depleted D6P2T cells.
Figure 4. Distribution of cholesterol and sphingomyelin in lipid raft compartments in D6P2T cells.

Cells were cultured as described in Methods and cell homogenates were prepared by solubilization of D6P2TL and D6P2TA cells in Triton X-100 and Lubrol WX in MBS buffer, followed by sucrose density gradient ultracentrifugation as described in Methods. Twelve fractions were recovered and probed by Western blot with antibodies to flotillin-1 (raft) and calnexin (non-raft). Cholesterol and sphingomyelin levels were determined on an aliquot of each fraction using fluorimetric assays as described in Methods.
Although ABCA2 expression level did not alter the cholesterol distribution in lipid raft compartments the association of cholesterol with membrane sphingomyelin may be a mechanism by which ABCA2 regulates cholesteryl ester formation in D6P2T cells. For example, an increase in sphingomyelin in ABCA2-depleted cells (D6P2TA) could account for the increased cholesteryl ester formation following bSMase depletion of plasma membrane sphingomyelin and release of associated cholesterol. To determine whether D6P2TA contained more sphingomyelin in lipid raft compartments, their levels in raft fractions (fractions 4, 5, 6) were measured using the Amplite fluorimetric sphingomyelin assay kit. Sphingomyelin distribution did not differ significantly in lipid raft compartments in control D6P2TL (54 %) and in ABCA2-depeleted D6P2TA cells (50 %), respectively (Fig. 4). These results indicate that the distribution of sphingomyelin in lipid raft compartments may not be the major mechanism regulating cholesterol esterfication in D6P2T cells.
3.6. Plasma membrane sphingomyelin-dependent lysenin toxicity in D6P2T cells
The sphingomyelin-specific pore forming toxin lysenin, from the coelomic fluid of the earthworm Eisenia foetida, has been used to study the organization of sphingomyelin in plasma membranes [26]. It selectively binds cell-surface sphingomyelin localized in clusters and induces cytolysis of sphingomyelin-containing cells [27]. A lysenin cell mortality assay was utilized to determine the relative levels of plasma membrane lysenin-sensitive sphingomyelin levels in control D6P2TL and ABCA2-depleted D6P2TA cells. Cells were treated with 200 ng/ml lysenin for 2 hours, counted and equal numbers of cells were incubated with the WST-1 cell proliferation reagent for 2 hours followed by measurement of absorbance by spectrophotometry. Cell survival in ABCA2-depleted D6P2TA cells was 80% of the control D6P2TL cells ± SD 1.79; p < 0.011 (*) (Fig. 5). These results indicate that ABCA2 depletion in D6P2TA cells renders them more susceptible to plasma membrane sphingomyelin-specific lysenin toxicity.
Figure 5. Plasma membrane sphingomyelin-dependent lysenin toxicity in D6P2T cells.
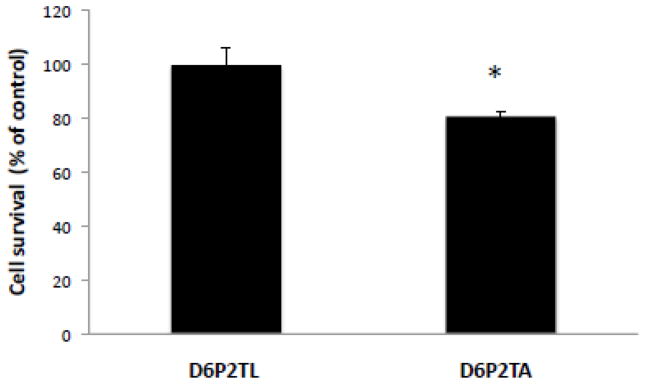
Cells were treated with 200 ng/ml of the plasma membrane sphingomyelin-specific toxin lysenin for 2 hours before cells were counted and re-plated at a density of 1.2 × 106 cells per ml. Cell viability was measured after addition of the WST-1 cell proliferation reagent, culture for 2 hours at 37° C and measurement of absorbance at 350 nm.
3.7. In vitro sphingomyelin synthase activity is modulated by ABCA2 in N2a and D6P2T cells
In another approach to determine whether sphingomyelin level is modulated by ABCA2 expression, an in vitro sphingomyelin synthase activity assay was performed, using the flurophore C6-NBD ceramide (N-[7-(4-nitrobenzo-2-oxa-1,3-diazole)] aminocaproyl sphingosine). In this assay, cell homogenates are incubated with the precursor C6-NBD ceramide (C6-NBD-Cer) that is converted to C6-NBD-sphingomyelin (C6-NBD-SM). In ABCA2-overexpressing A2.1 cells, the in vitro synthesis of C6-NBD-SM level declined to 71% of N2a control cells (Fig. 6). In ABCA2-depleted D6P2TA cells, in vitro synthesis of C6-NBD-SM increased nearly 125% ± SD 0.038 of control D6P2T cells. These results are consistent with the hypothesis that ABCA2 expression level modulates sphingomyelin synthesis.
Figure 6. In vitro sphingomyelin synthase activity is modulated by ABCA2 expression.

Cells were cultured as described in Methods. Cell pellets were lysed by N2 cavitation and 100 μg of cell homogenate were incubated with 2 μg/ml of C6-NBD ceramide and 10 μg of L-a-phosphatidylcholine for 2 hours at 37° C. Lipids were extracted by method of Bligh and Dyer and TLC was performed in chloroform/methanol/ammonium hydroxide (70:30:5). Fluorescent NBD-lipids were detected using a Storm phosphorimager and quantified using ImageQuant software.
