

Abstract
Therapeutic drug delivery across the blood-brain barrier (BBB) is not only inefficient, but also nonspecific to brain stroma. These are major limitations in the effective treatment of brain cancer. Transferrin peptide (Tfpep) targeted gold nanoparticles (Tfpep-Au NPs) loaded with the photodynamic pro-drug, Pc 4, have been designed and compared with untargeted Au NPs for delivery of the photosensitizer to brain cancer cell lines. In vitro studies of human glioma cancer lines (LN229 and U87) overexpressing the transferrin receptor (TfR) show a significant increase in cellular uptake for targeted conjugates as compared to un-targeted particles. Pc 4 delivered from Tfpep-Au NPs clusters within vesicles after targeting with the Tfpep. Pc 4 continues to accumulate over a 4 hour period. Our work suggests that TfR-targeted Au NPs may have important therapeutic implications for delivering brain tumor therapies and/or providing a platform for noninvasive imaging.
Introduction
Effective cancer therapy for the treatment of Glioblastoma Multiform (GBM, primary malignant brain tumor) still remains one of the most challenging areas in brain cancer research with little progress in GBM patient survival rate in the last few decades.1,2 One of the major limitations of chemotherapeutics for GBM in the clinic is the lack of tumor selectivity thereby inflicting cell death on healthy surrounding tissues and cells.3 In addition, clinicians are unable to take advantage of other chemotherapeutics that have been successful in treating cancers in more accessible organs due to their inability to cross the physical obstacle of the blood brain barrier (BBB).4
Recently, solid metal-based nanoparticles have shown potential as novel therapeutic platforms in GBM research and therapies.5 This can be attributed to their physical properties, such as small size, increased physiological stability, and bio-compatibility.6,7 More importantly, the surface of the nanoparticle can be tailored with tumor specific targeting moieties like antibodies, ligands or peptides and can encapsulate and deliver hydrophobic anti-cancer drugs through the BBB without altering drug functionalities.8–10 Nanoparticles such as iron oxide,11–13 gadolinium oxide14,15 or manganese oxide16,17 have been used previously to image brain glioma and deliver drugs. Other examples include super paramagnetic iron oxide particles (SPIONs) to target and image gliomas in a rat model18 and PLGA nanoparticles19 loaded with doxorubicin and paclitaxel for tumor inhibition. To enhance the active targeting of nanoparticles, they have been conjugated with cell surface markers which increase the efficacy of nanoparticles for transporting several agents into the tumor region while reducing toxicity in healthy cells.12,20 Despite these modifications, the nanoparticles suffer from in vitro and in vivo cyto-toxicity due to degradation and release of toxic metal ions.21 This necessitates the development of selectively targeted, bio-compatible nanoparticles for long-term monitoring and drug delivery in tumor cells.
Gold nanoparticles (Au NPs) have been used as efficient drug delivery systems10,22–24 due to their low cyto-toxicity (more inert causing less degradation), tunable sizes, and well-studied surface chemistries for stable attachment of ligands and bio-molecules. Polyethylene glycol (PEG)-coated Au NPs25–28 have been used for drug delivery in cancer cells. The PEG25,27 moiety on the nanoparticles provides two major advantages –(1) it ensures long term circulation in the blood since PEG prevents nonspecific interactions with the cellular milieu and (2) PEG also acts as a corona for encapsulating hydrophobic drugs due to its spiral mushroom like structure. In addition, functional groups on the PEG can be coupled to ligands specific for cancer biomarkers overexpressed by tumor cells, providing targeting abilities to the nanoparticles.
Ligand-conjugated PEGylated Au NPs that utilize cancer biomarkers overexpressed by tumor cells as targets are attractive, clinically relevant delivery vehicles for therapeutics.28 In previous studies from our lab,10 we utilized this strategy using EGF-coated PEGylated Au NPs to deliver the photosensitizer Pc 4 (phthalocyanine 4) to brain tumor cells and observed that targeted Au NPs were more efficient for drug delivery than untargeted ones. However, only 3% of the Au NPs were shuttled across the BBB. In an effort to improve the number of Au NPs traversing the BBB, we examined other targeting moieties. Transferrin (Tf) is one potential target that has been exploited.29,30 In brain tumors, GBM cell lines show the highest TfR expression.31 TfR in brain gliomas cause amplified iron accumulation and facilitates tumor progression, a requirement for the rapidly proliferating cells. Therefore, TfR is an attractive target for delivering brain tumor therapies.
Our group has pioneered the use of Au NPs to deliver Pc 4. We have shown in vivo Pc 4 delivery and therapy using untargeted PEGylated Au NPs and were the first to show that these NPs could target Pc 4 to orthotopic brain tumors in a mouse model of GBM.10 In this study, a novel Tfpep-coated Au NP system loaded with Pc 4 is designed and tested for in vitro cellular uptake and in vivo mice studies. Tfpep-conjugated Au NPs show drastic improvement in drug uptake as compared to untargeted Au NPs due to the targeting capabilities of Tfpep ligand. By employing Tfpep-coated Au NPs in combination with Pc 4 loading, we are able to achieve specificity and deliver therapeutic quantities of drug faster and more efficiently.
