

Abstract
The histone chaperone anti-silencing function 1 (Asf1) has emerged as a promising target for therapeutic intervention for multiple cancers.1 Asf1 is involved in the packaging of the eukaryotic genome into chromatin, which is essential for normal growth, development, and differentiation, as this regulates all nuclear processes that use DNA as a substrate. Starting from a collection of HTS leads, we identified a series of N-acyl hydrazones as novel inhibitors of the Asf-histone H3/H4 interaction. These compounds represent the first example of inhibitors capable of disrupting the Asf1-H3/H4 complex.
Keywords: Asf1, Histone chaperone, hydrazone, H3/H4, Drug discovery
Graphical abstract
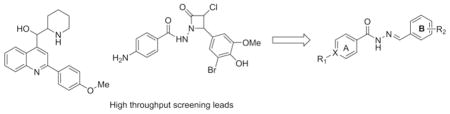
Introduction
The histone chaperone anti-silencing function 1 (Asf1) has emerged as a promising target for therapeutic intervention for multiple cancers.1,2,3,4 Asf1 is involved in the packaging of the eukaryotic genome into chromatin. Correct chromatin structure is essential for normal growth, development, and differentiation, as this regulates all nuclear processes that use DNA as a substrate. Asf1 binds directly to H3/H4 dimers and promotes acetylation of histone H3 lysine 56,5 which has been positively linked to genomic stability, DNA replication and repair, whereas decreased acetylation sensitizes cells to DNA damaging agents. Therefore, identification of compounds that disrupt this process may represent a new therapeutic approach. The goal of our program was to identify small molecules that are capable of disrupting the Asf1-H3/H4 interaction in order to validate the biological target.
Recently crystal structures of Asf1 from Saccharomyces cerevisiae and human Asf1a in complex with a dimer of H3/H4 were reported.6,7 Interestingly, as depicted in Figure 1, the structures revealed one molecule of Asf1 binds to the H3/H4 dimer via a small pocket on the concave surface of the Asf1 that binds the leucine-arginine-isoleuucine (leu-arg-ile) Ig-like region at the histone N-terminus that is highly conserved among eukaryotes.
Figure 1.

Ribbon diagram of Asf-1/histones H3/H4 complex along with expanded view of purported binding interaction of Asf-1 (purple) with Leu-Arg-Ile of histone H3 (blue).
Our search for inhibitors began by screening a 139,735 compound library (the National Cancer Institute Developmental Therapeutics Program [NCI–DTP] 2007 plated set) to identify candidates that may interact with residues on Asf1 that participate in binding H3/H4. More than 800 Å2 of surface area is buried at the H3/Asf1 interface whereas approximately 400 Å2 of surface area is buried at the H4/Asf1 interface.8 Asf1 Tyr112 contributes a significant fraction of surface area buried at the interface with H3 compared to other residues. Spheres depicting the sites of potential ligand atoms were selected within 8 Å of Asf1 Tyr112 using PDB code 2HUE. Molecular docking of each compound in the library (using DOCK6.5, UCSF) to this selected site resulted in a ranked list of compounds predicted to bind Asf1 and prevent H3/H4 interactions. Samples of the top scoring compounds (0.03%) were obtained from the NCI–DTP, from which 6 examples showed promising binding activity in an Asf1-H3/H4 ELISA assay.9 The set of HTS lead compounds was further refined to two chemical series exemplified by 1 (NSC23925) and 2 (DTP-35), both of which became the focus of medicinal chemistry efforts (Figure 2).
Figure 2.