3.8. ABCA2 expression modulates de novo sphingomyelin synthesis in N2a and D6P2T cells
To determine the relative levels of de novo sphingomyelin synthesis from radioactive precursors in intact cells, D6P2TL and D6P2TA cells were incubated with [14C-methyl]choline chloride for 4 hours followed by lipid extraction and thin layer chromatography. The level of [14C]sphingomyelin synthesized was 0.05 nmol/mg protein in control D6P2TL cells ± SD 0.01 and increased to 0.20 nmol/mg protein in ABCA2-depleted D6P2TA cells ± SD 0.02; p < 0.001 (*) (Fig. 7). In N2a cells the level of [14C]sphingomyelin was relatively unchanged at 0.012 nmol/mg protein ± SD 0.001 and in A2.1 cells, 0.015 nmole/mg protein ± SD 0.001; p < 0.186. These results indicate that ABCA2-depletion in D6P2TA cells increases the de novo synthesis of sphingomyelin.
Figure 7. ABCA2 expression modulates de novo sphingomyelin synthesis in intact cells.
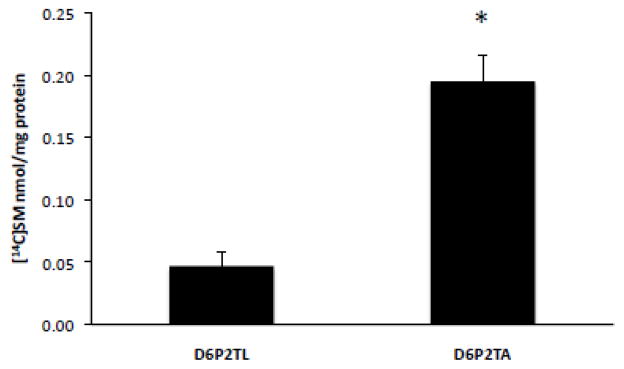
Cells were cultured as described in Methods. After addition of 2 μCi of [methyl-14C]chloride, the cells were cultured for 4 hours before lipid extraction in hexane/isopropanol (3:2) and TLC performed in chloroform/methanol/ammonium hydroxide (70:30:5). Formation of [14C]sphingomyelin was quantified using a Bioscan 2000 thin layer chromatography scanner and normalized to total cell protein.
3.9. ABCA2 level modulates SMS2 expression in N2a and D6P2T cells
The enzymes sphingomyelin synthase-1 and -2 (SMS1, SMS2) regulate de novo sphingomyelin synthesis. SMS1 is localized in the lumen of the Golgi complex and SMS2 is primarily localized in the exofacial leaflet of the plasma membrane and is the principal isoform responsible for synthesis of plasma membrane sphingomyelin [28, 29]. Since the bacterial sphingomyelinase-sensitive plasma membrane pool of sphingomyelin was responsible for elevation of cholesteryl ester formation in ABCA2-depleted D6P2TA cells, the level of SMS2 was measured by Western blot of cell homogenates. SMS2 expression was elevated in ABCA2-depleted D6P2TA cells compared to D6P2TL control cells (Fig. 8.). N2a and A2.1 cells expressed considerably less SMS2 than D6P2T cells but notably, ABCA2-overexpressing A2.1 cells expressed less SMS2 than in N2a control cells.
Figure 8. Plasma membrane sphingomyelin synthase-2 expression in N2a and D6P2T cells.
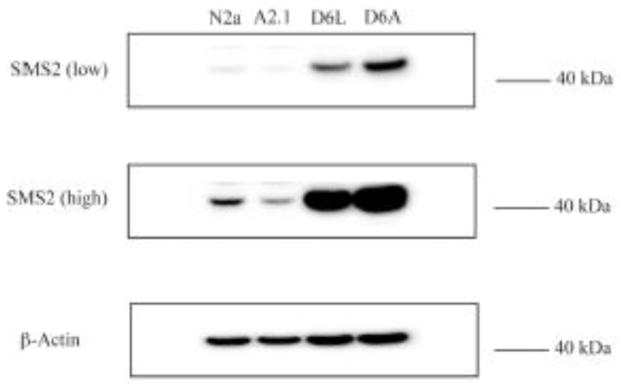
Whole cell homogenates were prepared from N2a and A2.1 cells or D6P2TL and D6P2TA cells and probed for SMS2 expression. Shown is a representative Western blot with a low-amplification signal to detect SMS2 expression in D6P2T cells and high-amplification signal to detect SMS2 expression in N2a cells.
3.10. ABCA2 modulates the transbilayer transport of ceramide in the plasma membrane of N2a cells
The results described above suggest that sphingomyelin level may be a major mechanism modulating cholesterol esterification in ABCA2-depleted D6P2TA cells but not in ABCA2-overexpressing A2.1 cells. Experiments were undertaken to evaluate whether the transbilayer distribution of ceramide in the plasma membrane of N2a and ABCA2-overexpressing A2.1 cells may be a mechanism for regulation of cholesterol esterification. ABCA2 overexpression in A2.1 cells may function to modulate the transport of ceramide across lipid bilayers and may function to decrease the movement of ceramide from the exofacial/outer leaflet to the cytofacial/inner leaflet. This ceramide may decrease esterification of plasma membrane-derived cholesterol by direct inhibition of ACAT activity. The transbilayer movement of ceramide in the plasma membrane of live cells may be determined by measuring ceramide kinase activity (CERK) [30]. All conversion of ceramide to ceramide-1-phosphate (C1P) by CERK occurs in intracellular compartments. CERK is localized to the Golgi complex, cytoplasmic vesicles and plasma membrane [31, 32]. To determine whether ABCA2 overexpression in N2a cells alters the transbilayer distribution of ceramide in the plasma membrane, cells were treated with C6-NBD-ceramide and the production of NBD-ceramide-1-phosphate was determined (NBD-C1P). The level of conversion of NBD-ceramide to NBD-C1P by was determined after differential partitioning of substrate and product, following chloroform/methanol extraction. The NBD-C1P product of ceramide kinase partitions to the aqueous phase and can be quantitated in a fluorescent plate reader [33]. ABCA2 overexpression in A2.1 cells decreased NBD-C1P production by ~ 20% ± SD 0.06; p < 0.001 (*) (Fig. 9). These results suggest that ABCA2 overexpression may alter the transbilayer distribution of ceramide in the plasma membrane favoring its retention in the exofacial/outer leaflet.