Experimental methods
Chemicals used in the experiments described below were purchased from Thermo Fisher Scientific, WA. Cell media was purchased from Life Technologies, NY.
Synthesis Tfpep-PEGylated Au NPs-Pc 4
Hydrophobic tetraoctylammonium bromide (TOAB)-coated Au NPs were synthesized via a modified Brust–Schiffrin method as previously described in detail.29,40,41 PEGylated Au NPs were conjugated to Tfpep (Peptides International, KY; 1: 10 molar ratio of carboxyl groups on Au NPs micelle to peptide) via the EDC/NHS protocol42 via the formation of covalent amide bond. The excess reagents were removed using a 10 K MWCO ultracentrifugal centrifugation device (EMD Millipore, MA) at 4000 rpm for 15 minutes. Solutions of untargeted Au NPs and targeted Au NPs were vacuum dried and resuspended in 2 ml of chloroform. Pc 4 drug was added to the reaction solution at a ratio of 40: 1 (Pc 4 molecules to Au NPs). The reaction mixture was left stirring for 2 days at room temperature After drug incubation, chloroform was evaporated in a vacuum and the pellet resuspended in nanopure water and sonicated for 2 hours at room temperature. Excess Pc 4 was removed by purification with 10 K MWCO ultracentrifugal device at 4000 rpm at 4 °C.
Characterization of nanoparticles
The concentrations of Au NPs, PEGylated Au NPs, Tfpep-PEGy-lated Au NPs and Pc 4 loaded Au NPs (targeted and untargeted) were determined by UV-VIS absorption using a Biotek plate reader (Biotek, VT).
Transmission electron micrographs (TEM) of Au NPs and PEGylated Au NPs were taken by spreading 10 μl of organic and aqueous master solution (1 mg ml−1) on a carbon-coated copper grid (Sigma Aldrich, MO). The excess solution was removed with filter paper after ten minutes followed by air drying for 1 hour. The sample was visualized with a transmission electron microscope equipped with a digital camera (JEOL, MA) at 80 kV.
Dynamic Light Scattering (DLS) of Au NPs, PEGylated Au NPs, Tfpep-Au NPs and Pc 4 loaded Au NPs (targeted and untargeted) in aqueous solution was performed on a Brookhaven Zetapals particle analyzer (Brookhaven Instruments, NY). To prepare DLS samples, the respective organic and aqueous master solution (1 mg ml−1) was diluted five-fold and sonicated for 1 h to prevent aggregation. The solution was subsequently filtered using a 0.2 μm syringe filter (EMD Millipore, MA) before taking the measurements.
Gel electrophoresis was used to provide evidence for the anionic functional group attachment and Pc 4 attachment to the Au NPs, PEGylated Au NPs, Tfpep-Au NPs and Pc 4 loaded Au NPs (targeted and untargeted). Electrophoretic matrix was made of agarose gel (1%) in a TBE buffer [Tris base (0.4 M), boric acid (0.45 M), EDTA (10 mM)]. The sample was run at 90 V, and the mobility of the sample was determined by measuring the relative displacements with a CCD-equipped stereomicroscope. The Pc 4 fluorescence was observed at 670 nm on a Maestro small animal imaging system (PerkinElmer, MA).
Drug leaching or release from Au NPs-Pc 4 and Tfpep-Au NPs-Pc 4 was analyzed via UV-Vis spectroscopy. The samples (1 μM) were mixed with PBS buffer in a 96 well plate. UV-Vis measurements were taken every hour for a total of 24 hours at 630 nm (Pc 4) and 505 nm (Au NPs) using Biotek plate reader (Biotek, VT).
Immunofluorescence imaging
LN229 and U87 cells (American Type Culture Collection, VA) were plated on a 25 × 25 mm coverslip at a density of 30 000 cells per coverslip and maintained overnight in cDMEM at 37 °C in an incubator supplied with 5% CO2. After plating, cells were treated with increasing concentrations of Pc 4-loaded Au NPs (targeted and untargeted) with a final volume of 250 μl for 1–24 hours. After the incubation of the particles with the cells, the cells were fixed with 4% paraformaldehyde for 10 minutes followed by washing with PBS. The fixed cells were blocked with 3% goat serum for 1 hour. Then the cells were incubated with mouse anti-TfR (T8199-54; 1: 500; Invitrogen, NY) for 2 hours. The cells were washed with PBS buffer followed by incubation with secondary goat anti-mouse Alexafluor 594-conjugated antibody (A11032; 1: 1000; Invitrogen, NY). For nuclei staining, cells were incubated with DAPI (1: 7500 v/v; Invitrogen, NY) for 15 minutes followed by PBS washing. The uptake of the NPs was visualized by fluorescence microscopy at 40× magnification using a Leica DM 4000B microscope (Leica Microsystems, IL).
Co-localization immunofluorescence
Human glioma U87 cells were plated at a density of 30 000 cells per coverslip and treated with 500 nM Au NPs-Pc 4 or Tfpep-Au NPs-Pc 4 for 0.5–4 hours. The cells were concomitantly incubated with Tf-Alexa 488 (T13342; 0.5 μM; Invitrogen, NY) to identify Tf endosomes, MitoTracker (M7514; 200 nM; Invitrogen, NY) for mitochondria, and LysoTracker (L7526; 75 nM; Invitrogen, NY) for lysosomes in a final volume of 250 μl of cell media. The cells were washed with media and fixed with 4% paraformaldehyde. The stained, fixed cells were counterstained with DAPI nuclear stain. The location of Pc 4 was visualized using a Leica DM 4000B microscope at 40× magnification. The images were analyzed using ImageJ software (NIH, MA) for co-localization analysis.