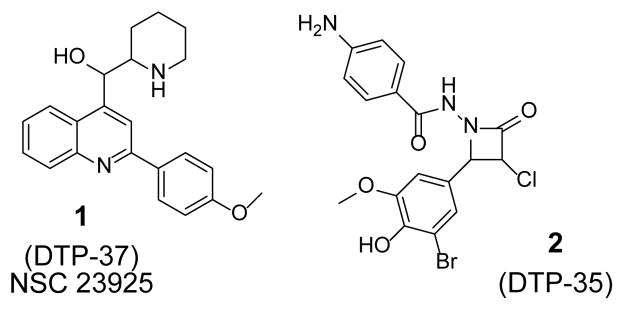
Lead series from HTS
Based on analysis of drug-like properties, initial medicinal chemistry efforts were carried out on the quinolone series 1. These compounds have been reported to possess antiviral10 and more recently have been reported to possess MDR activity.11 The proposed binding orientation of quinolone 1 in the Asf1 binding pocket is shown in Figure 3.
Figure 3.
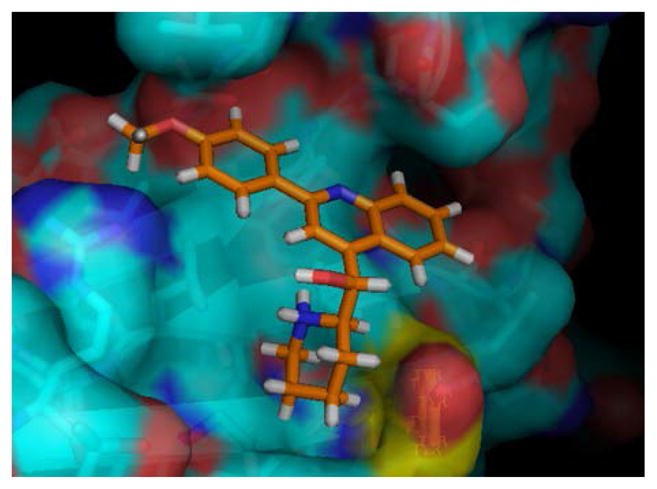
Proposed binding orientation of quinolone derivative 1 docked into AsF-1 binding pocket
Preparation of 1 (NSC23925) has been reported previously.12 Although we investigated this approach, we opted to develop an alternative route shown in Scheme 1. Pfitzinger reaction of isatin 3 with acetephenones 4 under basic conditions provided the quinolone carboxylic 5 which was converted to the hydroxamide 6. Lithiation of the Boc-piperidine 7 using conditions develop by Beak et.al13 followed by condensation with the hydroxamic ester afforded the amino ketone 8.
Scheme 1.

Preparation of quinolone derivatives
Fortuitously we discovered by simply changing the sequence of the subsequent reduction and Boc deprotection steps, we were able prepare both diastereomers. For example removal of the Boc protecting group from 8 followed by amide reduction afforded an authentic sample of DTP-37 that was identical to the sample provided by NCI (as judged by comparing proton NMR spectra). Interestingly, reduction of amide 8 followed by Boc deprotection appeared to provide the opposite diastereomer (9,Threo). Additional conformation of the structural assignment was later obtained by comparing NMR data with data recently reported by Duan et al.11
In addition to preparing an authentic sample of 1, we prepared a series of closely related amide analogs (e.g. 10–15) designed to explore the role of the amino alcohol moiety and the results summarized in Figure 4.
Figure 4.

Summary of cytotoxicity activity of quinolone analogs9
The original NCI sample (DTP-37), as well as the synthetic variants 1 and 9 appeared to be potently inhibit cell viability (Figure 4). However, all closely related amide-containing analogs 10–15 were inactive suggesting the hydroxyl-piperidine group is essential for activity. Unfortunately, in subsequent binding studies with Asf1 using two different versions of the Amplified Luminescent Proximity Homogeneous Assay (ALPHA)9 compound 1 and analogs were shown not to bind to untagged Asf1 (data not shown). Based on this result, we felt the cytotoxic effect was previously observed was most likely not due to disruption of Asf1-H3/H4 complex, and this series was halted.