Figure 9. Transbilayer transport of ceramide in the plasma membrane in N2a cells.
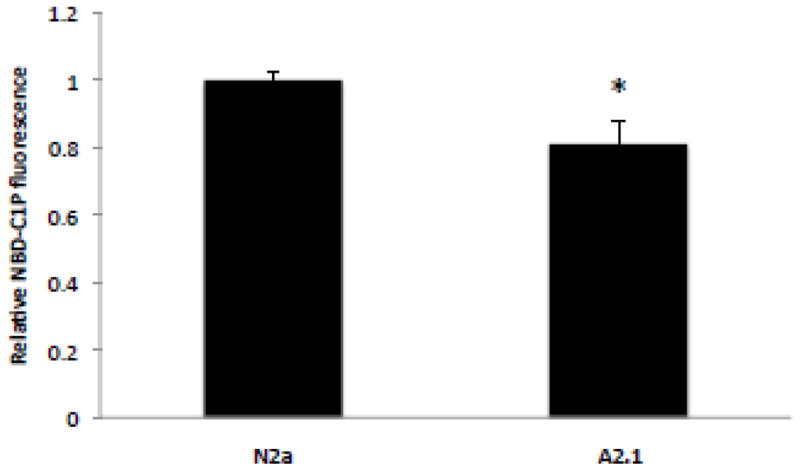
Cells were cultured as described in Methods. C6-NBD-ceramide complexed to BSA was added to a final concentration of 5 μM and the cells were incubated at 4° C for 30 min. in the dark and then warmed to 37° C for 2 hours. Cells were counted and lipid extraction was performed on equal numbers of cells with chloroform/methanol/50 mM Tris-HCl, pH 8.5 (2:1:0.3) as described in Methods. Fluorescent NBD-C1P was recovered from the aqueous phase and measured in a plate reader at ex:em 460:515–580. TLC confirmed that the fluorescent product was NBD-C1P.
3.11. Back-extraction of NBD-lipids from the plasma membrane in N2a cells
In another approach to evaluate the transport of C6-NBD-ceramide, cells were incubated with C6-NBD ceramide and cell surface NBD-metabolites were selectively removed from the outer surface of the plasma membrane with bovine serum albumin. The remaining cell-associated NBD-lipids were measured following lipid extraction and fluorography [34, 35]. If ABCA2 overexpression decreases C6-NBD-ceramide translocation across the plasma membrane from the outer leaflet to the inner cytofacial leaflet then BSA would extract more C6-NBD-ceramide from the plasma membrane and less would be cell-associated. Translocation of precursor NBD-ceramide is required for NBD-metabolite synthesis (NBD-glucosylceramide/NBDGlcCer and NBD-sphingomyelin/NBD-SM), from the outer leaflet to the cytosolic side across the lipid bilayer of the plasma membrane, or across the lipid bilayer of vesicles derived from the plasma membrane following internalization. Following internalization, the synthesis of the NBD-metabolites requires translocation of the precursor NBD-ceramide from the cytosolic side of the Golgi complex into the Golgi lumen; followed by recycling of vesicles containing NBD-metabolites from the Golgi back to the plasma membrane (BSA-extractable). To evaluate transport and metabolism of C6-NBD-ceramide, cells were incubated with 5 μM C6-NBD-ceramide for 30 min at 4° C and shifted to 37° C to for 2 hours to promote internalization and recycling. In N2a control cells, the levels of cell-associated NBD-ceramide after BSA back-extraction did not change dramatically, decreasing to 80 % of untreated N2a cells ± SD 12.4; p < 0.06 (Fig. 10). The levels of cell-associated NBD-GlcCer after back-extraction were decreased to 87 % ± SD 10.2; p < 0.11 and the levels of NBD-SM after back-extraction were decreased to 91 % ± SD 7.8; p < 0.283. In contrast, in ABCA2-overexpressing A2.1 cells, in the absence of back extraction, the reduction of cell-associated NBD-ceramide and NBD-metabolites was more significant compared to control N2a cells not subjected to BSA back-extraction. The levels of cell-associated NBD-ceramide decreased to 57 % ± SD 6.03; p < 0.004 % (*). The levels of cell-associated NBD-GlcCer decreased to 77.84 % ± SD 7.62; p < 0.02 (*) and the levels of NBD-SM decreased to 76 % ± SD 10.3; p < 0.03 (*). Back-extraction of surface NBD-lipids produced the greatest reduction in the levels of cell-associated NBD-lipids in ABCA2-overexpressing A2.1 cells. The levels of cell-associated NBD-ceramide decreased to 32.5% ± SD 2.03; p < 0.002 % (**). The levels of cell-associated NBD-GlcCer decreased to 44.05 % ± SD 7.05; p < 0.001 (**) and the levels of NBD-SM decreased to 64 % ± SD 14.62; p < 0.01 (**). These results provide further evidence that ABCA2 overexpression may alter the transbilayer distribution of ceramide in the plasma membrane favoring its retention in the exofacial/outer leaflet where it is available for BSA back-extraction, reducing the amount of cell-associated NBD-ceramide for conversion into NBD-metabolites.
Figure 10. Back-extraction of NBD-lipids from the plasma membrane in N2a cells.