Photodynamic therapy (PDT) assay
For dark and light assay, 30 000 U87 cells per well were plated in a 96 well plate. The cells were incubated for 4 hours at 37 °C with Tfpep-Au NPs-Pc 4 (10, 100 and 1000 nM) and compared to free Pc 4. The cells were then washed with fresh media prior to drug activation. The dark assay was kept in the incubator for another 24 hours without exposure to the 670 nm Intense-HPD 7404 diode laser (North Brunswick, NJ). The light assay was irradiated with a 670 nm laser (500 J cm−2) for 10 minutes at a fluence rate of 0.83. The cells were incubated for an additional 24 hours. Phase contrast images of the dark and light assay were taken using a Leica DMIL LED microscope (Leica Microsystems, IL). Cell viability studies were carried out using Guava Easycyte 8HT (EMD Millipore, MA). The cells were typsinized and stained with Guava Viacount Reagent (EMD Millipore, MA) for 15 minutes prior to the analysis. The viability assay collected 1000 cellular events per well at a steady flow rate of 0.59 μL s−1.
Orthotopic tumor implantation
For brain tumor implants, mice were anesthetized by intraperitoneal injection of 50 mg kg−1 ketamine/xylazine and fitted into a stereotaxic rodent frame (David Kopf Instruments, CA). A small incision was made just lateral to midline to expose the bregma suture. A small (1.0 mm) burr hole was drilled at AP = +1, ML = −2.5 from bregma. Glioblastoma cells (U87, 300 000 cells in 3 μl) were slowly deposited at a rate of 1 μL per minute in the right striatum at a depth of −3 mm from dura with a 10 μL Hamilton syringe (26G blunt needle, Fisher Scientific, PA). The needle was slowly withdrawn and the incision was closed with 2–3 sutures. The tumors developed for 9 days prior to tail vein injection. Animals were fed exclusively on a special rodent diet (Tekland 2018S; Harlan Laboratories, Inc., IN) to reduce auto-fluorescence. Animal experiments were performed according to policies and guidelines of the animal care and use committee at Medical University of South Carolina (IACUC).
In vivo fluorescence imaging
Mice with orthotopic tumors were anaesthetized with isoflurane and injected intravenously via the tail with either Tfpep-Au NP-Pc 4 or Au NP-Pc 4 at a dosage of 1 mg kg−1 of Pc 4 per total mouse body weight. Mice were euthanized at 24 hours. Excised organs were imaged after necropsy. Fluorescent multi-spectral images were obtained using the Maestro In Vivo Imaging System (PerkinElmer, MA). Multispectral in vivo images were acquired under a constant exposure of 2000 ms with an orange filter acquisition setting of 630–850 nm in 2 nm increments. A Cy5 excitation filter (575–605 nm bandpass) and emission filter (645 nm long-pass) combination was used. Multispectral images were unmixed into their component spectra (Pc 4, autofluorescence, and background) and these component images were used to gain quantitative information in terms of average fluorescence intensity by creating regions of interest (ROIs) around the organs in the Pc 4 component images.
Statistical analysis
All data are expressed as mean ± SD. All data analysis was performed using GraphPad Prism software version 6 (CA) unless specified. Multiple variables were analyzed via analysis of variance techniques, p value <0.05 was considered statistically significant.
Results and discussion
Various nanoparticles have been used to deliver drugs and image brain gliomas across the BBB. A major concern with approaches using these nanocarriers is the fate of these agents and their degradation byproducts as well as the risk of immunological responses associated with long-term or repetitive treatment of the base nanocarrier, especially on healthy tissues. Thus, our rationale was to design a biocompatible, inert multifunctional Tf-coated Au NP loaded with Pc 4 to increase the drug delivery efficiency and specificity from untargeted Au NPs and reduce exposure of healthy tissues to the NPs.
Hydrophobic Au NPs were synthesized to facilitate the loading of Pc 4 drug in the final step. The Au NPs were made water soluble via PEGylation for applications in physiological systems followed by attachment of Tfpep and loading with Pc 4 in organic solvent. To produce hydrophophic Au NPs, we used DDA and TOAB as capping ligands and the size of the Au NP was characterized by TEM. A representative TEM image exhibited negligible contrast from the capped TOAB layer; the particles were well dispersed and not aggregated (Fig. 1a). The core diameter of Au NPs (~1500 particles) calculated from the TEM was found to be 5.1 ± 0.6 nm (Fig. 1b). PEGylation of the Au NPs was carried out with a mixture of 20% hetero-bifunctional COOH-PEG-SH and mono-functional mPEG-SH.
Fig. 1.