We next turned our attention turned to exploring the β-lactam-based series exemplified by 2 (DTP-35) since binding data suggested the compound acted upon Asf1. Azetidinones have been reported to display a wide variety of biological activities.14,15 During QC evaluation of the sample from NCI, (LCMS, and 1H NMR) we became concerned regarding the chemical identity. For example, β-lactam compounds as 2 are reported to be prepared via a Staudinger reaction involving N-acyl hydrazines and chloroketene.16 However, after repeated attempts we were unsuccessful in preparing an azetidinone. Upon further scrutiny, we discovered the original NCI material appeared to be comprised predominately of an N-acyl hydrazone 17, not the β-lactam 2 (Equation 1).
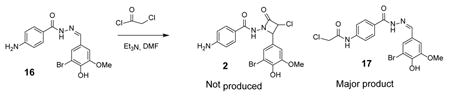 |
Eq. 1) |
This finding was confirmed through independent synthesis of the α-chloro keto N-acyl hydrazone 17 from hydrazone 16. Comparison of 1H NMR spectrum of 16 with samples obtained from the NCI collection confirmed the structural assignment.17
Having serendipitously uncovered a new chemotype of Asf1 inhibitors, we explored a small set of N-acyl hydrazone derivatives as chemical probes that could be used to further validate the biological mechanism of action of the target. Hydrazones have been reported to exhibit a wide variety of biological activity18 although their use as inhibitors of histone chaperones is novel. Moreover, we felt the ease of synthesis would allow rapid analog preparation thus expediting exploration of SAR (Scheme 2). Moreover, it was anticipated that once we had identified more potent derivatives it might be possible to replace the hydrazone portion therefore improving the overall drug-like nature of the compounds.19
Scheme 2.
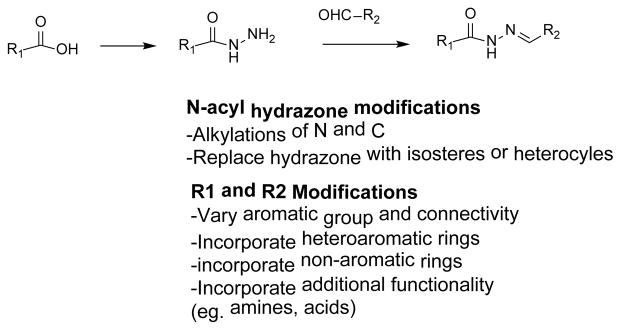
Proposed optimization of N-acyl hydrazines
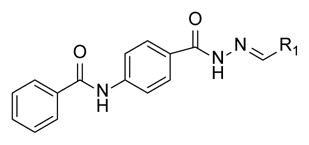
Our approach to explore SAR included dividing the molecule into three sections, which included the carboxylic acid (R1), the hydrazone linker and the group derived from aldehydes/ketones (R2). N-acyl hydrazones were readily prepared using known chemistry starting and all compounds were evaluated for Asf1-H3/H4 binding. Results from a variety of 5-bromovanillin derivatives are shown in Table 1.
Table 1.
SAR of Phenyl acetamide derivatives
| ||
|---|---|---|
| Cmpd | R1 | IC50, um |
| 20 |
|
12 |
| 35 |
|
>100 |
| 36 |
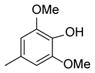
|
50 |
| 37 |
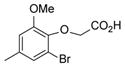
|
30 |
| 38 |
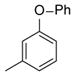
|
25 |
| 39 |
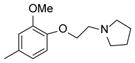
|
10 |
| 40 |
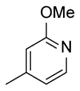
|
>100 |
| 41 |
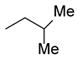
|
11 |
| 42 |
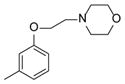
|
>100 |
|
43, 4-py 44, 3-py 45, 2-py |
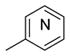
|
>100 85 55 |
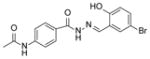 46 I C50 = 3 uM | ||
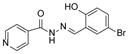 47 IC50 = 0.6 uM | ||
The biological activity of the synthesized α-chloro ketone hydrazone 17 recapitulated the biological activity observed with the original NCI sample. As expected, removal of the α-chloro group 18, resulted in a significant loss of potency. However we discovered potency could be retained by incorporating a phenyl group (20) or other bulky substituent (e.g. 21). Incorporation of non-aromatic groups such as the piperidine 25 or pyran 26 was ineffective. Attempts to incorporate branched groups or inclusion of solubilizing groups (eg. 33) did not improve potency.