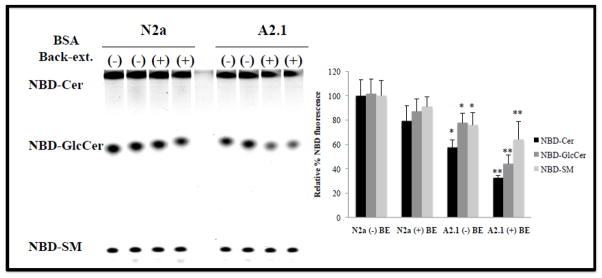
Cells were cultured as described in Methods. C6-NBD-ceramide, complexed to BSA was added to a final concentration of 5 μM and the cells were incubated at 4° C for 30 min. in the dark and then warmed to 37° C for 2 hours. After the incubation, period cells were washed and NBD-lipids were removed from the outer surface of the plasma membrane with ± 3% BSA at 4° C. Cells were counted and lipid extraction was performed on equal numbers of cells with chloroform/methanol/0.01 N HCl (1:2:0.6). Cell-associated NBD-lipids were dried under N2 and TLC performed in chloroform/methanol/ammonium hydroxide (70:30:5) followed by fluorescence imaging using a Storm phosphorimager as described above.
3.12. Elevation of endogenous ceramide by D-erythro MAPP decreases cholesterol esterification in N2a cells
In the esterification of plasma membrane cholesterol experiments described above, treatment of cells with sphingomyelinase to decrease plasma membrane sphingomyelin level, increased cholesterol esterification in N2a control cells but not to the same extent as in ABCA2 overexpressing A2.1 cells. A plausible explanation for the reduction in cholesteryl ester formation in ABCA2 overexpressing A2.1 cells may be that bacterial sphingomyelinase hydrolysis of sphingomyelin in the outer/exofacial leaflet of the plasma membrane increases ceramide production that may promote direct inhibition of ACAT enzyme activity. Exogenous ceramide can inhibit ACAT in both cell-free homogenates and intact cells [36, 37]. To test whether elevation of endogenous ceramide alone without loss of plasma membrane sphingomyelin can modulate cholesterol esterification, cells were treated with an inhibitor of ceramidase activity 1S,2R-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol (MAPP) [38] for 4 hours before lipid extraction and TLC. Ceramidase is localized to the outer/exofacial leaflet of the plasma membrane [39] and elevates ceramide in this compartment. In control N2a cells, MAPP decreased cholesteryl ester formation from 11.3% to 5.6% ± SD 0.19; p < 0.001 (*). The effect of was more pronounced in ABCA2-overexpressing A2.1 cells; MAPP decreased cholesteryl ester formation from 5.7 % to 3.4 % ± SD 0.47; p < 0.005 (**) (Fig. 11). The repressive of MAPP on cholesterol esterification in control N2a and A2.1 cells was similar, approximately 40%; however, comparing MAPP-treated N2a cells and MAPP-treated ABCA2 overexpressing A2.1 cells a further 40% decrease in cholesterol esterification was observed in the latter.
Figure 11. Elevation of endogenous ceramide by D-erythro MAPP decreases cholesterol esterification in N2a cells.
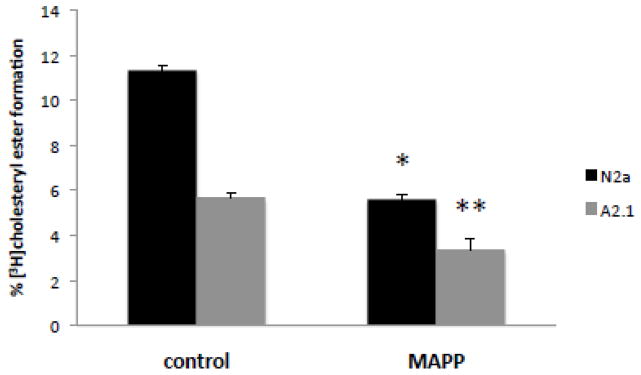
Cells were cultured as described in Methods. D-erythro-MAPP (50 μg/ml) was added and cells were cultured for 5 hours. Lipids were extracted in hexane/isopropanol (3:2) and TLC performed in hexane/ethyl ether/acetic acid (80:18.5:1.5) to determine [3H]cholesteryl ester levels.
3.13. Elevation of endogenous ceramides does not directly inhibit in vitro ACAT activity in N2a cells
To evaluate whether ABCA2 expression directly inhibited ACAT activity in cell-free homogenates of control N2a and ABCA2-overexpressing A2.1, cells treated with MAPP, an in vitro ACAT activity assay was performed with [1-14C]oleoyl coenzyme A. MAPP treatment did not inhibit ACAT activity in cell-free homogenates. MAPP treatment slightly increased [14C] cholesteryl ester formation from 0.12 nmol/mg protein ± SD 0.019 to 0.14 nmol/mg ± SD 0.018. In ABCA2-overexpressing A2.1 cells, MAPP treatment slightly increased [14C]cholesteryl ester formation from 0.17 nmol/mg ± SD 0.008 protein to 0.18 nmol/mg ± SD 0.01. These results indicate that elevation of endogenous ceramides with MAPP and inhibition of esterification of plasma membrane derived cholesterol in ABCA2 overexpressing A2.1 cells requires intact cells for inhibition of ACAT activity and esterification of plasma membrane-derived cholesterol.