Characterization of Au NPs. (a) TEM of hydrophobic Au NPs and corresponding histogram (b), show uniformity of Au NP size, (c) Au NP size increases upon each additional conjugation as measured by DLS, (d) Absorbance spectra of Au NPs shows adsorption of Pc 4, (e) Electrophoretic mobility of negatively charged Au NPs in 1% agarose gel. From left to right: (1) Au NPs-Pc 4, (2) Tfpep-Au NPs-Pc 4. Left panel, Au NPs migration; right panel, Pc 4 fluorescence, (f) Loading of Pc 4 onto Tfpep-Au NPs and Au NPs is stable over 24 hours time.
Full length Tf has been used to target NPs across the BBB.32,33 The large size of the full length Tf (8 × 10 nm2)29,34 attached to a NP, however, could prevent or reduce the amount of functionalized NP crossing the BBB.35 This has focused researchers’ attention on the use of small peptides that have several advantages over metabolically active full-length growth factors, such as easy conjugation chemistries without sacrificing binding efficiency, high receptor specificity, and low metabolic consequences.36 We utilized the seven amino acid sequence (HAIYPRH, Tfpep) identified by Engler et al. that binds to TfR in a dose-dependent manner comparable to that of full length Tf.37 The Tfpep with a free amine group was linked to the carboxyl group on bifunctional PEG layer via the amide bond. A minimum number of targeting ligands on the NP is required to target the cells and low accumulation is attributed to low ligand ratios.38 To improve the binding capacity of the Au NPs, a higher Tf ligand per NP ratio is necessary. Therefore in our work, by controlling the ratio of bifunctional PEG to monofunctional PEG, we bioengineered the Au NP to have a theoretical number of 39 Tf per Au NP which falls between the low (17 Tf per NP) and high (144 Tf per NP) range requirement to improve both targeting and accumulation over background.38
Pc 4 was then adsorbed onto the Au NP surface via N–Au bonding by the terminal amine group on the Pc 4 axial ligand in the last step.10 Noncovalent linkage is preferred for adsorption of the drug since it does not require modifications to FDA approved drug compositions and the drug is maintained in its active formulation. Noncovalent attachment of the drug also facilitates ease of loading and offloading of the drug as compared to covalent linkages or modifications of the drug used in the past. We previously demonstrated noncovalent drug loading on Au NPs for delivery in cancer cells.25 Since PEG does not contain any unconjugated reactive groups to assist in amine attachment, it is likely that the Pc 4 was adsorbed to the surface of the Au NP as a result of both hydrophobic and electrostatic interactions in the PEG corona in proximity to the gold surface.10,39
DLS data showed that hydrophobic Au NPs, PEGylated Au NPs, and Tfpep-Au NPs have an average hydrodynamic diameter of 8.2 nm, 10.1 nm, and 12.3 nm, respectively, with a low polydispersity index (PDI) of 0.02 (Fig. 1c). The DLS size distribution is identical to the instrumental response function corresponding to a monodispersed sample, indicating that aggregation was negligible. To confirm loading of Pc 4 on the targeted Au NPs, Pc 4 absorption measurements at 679 nm in the UV-Vis spectra (Fig. 1d) determined that around twenty (20) Pc 4 molecules were adsorbed per Au NP along with surface plasmon of the Au NPs at 520 nm. Loading of Pc 4 onto the Tfpep-Au NPs in the last step increased the total NP size to 41 nm (Fig. 1c). These measurements are in accord with theoretical calculations of the change in NP surface area with and without loaded Pc 4. The increase in size of approximately 30 nm equates to the 20 Pc 4 molecules loaded, assuming a Pc 4 spherical shape, which would displace a surface area corresponding to about 32 nm in diameter. It is noteworthy that the hydrodynamic value is expected to be slightly larger than the actual diameter because of the counter-ion cloud contributions to particle mobility.40
Gel electrophoresis studies illustrated the surface charges of the various Au NPs, demonstrating successful conjugation with the particles moving towards the positive electrode in an agarose gel electrophoresis experiment. (Fig. 1e, left). These particles are negatively charged which reinforces the fact that Au NP uptake into the cells is driven specifically by the targeting peptide. This is in contrast to positively charged particles, which are more effectively and nonspecifically taken up by cells during endocytosis.41 In addition, Pc 4 adsorption onto the Au NPs is demonstrated by the Pc 4’s intrinsic fluorescence at 670 nm (Fig. 1e, right). Fluorescence leaching of Pc 4 drug from the particle reinforced the basis of hydrophobic interactions between the drug and the PEGylated nanoparticle.
Drug-loaded Au NPs were evaluated for loading stability using UV-vis spectroscopy (Fig. 1f). Au NPs-Pc 4 incubated in buffered solution over a 24 hours period lost on average one Pc 4 molecule per Au NP. In comparison, Tfpep-Au NPs-Pc 4 did not lose Pc 4 over the same time period. Under these buffered conditions, Pc 4 does not leach from the Au NPs.
Glioma cells overexpress TfR to varying degrees making them excellent candidates for targeted therapeutics. The human glioma cell lines, U87 and LN229, overexpress TfR as shown by Western blot analysis (Fig. 2a). Immunofluorescence confirms the overexpression of TfR in U87 and LN229 cells (Fig. 2b).
Fig. 2.