Based on these results, we kept the phenylacetamide intact and explored modifications to the vanillin and hydrazone motifs. Dramatic changes in potency were observed when modifications were made to the vanillin portion of the molecules (Table 2). Although several attempts were made, the vanillin substitution appeared to be favored. Interestingly, a significant boost in potency was observed in a series of 2-hydroxy substituted compounds exemplified by compounds 46 and 47.
Table 2.
SAR of vanillin derivatives
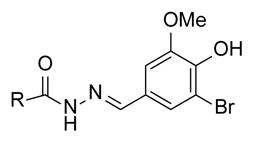
| ||
|---|---|---|
| Cmpd | R | IC50, um |
| 17 |
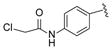
|
32 |
| 18 |
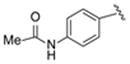
|
>100 |
| 19 |
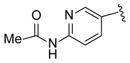
|
>100 |
| 20 |
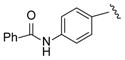
|
12 |
| 21 |
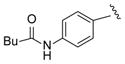
|
14 |
| 22 |
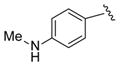
|
27 |
| 23 |
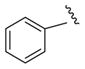
|
>100 |
| 24 |
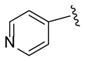
|
>100 |
| 25 |
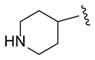
|
>100 |
| 26 |
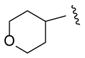
|
>100 |
|
27, 2-OMe 28, 3-OMe 29, 4-OMe |
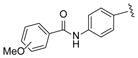
|
60 30 45 |
|
30, 2-Py 31, 3-Py 32, 4-Py |
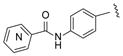
|
15 15 26 |
| 33 |
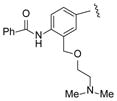
|
>100 |
| 34 |
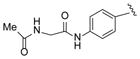
|
48 |
However, upon further analysis we discovered compounds such as 46/47 were acting as metal scavengers, leading to false positive results. Similar compounds have been reported to be potent metal chelators.20
Lastly, efforts were also directed at modification or replacement of the hydrazine linkage (48–52, Figure 5). All attempts to modify or remove the acyl hydrazine moiety resulted in significant loss of activity. Only N-methylation of the hydrazine 52 was tolerated. Interestingly, this outcome is similar to reports regarding other N-acyl hydrazone derivatives.21,22 For reasons that are unexplained at this time, the acyl hydrazone is required for biological activity.
Figure 5.

Summary of hydrazine modifications
Conclusion
Starting from a computational high throughput screen of the NCI compound collection, a series of N-acyl hydrazones that appear to inhibit the Asf1-H3/H4 interaction has been discovered. This is the first reported example of compounds that potentially inhibit Asf-1/histone interactions.
Acknowledgments
Program was supported through a grant from the State of Colorado Economic Development BDEGP Infrastructure grant, and a pilot grant from the University of Colorado Cancer Center (M.E.A.C.).