3.14. Ceramide and sphingomyelin mass in N2a, A2.1, D6P2TL and D6P2TA cells
In order to establish whether ABCA2 expression affects overall cellular levels of ceramide and sphingomyelin in the various cells used in this study, a comprehensive lipidomic analysis of metabolites was performed using Electrospray Ionization/Mass Spectrometry. In N2a and A2.1 cells, the levels of most ceramide species were not significantly different with the exceptions of abundant species C16-ceramide, and long-chain C24:1 ceramide (Fig. 12A). C16-ceramide levels declined from 4.45 pmol/nmol Pi in N2a cells to 1.92 pmol/nmol Pi in A2.1 cells (* p < 0.02). Long-chain C24:1 ceramide levels declined from 2.96 pmol/nmol Pi in N2a cells to 1.86 pmol/nmol Pi in A2.1 cells (* p < 0.006). In D6P2TL and D6P2TA cells the levels of most ceramide species were not significantly different with the exceptions of abundant species long-chain C24-ceramide and C24:1 ceramide. C24-ceramide levels increased from 1.24 pmol/nmol Pi in D6P2TL cells to 1.83 pmol/nmol Pi in D6P2TA cells (*p < 0.004). C24:1 ceramide levels increased from 1.38 pmol/nmol Pi to 1.9 pmol/nmol Pi (*p < 0.02). These results suggest that ABCA2 expression level modulates the abundance of individual ceramide species, particularly long-chain C24:1 ceramide. Interestingly, measurements of sphingomyelin mass in N2a and A2.1 did not differ significantly in any particular species. Similarly, D6P2TL and D6P2TA cells displayed similar levels of all sphingomyelin species, although the total mass of individual species was greater than in N2a and A2.1 cells (Fig. 12B). These results indicate that ABCA2 expression does not modulate the steady-state level of sphingomyelin mass under these conditions.
Fig. 12. Analysis of ceramide and sphingomyelin levels in N2a, A2.1, D6P2TL and D6P2TA cells.

Total lipids were extracted from the cells and subjected to electrospray ionization/mass spectrometry analysis as described in Methods. The levels of ceramide (A) and sphingomyelin B were determined and normalized to Pi concentrations. Results are the mean ± SD. Statistical significance was determined using the Student’s t test, p < 0.05.
4. Discussion
The key findings in this report indicate that ABCA2 expression level modulates esterification of plasma membrane cholesterol through modulation of sphingolipid metabolism. Two model systems were employed, human ABCA2 overexpression in mouse neuroblastoma N2a cells and depletion of endogenous ABCA2 in rat schwannoma cells. Treatment of cells with bacterial sphingomyelinase hydrolyzes plasma membrane sphingomyelin to ceramide and phosphocholine. The loss of membrane sphingomyelin following sphingomyelinase treatment is reported to increase the mobility of plasma membrane cholesterol and promotes its trafficking to the endoplasmic reticulum for esterification by ACAT [19].
Esterification of plasma membrane cholesterol in these model systems correlates with ABCA2 expression level. In ABCA2-overexpressing A2.1 neuroblastoma cells, the basal level cholesteryl ester formation was lower than in control N2a cells. Following bSMase treatment to hydrolyze plasma membrane sphingomyelin, ABCA2-overexpressing A2.1 cells generated significantly less cholesteryl ester than N2a cells (Fig. 2A). Interestingly, treatment of A2.1 cells with 25-hydroxycholesterol, which is reported to mobilize cholesterol from membrane lipids [21] restored cholesteryl ester formation to the level produced in control N2a cells. The source of cholesterol mobilized by 25-hydroxycholesterol for esterification was sensitive to the cholesterol trafficking inhibitor U18666a, which blocks transport of free cholesterol from late-endosomal/lysosomal compartments to the plasma membrane and from the plasma membrane to the endoplasmic reticulum [22, 23]. Treatment with both 25-HC and U18666a demonstrated that the effect of U18666a-mediated cholesterol trafficking inhibition was predominant to the effect of 25-HC-mediated cholesterol mobilization in reducing cholesteryl ester formation and this effect was independent of ABCA2 expression level (Fig. 2B).
In contrast, in ABCA2-depleted D6P2TA cells, the basal level of cholesteryl ester formation was higher than in control D6P2TL cells and following bSMase treatment the level of cholesteryl ester formation increased further relative to D6P2TL control cells (Fig. 3). These data suggest that bSMase depletion of membrane sphingomyelin results in opposing effects on cholesterol esterification in N2a and D6P2T cells that correlate with ABCA2 expression level. Cholesterol and sphingomyelin compartmentation in lateral domains of lipid rafts did not significantly vary with ABCA2 depletion in D6P2T cells and do not appear to be a major mechanism for ABCA2 modulation of esterification of plasma membrane cholesterol (Fig. 4).
Several lines of evidence suggest that membrane sphingomyelin level may be a locus of regulation for modulation of esterfication of plasma membrane cholesterol in D6P2T schwannoma cells. The plasma membrane sphingomyelin-specific toxin lysenin is commonly utilized to determine the relative amount of plasma membrane sphingomyelin [40]. Lysenin specifically binds sphingomyelin and induces cell lysis [41]. In ABCA2-depleted D6P2TA cells, exposure to lysenin resulted in increased toxicity compared to control D6P2TL cells (Fig. 5). These data suggest that ABCA2 depletion in D6P2TA cells renders cells more susceptible to plasma membrane sphingomyelin-specific lysenin toxicity.
To determine the relative sphingomyelin synthetic activity in ABCA2-depleted D6P2TA and control D6P2TL, an in vitro sphingomyelin synthase assay was performed on cell homogenates. As shown in Fig. 6, ABCA2-depleted D6P2TA cells exhibited greater in vitro sphingomyelin synthase activity relative to control D6P2TL cells. As another measure of sphingomyelin synthetic activity, a de novo sphingomyelin synthesis assay in intact cells using radiolabeled precursor [methyl-14C]choline was performed. Consistent with the in vitro sphingomyelin synthase results, ABCA2 depletion in D6P2TA cells increased de novo synthesis of [14C]sphingomyelin in intact cells (Fig. 7). Sphingomyelin synthase-2 activity is the principal isoform regulating plasma membrane sphingomyelin synthesis. Western blot analysis detected greater SMS2 expression in ABCA2-depleted D6P2TA cells compared to control cells (Fig. 8). Also noteworthy was that SMS2 expression was lower in ABCA2-ovexpressing A2.1 cells than in N2a control cells.