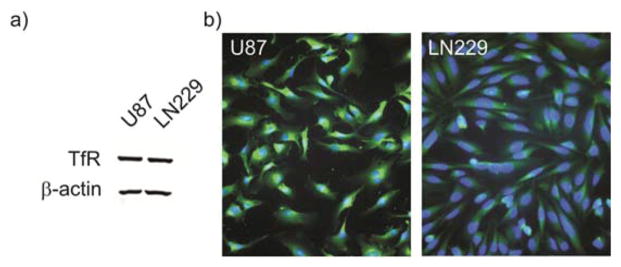
TfR overexpression in glioma cells. (a) Western blot showing TfR overexpression in glioma cell lines, (b) Immunofluorescence of over-expressed TfR in glioma cell lines, epifluorescence microscopy (40×) showing nuclei (blue, DAPI) and TfR (green, anti-TfR antibody).
Tfpep first binds to TfR and is then internalized.37,42 In vitro cell accumulation studies were used to demonstrate targeting specificity of Tfpep-Au NPs compared to that of untargeted Au NPs in glioma cell lines over time. Pc 4 fluorescence for Tfpep targeted Au NPs was compared to that of untargeted particles in U87 and LN229 cell lines (Fig. 3a). Quantification of Pc 4 fluorescence (682 nm) showed that uptake occurred as early as 1 hour in U87 (Fig. 3b) and LN229 cells (Fig. 3c) and continued over the 24 hours period. The increase in both cell lines was quantified as 2–3-fold greater than that of untargeted Au NP Pc 4 uptake. Pc 4 fluorescence continued to increase over time in U87 cells (Fig. 3b) while Pc 4 uptake plateaued after 1 hour in LN229 cells (Fig. 3c). Little Pc 4 fluorescence was observed in cells treated with Au NPs-Pc 4. These results suggest that Pc 4-loaded Au NPs are taken up by Tf receptor-mediated endocytosis of Tfpep-Au NPs instead of nonspecific phagocytic mechanisms.
Fig. 3.
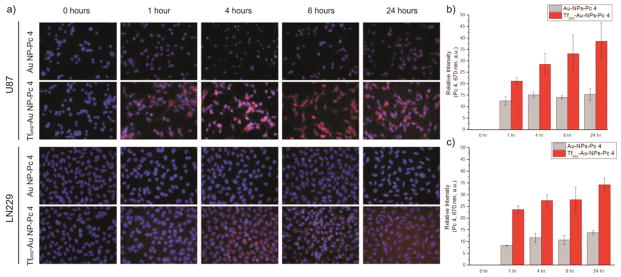
In vitro studies of the PDT drug accumulation. (a) Epi-fluorescence images of U87 and LN229 cells showing time-dependent uptake of Pc 4 after incubation with Tfpep-Au NPs-Pc 4 during 1–24 hours. Nuclei (DAPI, blue), Pc 4 (red), 40× magnification, (b), (c) Pc 4 delivery from targeted Au NPs was quantified and show significant accumulation of Pc 4 over time for U87 and LN229 cell lines compared to untargeted Au NPs.
To further demonstrate specificity of the targeting moiety conjugated to the Au NPs, we conducted a competition experiment of Au NPs-Pc 4 against the targeting peptide (Fig. 4). U87 cells were treated with either targeted or untargeted Au NPs in the absence or presence of Tfpep. After 4 hours of incubation, the accumulation of Pc 4 is inhibited by 61% when U87 cells are treated with Tfpep-Au NPs-Pc 4 and Tfpep at a 1: 4 ratio. The inhibition increases to 72% when the amount of Tfpep is doubled. Incubation of cells with the combination of Au NPs-Pc 4 and Tfpep results in minimal reduction of uptake (26%). This confirms the highly selective and specific nature of Tfpep- Au NPs-Pc 4 for targeting tumor cells with overexpressed receptors.
Fig. 4.

Specificity of peptide targeting. U87 cells incubated with untargeted (Au NPs-Pc 4) or targeted (Tfpep-Au NPs-Pc 4) nanoparticles were simultaneously treated without or with Tfpep for 4 hours. Tfpep inhibited Pc 4 accumulation at peptide ratios, 4: 1 (black and light gray) or 10: 1 (dark gray). Pc 4 was measured by fluorescence in the red channel.
Based on the above, it is expected that Pc 4 delivered by Tfpep-Au NPs would be found internalized within vesicles along a receptor-mediated route. First, we examined whether Tfpep-Au NPs-Pc 4 were specifically associated with TfR. U87 cells were treated with Tfpep-Au NPs-Pc 4 (500 nM) and stained with mouse anti-human TfR. After counterstaining with anti-mouse-Alexa 594 (orange), fluorescence micrographs showed that Tfpep-Au NPs-Pc 4 (red) co-localized with the TfR within 30 minutes with a Pearson’s correlation coeffcient (PCC) of 0.951 (Fig. 5a). In comparison, very few Au NPs-Pc 4 were found to be associated with TfR with a PCC of 0.322.
Fig. 5.