References and notes
- 1.Bao Y, Shen X. Cell. 2006;127:458. doi: 10.1016/j.cell.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 2.Das C, Tyler JK, Churchill MEA. Trends in Biochemical Sciences. 2010;35:476. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donham DC, Scorgie JK, Churchill MEA. Nucleic Acids Research. 2011;39(13):5449. doi: 10.1093/nar/gkr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lamaitre C, O’Sullivan RJ, Karlseder J, Barillot E, Asselain B, Sastre-Garau X, Almouzni G. EMBO J. 2011;30(3):480. doi: 10.1038/emboj.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das C, Lucia MS, Hansen KC, Tyler JK. Nature. 2009;459(7243):113. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.English CM, Adkins MW, Carson JJ, Churchill MEEA. Cell. 2006;127(3):495–508. doi: 10.1016/j.cell.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Natsume R, Eitoku M, Yusuke Akai Y, Sano N, Horikoshi M, Senda T. Nature. 2007;446:338. doi: 10.1038/nature05613. [DOI] [PubMed] [Google Scholar]
- 8.The atomic coordinates for H3/H4 from PDB code 2HUE were extracted to permit small molecule docking to a structural pocket on the surface of Asf1. The site for molecular docking was selected using DMS (UCSF) to generate a molecular surface, and SPHGEN_CPP to define spheres that represent sites of potential ligand atoms. Spheres within 8 Å of Asf1 position 112 (Tyr) were selected as the site for molecular docking. Grid-based scoring implemented in DOCK6.5 was used. This was accomplished using a scoring grid extending 5 Å in 3 dimensions from the selected spheres. DOCK6.5 was used to screen drug-like compounds (National Cancer Institute Developmental Therapeutics Program NCI plated 2007, 139,735 compounds, http://zinc.docking.org/catalogs/ncip). Each compound was docked in 1,000 orientations and scored for hydrophobic (van der Waals score) and electrostatic interactions (electrostatic score) at the UF High Performance Computing Center by parallel processing using 8 cpu. Compounds were ranked based on overall Energy Score (van der Waals score + electrostatic score). The top 40 scoring compounds (out of 103, 539 drug-like small molecules screened by molecular docking) were obtained from the NCI DTP to measure effects on Asf1 activity.
- 9.Biological evaluation of compounds was carried out using several assays. ELISA assay:50 μL H3/H4 dimer at 1 μg/mL in PBS-T (phosphate buffered saline and Tween 20) was used to coat the wells of a PVC microtiter plate and left at 4°C overnight. The wells were washed 3 times with 200 μL PBS-T. They were then blocked by incubating with 200 μL blocking buffer (5% non fat dry milk in PBS-T) for at least 2 h at room temperature, followed by 3 washes with PBS-T. The globular core domain of Asf1 at 5 μM was incubated with each compound at 100 μM (or various concentrations) for 1 h at 4°C. After diluting samples with buffer (10 mM Tris pH 7.9, 1 mM DTT), 50 μL of each sample was applied to the microtitre plate, and incubated for 1 h at room temperature, prior to washing 3 times with PBS-T. The presence of Asf1 was evaluated using the primary Asf1 antibody (sc-166482; Santa Cruz) and HRPT-conjugated secondary antibody following the manufactures protocol. 50 uL TMB (3,3′,5,5′-tetramethylbenzidine) was added to each well, and incubated for 15–30 min, prior to addition of stop solution (2 M H2SO4). The optical density was measured at 450 nm and Asf1 binding to H3/H4 was analyzed using Graphpad Prism software. Cell viability: MDA-MB-231 cells were cultured to reach 80% confluence. Cells were harvested, suspended in medium (1.5 × 105 cells/mL) and seeded into 12 well plates (1 ml/well) for overnight incubation. After overnight incubation, culture medium in each well was replaced by 1mL of fresh drug medium (medium without 10% FBS and drug dissolved as 24 μM). After 1 h drug treatment, 100 μL of FBS was added to each well. Trypan blue exclusion was used to quantify cell density after additional 24-hour treatment. Following data were reported as: Cell viability (%) = [cell density (drug-treated sample)/cell density (control sample – with 100%DMSO only)]*100. ALPHA assay: The ALPHA assays with biotin-labeled H3/H4 (50 nM) and either His-tagged human Asf1a (1–155 aa) at 50 nM or GST-tagged human Asf1a (1–155 aa) at 50 nM were performed according to manufacturers instructions. Briefly, H3/H4 dimer and Asf1 were incubated together for 15 min at room temperature. The compounds (at 100 μM or lower concentrations in 2 % DMSO) were then added and incubated for 30 min. The appropriate donor and acceptor beads were then added, and the 96-well tray incubated at 4 °C overnight in the dark. The fluorescence intensity of each sample was then measured using an EnVision Plate Reader (Perkin Elmer). The values for IC50 or % inhibition at 100 μM compound were determined from curves fitted using Graphpad Prism software.