Whereas ABCA2-depletion and elevation of plasma membrane sphingomyelin in D6PTA cells may be a mechanism for modulation of esterification of plasma membrane cholesterol, altering the transbilayer transport of ceramide distribution across the plasma membrane and the synthesis of ceramide-derived metabolites in the Golgi complex may define ABCA2 function in ABCA2 overexpressing A2.1 neuroblastoma cells. Ceramide is reported to undergo rapid transbilayer movement across artificial and cell membranes [42, 43] and ABCA2 may function to alter this equilibrium, favoring retention in the exofacial/outer plasma membrane leaflet. As a measure of transbilayer ceramide transport, the precursor C6-NBD-ceramide was utilized in a ceramide kinase assay (CERK). CERK converts C6-NBD-ceramide to NBD-ceramide-1 phosphate (NBD-C1P). CERK activity requires transport of C6-NBD-ceramide from the outer/exofacial leaflet to the inner cytofacial leaflet of the plasma membrane to generate NBD-C1P [30]. ABCA2-overexpression in A2.1 cells altered the transbilayer transport of fluorescent C6-NBD-ceramide, as evidenced by decreased production of (NBD-C1P) (Fig. 9).
In another approach to evaluate the transport of plasma membrane-bound C6-NBD-ceramide, NBD-lipids were selectively extracted from the outer/exofacial leaflet with bovine serum albumin. In ABCA2 overexpressing A2.1 cells, greater levels of the precursor C6-NBD-ceramide were removed by back-extraction from the outer/exofacial leaflet of the plasma membrane and less was cell-associated (Fig. 10). The decrease in cell-associated precursor C6-NBD-ceramide in ABCA2-overexpressing A2.1 cells also resulted in reduced synthesis of the NBD-metabolites, NBD-SM and NBD-GlcCer. These data provide additional evidence that suggest that ABCA2 may function to alter the transbilayer transport of ceramide across the plasma membrane. The decrease in transport of ceramide for metabolite synthesis may increase the effective ceramide concentration for the direct inhibition of ACAT and reduce esterification of plasma membrane-derived cholesterol.
Bacterial sphingomyelinase-mediated degradation of plasma membrane sphingomyelin and production of ceramide that could directly inhibit ACAT activity may be a possible mechanism for reducing esterification of plasma membrane-derived cholesterol in ABCA2-overexpressing A2.1 cells. To examine the consequences of elevation of endogenous ceramides, independent of sphingomyelin degradation, on cholesterol esterification the ceramidase inhibitor, D-erythro-MAPP [38] was employed. MAPP treatment of intact cells suppressed cholesterol esterification relative to untreated samples and the inhibition was greater in ABCA2-overexpressing A2.1 cells than control N2a cells. To determine whether MAPP directly inhibited ACAT activity, an in vitro ACAT activity assay on cell homogenates was performed. Elevation of endogenous ceramides by MAPP in cell homogenates of N2a and ABCA2-overexpressing A2.1 cells did not inhibit in vitro ACAT activity; therefore, intact cells are required for MAPP inhibition of cholesterol esterification in N2a and A2.1 cells.
The significance of understanding the function of key regulators of sphingolipid metabolism and intracellular cholesterol trafficking may aid in understanding disease mechanisms whose etiology is linked to defects in these processes. In these experiments, plasma membrane sphingomyelin was degraded with exogenous sphingomyelinase. Physiological degradation of sphingomyelin by neutral and acidic sphingomyelinases has been described for a number of disease states, including inflammation, cardiovascular and neurological diseases [39]. ABCA2 function may be a key modulator in the etiology of these diseases. Cross talk between sphingolipid and intracellular cholesterol trafficking pathways in normal and disease states are areas requiring further study. Continuing studies of ABCA2 and its coordination with other ABC-transporters in regulating intracellular lipid metabolism may provide strategies for therapeutic intervention to ameliorate disease outcomes.
Fig. 13. Model of ABCA2 function in esterification of plasma membrane cholesterol.

A. Ceramide moves between inner and outer leaflet of the lipid bilayer. ABCA2 overexpression in A2.1 cells alters the bilayer distribution, favoring the outer leaflet. ABCA2 promotes NBD-ceramide distribution in the outer leaflet and BSA extraction from cells. Sphingomyelinase hydrolysis of membrane sphingomyelin or inhibition of ceramidase activity, both elevate membrane ceramide levels. In ABCA2-overexpressing cells, elevated ceramide in the outer leaflet inhibits ACAT activity and conversion of plasma membrane cholesterol to cholesteryl esters. B. Depletion of endogenous ABCA2 by RNAi in D6P2TA cells is associated with decrease sphingomyelin synthase-2 (SMS2) expression, the enzyme responsible for synthesis of plasma membrane sphingomyelin. D6P2TA cells also show increased lysenin toxicity, an agent that specifically binds sphingomyelin in the outer leaflet of the plasma membrane. Sphingomyelinase hydrolysis of plasma membrane sphingomyelin releases associated cholesterol for trafficking to the endoplasmic reticulum for esterification.
Research Highlights.
Overexpression of human ABCA2 in N2a cells decreased cholesterol esterification
RNAi-depletion of rat ABCA2 in D6P2T cells increased cholesterol esterification
ABCA2-depletion in D6P2T cells increased de novo sphingomyelin synthesis
ABCA2 overexpression in N2a cells increased ceramide inhibition of ACAT
Acknowledgments
This research was supported by Grant Number 1K01NS062113-01A2 (Warren Davis) from the National Institute of Neurological Disorders and Stroke.