Internal locations of the drug after in vitro uptake. (a) Tfpep-Au NPs-Pc 4 associate with TfR in U87 cells (magenta). Pc 4 (red); TfR (orange). Images acquired at 40× magnification, (b) U87 cells treated with Tfpep-Au NPs-Pc 4 and Tf-Alexa 488 at 5, 30 and 60 minutes show early co-localization with Tf-containing vesicles (Pc 4, red; Tf-Alexa 488, green; DAPI, blue). Images acquired at 100× magnification, (c) Co-localization graph of Tf-Alexa 488 with Tfpep-Au NPs-Pc 4 shows Pc 4 leaving Tf vesicles after 30 minutes and increasing in mitochondria, (d) Pc 4 (red) co-localizes (yellow) with mitochondria in U87 cells treated with Tfpep-Au NPs-Pc and Mitotracker (green). Nuclei stained with DAPI (blue). Images acquired at 100× magnification.
Next, to quantify the degree of co-localization of Tfpep-Au NPs-Pc 4 within TfR specific endosomes, U87 cells were treated with a combination of Tfpep-Au NPs-Pc 4 (500 nM) and Tf-Alexa 488 (500 nM) for 0–60 minutes (Fig. 5b). The Tfpep targeting moiety binds to a region within the TfR that does not compete with full-length Tf binding but is still internalized; therefore, Tf-Alexa 488 delineates the TfR-containing endosomes allowing for precise identification. Two-channel overlay RGB images of TfR endosomes (green) and Pc 4 (red) were analyzed. At early time points (5 and 15 minutes), Pc 4 from Tfpep-Au NPs is internalized within the TfR endosomes. Quantification of the co-localization between the two fluorophores (Pc 4, 670 nm and Tf-Alexa 488, 488 nm) and an analysis of PCC suggest that the Pc 4 correlated well with the TfR endosomes over time (Fig. 5c). After 30 minutes, however, the Pc 4 moved out of the TfR endosomes into the cytoplasm or into other organelles. Quantification was completed comparing Pc 4 channel with Mitotracker channel (488 nm). After 1 hour, the Pc 4 was found primarily in mitochondria with a PCC of 0.9 ± 0.045 (Fig. 5c and d) and not within lysosomes (not shown). As a control, the Pc4 channel was also overlaid with the DAPI nuclear (blue) channel. No correlation was found between Pc 4 and the nucleus (PCC = −0.58 ± 0.106).
Although Pc 4 is delivered to the tumor cells, cellular cyto-toxicity is not induced until the drug is activated with light at 670 nm. The current standard of care (SOC) therapeutic dose for systemically delivered (free) Pc 4 is 1000 nM. To illustrate the efficiency of our Au NPs to induce cell death after drug delivery, we compared decreasing concentrations of Tfpep-Au NPs delivered Pc 4 to the SOC concentration of Pc 4. In a dark assay, in which Pc 4 is not activated by light, cells incubated with Tfpep-Au NPs-Pc 4 (Fig. 6a, left column, right panel) or free Pc 4 (Fig. 6a, left column, left panel) show intact cellular morphology for all the concentrations. Cellular viability is maintained at greater than ~90% for cells treated with targeted, untargeted or free Pc 4 (Fig. 6b). In a light assay, however, Pc 4 delivered by Tfpep-Au NPs-Pc 4 is activated by 670 nm light and significant cell death is observed at the SOC dose of 1000 nM (57% reduction) and also at 100 nM (42% reduction). In contrast, free Pc 4 or untargeted Au NPs delivered Pc 4 does not show a significant cellular cyto-toxicity until the SOC dose of 1000 nM (63% reduction) is reached. Overall, it is noted that targeted Tfpep-Au NPs-Pc 4 is more efficacious than free Pc 4 and Au NPs-Pc 4 at lower concentrations (100 nM). This results in a reduced therapeutic dose for glioma treatments, which could potentially decrease unintended side effects of the drug in vivo.
Fig. 6.

Toxicity studies of Au NPs-Pc 4 conjugates under dark and light activation in U87 cells. (a) Cell morphology of U87 cells treated with Tfpep-Au NPs-Pc 4 versus free Pc 4 treatment for 10–1000 nM after 4 hours show better cell killing at lower concentrations after light activation of the drug, (b) Tfpep-Au NPs-Pc 4 activation significantly kills U87 cells at 100 nM drug versus free Pc 4 at a similar concentration.
To examine the accumulation and biodistribution of Tfpep-Au NPs-Pc 4 versus untargeted Au NPs-Pc 4, mice (N = 3 per group) were implanted orthotopically with glioma cells overexpressing TfR, U87 cells, and injected via the tail vein with either Au NPs-Pc 4 or Tfpep-Au NPs-Pc 4 [1 mg kg−1 Pc 4] (Fig. 7). The Au NPs loaded with Pc 4 were allowed to circulate over 24 hours and mice craniums were imaged noninvasively from 1 to 24 hours (Fig. 7a). Spectral analysis revealed that the amount of Pc 4 increased from 1 to 6 hours in the brains of glioma-implanted mice administered targeted Au NPs-Pc 4 as compared to untargeted Au NPs. After 24 hours, residual Pc 4 washed away from the brain. Tfpep-Au NPs-Pc 4 injected mice accumulated significant amounts of Pc 4 at 6 hours while Au NPs-Pc 4 injected mice took up little drug (Fig. 7b). Quantification of the Pc 4 delivered revealed that the accumulation resulting from targeting to the TfR increased drug accumulation by 6 ± 2.2 fold (Fig. 7b). When the brains of these mice were removed and imaged ex vivo, Pc 4 fluorescence was greater for targeted Au NPs compared to untargeted Au NPs (Fig. 7c). However, with the additional manipulation and removal from the skull, some fluorescence was lost, resulting in a reduced (1–2 fold) difference.