- 10.Kim KH, Hansch C, Fukunaga JY, Edward E, Steller EE, Jow PYC. J Med Chem. 1979;22(4):366–391. doi: 10.1021/jm00190a007. [DOI] [PubMed] [Google Scholar]
- 11.Duan Z, Xin L, Huang H, Yuan W, Zheng S, Liu X, Zhang Z, Choy E, Harmon D, Mankin H, Hornicek F. J Med Chem. 2012;55:3113–3121. doi: 10.1021/jm300117u. [DOI] [PubMed] [Google Scholar]
- 12.Brown RF, Jacobs TL, Winstein S, Kloetzel MC, Spaeth EC, Florsheim WH, Robson JH, Levy EF, Bryan GM, Magnusson AB, Miller SJ, Ott ML, Terek JA. J Am Chem Soc. 1946:2705. [Google Scholar]
- 13.Beak P, Lee WK. J Org Chem. 1990;55(9):2578–2580. [Google Scholar]
- 14.Mehta PD, Sengar NPS, Pathak AK. Eur J Medchem. 2010;45:5541. doi: 10.1016/j.ejmech.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 15.Hasan H, Akhter M, Ali I, Zaheen M, Ahsan I, Mahmood D. Med Chem Res. 2011;20:1357. [Google Scholar]
- 16.Cossio FP, Arrieta A, Sierra A. Acc Chem Res. 2008;41(8):925. doi: 10.1021/ar800033j. [DOI] [PubMed] [Google Scholar]
- 17.Diagnostic proton NMR signals for the acyl hydrazone was the methylene group which appears in the NMR as a singlet. In comparison, the beta lactam protons appear as a doublet of doublets due to germinal coupling. See for example ref 15 or Upadhyay A, Srivastava SK, Srivastava SD. Synthetic Communications. 2011;41:2544–2556.
- 18.For a review of acyl hydrazone biological activity see Padmini K, Preethi PJ, Divya M, Rohini P, Lohita M, Swetha K, Kaladar P. Int J Pharma Res Review. 2013 Aug;2(8):43–58.
- 19.Kümmerle AE, Schmitt M, Cardozo SVS, Lugnier C, Villa P, Lopes AB, Romeiro NC, Justiniano H, Martins MA, Fraga CAM, Bourguignon J, Barreiro EJ. J Med Chem. 2012;55:7525–7545. doi: 10.1021/jm300514y. [DOI] [PubMed] [Google Scholar]
- 20.Melnyk P, Leroux V, Sergheraerta C, Grellier P. Bioorg Med Chem Letters. 2006;16:31–35. doi: 10.1016/j.bmcl.2005.09.058. and references therein. [DOI] [PubMed] [Google Scholar]
- 21.Ling A, Hong Y, Gonzalez J, Gregor V, Polinsky A, Kuki A, Shi S, Teston K, Murphy D, Porter J, Kiel D, Lakis J, Anderes K, May J. J Med Chem. 2001;44:3141–3149. doi: 10.1021/jm000547o. [DOI] [PubMed] [Google Scholar]
- 22.Beebe X, Darczak D, Davis-Taber RA, Uchic ME, Scott VE, Jarvis MF, Stewart AO. Bioorg Med Chem Letters. 2008;18:2162–2166. doi: 10.1016/j.bmcl.2008.01.052. [DOI] [PubMed] [Google Scholar]