This research was supported in part by the Lipidomics Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313) and the Lipidomics Core in the SC Lipidomics and Pathobiology COBRE (P20 RR017677).
The stable RNAi rat schwannoma cell lines, D6P2TL (rat lamin-specific RNAi) and D6P2TA (rat ABCA2-specific RNAi) were the kind gift of Dr. Kenneth Tew, Medical University of South Carolina, Charleston, SC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voloshyna I, Reiss AB. The ABC transporters in lipid flux and atherosclerosis. Prog Lipid Res. 2011;50:213–224. doi: 10.1016/j.plipres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. 2012;5:746–755. doi: 10.1242/dmm.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Kimura Y, Nagata K, Yamamoto A, Matsuo M, Ueda K. ABC proteins: key molecules for lipid homeostasis. Med Mol Morphol. 2005;38:2–12. doi: 10.1007/s00795-004-0278-8. [DOI] [PubMed] [Google Scholar]
- 6.Wenzel JJ, Piehler A, Kaminski WE. ABC A-subclass proteins: gatekeepers of cellular phospho- and sphingolipid transport. Front Biosci. 2007;12:3177–3193. doi: 10.2741/2305. [DOI] [PubMed] [Google Scholar]
- 7.Kim WS, Guillemin GJ, Glaros EN, Lim CK, Garner B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport. 2006;17:891–896. doi: 10.1097/01.wnr.0000221833.41340.cd. [DOI] [PubMed] [Google Scholar]
- 8.Mack JT, Beljanski V, Soulika AM, Townsend DM, Brown CB, Davis W, Tew KD. “Skittish” Abca2 knockout mice display tremor, hyperactivity, and abnormal myelin ultrastructure in the central nervous system. Mol Cell Biol. 2007;27:44–53. doi: 10.1128/MCB.01824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai H, Tanaka Y, Tanaka M, Ban N, Yamada K, Matsumura Y, Watanabe D, Sasaki M, Kita T, Inagaki N. ABCA2 deficiency results in abnormal sphingolipid metabolism in mouse brain. J Biol Chem. 2007;282:19692–19699. doi: 10.1074/jbc.M611056200. [DOI] [PubMed] [Google Scholar]
- 10.Mace S, Cousin E, Ricard S, Genin E, Spanakis E, Lafargue-Soubigou C, Genin B, Fournel R, Roche S, Haussy G, Massey F, Soubigou S, Brefort G, Benoit P, Brice A, Campion D, Hollis M, Pradier L, Benavides J, Deleuze JF. ABCA2 is a strong genetic risk factor for early-onset Alzheimer’s disease. Neurobiol Dis. 2005;18:119–125. doi: 10.1016/j.nbd.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Wollmer MA, Kapaki E, Hersberger M, Muntwyler J, Brunner F, Tsolaki M, Akatsu H, Kosaka K, Michikawa M, Molyva D, Paraskevas GP, Lutjohann D, von Eckardstein A, Hock C, Nitsch RM, Papassotiropoulos A. Ethnicity-dependent genetic association of ABCA2 with sporadic Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:534–536. doi: 10.1002/ajmg.b.30345. [DOI] [PubMed] [Google Scholar]
- 12.Chen ZJ, Vulevic B, Ile KE, Soulika A, Davis W, Jr, Reiner PB, Connop BP, Nathwani P, Trojanowski JQ, Tew KD. Association of ABCA2 expression with determinants of Alzheimer’s disease. FASEB J. 2004;18:1129–1131. doi: 10.1096/fj.03-1490fje. [DOI] [PubMed] [Google Scholar]
- 13.Davis W., Jr The ATP-binding cassette transporter-2 (ABCA2) increases endogenous amyloid precursor protein expression and Abeta fragment generation. Curr Alzheimer Res. 2010;7:566–577. doi: 10.2174/156720510793499002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michaki V, Guix FX, Vennekens K, Munck S, Dingwall C, Davis JB, Townsend DM, Tew KD, Feiguin F, De Strooper B, Dotti CG, Wahle T. Down-regulation of the ATP-binding cassette transporter 2 (Abca2) reduces amyloid-beta production by altering Nicastrin maturation and intracellular localization. J Biol Chem. 2011;287:1100–1111. doi: 10.1074/jbc.M111.288258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calpe-Berdiel L, Zhao Y, de Graauw M, Ye D, van Santbrink PJ, Mommaas AM, Foks A, Bot M, Meurs I, Kuiper J, Mack JT, Van Eck M, Tew KD, van Berkel TJ. Macrophage ABCA2 deletion modulates intracellular cholesterol deposition, affects macrophage apoptosis, and decreases early atherosclerosis in LDL receptor knockout mice. Atherosclerosis. 2012;223:332–341. doi: 10.1016/j.atherosclerosis.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 16.Davis W, Jr, Boyd JT, Ile KE, Tew KD. Human ATP-binding cassette transporter-2 (ABCA2) positively regulates low-density lipoprotein receptor expression and negatively regulates cholesterol esterification in Chinese hamster ovary cells. Biochim Biophys Acta. 2004;1683:89–100. doi: 10.1016/j.bbalip.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Davis W., Jr The ATP-binding cassette transporter-2 (ABCA2) regulates cholesterol homeostasis and low-density lipoprotein receptor metabolism in N2a neuroblastoma cells. Biochim Biophys Acta. 2011;1811:1152–1164. doi: 10.1016/j.bbalip.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hai M, Muja N, DeVries GH, Quarles RH, Patel PI. Comparative analysis of Schwann cell lines as model systems for myelin gene transcription studies. J Neurosci Res. 2002;69:497–508. doi: 10.1002/jnr.10327. [DOI] [PubMed] [Google Scholar]