Fig. 7.

Tfpep-Au NPs-Pc 4 accumulate in orthotopic brain tumors in vivo. (a) Representative in vivo fluorescence hotmap images of a mouse implanted with an orthotopic glioma from original groups of N = 3 injected with Tfpep-Au NPs-Pc 4 over time. Bar graph quantifies uptake of Tfpep-Au NPs-Pc 4 versus Au NPs-Pc 4, (b) Mice were analysed for free Pc 4 fluorescence (RFU) 6 hours post injection. All graphical values are expressed as an average with error bars representing SD. There is statistical significance of RFU from Au NP-Pc 4 injected mice with P values <0.05 considered statistically significant, (c) Brains of glioma mice were excised and examined for ex vivo fluorescence (hotmap) after 6 hours, (d) Fluorescence imaging 24 hours post injection of ex vivo organs from mice injected with Au NP-Pc 4 or Tfpep-Au NP-Pc 4. Images were scaled evenly and overlayed on corresponding black and white pictures.
Finally, mice implanted with orthotopic gliomas were administered untargeted or targeted Au NPs as above and sacrificed at 24 hours to observe the biodistribution of Pc 4 (Fig. 7d). Several organs (skin, lung, heart, liver, spleen, and kidney) were excised and imaged using ex vivo fluorescence as shown in the representative image. Negligible Pc 4 fluorescence was observed in the liver, kidney, heart, lung, or spleen after 24 hours. In combination with the above studies, increased accumulation of Tfpep-Au NPs-Pc 4 in the tumor as compared to untargeted Au NPs as shown by noninvasive imaging and biodistribution studies clearly indicates the selectivity of these agents to target the tumor without affecting healthy tissue.
Conclusions
In conclusion, utilizing the Tfpep as the targeting moiety improves the Au NPs specificity for cells overexpressing the TfR. The Tfpep-Au NPs-Pc 4 are internalized in Tf-containing endosomes and later accumulate within the mitochondria of glioma cells. PDT killing efficacy is achieved at 1/10th the concentration of SOC free Pc 4. Tfpep-Au NPs-Pc 4 successfully accumulates in orthotopic brain tumors with little offsite accumulation. This work illustrates successful and logical design of a noncovalent drug delivery system to brain tumors using targeted Au NPs. Studies are underway to test the PDT efficiency of these NPs in in vivo biological models.
Acknowledgments
This work was supported by grant number R01EB012099 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB). The content and views are solely the responsibility of the authors and does not necessarily represent the official views of the NIBIB or the National Institutes of Health.
We would like to thank Mr Alfred Moore and Dr Xingju Nie for their technical support and animal handling during fluorescence imaging. We would also like to thank Mrs Emilie T. McKinnon for post image analysis of in vivo fluorescence images.
Contributor Information
Malcolm E. Kenney, Email: mek9@case.edu.
Ann-Marie Broome, Email: broomea@musc.edu.
Notes and references
- 1.Huse JT, Holland EC. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 2.Jansen M, de Witt Hamer PC, Witmer AN, Troost D, van Noorden CJ. Brain Res. 2004;45:143–163. doi: 10.1016/j.brainresrev.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Pardridge WM. Nat Rev Drug Discovery. 2002;1:131–139. doi: 10.1038/nrd725. [DOI] [PubMed] [Google Scholar]
- 4.Huynh GH, Deen DF, Szoka FC., Jr J Controlled Release. 2006;110:236–259. doi: 10.1016/j.jconrel.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Clin Pharmacol Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 6.Whitesides GM. Nat Biotechnol. 2003;21:1161–1165. doi: 10.1038/nbt872. [DOI] [PubMed] [Google Scholar]
- 7.Youns M, Hoheisel JD, Efferth T. Curr Drug Targets. 2011;12:357–365. doi: 10.2174/138945011794815257. [DOI] [PubMed] [Google Scholar]
- 8.Nune SK, Gunda P, Thallapally PK, Lin YY, Forrest ML, Berkland CJ. Exp Opin Drug Delivery. 2009;6:1175–1194. doi: 10.1517/17425240903229031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain KK. Expert Rev Neurother. 2007;7:363–372. doi: 10.1586/14737175.7.4.363. [DOI] [PubMed] [Google Scholar]
- 10.Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome AM, Burda C, Basilion JP. Small. 2011;7:2301–2306. doi: 10.1002/smll.201100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, Bhattarai N, Ellenbogen R, Sze R, Hallahan A, Olson J, Zhang M. Nano Lett. 2005;5:1003–1008. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- 12.Hadjipanayis CG, Machaidze R, Kaluzova M, Wang L, Schuette AJ, Chen H, Wu X, Mao H. Cancer Res. 2010;70:6303–6312. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dilnawaz F, Singh A, Mewar S, Sharma U, Jagannathan NR, Sahoo SK. Biomaterials. 2012;33:2936–2951. doi: 10.1016/j.biomaterials.2011.12.046. [DOI] [PubMed] [Google Scholar]
- 14.Faucher L, Guay-Begin AA, Lagueux J, Cote MF, Petitclerc E, Fortin MA. Contrast Media Mol Imaging. 2011;6:209–218. doi: 10.1002/cmmi.420. [DOI] [PubMed] [Google Scholar]
- 15.Park JY, Baek MJ, Choi ES, Woo S, Kim JH, Kim TJ, Jung JC, Chae KS, Chang Y, Lee GH. ACS Nano. 2009;3:3663–3669. doi: 10.1021/nn900761s. [DOI] [PubMed] [Google Scholar]
- 16.Huang J, Xie J, Chen K, Bu L, Lee S, Cheng Z, Li X, Chen X. Chem Commun. 2010;46:6684–6686. doi: 10.1039/c0cc01041c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennewitz MF, Lobo TL, Nkansah MK, Ulas G, Brudvig GW, Shapiro EM. ACS Nano. 2011;5:3438–3446. doi: 10.1021/nn1019779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang W, Xie H, Ghoorah D, Shang Y, Shi H, Liu F, Yang X, Xu H. PLoS One. 2012;7:e37376. doi: 10.1371/journal.pone.0037376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Y, Xu Q, Chow PK, Wang D, Wang CH. Biomaterials. 2013;34:8511–8520. doi: 10.1016/j.biomaterials.2013.07.075. [DOI] [PubMed] [Google Scholar]
- 20.Lee HY, Li Z, Chen K, Hsu AR, Xu C, Xie J, Sun S, Chen X. J Nucl Med. 2008;49:1371–1379. doi: 10.2967/jnumed.108.051243. [DOI] [PubMed] [Google Scholar]
- 21.Shubayev VI, Pisanic TR, 2nd, Jin S. Adv Drug Delivery Rev. 2009;61:467–477. doi: 10.1016/j.addr.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paciotti GF, Kingston DGI, Tamarkin L. Drug Dev Res. 2006;67:47–54. [Google Scholar]
- 23.Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv Drug Delivery Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652–14653. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Y, Samia CA, Meyers JD, Panagopoulos I, Fei B, Burda C. J Am Chem Soc. 2008;130:10643–10647. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Drug Delivery. 2004;11:169–183. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 27.Kim CK, Ghosh P, Pagliuca C, Zhu ZJ, Menichetti S, Rotello VM. J Am Chem Soc. 2009;131:1360–1361. doi: 10.1021/ja808137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi CH, Alabi CA, Webster P, Davis ME. Proc Natl Acad Sci U S A. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian ZM, Li H, Sun H, Ho K. Pharmacol Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- 30.Bell RD, Ehlers MD. Neuron. 2014;81:1–3. doi: 10.1016/j.neuron.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 31.Recht L, Torres CO, Smith TW, Raso V, Griffin TW. J Neurosurg. 1990;72:941–945. doi: 10.3171/jns.1990.72.6.0941. [DOI] [PubMed] [Google Scholar]
- 32.Gabathuler R. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 33.Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Eur J Pharm Biopharm. 2009;71:251–256. doi: 10.1016/j.ejpb.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh S, Mukherjee A, Sadler PJ, Verma S. Angew Chem, Int Ed. 2008;47:2217–2221. doi: 10.1002/anie.200705723. [DOI] [PubMed] [Google Scholar]
- 35.Sakhtianchi R, Minchin RF, Lee KB, Alkilany AM, Serpooshan V, Mahmoudi M. Adv Colloid Interface Sci. 2013;201–202:18–29. doi: 10.1016/j.cis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 36.Prades R, Guerrero S, Araya E, Molina C, Salas E, Zurita E, Selva J, Egea G, Lopez-Iglesias C, Teixido M, Kogan MJ, Giralt E. Biomaterials. 2012;33:7194–7205. doi: 10.1016/j.biomaterials.2012.06.063. [DOI] [PubMed] [Google Scholar]
- 37.Lee JH, Engler JA, Collawn JF, Moore BA. Eur J Biochem. 2001;268:2004–2012. doi: 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- 38.Wiley DT, Webster P, Gale A, Davis ME. Proc Natl Acad Sci U S A. 2013;110:8662–8667. doi: 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newland B, Aied A, Pinoncely AV, Zheng Y, Zhao T, Zhang H, Niemeier R, Dowd E, Pandit A, Wang W. Nanoscale. 2014;6:7526–7533. doi: 10.1039/c3nr06737h. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Nieves A, Fernandez-Barbero A, de las Nieves FJ. Phys Rev E: Stat, Nonlinear, Soft Matter Phys. 2001;63:041404. doi: 10.1103/PhysRevE.63.041404. [DOI] [PubMed] [Google Scholar]
- 41.Yu SS, Lau CM, Thomas SN, Jerome WG, Maron DJ, Dickerson JH, Hubbell JA, Giorgio TD. Int J Nanomed. 2012;7:799–813. doi: 10.2147/IJN.S28531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuang Y, An S, Guo Y, Huang S, Shao K, Liu Y, Li J, Ma H, Jiang C. Int J Pharm. 2013;454:11–20. doi: 10.1016/j.ijpharm.2013.07.019. [DOI] [PubMed] [Google Scholar]