- 19.Porn MI, Slotte JP. Reversible effects of sphingomyelin degradation on cholesterol distribution and metabolism in fibroblasts and transformed neuroblastoma cells. Biochem J. 1990;271:121–126. doi: 10.1042/bj2710121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller SC, Melnykovych G. Regulation of cholesterol biosynthesis and esterification by 25-hydroxycholesterol in a macrophage-like cell line: uncoupling by progesterone. J Lipid Res. 1984;25:991–999. [PubMed] [Google Scholar]
- 21.Du X, Pham YH, Brown AJ. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J Biol Chem. 2004;279:47010–47016. doi: 10.1074/jbc.M408690200. [DOI] [PubMed] [Google Scholar]
- 22.Underwood KW, Andemariam B, McWilliams GL, Liscum L. Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J Lipid Res. 1996;37:1556–1568. [PubMed] [Google Scholar]
- 23.Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–2270. [PubMed] [Google Scholar]
- 24.Lange Y. Tracking cell cholesterol with cholesterol oxidase. J Lipid Res. 1992;33:315–321. [PubMed] [Google Scholar]
- 25.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 26.Shogomori H, Kobayashi T. Lysenin: a sphingomyelin specific pore-forming toxin. Biochim Biophys Acta. 2008;1780:612–618. doi: 10.1016/j.bbagen.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Ishitsuka R, Yamaji-Hasegawa A, Makino A, Hirabayashi Y, Kobayashi T. A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys J. 2004;86:296–307. doi: 10.1016/S0006-3495(04)74105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tafesse FG, Huitema K, Hermansson M, van der Poel S, van den Dikkenberg J, Uphoff A, Somerharju P, Holthuis JC. Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J Biol Chem. 2007;282:17537–17547. doi: 10.1074/jbc.M702423200. [DOI] [PubMed] [Google Scholar]
- 30.Mitsutake S, Igarashi Y. Transbilayer movement of ceramide in the plasma membrane of live cells. Biochem Biophys Res Commun. 2007;359:622–627. doi: 10.1016/j.bbrc.2007.05.160. [DOI] [PubMed] [Google Scholar]
- 31.Rovina P, Schanzer A, Graf C, Mechtcheriakova D, Jaritz M, Bornancin F. Subcellular localization of ceramide kinase and ceramide kinase-like protein requires interplay of their Pleckstrin Homology domain-containing N-terminal regions together with C-terminal domains. Biochim Biophys Acta. 2009;1791:1023–1030. doi: 10.1016/j.bbalip.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Carre A, Graf C, Stora S, Mechtcheriakova D, Csonga R, Urtz N, Billich A, Baumruker T, Bornancin F. Ceramide kinase targeting and activity determined by its N-terminal pleckstrin homology domain. Biochem Biophys Res Commun. 2004;324:1215–1219. doi: 10.1016/j.bbrc.2004.09.181. [DOI] [PubMed] [Google Scholar]
- 33.Don AS, Rosen H. A fluorescent plate reader assay for ceramide kinase. Anal Biochem. 2008;375:265–271. doi: 10.1016/j.ab.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marx U, Lassmann G, Holzhutter HG, Wustner D, Muller P, Hohlig A, Kubelt J, Herrmann A. Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys J. 2000;78:2628–2640. doi: 10.1016/S0006-3495(00)76807-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipsky NG, Pagano RE. Intracellular translocation of fluorescent sphingolipids in cultured fibroblasts: endogenously synthesized sphingomyelin and glucocerebroside analogues pass through the Golgi apparatus en route to the plasma membrane. J Cell Biol. 1985;100:27–34. doi: 10.1083/jcb.100.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridgway ND. Inhibition of acyl-CoA:cholesterol acyltransferase in Chinese hamster ovary (CHO) cells by short-chain ceramide and dihydroceramide. Biochim Biophys Acta. 1995;1256:39–46. doi: 10.1016/0005-2760(95)00009-2. [DOI] [PubMed] [Google Scholar]
- 37.Subbaiah PV, Billington SJ, Jost BH, Songer JG, Lange Y. Sphingomyelinase D, a novel probe for cellular sphingomyelin: effects on cholesterol homeostasis in human skin fibroblasts. J Lipid Res. 2003;44:1574–1580. doi: 10.1194/jlr.M300103-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Venable ME, Bielawska A, Obeid LM. Ceramide inhibits phospholipase D in a cell-free system. J Biol Chem. 1996;271:24800–24805. doi: 10.1074/jbc.271.40.24800. [DOI] [PubMed] [Google Scholar]
- 39.Milhas D, Clarke CJ, Hannun YA. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett. 2010;584:1887–1894. doi: 10.1016/j.febslet.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamaji A, Sekizawa Y, Emoto K, Sakuraba H, Inoue K, Kobayashi H, Umeda M. Lysenin, a novel sphingomyelin-specific binding protein. J Biol Chem. 1998;273:5300–5306. doi: 10.1074/jbc.273.9.5300. [DOI] [PubMed] [Google Scholar]
- 41.Hanada K, Hara T, Fukasawa M, Yamaji A, Umeda M, Nishijima M. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J Biol Chem. 1998;273:33787–33794. doi: 10.1074/jbc.273.50.33787. [DOI] [PubMed] [Google Scholar]
- 42.Bai J, Pagano RE. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 1997;36:8840–8848. doi: 10.1021/bi970145r. [DOI] [PubMed] [Google Scholar]
- 43.Lopez-Montero I, Rodriguez N, Cribier S, Pohl A, Velez M, Devaux PF. Rapid transbilayer movement of ceramides in phospholipid vesicles and in human erythrocytes. J Biol Chem. 2005;280:25811–25819. doi: 10.1074/jbc.M412052200. [DOI] [PubMed] [Google Scholar]